

Abstract
To assess the role of lymphotoxin-β receptor (LTβR) in diabetes pathogenesis, we expressed an LTβR–Fc fusion protein in nonobese diabetic (NOD) mice. The fusion protein was expressed in the embryo, reached high levels for the first 2 wk after birth, and then declined progressively with age. High expression of LTβR–Fc blocked diabetes development but not insulitis. After the decline in chimeric protein concentration, mice became diabetic with kinetics similar to the controls. Early expression of fusion protein resulted in disrupted splenic architecture. However, primary follicles and follicular dendritic cells, but not marginal zones, developed in aged mice. Hence, LTβR signaling is required for diabetes development and regulates follicular and marginal zone structures via qualitatively or quantitatively distinct mechanisms.
Keywords: autoimmune disease, tumor necrosis factor, LIGHT, lymphoid development, marginal zone
Introduction
The lymphotoxin-β receptor (LTβR) is a member of the TNF receptor family and is required for normal development of the secondary lymphoid system 1. To date, there are two known cytokines capable of signaling through LTβR. The first is lymphotoxin-β (LTβ), a cell-bound heterotrimeric complex, whose genes, LTα and LTβ (along with TNF), are clustered within the MHC gene complex 2. The second cytokine, LIGHT, is a membrane-anchored homotrimeric complex, capable of binding LTβR, and an orphan member of the TNF receptor family, herpes simplex virus entry mediator 3 4 5.
Blockade of LTβR signaling through selective gene targeting or neutralizing fusion protein results in loss of lymph nodes and Peyer's patches 6 7 8. Moreover, these mice exhibit disrupted splenic architecture, lacking marginal zones, primary follicles, follicular dendritic cells (FDCs), and germinal centers 1. The role of LTβR signaling in autoimmune processes has been difficult to evaluate due to the lack of major lymphoid organ development in LTβR-, LTβ-, and LTα-deficient mice.
To address this issue, we backcrossed mice that express a neutralizing LTβR–Fc chimeric protein onto the nonobese diabetic (NOD) background. Like mice deficient in LTβR expression, these mice display disrupted splenic architecture 7 9. However, most lymph nodes develop normally 10. These animals provide an important opportunity to study the role of LTβR signaling in diabetes development in an intact lymphoid environment. Here we report that LTβR signaling is a critical element in type 1 diabetes mellitus and that temporal variation in expression of the LTβR–Fc fusion protein reveals novel aspects of B cell localization and migration.
Materials and Methods
Mice.
All mice were maintained and bred at Stanford University Animal Facility under barrier isolation conditions. BALB/c mice expressing a soluble murine LTβR–Fc fusion protein have previously been described 9 10. Transgene-positive male mice were bred for seven generations to female NOD mice obtained from the Stanford/McD NOD colony. These mice were then bred for two to six additional generations to NOD.Lt mice obtained directly from The Jackson Laboratory. Mice containing the integrated transgene were identified by PCR analysis, and soluble chimeric protein in the serum was quantified by immunoassay as described previously 10.
Detection of T Cells Autoreactive to Islet Cell Antigens.
106 splenocytes per well of 11-wk-old female transgene-positive and littermate control NOD mice were cultured in 96-well dishes in RPMI medium containing 0.5% Neutridoma-SP (Boehringer Mannheim) and 0.5% autologous NOD serum with 10 μg/ml of purified recombinant murine glutamic acid decarboxylase 65 (GAD) 11. Response to GAD was detected with [3H]thymidine incorporation and cytokine production after 72 h.
Production of IFN-γ was detected by immunoassay. 96-well plates were coated with rat anti–mouse IFN-γ antibodies (PharMingen). Dilutions of supernatant or control cytokine were detected with biotin-conjugated anti–mouse IFN-γ (PharMingen) and streptavidin (SA)-conjugated europium (Wallac).
Assessment of Insulitis, Sialitis, and Diabetes.
Insulitis and sialitis were determined from hematoxylin and eosin–stained pancreas and salivary gland tissue from mice 11–52 wk of age. The severity of insulitis was scored in blinded fashion from pancreata isolated from 11–13- and 17–19-wk-old female LTβR–Fc-expressing NOD mice or littermate controls: intact islets, no infiltrating cells; periinsulitis, <50% islets infiltrated with intact β cells; insulitis, >50% islets infiltrated with sparse β cell damage; and destructive insulitis, >50% islets infiltrated with few intact β cells present. Mouse urine glucose levels were measured weekly using Chemstrips (Boehringer Mannheim), and mice were considered diabetic on the first of three consecutive weeks of glycosuria.
Immunohistology.
Spleen, pancreas, lymph nodes (axillary, brachial, inguinal, popliteal, and pancreatic), and salivary glands from female NOD transgene-positive and littermate control mice were formalin fixed and stained with hematoxylin and eosin. Acetone-fixed frozen lymphoid tissue was stained with biotinylated CD3 (PharMingen), biotinylated CD11c, biotinylated MECA-367, biotinylated CR-1 antibodies, biotinylated peanut agglutinin (PNA) (Vector Laboratories), or rat antibodies specific for IgD (1126C) or FDC (FDC-M2). Bound antibodies were detected with a combination of the following: SA-conjugated Alexa-350 (blue; Molecular Probes), SA-conjugated Oregon Green (green; Molecular Probes), Neutralite–avidin-conjugated Texas Red (red; Southern Biotechnology), and goat anti–rat Oregon Green. IgM was detected with goat anti–mouse IgM–Texas Red (red; Southern Biotechnology), and CD8 (53.6.7) was detected with directly conjugated Alexa-350 antibody (blue).
Flow Cytometry.
Splenocytes were stained with mAbs specific for T cell, B cell, and macrophage surface antigens as described previously 9.
Results and Discussion
The LTβR–Fc Fusion Protein Is Expressed Transiently on the NOD Background.
To determine the role of LTβR signaling in diabetes development, mice expressing a neutralizing LTβR–human IgG1 (LTβR–Fc) fusion protein under control of the human CMV promoter 9 10 were bred onto the NOD background. Two independently generated lines of mice were established. Line 1610 expressed high concentrations of LTβR–Fc (“high expressers”; 0.8–30 μg/ml [mean 4] at 5–10 wk of age), while line 201 expressed low levels of this molecule (“low expressers”; 0.38–1.1 μg/ml [mean 0.6] at 5–10 wk of age).
To assess onset of transgene expression, 1610 NOD male mice were mated to NOD females. Serum was collected on multiple days before and after birth and analyzed for LTβR–Fc protein. As shown in Fig. 1, the transgene was expressed at high levels (2.3–3.9 μg/ml) on or before E16.5. Moreover, significant concentrations of fusion protein were also observed in transgene-negative littermates (∼2 μg/ml), and low amounts were detected in transgene-negative mothers (∼0.5 μg/ml). In transgene-negative mice, serum fusion protein concentration declined to insignificant levels by 14 d of age, while transgene-positive mice expressed very high levels at this age (7–18 μg/ml). Interestingly, the LTβR–Fc serum concentration from transgene-positive NOD mice was 3–10 times higher than those observed in BALB/c mice (data not shown).
Figure 1.

Both transgene-negative and -positive embryonic mice contain circulating LTβR–Fc fusion protein. The concentration of soluble LTβR–Fc chimeric protein was determined from embryos at E16.5, E18.5, and E19.5, neonates at 4, 7, and 14 d of age, and from their transgene-negative mothers (black bars*) that were bred to male 1610 NOD mice. NOD negative control serum determined baseline levels (first bar). Transgene-negative (black bars) and transgene-positive mice (gray bars) were identified by PCR analysis.
Although the Fc portion of the human IgG1 was mutated to inhibit Fc binding and complement fixation, the region required for placental transfer was not affected 12. Thus, the fusion protein presumably crossed the placenta from the transgene-positive fetus to the mother and hence into the transgene-negative littermates.
Serial blood samples were collected from single mice and individual litters to determine LTβR–Fc levels over many months. We were surprised to find that while the fusion protein was expressed at very high concentrations in the first few weeks of life, these levels declined with age. By 22 wk, circulating fusion protein concentrations in all NOD mice tested were reduced by as much as 90% (Table ; see Fig. 4 C). It is of interest that expression of the same transgene on the BALB/c background did not decrease with age (data not shown).
Table 1.
Early LTβR Blockade Inhibits Marginal Zone B Cell Development
| Mouse | Age | LTβR–Fc | MZB | FOB |
|---|---|---|---|---|
| wk | μg/ml | % | % | |
| Line 201 | ||||
| 33 | 25 | – | 5.3 | 22.4 |
| 46 | 30 | – | 4.1 | 24.1 |
| (4.7) | (23.25) | |||
| 30 | 25 | 0.2 | 1.2 | 24.6 |
| 42 | 30 | 0.2 | 1.9 | 21.1 |
| 44 | 30 | 0.1 | 2.9 | 32.6 |
| (2.0 ± 0.9) | (26.1 ± 5.9) | |||
| Line 1610 | ||||
| 70 | 11 | – | 4.5 | 23.2 |
| 71 | 11 | – | 3.8 | 24.2 |
| 73 | 11 | – | 3.9 | 25.6 |
| 95 | 11 | – | 3.9 | 26.5 |
| (4.0 ± 0.3) | (24.9 ± 1.5) | |||
| 69 | 11 | 2.8 | 1.4 | 23.5 |
| 72 | 11 | 7.1 | 0.6 | 23.9 |
| 94 | 11 | 2.8 | 0.8 | 22.1 |
| 96 | 11 | 1.4 | 1.75 | 26.8 |
| (1.1 ± 0.5) | (24.1 ± 2.0) | |||
| 6 | 26 | – | 8.5 | 24.0 |
| 22 | 26 | – | 5.3 | 27.7 |
| 82 | 31 | – | 7.2 | 25.2 |
| (7.0 ± 1.6) | (25.6 ± 1.9) | |||
| 15 | 26 | 0.8 | 0.4 | 35.1 |
| 21 | 26 | 0.6 | 1.1 | 28.3 |
| 76 | 31 | 0.5 | 1.6 | 38.6 |
| (1.0 ± 0.6) | (34.0 ± 5.2) |
Figure 4.
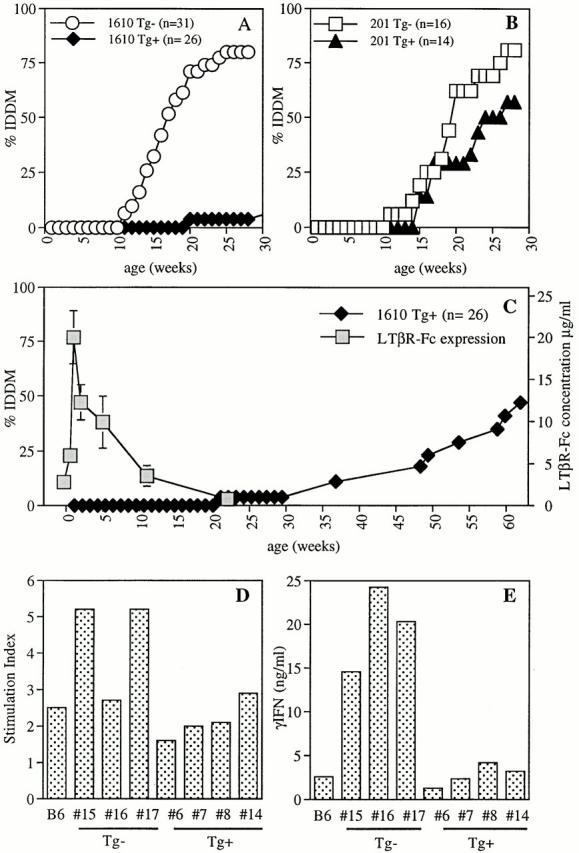
High LTβR–Fc expression inhibits diabetes and autoreactive T cell development. Diabetes incidence is shown for female mice expressing (A) high LTβR–Fc levels (line 1610, ♦) and littermate controls (○) or (B) low LTβR–Fc levels (line 201, ▴) and littermate controls (□). (C) Diabetes incidence for over one year (♦), as well as soluble serum fusion protein concentrations (light gray squares) are shown for mouse line 1610. Each time point represents the mean and SEM of circulating fusion protein levels from individual litters of three to seven mice. In data not shown, monthly serial blood samples of an individual female 1610 NOD mouse mirrored the mean serum LTβR–Fc concentrations from individual litters. (D) Stimulation index and (E) IFN-γ cytokine production of splenocytes from 12-wk-old female 1610 NOD mice stimulated with GAD 65. [3H]thymidine incorporation (D) was determined from triplicate wells of individual mice, and IFN-γ (E) was determined from pooled wells of individual mice. Mice #15, 16, and 17 were transgene (Tg) negative, verified by both ELISA and PCR analysis. Soluble LTβR–Fc fusion protein concentration at time of sacrifice was: #6, 3.3 μg/ml; #7, 1.8 μg/ml; #8, 2.0 μg/ml, and #14, 3.5 μg/ml. Data in D and E depicts one of two representative experiments.
Declining expression of the fusion protein may reflect the availability of transcription factors able to bind the CMV promoter or epigenetic modifications such as differential methylation patterns. This modulated expression of the chimeric fusion protein provides a unique opportunity to explore the relationship between LTβR signaling, lymphoid architecture, and diabetes development.
Early LTβR Blockade Results in Sustained Arrest of Marginal Zone Cell Development.
Given the declining expression of blocking fusion protein, we examined lymphoid tissue from NOD mice at multiple ages. Phenotypic characterization determined by flow cytometry revealed no significant differences between LTβR–Fc-expressing mice and littermate controls with respect to percentages of cells that express follicular B cell (Table ) and T cell and macrophage (data not shown) surface antigens. However, marginal zone B cell populations were significantly reduced in both NOD line 201 and 1610 as determined by CD21highCD23low/neg expression (Table ). By immunohistological analysis, it was determined that spleens isolated from 11–13-wk-old “high expressing” 1610 mice developed poorly defined primary follicles, FDC networks, and marginal zones (compare panels A and D with B and E in Fig. 2) and had diminished or no expression of MOMA-1 on marginal zone metallophilic macrophages or ER-TR9 on marginal zone macrophages (data not shown) or marginal zone B cells (Table ), and no mucosal addressin cell adhesion molecule (MAdCAM)-1 on marginal sinus (compare Fig. 2C and Fig. I) was observed in aged mice. However, the extent of splenic disruption from mouse to mouse (and within the same spleen) varied, with occasional secondary follicles containing FDC and germinal centers observed in young 1610 NOD mice.
Figure 2.
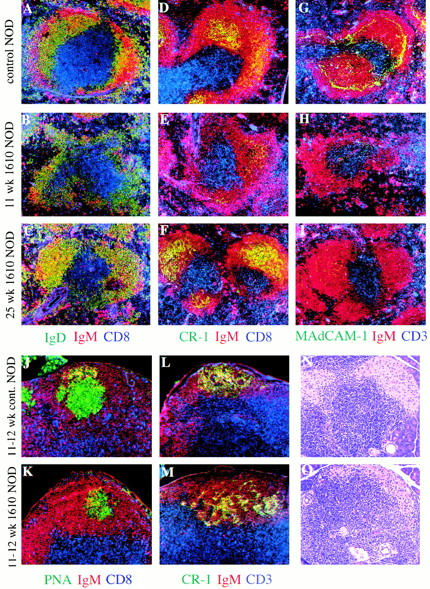
Formation of primary follicles and FDCs, but not marginal zones, after a decline of LTβR–Fc in aged mice. Spleens were removed from either 11-wk-old (B, E, H) or 26-wk-old (C, F, I) female NOD mice that expressed the LTβR–Fc (line 1610) fusion protein or 11-wk-old littermate controls (A, D, G). Frozen tissue sections were stained for: (A–C) B cells (red, IgM; green, IgD) and T cells (blue, CD8); (D–F) B cells (red, IgM), FDCs (green, CR-1), and T cells (blue, CD8); (G–I) B cells (red, IgM), MAdCAM-1 (green, MECA367), and T cells (blue, CD3) (original magnification ×100). Yellow color indicates red and green colocalized. Circulating fusion protein concentrations at time of sacrifice in 11-wk-old mice (B, E, H) were between 1.4 and 7.1 μg/ml (mean 3.4) and in 26-wk-old mice (C, F, I) were 0.5–0.8 μg/ml (mean 0.63). In the latter group, LTβR–Fc levels at 11 wk of age were 4.4–21.8 μg/ml (mean 11.5). Draining pancreatic lymph nodes from either 12-wk-old transgene-negative NOD mice (J and L) or transgene-positive littermates (K and M) were stained for: (J–K) B cells (red, IgM), germinal centers (green, PNA), and T cells (blue, CD8); (L–M) B cells (red, IgM), FDCs (green, CR-1), and T cells (blue, CD3). The concentration of fusion protein at time they were killed (mouse #6, 7, 8, and 14) is described in Fig. 4D and Fig. E. Pancreata from 11-wk-old high expressing (line 1610) LTβR–Fc female NOD mice (O) or female transgene-negative littermates (N) were stained with hematoxylin and eosin. The data is representative of numerous mice examined (original magnification ×250). Circulating fusion protein concentrations at time of sacrifice was 2.8 μg/ml.
As circulating levels of fusion protein declined with age, normalization of splenic architecture was observed. As early as 17 wk of age, splenic primary follicles and FDC networks were present (Fig. 2C and Fig. F). However, this reduction of LTβR–Fc levels did not lead to well defined marginal zone development (as determined up to 31 wk of age). Few MOMA-1+ or ER-TR9+ marginal zone metallophilic macrophages (data not shown) or marginal zone B cells (Table ), and no MAdCAM-1 (Fig. 2C and Fig. I) was observed in aged mice. As shown in Fig. 2 C, although IgM+IgD+ follicular B cells (yellow) formed polarized follicles, few IgMhighIgD− marginal zone B cells (red) were observed beyond the B cell follicle boundary in aged transgene-positive mice. Although conventional marginal zone structures were absent or greatly reduced in aged high expressing 1610 mice, some B cells did appear to reside in the marginal zone areas. These data suggest that “follicular” B cell populations localized to the marginal zone area but did not express the marginal zone B cell phenotype.
The events that control B cell compartmentalization may require less LTβR engagement than required for marginal zone cell development. Notably, mouse line 201 does not express sufficient concentrations (at any time) to affect primary follicles and FDC networks. However, marginal zones are reduced in these mice (Table ). This result suggests that LTβR differentially controls follicle/FDC formation and marginal zone development. The low levels of circulating LTβR–Fc fusion protein in aged high expressing 1610 mice may be sufficient to allow for B cell polarization and FDC development but not for marginal zone formation.
Draining Pancreatic Lymph Nodes Develop Germinal Centers.
Due to early expression of fusion protein in both transgene-positive and -negative neonates, we evaluated lymph node development in adult mice. Only 1/16 high expressing 1610 mice and 7/11 transgene-negative mice possessed inguinal lymph nodes. Popliteal lymph nodes were not present in any of seven transgene-positive mice and were found in two of four transgene-negative mice, as determined with India ink. However, six of six transgene-negative mice and six of eight transgene-positive mice contained at least one draining popliteal and inguinal lymph node after footpad immunization. The draining lymph nodes contained secondary follicles, FDC networks, and germinal centers (data not shown). These data suggest that poorly developed, small popliteal and inguinal lymph nodes are present and can recruit lymphocytes after immunization.
Recently, it has been reported that draining pancreatic lymph nodes are an important site where autoreactive T cells first encounter self-antigen on APCs and become activated 13. Thus, it was important to determine if draining pancreatic lymph nodes were present in LTβR–Fc-expressing mice. As shown in Fig. 2, mice expressing high concentrations of fusion protein did not develop well defined splenic primary follicles or FDC networks. However, all 14 high expressing LTβR–Fc 1610 mice examined contained draining pancreatic lymph nodes. These pancreatic lymph nodes contained PNA+ and CR-1+ germinal centers in B cell follicles (compare panels J and L to K and M in Fig. 2). In addition, axillary, brachial, mesenteric, facial, and sub-aortic lymph nodes were all present and appeared normal with regard to primary follicles, FDCs, T cell areas, and dendritic cells (data not shown). These experiments suggest that once these structures are formed, LTβR engagement regulates lymphocyte migration within the splenic white pulp, but not lymph nodes. Although the splenic microenvironment was disrupted in 11-wk-old NOD mice, lymphocytes migrated properly within most peripheral lymph nodes.
In contrast to LTα, LTβ, or LTβR gene–targeted mice, which lack most peripheral lymph nodes, these animals provide an important opportunity to study the role of LTβR in diabetes pathogenesis in an environment in which the majority of lymph nodes develop and respond normally. Thus, it is now possible to discriminate between developmental and functional effects due to loss of LTβR signaling.
LTβR–Fc Expression Does Not Inhibit Insulitis or Sialitis.
Insulitis is an essential step toward type 1 diabetes mellitus and is characterized by intraislet infiltration of lymphocytes that enter via high endothelial venules expressing MAdCAM-1 and peripheral lymph node addressin (PNAd) 14 15.
To assess if fusion protein expression affects diabetic processes, a total of 31 mice expressing the antagonist chimeric protein (as well as 23 nondiabetic transgene-negative littermates) were examined at 11, 13, 23, 28, 36, 43, and 52 wk of age for lymphocytic infiltration of islets. As determined by hematoxylin and eosin–stained tissue, neither insulitis (compare Fig. 2N and Fig. O) nor sialitis (data not shown) was inhibited by transgene expression in either low expressing 201 mice or high expressing 1610 mice. As early as 11 wk of age, lymphocytic infiltration was observed in both islets and salivary glands of control and LTβR–Fc-expressing mice.
To determine whether the fusion protein qualitatively altered the type of lymphocytic infiltrate, pancreatic tissue from 12–19-wk-old mice was stained with antibodies specific for T cell, B cell, macrophage, and dendritic cell surface antigens, as well as for MAdCAM-1, PNAd, vascular cell adhesion molecule (VCAM)-1, intercellular adhesion molecule (ICAM)-1, very late antigen (VLA)-4, and l-selectin (MEL-14). No difference was observed between NOD mice expressing high concentrations of fusion protein and littermate controls with regard to type and amount of infiltrate (data not shown).
To determine the extent and severity of islet infiltration, insulitis was scored from pancreata of 11–13- and 17–19-wk-old nondiabetic, transgene-positive, and transgene-negative female littermates. As shown in Fig. 3, transgene-positive and -negative mice were similar with regard to level and severity of insulitis. Only minor infiltration was observed in tissue other than endocrine organs in 6-mo-old transgene-positive and -negative NOD mice (which occurs in LTβR-deficient mice) 7.
Figure 3.

LTβR blockade does not affect the severity of insulitis. Severity of pancreatic infiltration in high expressing mice (line 1610) or littermate controls was determined in 17–19-wk-old nondiabetic female mice. Insulitis was classified as: intact islets, periinsulitis, insulitis, or destructive insulitis as described in Materials and Methods. A total of 248 islets (average of 62 islets/mouse) from transgene (Tg)-negative animals and 160 islets (average of 53 islets/mouse) from transgene-positive mice was scored. Circulating fusion protein concentrations at time of islet scoring were: #2, 0.47 μg/ml; #9, 1.18 μg/ml; and #10, 0.53 μg/ml. Data is representative of one of three similar experiments.
Although fusion protein levels were sufficient to disrupt splenic lymphoid tissue architecture, pancreatic islet lymphocytic infiltration was unaffected. These findings suggest that this step in the progression toward insulin-dependent diabetes mellitus (IDDM) is LTβR independent or is quantitatively different from lymphocyte migration within splenic tissues.
High Concentrations of LTβR–Fc Inhibit Diabetes Development.
To determine the effect of transgene expression on diabetes development, LTβR–Fc-expressing mice were followed for over 1 yr for glycosuria. As shown in Fig. 4 A, 80% of control transgene-negative female NOD mice developed diabetes by 25 wk of age. In striking contrast, only 1 of 26 female mice from the high expressing 1610 line developed diabetes by this age. The low expressing 201 mouse line was also followed for disease onset. IDDM pathogenesis was inversely related to circulating LTβR–Fc concentrations, as these mice developed somewhat reduced IDDM compared with littermate controls (which was not significantly different [P = 0.09; Fig. 4 B]).
As shown in Fig. 4 C and described above, LTβR–Fc blocking protein levels declined with age. As LTβR–Fc concentrations fell, the incidence of IDDM began to rise, so that by 59 wk of age, 47% of the transgene-positive mice became diabetic (Fig. 4 C). The kinetics of diabetes onset after the decline in chimeric protein activity closely mirrored that of the control group.
These data demonstrate that LTβR engagement is a crucial component in diabetic pathogenesis. This was demonstrated in two ways: first, high expression of fusion protein inhibited disease onset; second, decline in blocking protein activity in aged mice was followed by diabetes development. We conclude from these experiments that tissue destruction characteristic of diabetes requires LTβR signaling.
Mice Expressing LTβR–Fc Lack Anti-GAD Autoreactive T Cells.
Although mice expressing high concentrations of LTβR–Fc did not begin to develop diabetes until ∼24 wk after the control mice, we observed no difference in pancreatic infiltrates between the two groups at early (as well as late) time points. These data suggest that autoreactive T cells were present in inflamed tissues but unable to cause tissue destruction due to loss of LTβR signaling.
To test whether autoreactive T cells against islet cell antigens were present, splenocytes were tested for reactivity to the pancreatic autoantigen, GAD. As shown in Fig. 4, splenocytes isolated from high expressing 1610 mice displayed reduced reactivity toward GAD autoantigen compared with their transgene-negative littermates. Both stimulation indices as well as IFN-γ (Fig. 4D and Fig. E) and IL-10 (data not shown) production in response to GAD were equivalent to those of C57BL/6 control mice. IL-5 was not detected in any of the cultures (data not shown), suggesting that the lack of IDDM was not simply due to a shift from a Th1- to a Th2-type response.
These data suggest that the absence of glycosuria in LTβR–Fc-expressing, 30-wk-old NOD mice was due either to a lack of activation of or an absence of autoreactive T cells specific for the islet cell antigen GAD. However, these findings do not rule out that T cell reactivity to pancreatic autoantigens other than GAD was present in these mice.
Summary.
This study demonstrates that a neutralizing LTβR–Fc fusion protein has the ability to block autoreactive T cell induction of diabetes in NOD mice. The diabetic T cell potential remains latent in the presence of high concentrations of the fusion protein. Remarkably, the diabetic process spontaneously activates after the decline of LTβR–Fc fusion protein levels below a critical threshold. High levels of fusion protein expression are also associated with disrupted splenic architecture. However, splenic primary follicles and FDCs formed in aged mice, demonstrating that the maintenance of splenic structure is a dynamic process. Possible mechanisms by which LTβR–Fc affects diabetes development include altered T cell localization, migration, and/or defective cellular interactions. The results reported here demonstrate that LTβR signaling is required for progression to autoimmune diabetes.
Acknowledgments
We thank Dr. Sara A. Michie for help with immunohistology and Dr. Marie Kosco-Vilbois for the FDC-M2 antibody. We also thank Leslie Nicklin and Drs. Paul Kincade, Ursula Gunthert, and Marco Colonna for critical reading of the manuscript, Beatrice Pfeiffer and Allison Dwileski for assistance with illustrations, and Christian Mueller for statistical analysis.
This work was supported in part by the Walter V. and Idun Y. Berry Foundation, Juvenile Diabetes Foundation International, and by National Institutes of Health grants AI-36535 (National Institute of Allergy and Infectious Diseases) and DK-51705 (National Institute of Diabetes and Digestive and Kidney Diseases). The Basel Institute for Immunology was founded and supported by Hoffmann-La Roche, Inc., Basel, Switzerland.
Footnotes
R. Ettinger and S. Munson contributed equally to this study.
References
- Ettinger R. The role of tumor necrosis factor and lymphotoxin in lymphoid organ development. Curr. Top. Microbiol. Immunol. 2000;251:203–210. doi: 10.1007/978-3-642-57276-0_26. [DOI] [PubMed] [Google Scholar]
- Ware C.F., VanArsdale T.L., Crowe P.D., Browning J.L. The ligands and receptors of the lymphotoxin system. Curr. Top. Microbiol. Immunol. 1995;198:175–218. doi: 10.1007/978-3-642-79414-8_11. [DOI] [PubMed] [Google Scholar]
- Kwon B.S., Tan K.B., Ni J., Oh K.O., Lee Z.H., Kim K.K., Kim Y.J., Wang S., Gentz R., Yu G.L. A newly identified member of the tumor necrosis factor receptor superfamily with a wide tissue distribution and involvement in lymphocyte activation. J. Biol. Chem. 1997;272:14272–14276. doi: 10.1074/jbc.272.22.14272. [DOI] [PubMed] [Google Scholar]
- Montgomery R.I., Warner M.S., Lum B.J., Spear P.G. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87:427–436. doi: 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- Mauri D.N., Ebner R., Montgomery R.I., Kochel K.D., Cheung T.C., Yu G.L., Ruben S., Murphy M., Eisenberg R.J., Cohen G.H. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity. 1998;8:21–30. doi: 10.1016/s1074-7613(00)80455-0. [DOI] [PubMed] [Google Scholar]
- Rennert P.D., Browning J.L., Mebius R., Mackay F., Hochman P.S. Surface lymphotoxin alpha/beta complex is required for the development of peripheral lymphoid organs. J. Exp. Med. 1996;184:1999–2006. doi: 10.1084/jem.184.5.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futterer A., Mink K., Luz A., Kosco-Vilbois M.H., Pfeffer K. The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. 1998;9:59–70. doi: 10.1016/s1074-7613(00)80588-9. [DOI] [PubMed] [Google Scholar]
- Koni P.A., Sacca R., Lawton P., Browning J.L., Ruddle N.H., Flavell R.A. Distinct roles in lymphoid organogenesis for lymphotoxins alpha and beta revealed in lymphotoxin beta-deficient mice. Immunity. 1997;6:491–500. doi: 10.1016/s1074-7613(00)80292-7. [DOI] [PubMed] [Google Scholar]
- Ettinger R., Mebius R., Browning J.L., Michie S.A., van Tuijl S., Kraal G., van Ewijk W., McDevitt H.O. Effects of tumor necrosis factor and lymphotoxin on peripheral lymphoid tissue development. Int. Immunol. 1998;10:727–741. doi: 10.1093/intimm/10.6.727. [DOI] [PubMed] [Google Scholar]
- Ettinger R., Browning J.L., Michie S.A., van Ewijk W., McDevitt H.O. Disrupted splenic architecture, but normal lymph node development in mice expressing a soluble lymphotoxin-beta receptor-IgG1 fusion protein. Proc. Natl. Acad. Sci. USA. 1996;93:13102–13107. doi: 10.1073/pnas.93.23.13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisch R., Yang X.D., Singer S.M., Liblau R.S., Fugger L., McDevitt H.O. Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic mice. Nature. 1993;366:72–75. doi: 10.1038/366072a0. [DOI] [PubMed] [Google Scholar]
- Clark M.R. IgG effector mechanisms. Chem. Immunol. 1997;65:88–110. [PubMed] [Google Scholar]
- Hoglund P., Mintern J., Waltzinger C., Heath W., Benoist C., Mathis D. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J. Exp. Med. 1999;189:331–339. doi: 10.1084/jem.189.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.D., Sytwu H.K., McDevitt H.O., Michie S.A. Involvement of beta 7 integrin and mucosal addressin cell adhesion molecule-1 (MAdCAM-1) in the development of diabetes in obese diabetic mice. Diabetes. 1997;46:1542–1547. doi: 10.2337/diacare.46.10.1542. [DOI] [PubMed] [Google Scholar]
- Hanninen A., Taylor C., Streeter P.R., Stark L.S., Sarte J.M., Shizuru J.A., Simell O., Michie S.A. Vascular addressins are induced on islet vessels during insulitis in nonobese diabetic mice and are involved in lymphoid cell binding to islet endothelium. J. Clin. Invest. 1993;92:2509–2515. doi: 10.1172/JCI116859. [DOI] [PMC free article] [PubMed] [Google Scholar]