

Abstract
Rodents immunized with complete Freund's adjuvant (CFA) are resistant to subsequent attempts to induce autoimmune disease, while animals immunized with incomplete Freund's adjuvant (IFA) remain susceptible. Mycobacterial extracts can stimulate inducible nitric oxide synthase (NOS2) gene transcription. Robust expression of NOS2 has been linked to suppression of T cell proliferation and alterations in immune responses. Our studies investigated the hypothesis that the immunoprotective effect of CFA before immunization requires functional NOS2. NOS2 gene expression is chronically elevated in lymph nodes and spleens of CFA-immunized mice. Maximal expression of NOS2 after CFA immunization requires the presence of functional type I tumor necrosis factor α receptor (TNFR1) and interferon γ. Groups of nontreated and CFA-preimmunized male C57BL/6J or C57BL/6NOS2−/− mice were immunized with myelin oligodendrocyte glycoprotein (MOG) peptide 35–55 in CFA to induce experimental allergic encephalomyelitis (EAE). Wild-type C57BL/6J mice were protected from the development of symptoms of EAE, while the NOS2−/− mice failed to be protected. NOS2-dependent effects of CFA included an augmentation of the MOG-specific IgG1 response, a decrease in interleukin 6 production by MOG-reactive lymphocytes, and a marked decrease in mononuclear cell infiltrates in the central nervous system. These studies support the hypothesis that CFA immunization modulates immune responses through a nitric oxide–dependent mechanism.
Keywords: experimental allergic encephalomyelitis, Freund's adjuvant, immunosuppression, interleukin 6, tumor necrosis factor α
Introduction
Exposure to CFA can impair the subsequent expression of autoimmune disease in small rodents. This observation, also referred to as “adjuvant immunotherapy,” was first described over 40 years ago and found to be dependent on the presence of Mycobacterium tuberculosis within the adjuvant 1. This immunoprotective effect of CFA has been subsequently demonstrated in multiple autoimmune disease models, both spontaneous and induced, and in multiple species, including rats 2 3 4 5, mice 6, and guinea pigs 1 7 8. In each case, preimmunization with CFA alone 21–40 d before attempted disease induction by immunization resulted in a decreased incidence and severity of disease 1 2 3 7 8.
Much of our current understanding of the CFA effect is derived from rodent models of spontaneous autoimmune diabetes mellitus. A single injection of CFA into nonobese diabetic (NOD) mice between the ages of 4 and 12 wk prevents the development of clinical diabetes indefinitely and extends NOD life span significantly 9 10 11. CFA immunization of recipient NOD mice blocks the ability of diabetogenic T cells to transfer disease 9. Treatment of NOD mice with CFA at the time of islet cell transplantation blocks rejection and promotes the functional survival of the graft 12 13. CFA did not result in a generalized state of immunosuppression, as evidenced by the ability of the recipient to reject an allograft 12 13 14.
Cotransfer of diabetogenic T cell clones with splenocytes from a CFA-treated mouse blocks the adoptive transfer of disease 9. T cell or CD4+ cell depletion in the CFA-treated spleen eliminates the inhibitory effect 9. Enrichment of the CFA treated splenocytes for Mac-1+ cells abrogates the ability of splenocytes from a diabetic mouse to cause disease in a prediabetic NOD mouse 15. These findings may reflect a dual requirement for both T cells and macrophages in mediating adjuvant immunotherapy. Concomitant treatment of BioBreeding rats with CFA and anti–TNF-α antibody eliminates the inhibitory effect of CFA, suggesting that TNF-α is a required mediator 16.
Nitric oxide (NO) generated through the cytokine-inducible pathway has been increasingly recognized to play an important role in immune regulation in models of autoimmune disease and T cell tolerance 17 18 19 20 21. Immune responses induced with Ag in CFA are markedly augmented in the presence of inducible NO synthase (NOS2) inhibitors or when using NOS2−/− mice. In this study, we examined whether functional NOS2 was required for the protective effect of CFA.
Materials and Methods
Experimental Animals.
Male C57BL/6, C57BL/6NOS2−/−, C57BL/6IFN-γ2/−, and C57BL/6TNFR1−/− mice were obtained from The Jackson Laboratory. The mice were used between 4 and 6 wk of age. Mice were housed and handled in accordance with Veteran's Affairs and National Institutes of Health guidelines under Institutional Animal Care and Use Committee–approved protocols.
Reagents.
Myelin oligodendrocyte glycoprotein (MOG) peptide 35–55 N-MEVGWYRSPFSRVVHLYRNGK-C was prepared by custom solid phase synthesis (Research Genetics). IFA and Mycobacterium tuberculosis H37RA was obtained from Difco Labs. Purified pertussis toxin was obtained from List Biochemicals.
CFA Pretreatment and Experimental Allergic Encephalomyelitis Induction.
Mice to be evaluated for CFA-mediated protection were immunized i.p. 28 d before pMOG35–55 immunization with 100 μl of CFA (prepared as 0.5 mg/ml M. tuberculosis H37RA in a 1:1 (vol/vol) emulsion of Freund's adjuvant and PBS). To induce experimental allergic encephalomyelitis (EAE), mice were immunized s.c. with 300 μg of pMOG35–55 and 400 μg of M. tuberculosis H37RA in 200 μl of Freund's adjuvant. Additionally, mice were injected i.p. with 500 ng of pertussis toxin in 50 μl of PBS at the time of pMOG35–55 immunization and 48 h later. Mice were evaluated daily for signs of disease. The clinical course of the EAE was scored according to the severity as follows: 0 = no obvious signs of disease, 0.5 = partial tail weakness, 1 = limp tail, 1.5 = limp tail and hindlimb weakness, 2 = limp tail and impairment in righting reflex, 2.5 = limp tail and hindlimb paresis, 3 = bilateral hindlimb paralysis, 3.5 = bilateral hindlimb paralysis and forelimb paresis, 4 = moribund, and 5 = dead. The mice were followed for a total of 28 d, at which point the mice were killed. The brains and spinal cords were prepared for histologic evaluation by careful dissection and fixation in 10% neutral buffered formalin.
CFA Treatment to Induce NOS2 Expression.
Mice were immunized s.c. at the base of the tail with CFA. At the indicated day (see Fig. 1 legend) after CFA immunization, the mice were killed, and draining lymph nodes, spleen, liver, and kidney were harvested. Of these tissues, half was flash frozen in liquid N2, and total RNA was prepared from the remaining half.
Figure 1.
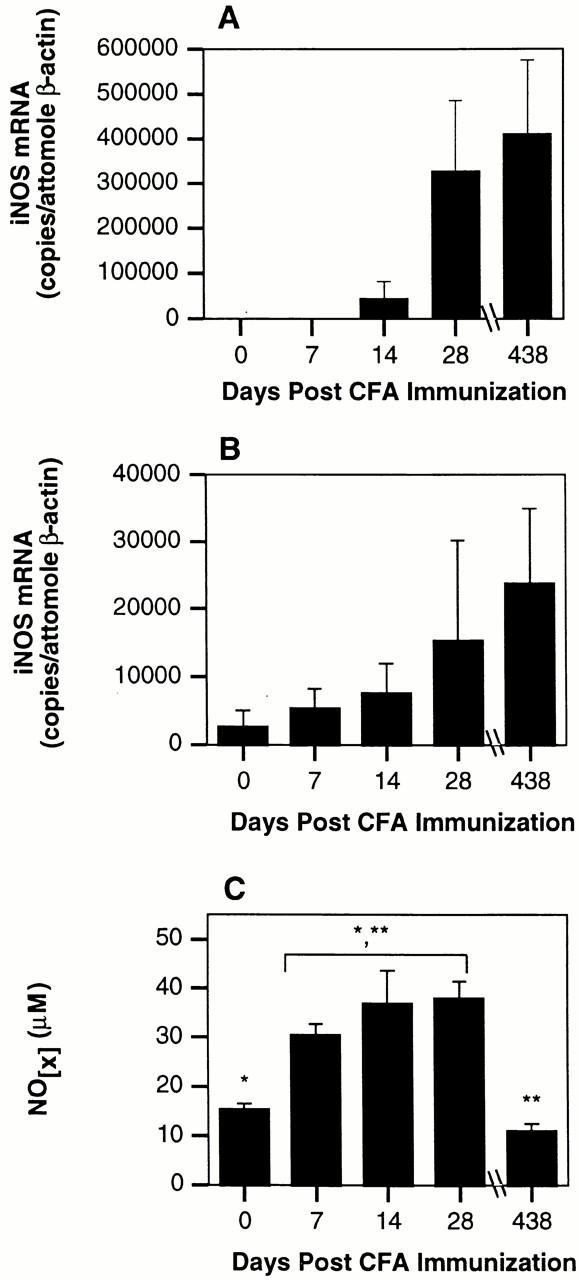
CFA-immunized C57BL/6 mice have long-term augmented basal NOS2 expression in lymphoid tissues. (A and B) C57BL/6J mice (n = 6) were immunized s.c. with 200 μl of CFA. On the indicated days, mice were killed and total RNA prepared from draining lymph nodes (A) and spleen (B). Samples were evaluated for NOS2 and β-actin mRNA concentrations by competitive reverse transcription (RT)-PCR. (C) Serum was isolated from the mice described above. Total nitrate and nitrite was determined by complete Griess assay. Analysis of variance (ANOVA) with Bonferroni/Dunn post-test: *P = ≤ 0.003, **P = ≤ 0.0002.
Histology.
Paraffin blocks of the fixed spinal cords were prepared. Transverse and longitudinal sections were cut and placed on a frost-free glass slide. Sections were stained for myelin with Luxol Fast Blue. The cellular infiltrate was evaluated by hematoxylin and eosin staining. Sections were scored according to the following system: 0 = no sign of infiltrate, 1 = perivascular congestion (light), 2 = perivascular congestion (heavy), 3 = perivascular congestion (heavy) and parenchymal infiltrate, 4 = focal meningeal lymphocytosis, and 5 = extensive sclerosis.
Proliferation Assays.
Spleens were harvested and prepared as single-cell suspensions with a metal screen. RBCs were lysed by a room temperature incubation in a hypotonic solution (0.83% NH4Cl, 0.02 mM Tris, pH 7.6). Proliferation assays were set up as described previously 22. Cells assayed for proliferation were pulsed with 1 μCi/well [3H]thymidine (Amersham Pharmacia Biotech) at 48 h and harvested 18 h later.
Cytokine ELISAs.
Culture supernatant concentrations of IL-4, IL-6, IFN-γ, and TNF-α were determined by sandwich ELISA with antibodies purchased from PharMingen. IL-4, IL-6, IFN-γ, and TNF-α concentrations were determined from culture supernatants after 66 h of activation with soluble MOG peptide. Antibody dilutions that maximized signal to noise were determined for each antibody pair, and ELISAs were performed as described previously 22.
Serum MOG-specific Antibody Titer Determination. Serum was collected from mice by terminal cardiac puncture. Samples of serum were analyzed at various dilutions. Briefly, 96-well MAXISORP™ microtiter plates (Nunc-Nalgene) were treated with 0.2% glutaraldehyde (Sigma-Aldrich) for 2 h at 37°C. Plates were coated with pMOG35–55 (1 μg/ml in 0.1 M carbonate buffer, pH 9.6) for 3 h at 37°C and washed three times with TBS–0.1% Tween-20 (TBST). Plates were blocked overnight at 4°C with PBS containing 2% BSA (Sigma-Aldrich) and 0.05% Tween-20 and washed once with TBST. Plates were incubated with serum samples for 1 h at 37°C and washed three times with TBST. Plates were developed with anti-IgG (Calbiochem), anti-IgG1 (Caltag Laboratories), or anti-IgG2a (PharMingen) alkaline phosphatase conjugates at 37°C for 1 h and washed five times with TBST. Color was developed by incubating plates with p-nitrophenylphosphate disodium purchased from Sigma-Aldrich (1 mg/ml in 1 M carbonate buffer, pH 9.6) at room temperature for equal amounts of time. Color development was evaluated in a microplate reader at 605 nm.
Competitive Reverse Transcription PCR.
Total RNA was prepared from draining lymph node, spleen, liver, and kidney with the RNeasy Mini™ kit (QIAGEN) and stored at −70°C with 40 U of RNase-OUT™ inhibitor purchased from GIBCO BRL. cDNA was prepared from 2 μg of each sample using a kit from GIBCO BRL (Superscript II preamp™) according to the manufacturer's instructions. The cDNA was used in a PCR reaction with serial dilutions of a known molar amount of a competitive template for NOS2 (CLONTECH Laboratories, Inc.) and β-actin and performed as described previously 22.
Statistics.
Differences were statistically analyzed using unpaired Student's t test. Analysis was accomplished with STATVIEW™ (v4.5; Abacus Concepts).
Results and Discussion
CFA Immunization Induces Chronic NOS2 Expression in Lymphoid Tissues.
Immune responses induced by immunizations with Ag in CFA are augmented in the absence of functional NOS2 20 21 22. We examined whether NOS2 expression is induced in lymphoid tissue after immunization with CFA alone. Fig. 1 A and B depict a time course of NOS2 mRNA expression levels in draining lymph nodes and spleen after a single CFA immunization. Levels are increased by 28 d after immunization. Curiously, this increase in NOS2 mRNA expression is maintained for up to 14 mo after immunization. The kinetics of expression appear to be similar in lymph node and spleen. Absolute levels are higher in draining lymph node, and the increase in NOS2 mRNA levels occurs slightly earlier in the spleen. The relative levels of NOS2 mRNA in spleen and lymph node depend on the site of CFA immunization: intraperitoneal injections result in heightened splenic mRNA expression relative to lymph node expression (data not shown). Sera from CFA-immunized mice display augmented levels of NOx (sum of nitrite and nitrate measured by Griess reaction) starting 1 wk after immunization. The dissociation between NOS2 mRNA and serum NOx at 438 d is unexplained but may reflect altered clearance of NOx. Thus, a single immunization with CFA induces chronic NOS2 expression in lymphoid organs. IFA immunization does not elicit an increase in serum NOx or NOS2 mRNA expression (data not shown).
CFA-induced NOS2 Expression Is Partially Dependent on the Production of IFN-γ and the Expression of TNFR1.
The NOS2 gene is transcriptionally activated by proinflammatory cytokines 23. We investigated whether IFN-γ and TNF-α were required for CFA induction of NOS2 by immunizing C57BL/6, C57BL/6IFN-γ−/−, and C57BL/6TNFR1−/− mice with CFA and evaluating them 28 d later for NOS2 mRNA expression in lymph node and spleen. We used TNFR1−/− mice rather than TNF-α−/− mice because TNFR1 is the primary mediator of TNF-α–dependent nuclear factor (NF)-κB activation, and TNF-α−/− mice have abnormal lymphoid organization 24 25. As shown in Fig. 2, the NOS2 mRNA levels in lymph nodes of CFA-treated animals are significantly lower in both targeted mutant strains. Serum NOx concentrations are significantly lower as well. IFN-γ–deficient mice make only a partial response, while those mice lacking TNFR1 have undetectable levels of NOS2 mRNA in the lymph node and spleen. Similar to observations made in wild-type mice, CFA-induced NOS2 mRNA expression in IFN-γ–deficient mice was greater in the lymph node than in the spleen. While the difference between CFA-induced splenic NOS2 mRNA expression in wild-type and mutant mice failed to reach statistical significance, the pattern of hypoexpression is preserved (Fig. 2 B). The serum NOx levels in the CFA-immunized TNFR1−/− mice are elevated compared with unimmunized controls, although not to the extent seen in the wild type. Thus, both IFN-γ and TNFR1 are required for full induction of NOS2 in response to CFA.
Figure 2.
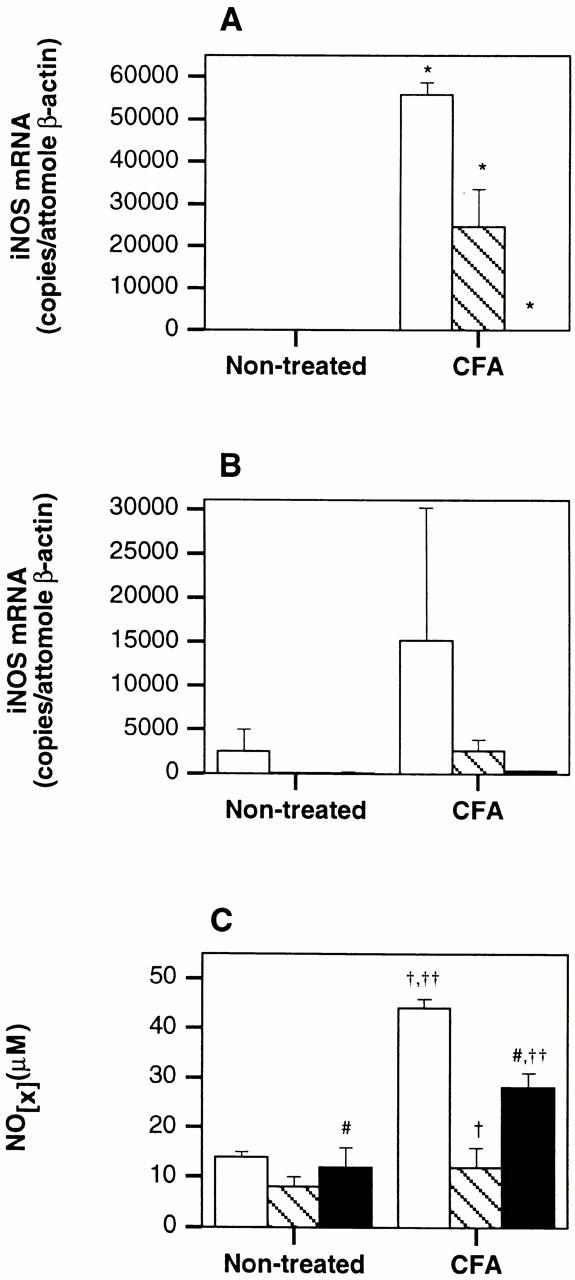
CFA-augmented basal NOS2 expression in lymphoid tissues is dependent on the expression of IFN-γ and TNFR1. (A and B) Wild-type, C57BL/6IFN-γ−/− (n = 6), and C57BL/6TNRF1−/− mice were immunized s.c. with CFA. On day 28, mice were killed and total RNA prepared from draining lymph nodes (A) and spleen (B). Samples were evaluated for NOS2 and β-actin mRNA concentrations by competitive RT-PCR. ANOVA with Bonferroni/Dunn post-test: *P < 0.0001. Open bars, wild-type mice; hatched bars, IFN-γ−/− mice; black bars, TNFR1−/− mice. (C) Serum was isolated from the mice described above. Total nitrate and nitrites were determined by complete Griess assay. ANOVA with Bonferroni/Dunn post-test: # P = 0.001, † P < 0.0001, †† P = 0.002.
Pretreatment of C57BL/6 Wild-Type but Not NOS2− /− Mice with CFA Results in Protection from MOG-induced EAE.
To examine whether CFA induction of NOS2 might account for the ability of CFA to modulate disease expression, we used the murine model of EAE induced by immunization with an encephalitogenic MOG peptide in CFA 18. Mice immunized with pMOG35–55 develop a severe, chronic, nonremitting form of EAE. This illness develops over 20–30 d as shown in Fig. 3 A. Consistent with previous reports in other model systems, C57BL/6 mice are protected from the development of MOG-induced EAE by a single injection of CFA i.p. 28 d before immunization with MOG/CFA. Of the 15 wild-type mice immunized with MOG/CFA 28 d after i.p. CFA, only 5 had any detectable clinical evidence of disease, and only 1 animal developed symptoms more severe than a symptom score of 2.
Figure 3.
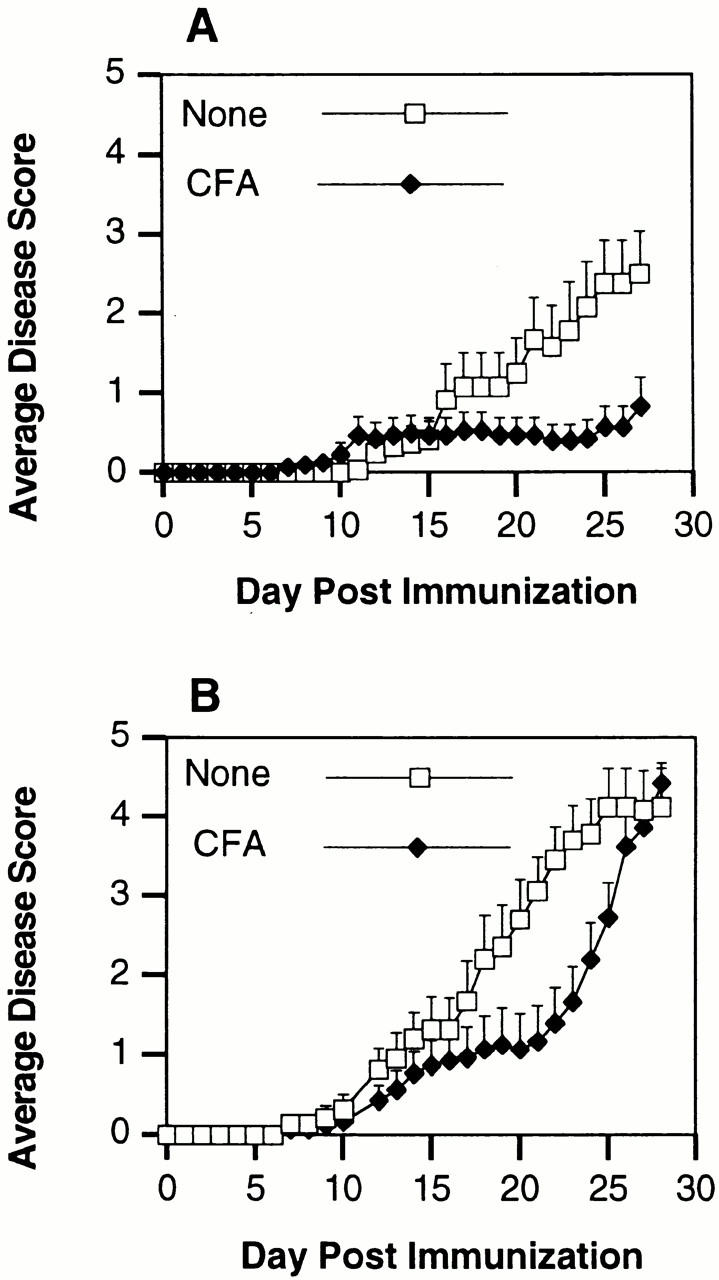
CFA protection of C57BL/6J mice from pMOG35–55-induced EAE requires NOS2 expression. (A) C57BL/6J mice (n = 15) were immunized i.p. with 100 μl of CFA containing 50 μg of M. tuberculosis H37Ra. On day 28, these mice, along with a group of nonimmunized C57BL/6J mice (n = 15), were immunized s.c. with 300 μg of pMOG35–55 to induce EAE. Mice were scored daily for signs of clinical disease. Day numbering in the figure corresponds to days after MOG/CFA immunization. (B) C57BL/6NOS2−/− mice (n = 15) were pretreated with CFA as above. On day 28, these mice, along with a group of nonimmunized C57BL/6NOS2−/− mice (n = 15), were immunized s.c. with 300 μg of pMOG35–55 to induce EAE. Mice were scored daily for signs of clinical disease.
In contemporaneous experiments, we examined the effect of CFA preimmunization on the course of EAE in MOG/CFA-immunized C57BL/6NOS2−/− mice. The disease course in these mutant mice is depicted in Fig. 3 B and confirms previous reports that this disease is more severe in the NOS2−/− mice. By day 27 after MOG/CFA immunization, the mean clinical scores are above 4.0 in the NOS2−/− mice, compared with ∼2.5 in the C57BL/6 wild-type mice. NOS2−/− mice preimmunized with CFA all develop significant clinical disease by day 27. Between days 17 and 21 after MOG/CFA immunization, the disease course appears to transiently stabilize in the CFA-preimmunized animals before accelerating to fulminant disease. Therefore, while CFA preimmunization may transiently alter the course of disease in the NOS2−/− mice, it does not impart the same level of protection seen in the C57BL/6 wild-type mice.
NO-dependent CFA Immunomodulation Alters the Humoral Response after Immunization with MOG.
In a second series of experiments, we examined whether preimmunization with CFA altered the subsequent immune response to MOG/CFA in both wild-type and NOS2−/− mice. Both strains were preimmunized with CFA and then 28 d later immunized with MOG/CFA as for disease induction. On day 10 after MOG/CFA immunization, the mice were killed. Sera from experimental groups were evaluated for anti-MOG total IgG, IgG1, and IgG2a antibody titers. Spleens from all animals were assayed for their proliferative responses and cytokine production in response to MOG.
As shown in Fig. 4 A, wild-type mice pretreated with CFA have significantly augmented levels of anti-MOG IgG compared with wild-type mice not preimmunized with CFA. Under the conditions of this assay, we do not detect anti-MOG IgG in the serum of naive mice (data not shown). The total anti-MOG IgG levels in NOS2−/− mice are not augmented with CFA pretreatment and are comparable to the wild-type mice immunized with MOG/CFA. The increase in total anti-MOG IgG in CFA-pretreated wild-type mice is mainly due to a substantial increase in anti-MOG IgG1 concentrations. Again, this does not occur in NOS2−/− mice (Fig. 4 B). The increases in MOG-specific IgG2a in both strains pretreated with CFA are small but statistically significant.
Figure 4.

NO-dependent CFA-induced alterations of immune response to pMOG35–55. Sera from C57BL/6 (n = 12) and C57BL/6NOS2−/− (n = 12) mice pretreated as indicated were collected on day 10 after immunization with pMOG35–55. Sera were evaluated by ELISA for titers of pMOG35–55-specific immunoglobulins. The OD values shown here are from a serum dilution determined to be on the linear part of the OD vs. dilution curve. A depicts total IgG, and B depicts IgG1 and IgG2a. The same dilution was used for all samples. Statistics determined from two-tailed unpaired Student's t test: in A, *P = 0.0193, **P = 0.3085. In B, *P = 0.0573, **P = 0.3990, † P = 0.0521, †† P = 0.0277. (C) CFA-induced alterations in splenocyte cellular proliferation. Splenocytes were taken from the mice described in Fig. 5 on day 10. Cellular proliferation assay was performed at 2 × 106 cells/ml to 50 μg/well pMOG35–55. Cultures were pulsed with 1 μCi/well [3H]TdR at 48 h and incorporation assayed at 64 h. Statistical differences determined by two-tailed unpaired Student's t test. In C, *P = 0.0583, **P = 0.6361. (D) NO-dependent CFA-induced alterations of splenic cytokine production. Cytokine concentrations were determined by ELISA on the supernatants from the splenic cultures in C. Hatched bars, IL-4; gray bars, IFN-γ; open bars, TNF-α; black bars, IL-6. Statistical differences determined by unpaired Student's t test. In D, *P = 0.0025, **P = 0.2677.
Splenocytes from the groups described above were assayed in vitro for proliferative responses to pMOG35–55. Consistent with a regulatory role for NO on T cell proliferation, NOS2−/− mice display greatly augmented proliferative responses to MOG compared with wild-type mice (Fig. 4 C). CFA pretreatment failed to significantly alter these proliferative responses in either strain. Fig. 4 D demonstrates the cytokine profiles produced in response to MOG from the various experimental groups. A screen of cytokines critical for either the development of EAE or of NOS2 regulation showed that pretreatment with CFA had no statistically significant effect on the production of IL-4, TNF-α, or IFN-γ. However, IL-6 production was reduced by pretreatment with CFA in an NO-dependent manner (Fig. 4 D).
CFA Pretreatment Causes an NO-dependent Reduction in Early Histologic Evidence of Inflammatory Central Nervous System Infiltrate after Immunization with MOG.
CFA-pretreated and nonpretreated mice were evaluated on day 10 after pMOG35–55 immunization for early histologic changes in the central nervous system (CNS). As shown in Fig. 5 A, wild-type mice pretreated with CFA lack the extensive infiltrate seen in CNS tissue from mice with active EAE. While CFA-pretreated wild-type mice have only minimal evidence of infiltrate, healthy NOS2−/− mice pretreated with CFA clearly show histologic changes associated with the development of EAE (Fig. 5 B). Scoring of the histology from the various groups additionally demonstrates CFA-mediated protection from early histologic disease in wild-type mice that is not observed in the absence of NOS2 (Fig. 5 C).
Figure 5.
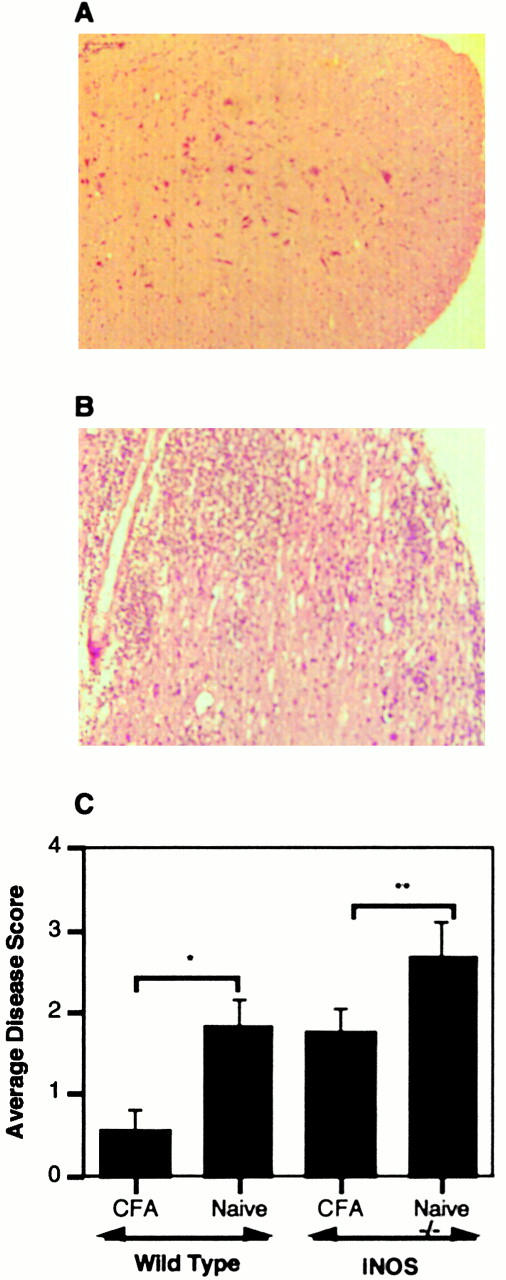
NO-dependent CFA-induced prevention of CNS infiltrate. Spinal cord tissue was harvested from the mice described in Fig. 4. Spinal cord tissue from a wild-type (A) or NOS2−/− mouse (B) preimmunized with CFA before MOG/CFA challenge. Panel C depicts disease scores from all of the animals studied in these groups. *P = 0.01, **P = 0.1.
These studies support the hypothesis that the protective effects of CFA preimmunization require functional NOS2. CFA-preimmunized NOS2 knockout mice fail to be protected from the development of EAE. Wild-type mice protected by CFA demonstrate several alterations in their immune response to MOG that may be relevant to the mechanism of protection. The augmentation of anti-MOG IgG1 titers is likely an epiphenomenon rather than responsible for disease protection, as antibodies are not major effector mechanisms of disease in the MOG peptide model of EAE 26. It is interesting that NOD mice treated with CFA demonstrate an augmented IgG1 response to GAD67, consistent with our observation 27. The NO-dependent inhibition of IL-6 production with CFA treatment is highly significant. IL-6–deficient mice are entirely resistant to MOG-induced EAE 28 29 30. Furthermore, the mice that have been genetically modified to overexpress IL-6 in the CNS develop EAE spontaneously 31. The mechanism by which NO inhibits the expression of IL-6 may well be due to inhibition of NF-κB activation, which has been previously attributed to NO-dependent stabilization of IκBα 32. We cannot rule out effects of NOS2-generated NO on adhesion molecule expression and egress of effector T cells from the vasculature into the CNS 33. Our results are consistent with previous studies in models of autoimmune diabetes that demonstrate important protective effects of TNF-α 16. These studies provide further support for the importance of NOS2 in immunoregulation and suggest that chronic stimulation of NOS2 in vivo may have profound effects on subsequent immune responses. Given the continued interest in bacillus Calmette-Guerin vaccines for individuals predisposed to autoimmune diseases such as type I diabetes mellitus, delineation of the mechanism of the CFA effect remains an important goal.
Acknowledgments
This work was supported in part by research grants from the National Institutes of Health (NIH; DK45346 and DK42155) and the Office of Research and Development, Medical Research Service, Department of Veterans Affairs. D.A. Kahn is supported by the NIH-supported Medical Scientist Training Program at the University of California at San Diego (GM07198).
Footnotes
Abbreviations used in this paper: CNS, central nervous system; EAE, experimental allergic encephalomyelitis; MOG, myelin oligodendrocyte glycoprotein; NOD, nonobese diabetic.
References
- Kies M.W., Alvord E.C. Prevention of allergic encephalomyelitis by prior injection of adjuvants. Nature. 1958;182:1106–1107. doi: 10.1038/1821106a0. [DOI] [PubMed] [Google Scholar]
- Hempel K., Freitag A., Freitag B., Endres B., Mai B., Liebaldt G. Unresponsiveness to experimental allergic encephalomyelitis in Lewis rats pretreated with complete Freund's adjuvant. Int. Arch. Allergy Appl. Immunol. 1985;76:193–199. doi: 10.1159/000233691. [DOI] [PubMed] [Google Scholar]
- Stevens D.B., Karpus W.J., Gould K.E., Swanborg R.H. Studies of V beta 8 T cell receptor peptide treatment in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 1992;37:123–129. doi: 10.1016/0165-5728(92)90163-f. [DOI] [PubMed] [Google Scholar]
- Raziuddin S., Kibler R.F., Morrison D.C. Experimental allergic encephalomyelitis in Lewis ratsinhibition by bacterial lipopolysaccharides and acquired resistance to reinduction by challenge with myelin basic protein. J. Immunol. 1981;127:16–20. [PubMed] [Google Scholar]
- Kawano Y., Sasamoto Y., Kotake S., Thurau S.R., Wiggert B., Gery I. Trials of vaccination against experimental autoimmune uveoretinitis with a T-cell receptor peptide. Curr. Eye Res. 1991;10:789–795. doi: 10.3109/02713689109013873. [DOI] [PubMed] [Google Scholar]
- Cua D.J., Hinton D.R., Kirkman L., Stohlman S.A. Macrophages regulate induction of delayed-type hypersensitivity and experimental allergic encephalomyelitis in SJL mice. Eur. J. Immunol. 1995;25:2318–2324. doi: 10.1002/eji.1830250830. [DOI] [PubMed] [Google Scholar]
- Lisak R.P., Zweiman B. Immune responses to myelin basic protein in mycobacterial-induced suppression of experimental allergic encephalomyelitis. Cell. Immunol. 1974;14:242–254. doi: 10.1016/0008-8749(74)90209-3. [DOI] [PubMed] [Google Scholar]
- Falk G.A., Kies M.W., Alvord E.C., Jr. Passive transfer of experimental allergic encephalomyelitismechanisms of suppression. J. Immunol. 1969;103:1248–1253. [PubMed] [Google Scholar]
- Qin H.Y., Sadelain M.W., Hitchon C., Lauzon J., Singh B. Complete Freund's adjuvant-induced T cells prevent the development and adoptive transfer of diabetes in nonobese diabetic mice. J. Immunol. 1993;150:2072–2080. [PubMed] [Google Scholar]
- Sadelain M.W., Qin H.Y., Lauzon J., Singh B. Prevention of type I diabetes in NOD mice by adjuvant immunotherapy. Diabetes. 1990;39:583–589. doi: 10.2337/diab.39.5.583. [DOI] [PubMed] [Google Scholar]
- Shehadeh N.N., LaRosa F., Lafferty K.J. Altered cytokine activity in adjuvant inhibition of autoimmune diabetes. J. Autoimmun. 1993;6:291–300. doi: 10.1006/jaut.1993.1025. [DOI] [PubMed] [Google Scholar]
- Lakey J.R., Wang T., Warnock G.L., Singh B., Rajotte R.V. Prevention of recurrence of insulin-dependent diabetes mellitus in islet cell-transplanted diabetic NOD mice using adjuvant therapy. Transplant Proc. 1992;24:2848. [PubMed] [Google Scholar]
- Wang T., Singh B., Warnock G.L., Rajotte R.V. Prevention of recurrence of IDDM in islet-transplanted diabetic NOD mice by adjuvant immunotherapy. Diabetes. 1992;41:114–117. doi: 10.2337/diab.41.1.114. [DOI] [PubMed] [Google Scholar]
- Lakey J.R., Singh B., Warnock G.L., Rajotte R.V. BCG immunotherapy prevents recurrence of diabetes in islet grafts transplanted into spontaneously diabetic NOD mice. Transplantation. 1994;57:1213–1217. doi: 10.1097/00007890-199404270-00013. [DOI] [PubMed] [Google Scholar]
- McInerney M.F., Pek S.B., Thomas D.W. Prevention of insulitis and diabetes onset by treatment with complete Freund's adjuvant in NOD mice. Diabetes. 1991;40:715–725. doi: 10.2337/diab.40.6.715. [DOI] [PubMed] [Google Scholar]
- Rabinovitch A., Suarez-Pinzon W.L., Lapchak P.H., Meager A., Power R.F. Tumor necrosis factor mediates the protective effect of Freund's adjuvant against autoimmune diabetes in BB rats. J. Autoimmun. 1995;8:357–366. doi: 10.1006/jaut.1995.0028. [DOI] [PubMed] [Google Scholar]
- Fenyk-Melody J.E., Garrison A.E., Brunnert S.R., Weidner J.R., Shen F., Shelton B.A., Mudgett J.S. Experimental autoimmune encephalomyelitis is exacerbated in mice lacking the NOS2 gene. J. Immunol. 1998;160:2940–2946. [PubMed] [Google Scholar]
- Sahrbacher U.C., Lechner F., Eugster H.P., Frei K., Lassmann H., Fontana A. Mice with an inactivation of the inducible nitric oxide synthase gene are susceptible to experimental autoimmune encephalomyelitis. Eur. J. Immunol. 1998;28:1332–1338. doi: 10.1002/(SICI)1521-4141(199804)28:04<1332::AID-IMMU1332>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Wei X.Q., Charles I.G., Smith A., Ure J., Feng G.J., Huang F.P., Xu D., Muller W., Moncada S., Liew F.Y. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature. 1995;375:408–411. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]
- Gabbai F.B., Boggiano C., Peter T., Khang S., Archer C., Gold D.P., Kelly C.J. Inhibition of inducible nitric oxide synthase intensifies injury and functional deterioration in autoimmune interstitial nephritis. J. Immunol. 1997;159:6266–6275. [PubMed] [Google Scholar]
- Gold D.P., Schroder K., Powell H.C., Kelly C.J. Nitric oxide and the immunomodulation of experimental allergic encephalomyelitis. Eur. J. Immunol. 1997;27:2863–2869. doi: 10.1002/eji.1830271118. [DOI] [PubMed] [Google Scholar]
- Kahn D.A., Archer D.C., Kelly C.J. Absence of functional inducible NO synthase enhances the efficacy of tolerance induced by high dose antigen feeding. J. Immunol. 2000;165:6116–6122. doi: 10.4049/jimmunol.165.11.6116. [DOI] [PubMed] [Google Scholar]
- MacMicking J., Xie Q.W., Nathan C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- Pasparakis M., Alexopoulou L., Episkopou V., Kollias G. Immune and inflammatory responses in TNF alpha-deficient micea critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med. 1996;184:1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer K., Matsuyama T., Kündig T.M., Wakeham A., Kishihara K., Shahinian A., Wiegmann K., Ohashi P.S., Krönke M., Mak T.W. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- Lyons J.A., San M., Happ M.P., Cross A.H. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur. J. Immunol. 1999;29:3432–3439. doi: 10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Qin H.Y., Elliott J.F., Lakey J.R.T., Rajotte R.V., Singh B. Endogenous immune response to glutamic acid decarboxylase (GAD67) in NOD mice is modulated by adjuvant immunotherapy. J. Autoimmun. 1998;11:591–601. doi: 10.1006/jaut.1998.0243. [DOI] [PubMed] [Google Scholar]
- Mendel I., Katz A., Kozak N., Ben-Nun A., Revel M. Interleukin-6 functions in autoimmune encephalomyelitisa study in gene-targeted mice. Eur. J. Immunol. 1998;28:1727–1737. doi: 10.1002/(SICI)1521-4141(199805)28:05<1727::AID-IMMU1727>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Eugster H.P., Frei K., Kopf M., Lassmann H., Fontana A. IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur. J. Immunol. 1998;28:2178–2187. doi: 10.1002/(SICI)1521-4141(199807)28:07<2178::AID-IMMU2178>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Okuda Y., Sakoda S., Bernard C.C., Fujimura H., Saeki Y., Kishimoto T., Yanagihara T. IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein. Int. Immunol. 1998;10:703–708. doi: 10.1093/intimm/10.5.703. [DOI] [PubMed] [Google Scholar]
- Campbell I.L., Abraham C.R., Masliah E., Kemper P., Inglis J.D., Oldstone M.B., Mucke L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. USA. 1993;90:10061–10065. doi: 10.1073/pnas.90.21.10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H.B., Libby P., Liao J.K. Induction and stabilization of I kappa B alpha by nitric oxide mediates inhibition of NF-kappa B. J. Biol. Chem. 1995;270:14214–14219. doi: 10.1074/jbc.270.23.14214. [DOI] [PubMed] [Google Scholar]
- Peng H.B., Spiecker M., Liao J.K. Inducible nitric oxidean autoregulatory feedback inhibitor of vascular inflammation. J. Immunol. 1998;161:1970–1976. [PubMed] [Google Scholar]