

Abstract
The peroxisome proliferator–activated receptor γ (PPARγ) is highly expressed in the colon mucosa and its activation has been reported to protect against colitis. We studied the involvement of PPARγ and its heterodimeric partner, the retinoid X receptor (RXR) in intestinal inflammatory responses. PPARγ1/− and RXRα1/− mice both displayed a significantly enhanced susceptibility to 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis compared with their wild-type littermates. A role for the RXR/PPARγ heterodimer in the protection against colon inflammation was explored by the use of selective RXR and PPARγ agonists. TNBS-induced colitis was significantly reduced by the administration of both PPARγ and RXR agonists. This beneficial effect was reflected by increased survival rates, an improvement of macroscopic and histologic scores, a decrease in tumor necrosis factor α and interleukin 1β mRNA levels, a diminished myeloperoxidase concentration, and reduction of nuclear factor κB DNA binding activity, c-Jun NH2-terminal kinase, and p38 activities in the colon. When coadministered, a significant synergistic effect of PPARγ and RXR ligands was observed. In combination, these data demonstrate that activation of the RXR/PPARγ heterodimer protects against colon inflammation and suggest that combination therapy with both RXR and PPARγ ligands might hold promise in the clinic due to their synergistic effects.
Keywords: colitis, inflammatory bowel disease, nuclear receptors, tumor necrosis factor α, signal transduction pathway
Introduction
The peroxisome proliferator–activated receptor γ (PPARγ) is a nuclear receptor that controls the expression of several genes involved in metabolic control. PPARγ is activated by fatty acid derivatives, thiazolidinediones, and certain nonsteroidal antiinflammatory drugs 1 2 3. Thiazolidinedione PPARγ agonists, such as troglitazone, rosiglitazone, and pioglitazone, have been or are currently being used as insulin-sensitizing drugs in the treatment of the type 2 diabetes 4. In addition to these effects on glucose homeostasis, in vitro studies have shown that PPARγ activators could have antiinflammatory effects. PPARγ activators were able to limit the production of inflammatory mediators such as inflammatory cytokines produced by human activated monocytes-macrophages and intestinal epithelial cells through an inhibition of transcription driven by nuclear factor (NF)-κB and activating protein 1 (AP-1) transcription factors 5 6 7. Recently, PPARγ agonists have been reported to attenuate colitis in a murine model in which inflammation was induced by administration of dextran sodium sulfate 7. This observation suggested that PPARγ activators may be useful in the treatment of patients with inflammatory bowel disease (IBD). IBD encompasses several chronic diseases, which are characterized at least in part by an overproduction of pathogenic inflammatory cytokines such as TNF-α and IL-1β, leading to the activation of the NF-κB and c-Jun NH2-terminal kinase (JNK)/p38 mitogen-activated protein kinase (MAPK) pathways 8 9 10.
To activate transcription, PPARγ requires heterodimerization with the retinoid X receptor (RXR). The RXR/PPARγ heterodimers are permissive to activation by both PPARγ and RXR ligands. Therefore, several of the biological effects of PPARγ activation, such as insulin sensitization, can be reproduced by specific RXR agonists or rexinoids. Proof of a role for the RXR/PPARγ heterodimer in intestinal inflammation has been limited to pharmacological studies demonstrating the efficacy of PPARγ agonists. In this study, we investigated the potential effects of both PPARγ and RXR in an experimental animal model in which colitis was induced by intrarectal administration of 2,4,6-trinitrobenzene sulfonic acid (TNBS). We provide genetic evidence of an involvement of both PPARγ and RXR in colon inflammation, by showing that PPARγ1/− and RXRα1/− mice were significantly more susceptible to inflammation than their wild-type littermates. Furthermore, we confirm the protective effects of PPARγ ligands in a different experimental colitis model than the one used by Su et al. 7. Most importantly, we demonstrate for the first time that RXR agonists were equally effective as PPARγ agonists in reducing intestinal inflammation. In addition, rexinoids have a marked synergistic effects with PPARγ agonists on inflammation. In combination, our data emphasize the importance of both partners of the RXR/PPARγ heterodimer in the regulation of colon inflammation. The synergistic antiinflammatory activities of RXR and PPARγ agonists suggests that coadministration of low doses of PPARγ and RXR agonists might be worth exploring in human IBD.
Materials and Methods
Materials
The PPARγ and RXR agonists were synthesized at Ligand Pharmaceuticals. TNBS was purchased at Fluka.
Induction of Colitis and Study Design
Animal experiments were performed in accredited establishments (N° B59-108 and B67-218-5) according to governmental guidelines N°86/609/CEE. Animals were group housed (6–8/cage) and had free access to regular rodent chow and tap water. For induction of colitis, mice, which were anesthesized for 90–120 min, received by intrarectal administration 40 μl of a solution of TNBS (150 mg/kg) dissolved in 0.9% NaCl and mixed with an equal volume of ethanol (50% ethanol). Control mice received 50% ethanol or a saline solution using the same technique. Animals were killed by cervical dislocation 2 or 5 d after TNBS administration. First, wild-type male Balb/c mice were used in the intervention studies with the PPARγ and RXR agonists, rosiglitazone (5–50 mg/kg/day), troglitazone (50–200 mg/kg/day), or LG101305 (5–50 mg/kg/day). These compounds were administered once daily by oral gavage 11, starting either 2 d before (preventive mode) or just after the induction of colitis (therapeutic mode). A general outline of the different intervention studies is represented in Fig. 1. In a second set of experiments, PPARγ1/− and RXRα1/− mice or their respective wild-type littermates (both on a 129/Sv background) were used. Heterozygote mice were used because homozygous PPARγ−/− and RXRα−/− mice are embryonic lethal. These 129/Sv mice were killed 2 d after the induction of colitis.
Figure 1.

Design of the animal intervention studies. TNBS (black ellipse) was administered intrarectally on day (D) 0 and mice were killed 2 or 5 d later. The rexinoid (LG101305), the PPARγ agonists, such as rosiglitazone and troglitazone, or the combination of a rexinoid and a PPARγ agonist were administered at the indicated doses 2 d before the induction of colitis (preventive mode). The therapeutic effects were evaluated in mice receiving the same dose of rosiglitazone just after TNBS administration (therapeutic mode). Treatment with PPARγ and/or RXR agonists was evaluated at day 2 or 5 by scoring for mortality, determination of macroscopic and histologic inflammation scores, and measurements of inflammatory parameters (MPO levels, TNF-α and IL-1β mRNA, NF-κB pathway, and MAPK activity).
Macroscopic and Histologic Assessment of Colitis
The colon was examined under a dissecting microscope (×5) to evaluate the macroscopic lesions according to the Wallace criteria. The Wallace score rates macroscopic colon lesions on a scale from 0 to 10 based on criteria reflecting inflammation, such as hyperemia, thickening of the bowel, and the extent of ulceration 12. A colon specimen located precisely 2 cm above the anal canal was cut into four parts. One part was fixed overnight in 4% paraformaldehyde acid and embedded in paraffin. Sections stained with hematoxylin and eosin were examined blindly by a pathologist and scored according to the Ameho criteria 13. This grading on a scale from 0 to 6 takes into account the degree of the inflammatory infiltrate, the presence of erosion, ulceration or necrosis, and the depth and surface extension of the lesions 13. The other parts of the colon were used for quantification of TNF-α and IL-1β mRNA, myeloperoxidase (MPO), and NF-κB DNA binding or MAPK activities.
Quantification of Protein and mRNA Expression in the Colon
Protein preparation and immunoblotting were performed as described 14. Total protein extracts were obtained by homogenization of tissues in an extraction buffer consisting of PBS with 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and a classical protease inhibitor cocktail. Total proteins were then separated by PAGE and electroblotted 14. Immunodetection with a secondary peroxidase-conjugated antibody and chemiluminescence was performed according to the manufacturer's protocol (ECL; Amersham Pharmacia Biotech). RNA was isolated from colon samples with the TRIzol reagent as described 15. After treatment at 37°C for 30 min with 20–50 units of DNase I RNase-free (Boehringer), total RNA (5–10 μg) was reverse transcribed into cDNA. The reverse transcription reaction mixture was amplified by PCR using sense and antisense primers specific for TNF-α and IL-1β (Table ; references 15 and 16). The samples were subjected to 40 PCR cycles (PerkinElmer). Quantification of cytokine cDNA was performed by electrophoresis in 2–3% agarose gel using an image analyzer (Gel Analyst; Clara Vision; reference 17). Results of cytokine measurements were expressed in proportion to the quantity of total RNA, in the same sample.
Table 1.
Mouse Oligonucleotide Sequences
| Primer | Sequence |
|---|---|
| TNF-α AS | 5′-GGG-AGT-AGA-CAA-GGT-ACA-AC-3′ |
| TNF-α S | 5′-TCT-CAT-CAG-TTC-TAT-GGC-CC-3′ |
| IL-1β AS | 5′-AGA-AGG-TGC-TCA-TGT-CCT-CAT-3′ |
| IL-1β S | 5′-TTG-ACG-GAC-CCC-AAA-AGA-TG-3′ |
AS, antisense; S, sense.
Quantification of NF-κB DNA Binding, JNK, and p38 Kinase Activities in the Colon
NF-κB Electromobility Shift Assay.
Cellular protein extracts were prepared by homogenizing the colon in the following buffer: 20 mM Hepes, pH 7.9, 350 mM NaCl, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM dithiothreitol, 0.1 mM EGTA, 20% glycerol, 1% NP-40, 1 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride (AEBSF), 2 μg/ml aprotinin, and 10 μM leupeptin. In vitro binding reactions of NF-κB in a total volume of 25 μl were initiated by incubation of 5 μg of nuclear protein extracts in a binding buffer containing 10 mM Hepes, pH 7.5, 50 mM NaCl, 1 mM dithiothreitol, 1 mM EDTA, 1 mM MgCl2, 5% glycerol, 2 μg poly(dI-dC), and 0.4 mg/ml salmon sperm DNA 18. After 10 min of preincubation on ice, 50,000–100,000 cpm of 32P end-labeled NF-κB oligonucleotide probe (5′-GATCCAAGGGACTTTCCATG-3′ corresponding to the NF-κB binding sequence of the human IL-2 promoter) were added and the binding reaction was allowed to proceed for 20 min at 21°C. Then, DNA–protein complexes were resolved by electrophoresis on Tris-acetic acid-EDTA (TAE)-4% polyacrylamide gels at 4°C, and analyzed by autoradiography. As control, a 50-fold molar excess of cold NF-κB competitor oligonucleotide was added during preincubation.
JNK or p38 Kinase Assay.
Colon was homogenized in TLB (triton lysis buffer): 20 mM Tris, pH 7.5, 137 mM NaCl, 2 mM EDTA, pH 7.5, 1% Triton X-100, 25 mM β-glycerophosphate, 1 mM Na orthovanadate, 2 mM NaPPi, 10% glycerol, 1 mM PMSF, and 10 μg/ml leupeptin. Soluble extracts were prepared by centrifugation at 12,000 rpm for 30 min at 4°C. The extracts (300 μg of protein) were incubated with 2 μl of anti-JNK or anti-p38 rabbit serum prebound to protein A-Sepharose 19. After 1 h of incubation at 4°C, the immunoprecipitates were washed twice with TLB and twice with kinase buffer: 25 mM Hepes, pH 7.5, 25 mM MgCl2, 25 mM β-glycerophosphate, 2 mM dithiothreitol (DTT), and 0.1 mM Na orthovanadate. Immune complex kinase assays were performed at 30°C for 20 min using 2 μg of glutathione-S-transferase (GST)-activating transcription factor (ATF)2(1-109), 50 μM ATP, and 3–10 μCi of [γ-32P]ATP as substrates in 20 μl of kinase buffer 20. The reactions were terminated with Laemmli sample buffer, the products were resolved by SDS-PAGE (12%), and quantified after autoradiographic analysis as described 20.
Statistics
All comparisons were analyzed by the nonparametric Kruskal-Wallis one-way analysis of variance (ANOVA) test. Differences were judged statistically significant if the P value was <0.05.
Results
TNBS-induced Colitis Is Improved by PPARγ Agonists.
First, we characterized the development of colitis in animals subjected to TNBS injection. Whereas control mice, killed 2 or 5 d after administration of 50% ethanol or a saline solution, had no macroscopic lesions in the colon, a severe colitis was induced as early as 2 d after administration of TNBS, resulting in death in 22 ± 6% of the animals (Fig. 2 A, and Table ). 5 d after induction of colitis, the lesions were more severe with necrosis of the colon leading to mortality in 68 ± 4% of the animals. On a histologic level, no abnormalities were detected in control mice (Fig. 3 A). In sharp contrast, 2 d after the administration of TNBS, colon histology was characterized by large areas of ulceration with a neutrophilic infiltrate, necrosis extending deeply into the muscular layer (Fig. 3B and Fig. C), and enhancement of MPO levels, a marker of neutrophil content (21; Fig. 4). Necrosis of the colon in mice surviving 5 d after TNBS administration involved ∼90% of the specimen, and was so severe that it precluded evaluation of other parameters such as MPO, TNF-α, and IL-1β mRNA levels, and activity of NF-κB.
Figure 2.
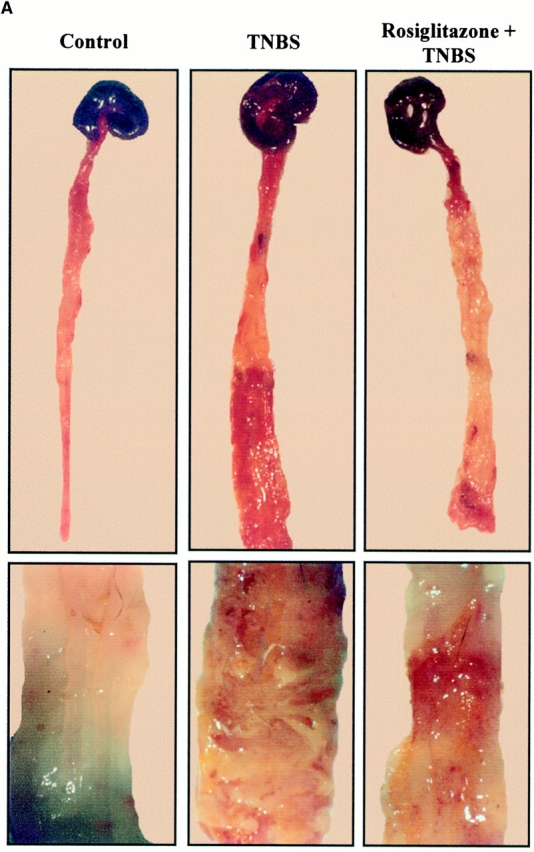
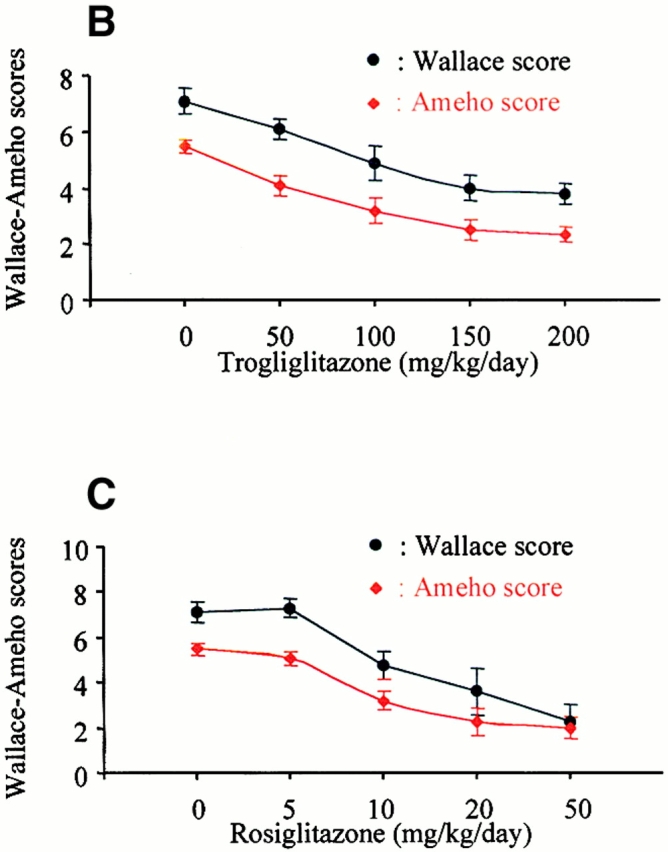
TNBS-induced colitis is dose-dependently improved by PPARγ agonists. (A) Macroscopic appearance of the colon of mice receiving vehicle only (Control), TNBS, or mice receiving rosiglitazone (20 mg/kg/d) 2 d before the administration of TNBS (Rosiglitazone + TNBS). The bottom shows a larger magnification of the colon at the injection site. (B and C) The antiinflammatory effects of different doses of troglitazone (B) and rosiglitazone (C) were assessed preventively in TNBS-induced colitis. The severity of the lesions was evaluated by macroscopic and histologic assessments using, respectively, the Wallace and Ameho scores in mice killed 2 d after colitis induction.
Table 2.
Preventive Administration of Rosiglitazone or Troglitazone Have Similar Therapeutic Effects on Colitis 2 d after TNBS Administration
| Control (n = 16) | TNBS (n = 23) | Rosiglitazone (n = 14)on TNBS-induced colitis | Troglitazone (n = 8)on TNBS-induced colitis | |
|---|---|---|---|---|
| Mortality | 0% | 22 ± 6% | 12.5 ± 12.5% | 10.2 ± 8% |
| Wallace score | 0 ± 0 | 7.14 ± 0.86 | 3.6 ± 2 | 3.8 ± 1.5 |
| Ameho score | 0 ± 0 | 5.5 ± 0.5 | 2.3 ± 1.2 | 2.5 ± 1 |
| MPO values | 2.5 ± 3.6 | 34 ± 16 | 14 ± 20 | 18 ± 14 |
| TNF-α mRNA | 0.84 ± 0.81 | 3.91 ± 3.94 | 0.58 ± 0.69 | 0.45 ± 0.74 |
| IL-1β mRNA | 0.32 ± 0.26 | 28.5 ± 31.1 | 4 ± 3 | 4 ± 4 |
Figure 3.
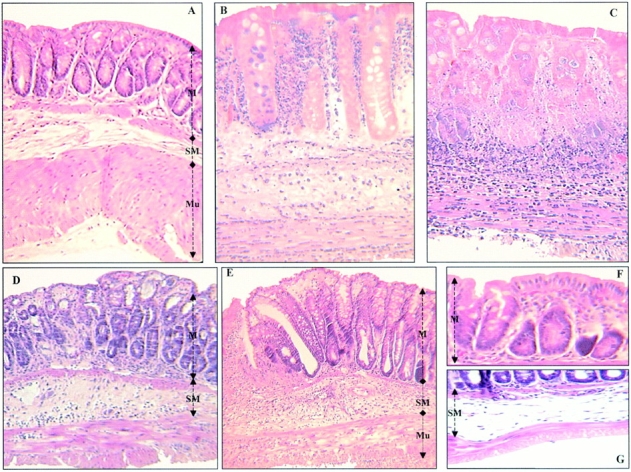
Representative histological sections of colon tissues of Balb/c mice. (A) Normal transparietal colon section of a vehicle-treated mouse with an Ameho score of 0 (×250). The different layers are indicated: M, mucosa; SM, submucosa; Mu, muscular layer. (B) Transparietal colon section (Ameho score 6) 2 d after the induction of colitis by TNBS. Thickening of the colon wall, with a predominant inflammatory infiltrate in the lamina propria, and necrosis extending deeply into the muscular and serosal layers are evident (×400). (C) Transparietal colon section (Ameho score 6) 5 d after the induction of colitis by TNBS. Parietal necrosis extending deeply into the muscular layer with the disappearance of cells in the mucosa is visible (×250). (D) Transparietal colon section of a mouse, which received rosiglitazone before TNBS administration. The mouse was killed 2 d after colitis induction. The Ameho score was graded 2. The picture shows a subepithelial edema with a diastasis of the crypts and a moderate inflammatory infiltrate in the mucosa and submucosa (×250). (E) Transparietal colonic section of mice treated with rosiglitazone after administration of TNBS. The pictures show mice killed 5 d after colitis induction. Ulceration extending into the submucosa, associated with a mucosal, submucosal, and muscular inflammatory infiltrate involving <50% of the specimen, is visible (×250). (F and G) Colon sections of mice that received rosiglitazone before TNBS administration. The mice were killed 2 d after colitis induction. In some cases, a total repair of the mucosa was observed (F; ×600) despite the persistence of an in-depth focal necrosis in the submucosal layer (G; ×400).
Figure 4.
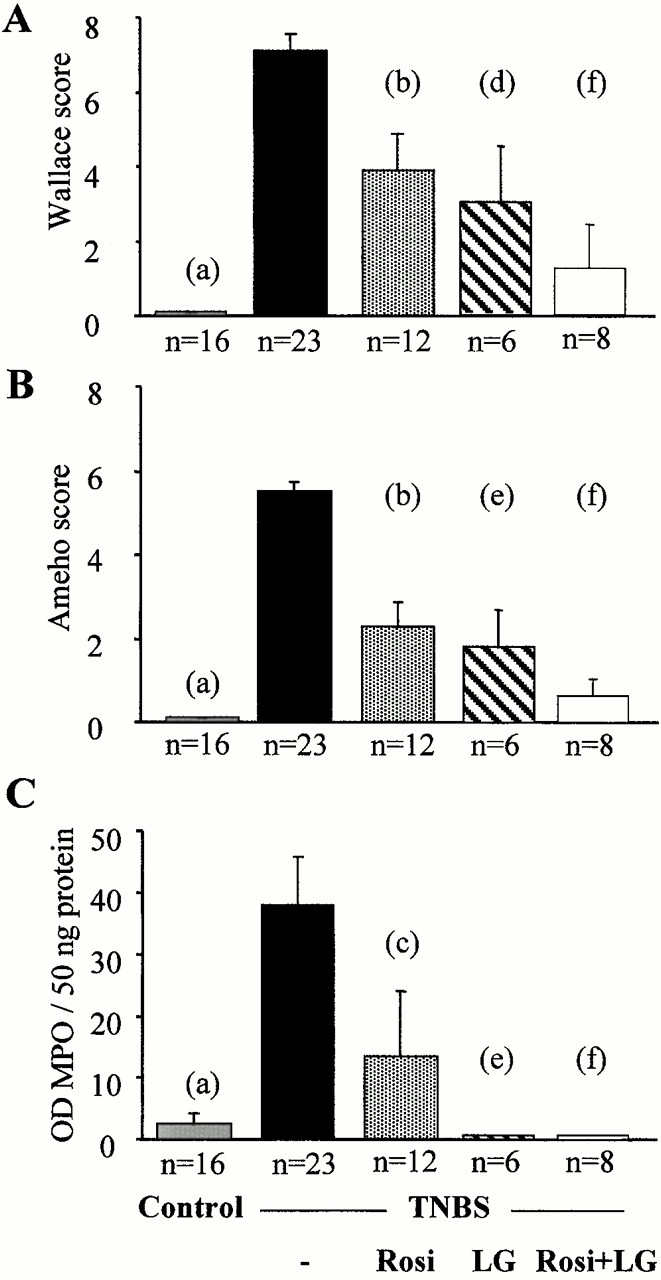
Effect of PPARγ and RXR agonists on TNBS-induced colitis. Wallace macroscopic inflammation score (A), Ameho histologic score (B), and colon MPO levels (C) of mice receiving vehicle only (Control), TNBS, rosiglitazone (Rosi at 20 mg/kg/d), LG101305 (LG at 20 mg/kg/d), or rosiglitazone and LG101305 simultaneously (Rosi + LG both at 20 mg/kg/d) 2 d before the administration of TNBS. The number of mice is indicated and results are expressed as the mean ± SEM. Animals were killed 2 d after TNBS treatment. (a) P < 0.001 in control mice vs. untreated TNBS colitis; (b) P < 0.001 and (c) P = 0.009 in untreated TNBS colitis vs. mice receiving rosiglitazone; (d) P = 0.002 and (e) P < 0.001 in untreated TNBS colitis vs. mice receiving LG101305; and (f) P < 0.001 in untreated TNBS colitis vs. mice receiving both rosiglitazone and LG101305.
We then evaluated the effects of PPARγ activation on TNBS-induced colon lesions by performing a detailed dose–response study, using two different synthetic PPARγ agon- ists, i.e., troglitazone (Fig. 2 B; from 50 to 200 mg/kg/day) and rosiglitazone (Fig. 2 C; from 5 to 50 mg/kg/day). In the first experiments, PPARγ agonists were used in a preventive mode and administered 2 d before colon lesions were induced. When animals were analyzed 2 d after the induction of colitis, both PPARγ agonists significantly reduced mortality compared with untreated mice with colitis and improved macroscopic and microscopic aspects of the TNBS-induced colon lesions (Fig. 2 and Table ). Optimal effects were obtained with a dose of 150–200 mg/kg/d of troglitazone and 20–50 mg/kg/d of rosiglitazone (Fig. 2B and Fig. C). Consistent with their efficacy and potency as PPARγ agonists in transfection assays, rosiglitazone was both a more potent and more efficacious antiinflammatory compound than troglitazone. These optimal doses were used in all following experiments. 5 d after induction of colitis, a significant decrease in both mortality and macroscopic lesion score was observed in mice that had received rosiglitazone preventively compared with untreated mice with colitis (Table ).
Table 3.
Rosiglitazone Used in a Preventive or Therapeutic Modes Improved Colitis 5 d after TNBS Administration
| Rosiglitazone on TNBS-induced colitis | ||||
|---|---|---|---|---|
| Control(n = 16) | TNBS(n = 14) | Preventive mode (n = 16) | Therapeutic mode (n = 16) | |
| Mortality | 0 ± 0% | 68 ± 4% | 19 ± 9% | 14 ± 8% |
| Wallace score | 0 ± 0 | 8.9 ± 1.4 | 3.5 ± 0.6 | 5.8 ± 0.9 |
| Ameho score | 0 ± 0 | 6 ± 0 | 2.1 ± 1.1 | 4.1 ± 0.7 |
Parallel to the gross macroscopic inflammation, histological analysis also revealed major differences between TNBS animals and animals receiving PPARγ agonists (Fig. 3B and Fig. D). This was reflected by a significant decrease of the Ameho inflammation score both at 2 (Fig. 4 B) and 5 d (Table ) after TNBS administration. Administration of PPARγ agonists dramatically reduced the inflammatory lesions, which consisted of smaller polymorphic inflammatory infiltrates, with mononuclear predominance, limited edema, and small focal necrotic lesions in close contact to regions with reepithelialization of the mucosal layer (Fig. 3 D). This improvement by the preventive administration of rosiglitazone was associated with a significant decrease of MPO levels 2 d after TNBS administration (Fig. 4 C). Interestingly, in certain cases, total repair of the mucosal and muscularis mucosa layers was observed despite the persistence of some in-depth lesions (Fig. 3 F). These lesions were either in the form of a mononuclear infiltrate in the submucosa and the muscular layers, or in some cases in the form of focal necrosis of the ischemic type in the muscular layer (Fig. 3 G).
In the experiments described above, administration of PPARγ agonists started before TNBS was administered. Therefore, we next analyzed whether the administration of rosiglitazone immediately after the induction of colitis was also effective in reducing lesion intensity. In this therapeutic mode, rosiglitazone also significantly improved the macroscopic lesions and mortality compared with untreated mice with colitis (Table ). This macroscopic improvement was again associated with a significant decrease of the histologic score (Table and Fig. 3 E). These data prove that PPARγ agonists can not only prevent lesion development, but are also effective in reducing established inflammatory lesions in the colon.
RXR Agonist Synergizes with PPARγ Agonists in Improving Colitis.
As PPARγ forms a “permissive” heterodimer with RXR, we next used the selective and potent RXR agonist LG101305 to analyze whether activation of RXR could mimic any of the beneficial effects of PPARγ activation on TNBS-induced colon lesions. In dose–response studies, LG101305 at a dose of 50 mg/kg/d achieved a maximal effect similar to that as observed with the maximally efficacious dose of rosiglitazone (Fig. 2 C and 5 A). This improvement was associated with a significant decrease in the histologic lesion score and a normalization of MPO levels (Fig. 4B and Fig. C).
As the above data demonstrated that both PPARγ and RXR agonists are effective in the treatment of colitis, we next assessed whether the combination of LG101305 with rosiglitazone could have synergistic beneficial effects on the TNBS-induced colitis. When both these agonists were given preventively at doses of 1–20 mg/kg/d, striking macroscopic and histologic improvements of TNBS-induced colitis were observed (Fig. 5 B), suggesting a synergistic effects of these two agonists. In fact, lesions were often almost absent and submucosal neutrophil infiltrate was modest, with very low MPO activity (Fig. 4 C). The synergism was best illustrated by the efficacy of the combination of PPARγ and RXR ligands at a dose of 1 mg/kg/d each, which is 1/20e of the maximally efficacious dose of each individual agonist, and which still retained a remarkable antiinflammatory effect (Fig. 4 and Fig. 5 B).
Figure 5.
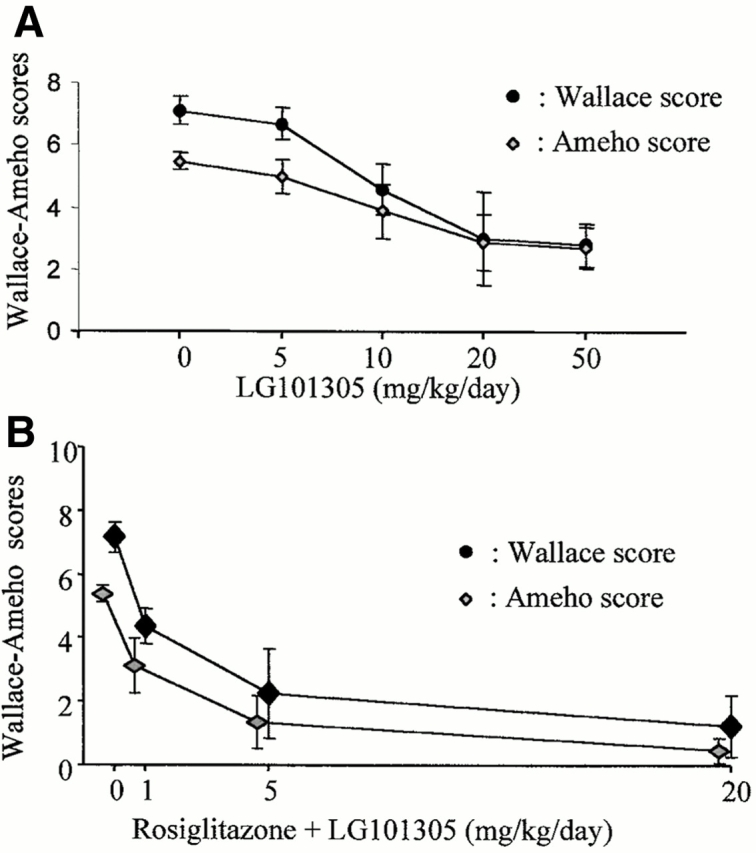
PPARγ-RXR agonists have a synergistic effect on colitis. The antiinflammatory effects of different doses of the RXR agonist LG101305 (A) and of the simultaneous administrations of rosiglitazone (from 1 to 20 mg/kg/d) and LG101305 (from 1 to 20 mg/kg/d)(B) were assessed in TNBS-induced colitis. The severity of the lesions was evaluated by macroscopic and histologic assessments using, respectively, the Wallace and Ameho scores in mice killed 2 d after colitis induction.
Increased Susceptibility of PPARγ1/− and RXRα1/− Mice to TNBS-induced Colitis.
As the above intervention studies using PPARγ and RXR agonists were suggestive of the involvement of the RXR/PPARγ heterodimer in improving colitis, we next tested whether mice heterozygous for a deficiency of PPARγ and/or RXRα were more susceptible to the development of TNBS-colitis. These heterozygous knockout mice and their wild-type littermates are on a 129/Sv background. Relative to Balb/c mice, TNBS induced a less severe colitis in 129/Sv mice (compare absolute macroscopic and histologic lesion scores in Fig. 4 and Fig. 6; P < 0.001). Relative to PPARγ1/+ littermates, PPARγ1/− mice had more pronounced macroscopic and microscopic lesions with an enhancement of MPO levels (data not shown) after a challenge with TNBS (Fig. 6). These lesions were characterized by large and deep ulcerations with a necrosis and inflammatory infiltrate extending deeply into the muscular layer (Fig. 6 D). Relative to the RXRα1/+ mice, the RXRα1/ − mice developed also significantly more intense macroscopic and histologic lesions after TNBS administration (Fig. 6B and Fig. E). Like for the PPARγ1/− mice, this increased susceptibility to lesion formation was associated with a dramatic induction of MPO levels (data not shown) in the colon and a necrosis which involved ∼90% of the specimen (Fig. 6B and Fig. E). In combination with the data obtained using PPARγ and RXR agonists (Fig. 2 Fig. 3 Fig. 4 Fig. 5), these studies in PPARγ and RXRα1/− mice implicate the RXR/PPARγ heterodimer in the protection against intestinal inflammation.
Figure 6.
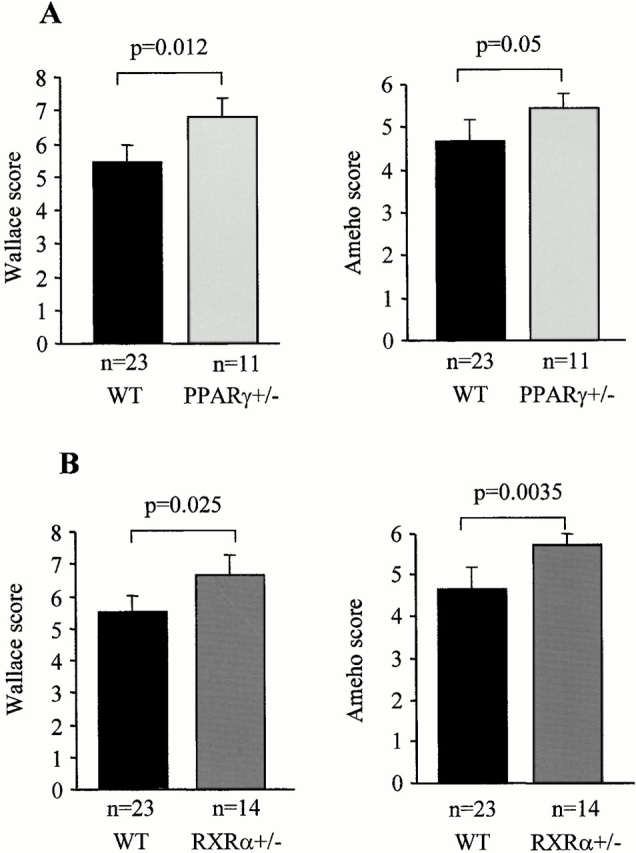
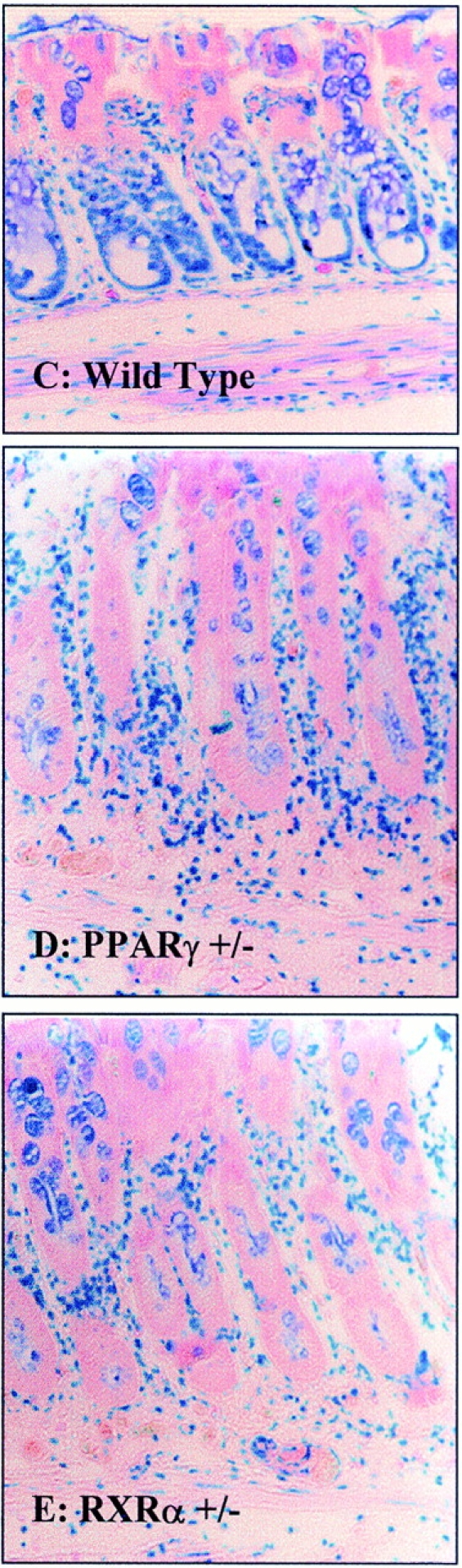
PPARγ1/− and RXRα1/− mice are more susceptible to TNBS-induced colitis. (A and B) Wallace macroscopic and Ameho histologic inflammation scores of 129/Sv wild-type (WT), PPARγ1/− (A) and RXRα1/− (B) mice 2 d after induction of colitis by TNBS administration. (C) Representative transparietal colon section of 129/Sv wild-type mice (Ameho score 5) 2 d after the induction of colitis by TNBS showing a moderate infiltrate with a necrosis limited to the superficial part of the mucosa (×200). (D and E) Transparietal colon sections in PPARγ1/− (D) and RXRα1/− (E) mice (both Ameho scores of 6) 2 d after the induction of colitis by TNBS. Thickening of the colon wall with a marked transparietal inflammatory infiltrate and necrosis (×200). Mean ± SEM are indicated, the number of mice, as well as the statistical significance are indicated.
PPARγ and RXR Agonists Inhibit Inflammatory Cytokine Expression and NF-κB and JNK/p38 MAPK Activities.
Although all the above data underscore the importance of the RXR/PPARγ heterodimer in colon inflammation, they did not address any of the possible downstream mechanisms involved in this protective effect. Intestinal inflammatory cytokines and the NF-κB and MAPK pathways were evaluated in mice killed 2 d after TNBS administration. Low concentrations of TNF-α and IL-1β mRNA were present in the colon of control mice (Fig. 7). 2 d after induction of colitis by TNBS, TNF-α and IL-1β mRNA were significantly induced, compatible with a major inflammatory reaction (Fig. 7). In contrast, the preventive administration of rosiglitazone (at 20 mg/kg/d), troglitazone (at 150 mg/kg/d), LG101305 (at 20 mg/kg/d), and the simultaneous administration of both rosiglitazone and LG101305 (both at the dose of 1 mg/kg/d) normalized TNF-α and IL-1β mRNA concentrations in the colon (Fig. 7 and Table ). Conversely, the more intense macroscopic and histologic colitis observed in PPARγ1/− and RXRα1/− mice were associated with a significant increase in the levels of TNF-α and IL-1β mRNA, compared with wild-type mice with colitis (data not shown).
Figure 7.


TNF-α and IL-1β mRNA levels are reduced by PPARγ and RXR agonists. TNF-α (A) and IL-1β (B) mRNA levels in the colon of mice receiving vehicle only (Control), TNBS, rosiglitazone (Rosi at 20 mg/kg/d), LG101305 (LG at 20 mg/kg/d), or simultaneous administration of rosiglitazone and LG101305 (Rosi + LG both at 1 mg/kg/d) 2 d before the administration of TNBS. Animals were killed 2 d after TNBS administration. Results are expressed as mean ± SEM. mol, molecules. (A): (a) P < 0.001 in control vs. TNBS; (b) P = 0.001 in untreated TNBS colitis vs. mice receiving rosiglitazone; (c) P = 0.076 in untreated TNBS colitis vs. mice receiving LG101305; and (d) P = 0.05 in untreated TNBS colitis vs. mice receiving both rosiglitazone and LG101305. (B): (a) P < 0.001 in control vs. TNBS; (b) P = 0.001 in untreated TNBS colitis vs. mice receiving rosiglitazone; (c) P = 0.003 in untreated TNBS colitis vs. mice receiving LG101305; and (d) P = 0.003 in untreated TNBS colitis vs. mice receiving both rosiglitazone and LG101305.
Furthermore, in control mice killed 2 d after administration of 50% ethanol or a saline solution, low levels of nuclear NF-κB DNA binding activity, as well as of JNK and p38 activities, were observed in colon protein extracts (Fig. 8). 2 d after TNBS administration, NF-κB DNA binding, JNK, and p38 kinase activities were strongly induced (Fig. 8). Preventive administration of rosiglitazone was associated both with an important decrease of the activity of NF-κB DNA binding, JNK, and p38 activities, suggesting the involvement of MAPKs in TNBS-induced intestinal inflammation in mice (Fig. 8).
Figure 8.

NF-κB, JNK, and p38 kinase activities 2 d after TNBS colitis induction in mice treated preventively with rosiglitazone. (A) NF-κB. Proteins in colon homogenates obtained from three control mice (Control), three mice receiving TNBS (TNBS), and three mice receiving TNBS and rosiglitazone (Rosi, 20 mg/kg/day) were used in electrophoretic mobility shift assay (EMSA) reactions as described in Materials and Methods. The top panel shows autoradiographs of the EMSA assay using a radiolabeled NF-κB consensus binding site. The bottom panel shows a graphical presentation obtained after scanning the corresponding autoradiographs. Compared with the most intense NF-κB complex radiolabeled signal obtained in mice receiving TNBS, control (Cont) corresponding to a competition with an excess of cold NF-κB showed a marked decrease of the radiolabeling. (B and C) JNK (B) and p38 kinase (C) activities in immunoprecipitates of colon extracts obtained from three control mice (Control), three mice receiving TNBS (TNBS), and three mice receiving TNBS and treated preventively with rosiglitazone (Rosi, 20 mg/kg/day). The top panel shows autoradiographs, showing the phosphorylation status of the GST-ATF2 fusion protein used as a substrate to measure the kinase activities in the JNK and p38 immunoprecipitates. The bottom panel shows a graphical presentation obtained after scanning the corresponding autoradiographs. AU, arbitrary units.
Discussion
Intrarectal administration of TNBS to mice provokes a severe colitis, which represents a well-validated model that has many macroscopic and histologic similarities to IBD in human. These similarities include the presence of granulomas, mucosal infiltration of neutrophils, mediated at least in part by TNF-α and IL-1β overexpression, and activation of the NF-κB pathway 22 23 24 25. Earlier studies have shown that TNBS-induced colitis responds favorably to many of the current therapies for IBD such as sulfasalazine or 5-aminosalicylic acid 26, glucocorticoids 27, cyclosporin 28, and anti–TNF-α antibodies 23. In view of the high level of expression of PPARγ in the colon 16 29 30 and the reported antiinflammatory effects of activation of PPARγ 5 6 31, we have analyzed the contribution of RXR/PPARγ heterodimers on intestinal inflammatory responses. As the RXR/PPARγ heterodimer is “permissive” and can be activated both by PPARγ or RXR ligands, we first analyzed whether administration of specific synthetic PPARγ or RXR agonists attenuated TNBS-induced colitis. Consistent with the work of Su et al. 7, administration of PPARγ agonists, such as rosiglitazone or troglitazone to TNBS-treated animals, attenuated the inflammatory response. These results confirm that PPARγ ligands have antiinflammatory effects in the intestine. Recently, two studies have shown unequivocally that PPARγ expression by macrophages is not required for PPARγ ligands to exert their antiinflammatory effects 32 33. This absence of direct effects of PPARγ has only been shown on macrophage biology and not in other cells involved in the inflammatory reaction. Therefore, these observations suggest that the ability of thiazolidinediones to suppress TNBS-induced colitis which involved numerous cells such as macrophages but also lymphocytes, neutrophils, mast cells, eosinophils, and epithelial cells may be mediated at least in part through a different target. The rexinoid LG101305 also had potent antiinflammatory effects in the intestine. Furthermore, simultaneous administration of both PPARγ and RXR ligands had a markedly synergistic beneficial effect on colitis, enabling a significant dose reduction for each agonist. Further evidence in support of the implication of the RXR/PPARγ heterodimer in the protection against colon inflammation came from the characterization of PPARγ1/− and RXRα1/− mice, both of which were more susceptible to TNBS-induced inflammation. Taken together, our studies, with synthetic RXR and PPARγ agonists and with heterozygous deficient mice, show that activators of the RXR/PPARγ heterodimer might exert a direct and indirect control of inflammatory responses in the intestine. Furthermore, these data suggest that the synergistic antiinflammatory effect of RXR and PPARγ agonists could be beneficial in a clinical setting, as it might avoid adverse events often encountered when these agonists are used in monotherapy at higher doses.
The persistence of inflammation in deeper layers of the colon in some animals treated with PPARγ and RXR agonists, despite the repair of mucosal lesions, is particularly interesting. The absence of histologic improvement in the deeper layers of the colon could be related at least in part to the preferential expression of PPARγ in the colonic mucosa 30 and its absence in the other layers of the colon (more particularly the muscular layer). Thus, activators of the RXR/PPARγ heterodimer might exert antiinflammatory effects in the mucosa which selectively expresses this heterodimer, and indirectly protects the deeper colon layers against damage by the preservation of the integrity of the mucosal barrier. Further confirmation of this possibility would require the study of animals in which activity of the RXR/PPARγ heterodimer was specifically eliminated in the intestinal mucosa.
The precise mechanisms by which activation of the RXR/PPARγ heterodimer negatively regulates intestinal inflammation remains to be elucidated 31, but our data suggest an involvement of both the NF-κB and stress kinase pathways. Many studies have suggested an important pathogenic role for TNF-α and IL-1β in the pathophysiology of TNBS-induced colitis through the recruitment of polymorphonuclear cells in the colon 34 35. This possibility is supported by the overexpression of these cytokines in TNBS-induced colitis, the improvement in disease severity after the neutralization of TNF-α and IL-1β in vivo, the absence of chronic TNBS-induced colitis in TNF-α knockout mice, and the development of lethal pan-colitis upon TNBS administration in TNF-α transgenic mice 23. In vivo, the predominant role of NF-κB during colonic inflammation has been demonstrated in TNBS-induced colitis and in spontaneous colitis of IL-10–deficient mice 22. It is generally believed that the signal transduction pathways activated in response to TNF-α and IL-1β initiate NF-κB activation through the NF-κB–inducing kinase (NIK) signaling pathway 36 37 and the activation of two IκB kinases, IKK-1 and IKK-2, which phosphorylate IκB, leading to its degradation 38. Nonetheless, several reports also demonstrate an involvement of members of the MAPK pathway such as MAPK kinase (MEKK)1 39 40 41 42 and JNK or p38 MAPK in the activation of NF-κB 8 9 10. In addition, concomitant activation of the JNK/p38 MAPK and NF-κB pathways has been observed in several cell types, including macrophages 43 44 45 46. Despite this suggestive role of stress kinase in NF-κB activation, the involvement of the MAPK pathway in chronic intestinal inflammation has been neglected. In this study, we demonstrated that NF-κB, JNK, and p38 are activated concomitantly during TNBS-induced intestinal inflammation in mice and that the activation of both pathways can be attenuated by activation of RXR/PPARγ heterodimers. Considering the ambiguous relationships between NF-κB activation and the MAPK signaling pathways 47 48 49 50, our observations suggest that activation of both signaling pathways can lead to colon inflammation.
In conclusion, the data obtained with TNBS-induced colitis in mice, an animal model having similarities with IBD in humans, support the existence of an important antiinflammatory action of RXR/PPARγ heterodimers in the intestine. Our data further suggest that the simultaneous administration of synthetic PPARγ and RXR agonists could represent an attractive therapeutic strategy for the treatment of IBD. In addition, they question the possible impact of changing dietary habits on the increased prevalence of IBD and suggest that beneficial effects could be expected from changes in diet.
Acknowledgments
We thank the various members of the Auwerx lab for discussions and suggestions, as well as C. Bourdon, C. Haby, V. Prima, V. Jéronimo, and R. Paris for their valuable technical assistance.
We enjoyed the support of grants from Institut de Recherche des Maladies de l'Appareil Digestif, the association F. Aupetit, the Centre Hospitalier et Universitaire de Lille (EA2687, CA14555, and PHRC1926), Hôpitaux de Strasbourg, Centre National de la Recherche Scientifique, Institut National de la Sante et de la Recherche Medicale, l'Association pour la Recherche contre le Cancer (ARC), the Juvenile Diabetes Foundation, the Human Frontiers Science Program, and from Ligand Pharmaceuticals.
Footnotes
P. Desreumaux and L. Dubuquoy contributed equally to this work.
Abbreviations used in this paper: IBD, inflammatory bowel disease; JNK, c-jun NH2-terminal kinase; MAPK, mitogen-activated protein kinase; MPO, myeloperoxidase; NF, nuclear factor; PPARγ, peroxisome proliferator–activated receptor γ; RXR, retinoid X receptor; TNBS, 2,4,6-trinitrobenzene sulfonic acid.
References
- Schoonjans K., Martin G., Staels B., Auwerx J. Peroxisome proliferator-activated receptors, orphans with ligands and functions. Curr. Opin. Lipidol. 1997;8:159–166. doi: 10.1097/00041433-199706000-00006. [DOI] [PubMed] [Google Scholar]
- Desvergne B., Wahli W. Peroxisome proliferator-activated receptorsnuclear control of metabolism. Endocr. Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- Willson T.M., Brown P.J., Sternbach D.D., Henke B.R. The PPARsfrom orphan receptors to drug discovery. J. Med. Chem. 2000;43:527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- Schoonjans K., Auwerx J. Thiazolidinedionesan update. Lancet. 2000;355:1008–1010. doi: 10.1016/S0140-6736(00)90002-3. [DOI] [PubMed] [Google Scholar]
- Jiang C., Ting A.T., Seed B. PPARγ agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- Ricote M., Li A.C., Willson T.M., Kelly C.J., Glass C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Su C.G., Wen X., Bailey S.T., Jiang W., Rangwala S.M., Keilbaugh S.A., Flanigan A., Murthy S., Lazar M.A., Wu G.D. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J. Clin. Invest. 1999;104:383–389. doi: 10.1172/JCI7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C., Wang X., Chang C., Templeton D., Tan T.H. Interaction between c-Rel and the mitogen-activated protein kinase kinase kinase 1 signaling cascade in mediating kappaB enhancer activation. J. Biol. Chem. 1996;271:8971–8976. doi: 10.1074/jbc.271.15.8971. [DOI] [PubMed] [Google Scholar]
- Hirano M., Osada S., Aoki T., Hirai S., Hosaka M., Inoue J., Ohno S. MEK kinase is involved in tumor necrosis factor alpha-induced NF-kappaB activation and degradation of IkappaB-alpha. J. Biol. Chem. 1996;271:13234–13238. doi: 10.1074/jbc.271.22.13234. [DOI] [PubMed] [Google Scholar]
- Carpentier I., Declercq W., Malinin N.L., Wallach D., Fiers W., Beyaert R. TRAF2 plays a dual role in NF-kappaB-dependent gene activation by mediating the TNF-induced activation of p38 MAPK and IkappaB kinase pathways. FEBS Lett. 1998;245:195–198. doi: 10.1016/s0014-5793(98)00226-9. [DOI] [PubMed] [Google Scholar]
- Lefebvre A.M., Chen I., Desreumaux P., Najib J., Fruchart J.C., Geboes K., Briggs M., Heyman R., Auwerx J. Activation of the peroxisome proliferator-activated receptor γ promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat. Med. 1998;4:1053–1057. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- Wallace J.L., MacNaughton W.K., Morris G.P., Beck P.L. Inhibition of leukotriene synthesis markedly accelerates healing in a rat model of inflammatory bowel disease. Gastroenterology. 1989;96:29–36. doi: 10.1016/0016-5085(89)90760-9. [DOI] [PubMed] [Google Scholar]
- Ameho C.K., Adjei A.A., Harrison E.K., Takeshita K., Morioka T., Arakaki Y., Ito E., Suzuki I., Kulkarni A.D., Kawajiri A., Yamamoto S. Prophylactic effect of dietary glutamine supplementation on interleukin 8 and tumor necrosis factor α production in trinitrobenzene sulfonic acid induced colitis. Gut. 1997;41:487–493. doi: 10.1136/gut.41.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajas L., Auboeuf D., Raspé E., Schoonjans K., Lefebvre A.M., Saladin R., Najib J., Laville M., Fruchart J.C., Deeb S. The organization, promoter analysis, and expression of the human PPARγ gene. J. Biol. Chem. 1997;272:18779–18789. doi: 10.1074/jbc.272.30.18779. [DOI] [PubMed] [Google Scholar]
- Desreumaux P., Ernst O., Geboes K., Gambiez L., Berreli D., Müller-Alouf H., Hafraoui S., Emilie D., Ectors N., Peuchmaur M. Inflammatory alterations in mesenteric adipose tissue in Crohn's disease. Gastroenterology. 1999;117:73–81. doi: 10.1016/s0016-5085(99)70552-4. [DOI] [PubMed] [Google Scholar]
- Müller-Alouf H., Gerlach D., Desreumaux P., Leportier C., Alouf J.E., Capron M. Streptococcal pyrogenic exotoxin A (SPE A) superantigen induced production of hematopoietic cytokines, IL-12 and IL-13 by human peripheral blood mononuclear cells. Microb. Pathog. 1997;23:265–272. doi: 10.1006/mpat.1997.0155. [DOI] [PubMed] [Google Scholar]
- Desreumaux P., Brandt E., Gambiez L., Emilie D., Geboes K., Klein O., Ectors N., Cortot A., Capron M., Colombel J.F. Distinct cytokine patterns in early and chronic ileal lesions of Crohn's disease. Gastroenterology. 1997;113:118–126. doi: 10.1016/s0016-5085(97)70116-1. [DOI] [PubMed] [Google Scholar]
- Englaro W., Bahadoran P., Bertolotto C., Busca R., Derijard B., Livolsi A., Peyron J.F., Ortonne J.P., Ballotti R. Tumor necrosis factor alpha-mediated inhibition of melanogenesis is dependent on nuclear factor kappa B activation. Oncogene. 1999;18:1553–1559. doi: 10.1038/sj.onc.1202446. [DOI] [PubMed] [Google Scholar]
- Dickens M., Rogers J.S., Cavanagh J., Raitano A., Xia Z., Halpern J.R., Greenberg M.E., Sawyers C.L., Davis R.J. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- Derijard B., Hibi M., Wu I.H., Barrett T., Su B., Deng T., Karin M., Davis R.J. JNK1a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Dykens J.A., Baginski T.J. Urinary 8-hydroxydeoxyguanosine excretion as a non-invasive marker of neutrophil activation in animal models of inflammatory bowel disease. Scand. J. Gastroenterol. 1998;33:628–636. doi: 10.1080/00365529850171918. [DOI] [PubMed] [Google Scholar]
- Neurath M. F., Petterson S., Meyer zum Buschenfelde K.H., Strober W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in mice. Nat. Med. 1996;2:998–1004. doi: 10.1038/nm0996-998. [DOI] [PubMed] [Google Scholar]
- Neurath M. F., Fuss I., Pasparakis M., Alexopoulou L., Haralambous S., Meyer zum Büschenfelde K.H., Strober W., Kollias G. Predominant pathogenic role of tumor necrosis factor in experimental colitis in mice. Eur. J. Immunol. 1997;27:1743–1750. doi: 10.1002/eji.1830270722. [DOI] [PubMed] [Google Scholar]
- Jacobson K., McHugh K., Collins S.M. The mechanism of altered neural function in a rat model of acute colitis. Gastroenterology. 1997;112:156–162. doi: 10.1016/s0016-5085(97)70230-0. [DOI] [PubMed] [Google Scholar]
- Wallace J., McKnight W., Asfaha S., Liu D.Y. Reduction of acute and reactivated colitis in rats by an inhibitor of neutrophil activation. Am. J. Physiol. 1998;274:G802–G808. doi: 10.1152/ajpgi.1998.274.5.G802. [DOI] [PubMed] [Google Scholar]
- Selve, N. 1992. Chronic intrajejunal TNBS application in TNBS-sensitized rats: a new model of chronic inflammatory bowel diseases. Agents Actions. Spec No:C15–C17. [PubMed]
- Palmen M.J., Dieleman L.A., Soesatyo M., Pena A.S., Meuwissen S.G., Van Rees E.P. Effects of local budesonide treatment on the cell-mediated immune response in acute and relapsing colitis in rats. Dig. Dis. Sci. 1998;43:2518–2525. doi: 10.1023/a:1026606904531. [DOI] [PubMed] [Google Scholar]
- Hoshino H., Sugiyama S., Ohara A., Goto H., Tsukamoto Y., Ozawa T. Mechanism and prevention of chronic colonic inflammation with trinitrobenzene sulfonic acid in rats. Clin. Exp. Pharmacol. Physiol. 1992;19:717–722. doi: 10.1111/j.1440-1681.1992.tb00409.x. [DOI] [PubMed] [Google Scholar]
- Auboeuf D., Rieusset J., Fajas L., Vallier P., Frering V., Riou J.P., Staels B., Auwerx J., Laville M., Vidal H. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and liver X receptor-α in humansno alteration in adipose tissue of obese and NIDDM patients. Diabetes. 1997;46:1319–1327. doi: 10.2337/diab.46.8.1319. [DOI] [PubMed] [Google Scholar]
- Lefebvre A. M., Paulweber B., Fajas L., Woods J., McCrary C., Colombel J.F., Najib J., Fruchart J.C., Datz C., Vidal H. Peroxisome proliferator-activated receptor gamma is induced during differentiation of colon epithelium cells. J. Endocrinol. 1999;162:331–340. doi: 10.1677/joe.0.1620331. [DOI] [PubMed] [Google Scholar]
- Gelman L., Fruchart J.C., Auwerx J. An update on the mechanisms of action of the peroxisome proliferator-activated receptors (PPARs) and their roles in inflammation and cancer. Cell. Mol. Life Sci. 1999;55:932–943. doi: 10.1007/s000180050345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A., Barak Y., Nagy L., Liao D., Tontonoz P., Evans R.M. PPAR-γ dependent and independent effects on macrophages gene expression in lipid metabolism and inflammation. Nat. Med. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- Moore J., Rosen E.D., Fitzgerald M.L., Randow F., Andersson L.P., Altshuler D., Milstone D.S., Mortensen R.M., Spiegelman B.M., Freeman M.W. The role of PPAR-γ in macrophage differentiation and cholesterol uptake. Nat. Med. 2001;7:41–47. doi: 10.1038/83328. [DOI] [PubMed] [Google Scholar]
- Palmen M.J.H.J., Dijkstra C.D., Van der Ende M.B., Peña A.S., Van Rees E.P. Anti-CD11b/CD18 antibodies reduce inflammation in acute colitis in rats. Clin. Exp. Immunol. 1995;101:351–356. doi: 10.1111/j.1365-2249.1995.tb08363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmen M.J.H.J., Dieleman L.A., Van der Ende M.B., Uyterlinde A., Peña A.S., Meuwissen S.G.M., Van Rees E.P. Non-lymphoid and lymphoid cells in acute, chronic and relapsing experimental colitis. Clin. Exp. Immunol. 1995;99:226–232. doi: 10.1111/j.1365-2249.1995.tb05537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercurio F., Manning A.M. Multiple signals converging on NF-kappaB. Curr. Opin. Cell Biol. 1999;11:226–232. doi: 10.1016/s0955-0674(99)80030-1. [DOI] [PubMed] [Google Scholar]
- Karin M., Ben-Neriah Y. Phosphorylation meets ubiquitinationthe control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Maniatis T. Catalysis by a multiprotein IkappaB kinase complex. Science. 1997;278:818–819. doi: 10.1126/science.278.5339.818. [DOI] [PubMed] [Google Scholar]
- Baud V., Liu Z.G., Bennett B., Suzuki N., Xia Y., Karin M. Signaling by proinflammatory cytokinesoligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev. 1999;13:1297–1308. doi: 10.1101/gad.13.10.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee F., Hagler J., Chen Z.J., Maniatis T. Activation of the IkappaB alpha kinase complex by MEKK1, a kinase of the JNK pathway. Cell. 1997;88:213–222. doi: 10.1016/s0092-8674(00)81842-5. [DOI] [PubMed] [Google Scholar]
- Lee F., Peters R.T., Dang L.C., Maniatis T. MEKK1 activates both IkappaB kinase alpha and IkappaB kinase beta. Proc. Natl. Acad. Sci. USA. 1998;95:9319–9324. doi: 10.1073/pnas.95.16.9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano H., Shindo M., Sakon S., Nishinaka S., Mihara M., Yagita H., Okumura K. Differential regulation of IkappaB kinase alpha and beta by two upstream kinases, NF-kappaB-inducing kinase and mitogen-activated protein kinase/ERK kinase kinase-1. Proc. Natl. Acad. Sci. USA. 1998;95:3537–3542. doi: 10.1073/pnas.95.7.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F., Demers L.M., Vallyathan V., Ding M., Lu Y., Castranova V., Shi X. Vanadate induction of NF-kappaB involves IkappaB kinase beta and SAPK/ERK kinase 1 in macrophages. J. Biol. Chem. 1999;274:20307–20312. doi: 10.1074/jbc.274.29.20307. [DOI] [PubMed] [Google Scholar]
- Darnay B., Ni J., Moore P.A., Aggarwal B.B. Activation of NF-kappaB by RANK requires tumor necrosis factor receptor-associated factor (TRAF) 6 and NF-kappaB-inducing kinase. Identification of a novel TRAF6 interaction motif. J. Biol. Chem. 1999;274:7724–7731. doi: 10.1074/jbc.274.12.7724. [DOI] [PubMed] [Google Scholar]
- Thome M., Martinon F., Hofmann K., Rubio V., Steiner V., Schneider P., Mattmann C., Tschopp J. Equine herpesvirus-2 E10 gene product, but not its cellular homologue, activates NF-kappaB transcription factor and c-Jun N-terminal kinase. J. Biol. Chem. 1999;274:9962–9968. doi: 10.1074/jbc.274.15.9962. [DOI] [PubMed] [Google Scholar]
- Wang Y., Seibenhener M.L., Vandenplas M.L., Wooten M.W. Atypical PKC zeta is activated by ceramide, resulting in coactivation of NF-kappaB/JNK kinase and cell survival. J. Neurosci. Res. 1999;55:293–302. doi: 10.1002/(SICI)1097-4547(19990201)55:3<293::AID-JNR4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Alpert D., Schwenger P., Han J., Vilcek J. Cell stress and MKK6b-mediated p38 MAP kinase activation inhibit tumor necrosis factor-induced IkappaB phosphorylation and NF-kappaB activation. J. Biol. Chem. 1999;274:22176–22183. doi: 10.1074/jbc.274.32.22176. [DOI] [PubMed] [Google Scholar]
- Karin M., Delhase M. JNK or IKK, AP-1 or NF-kappaB, which are the targets for MEK kinase 1 action? Proc. Natl. Acad. Sci. USA. 1998;95:9067–9069. doi: 10.1073/pnas.95.16.9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W., Huang C., Dong Z. Inhibition of ultraviolet C irradiation-induced AP-1 activity by aspirin is through inhibition of JNKs but not erks or P38 MAP kinase. Int. J. Oncol. 1998;12:565–568. doi: 10.3892/ijo.12.3.565. [DOI] [PubMed] [Google Scholar]
- Schwenger P., Alpert D., Skolnik E.Y., Vilcek J. Activation of p38 mitogen-activated protein kinase by sodium salicylate leads to inhibition of tumor necrosis factor-induced IkappaB alpha phosphorylation and degradation. Mol. Cell. Biol. 1998;18:78–84. doi: 10.1128/mcb.18.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]