

Abstract
Although it is clear that the function of CD40 on peripheral hematopoietic cells is pivotal to the development of autoimmunity, the function of CD40 in autoimmune disease outside this compartment is unresolved. In a model of experimental autoimmune encephalomyelitis (EAE), evidence is presented that CD40–CD154 interactions within the central nervous system (CNS) are critical determinants of disease development and progression. Using bone marrow (BM) chimeric mice, the data suggest that the lack of expression of CD40 by CNS-resident cells diminishes the intensity and duration of myelin oligodendrocyte glycoprotein (MOG)-induced EAE and also reduces the degree of inflammatory cell infiltrates into the CNS. Although CNS inflammation is compromised in the CD40+/+→CD40−/− BM chimeric mice, the restricted CD40 expression had no impact on peripheral T cell priming or recall responses. Analysis of RNA expression levels within the CNS demonstrated that encephalitogenic T cells, which entered a CNS environment in which CD40 was absent from parenchymal microglia, could not elicit the expression of chemokines within the CNS. These data provide evidence that CD40 functions outside of the systemic immune compartment to amplify organ-specific autoimmunity.
Keywords: autoimmunity, microglia, chemokines, multiple sclerosis, CD40–CD154
Introduction
It is now well established that the development of inflammatory diseases is dependent on interactions of CD40 with its ligand, CD154 (for reviews, see references 1 and 2). Numerous models of T cell–mediated autoimmunity in mice have been shown to be completely prevented by prophylactic blockade or by genetic deletion of this ligand–receptor pair 3 4 5 6 7 8. It is now known that CD40 blockade impairs the priming of T cells and prevents the differentiation of inflammatory T cells. This effect is due to the impairment of dendritic cell maturation and the lack of IL-12 production in the absence of CD154 function 8. The capacity of anti-CD154 mAbs to completely block the development of a central nervous system (CNS)-targeted autoimmune disease, experimental autoimmune encephalomyelitis (EAE), is likely due to the reduced frequency of inflammatory, encephalitogenic T cells induced in the absence of CD154 function 5 7 9. Although these observations explain why CD154–CD40 interactions are critical for the development of EAE, they do not answer why CD154 function is critical even after the priming, expansion, and differentiation of autoaggressive T cells. That is, in several T cell–mediated autoimmune models 10, including EAE, anti-CD154 treatment of mice with established disease ameliorates disease or prevents relapse formation 7. Although the mechanism(s) of this protection is poorly understood, CD40-bearing cells at the site of inflammation may play a critical role in the progression of autoimmunity.
There are several CD40-bearing cell types within the CNS that have demonstrated the capacity to be immunoregulatory and therefore may play an important role in the pathogenesis of inflammatory processes within the brain 11. For example, CD40-bearing macrophages located in the perivascular and meningial space may play a critical role in triggering autoreactive T cells to extravasate the blood–brain barrier, penetrate the parenchyma, and facilitate inflammation 12. In addition to perivascular cells, CNS-resident parenchymal cells have been shown to present antigen in vitro and express an array of molecules allowing them to potentially interact with T cells 13 14 15. CD40 expressed by parenchymal microglial cells has been implicated in playing a proinflammatory function, because its engagement triggers the release of IL-12 and thus could facilitate the generation of inflammatory T cells within the CNS 16 17 18. Although much circumstantial evidence exists as to the immunoregulatory function of CNS-resident cells in EAE, little or no in vivo data supporting their contribution to inflammation has been reported. This report provides in vivo data showing that CD40 expression by cells outside the systemic immune compartment functionally contributes to the progression of EAE.
The use of bone marrow (BM) chimeric mice has allowed studies to selectively manipulate the genotype of the peripheral hematopoietic immune system from that of the CNS resident cells, as CNS parenchymal microglia are not repopulated after BM reconstitution 19 20. In contrast to the CNS parenchyma, the genotype of the systemic immune compartment and that of perivascular cells can be exchanged with that of donor-derived hematopoietic cells 20 21. Using these approaches, several significant studies have provided valuable insights into the function of CNS-resident cells in the pathogenesis of EAE 12 19 22.
In this study, we provide in vivo evidence showing that CD40 expressed by CNS-endogenous cells controls the migration and retention of myelin oligodendrocyte glycoprotein (MOG)-reactive T cells in the CNS of mice during EAE. We show that in the absence of CD40 expression within the CNS, encephalitogenic T cells cannot manifest inflammatory responses even though they themselves appear to be fully differentiated inflammatory T cells.
Materials and Methods
Mice.
Female C57BL/6 and CD45 congenic C57BL/6-Ly5.2 mice were obtained from National Cancer Institute (Frederick, MD). CD40−/− C57BL/6 mice were originally purchased from The Jackson Laboratory and bred in house under pathogen-free conditions.
Irradiation BM Chimeric Mice.
BM donor mice were killed using CO2 and BM cells were isolated by flushing femur and tibia bones with balanced salt solution (BSS). BM was filtered through a 100-μm cell strainer and cells were washed with BSS. CD45 congenic recipient mice were lethally irradiated with 1,200 rads (split dose) and intravenously injected with 20 × 106 BM cells. Engraftment took place over 6–8 wk of recovery. Mice were bled retroorbitally to ensure >95% engraftment of blood leukocytes.
Peptides and Abs.
MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK) was obtained from Research Genetics. For cyto- fluorometric analysis, the following Abs were used: anti-CD45, CD45.1, CD45.2, CD11b (Mac1), CD4, CD62L, CD44, CD45RB, B7.1, B7.2, and MHC II. All Abs were obtained from BD PharMingen. For in vivo administration, anti-CD154 (MR-1; reference 23) was used at 350 μg/ml. As a control, hamster (H)-Ig was used at the same concentration. Mice were injected three times per week intraperitoneally.
Induction of EAE.
For active immunization, 5–8-wk-old female C57BL/6 or CD40−/− mice (13–16-wk-old BM chimeric mice) were immunized subcutaneously with 200 μg of MOG35–55 peptide emulsified in CFA supplemented with 500 μg of Mycobacterium tuberculosis (DIFCO). The mice received intraperitoneal injections with 250 ng pertussis toxin (Sigma-Aldrich) at the time of immunization and 48 h later. After 7 d, the mice received an identical boost immunization with MOG/CFA without pertussis toxin. Clinical disease usually commences between day 13 and 18 after immunization.
For adoptive transfer, donor mice were immunized subcutaneously with 200 μg MOG35–55 in CFA supplemented with 500 μg Mycobacterium tuberculosis. Immediately after and 2 d later the mice received 250 ng of pertussis toxin. 11 d after immunization, the mice were killed and spleens were removed, homogenized, and RBCs were lysed. The cells were cultured for 4 d in RPMI 1640 supplemented with 10% FCS (both obtained from BioWhitaker), 10 μg/ml of MOG peptide, and 2.5 ng/ml of recombinant IL-12 (PeproTech). The cells were harvested, and dead cells were removed by Ficoll (Sigma-Aldrich) centrifugation. Cells are then washed and injected into recipient mice (5–25 × 106 cells/mouse). Animals received 250 ng/mouse pertussis toxin on day 0 and 2 after transfer. Clinical disease usually begins 6–10 d after cell transfer.
Clinical Evaluation.
The mice were scored four times per week as follows: 0, no detectable signs of EAE; 0.5, distal limp tail; 1, complete limp tail; 1.5, limp tail and hind limb weakness; 2, unilateral partial hind limb paralysis; 2.5, bilateral partial hind limb paralysis; 3, complete bilateral hind limb paralysis; 3.5, complete hind limb paralysis and unilateral forelimb paralysis; 4, total paralysis of fore and hindlimbs (score >4 to be killed); 5, death.
Histology.
Mice were killed with CO2. Spinal column was removed and fixed in 10% buffered formalin. The spinal cord was dissected and paraffin embedded before staining with either hematoxylin and eosin to assess infiltration or luxol fast blue to assess the degree of demyelination.
Flow Cytometry.
Mice were killed with CO2 and spinal cords were removed by flushing the spinal column with sterile BSS. The brain was dissected to isolate the brain stem. Both tissues were homogenized and strained through a 100-μm nylon filter (Fisher Scientific). After centrifugation, the cell suspension was resuspended in 37% isotonic Percoll and underlayed with 70% isotonic Percoll. The gradient was centrifuged at 600 g for 25 min at room temperature. The interphase cells were collected and extensively washed before staining. For flow cytometry, the cells were stained with primary Abs for 30 min at 4°C, washed, and incubated with streptavidin-allophycocyanin (APC) or peridinine chlorophyll protein (PerCP; BD PharMingen) for 15 min. The cells were washed and analyzed using a FACSCalibur™ using CELLQuest™ software (Becton Dickinson). Postacquisition analysis was performed using WinMDI 2.8 software (Scripps-Research Institute).
Recall Responses.
Mice were primed by flank injections of MOG/CFA. After 5 d, the axillary and inguinal LNs were removed and homogenized. 5 × 105 LN cells were placed as triplicates in a 96-well plate and pulsed with different amounts of MOG peptide or irrelevant peptide (myelin proteolipid protein [PLP139–151]; Research Genetics) as a control. After 48 h, cells were pulsed with [3H]thymidine (NEN Life Science Products/Dupont) and incubated for an additional 15 h before cells were harvested and thymidine incorporation was assessed using a Filtermate harvester (Packard Instrument Co.) and TopCount-NXT Microplate scintillation and luminescence counter (Packard Instrument Co.). For cytokine analysis, sister cultures were harvested 48 h after culture supernatants were analyzed by ELISA for IFN-γ and IL-4 (BD PharMingen).
RNase Protection Assays.
Mice were killed with CO2 and spinal cords were removed by flushing with sterile BSS. Total RNA was isolated using TRIzol reagent (Life Technologies) following the manufacturer's recommendations. Chemokine mRNA levels were determined using the Riboquant Multiprobe RNase Protection Assay system (BD PharMingen). 32P-labeled riboprobes were synthesized from a customized plasmid template set using T7 polymerase. The DNA template was digested with DNase and total RNA (15 μg) was hybridized with the riboprobes overnight at 56°C. ssRNA species were removed by digestion with RNase A. The protected RNA species were phenol/chloroform extracted, ethanol precipitated, and electrophoresed on a 5% polyacrylamide gel. Protected chemokine probes were visualized by autoradiography of the dried gel.
Statistical Analysis.
Statistical differences of clinical diseases scores were evaluated using a nonpaired Student's t test. ELISA and proliferation assays are expressed as average of triplicate wells ± SD. Significance was determined by single-factor analysis of variance (ANOVA) and Bonferroni posttest. Differences were considered significant when P value < 0.05.
Results and Discussion
CD40–CD154 Function Is Essential for the Development of EAE in Response to MOG Peptide.
Data presented in Fig. 1 establish that CD40–CD154 interactions are essential for the development of MOG-induced EAE in C57BL/6 mice. These data are consistent with EAE models in both SJL and B10.PL mouse strains to other CNS peptides 7. Treatment of mice with anti-CD154 mAb at the time of immunization completely prevents clinical EAE (Fig. 1 A). Consistent with the function of this ligand receptor pair in the development of EAE are data establishing that CD40−/− mice are resistant to the induction of EAE via active immunization (Fig. 1 B). In this experiment, one of the nine CD40−/− mice developed a limp tail. Treatment of mice with established disease (day 13) causes a significant alleviation of clinical symptoms over time, compared with control mice (Fig. 1 C), indicating that even after the encephalitogenic T cells infiltrate the CNS, anti-CD154 can interfere with the effector phase of the disease.
Figure 1.

CD40 is critical for the induction of active MOG-EAE in C57BL/6 mice and T cell priming. (A) Early administration of anti-CD154 blocks the development of MOG-induced EAE. Mice were immunized with MOG35–55 in CFA and given 350 μg/mouse of anti-CD154 mAbs three times per week starting at the time of immunization (▵). Control mice were treated with H-Ig mAbs (♦; n = 6/group). (B) CD40−/− mice are not susceptible to MOG-induced EAE. CD40−/− (□) and CD40+/+ (♦) mice were immunized as described in panel A, and disease was monitored (n = 9/group). (C) Treatment of mice with anti-CD154 at disease onset ameliorated MOG-induced EAE. Mice were treated at the first sign of clinical disease with either anti-CD154 (▵) or control H-Ig (♦) (n = 9/group). (D) T cell priming to MOG is reduced in CD40−/− mice. To assess the ability of CD40−/− mice to prime T cells against MOG35–55, CD40+/+ (+/+) or CD40−/− (−/−) mice were immunized with MOG35–55 in CFA. LN cells were harvested after 5 d and stimulated with either no antigen, 100 μg of MOG35–55, or irrelevant PLP peptide. (E) MOG-induced IFN-γ secretion is reduced in CD40−/− mice. IFN-γ production from LN cells primed to MOG35–55 in CD40+/+ and CD40−/− was assessed in vitro as described in D. Displayed is a representative of at least three experiments.
To determine whether encephalitogenic T cells were generated, CD40−/− as well as CD40+/+ mice were immunized with MOG35–55 in CFA, and 5 d later LN cells were isolated. LN cells were cultured in the presence or absence of irradiated syngeneic splenocytes with either MOG peptide or irrelevant PLP peptide. Fig. 1D and Fig. E, shows by recall assay that priming of naive T cells to MOG peptide is drastically reduced in CD40−/− animals. Even though low level antigen-induced proliferative activity was measured in T cells from CD40−/− mice (Fig. 1 D), IFN-γ secretion by these T cells is completely blocked (Fig. 1 E). Although IFN-γ production was blocked, no IL-4 was detected upon recall, indicating that there is no overt emergence of Th2-like cells in the absence of CD40 (not shown).
Fig. 2 shows that neither CD40−/− (Fig. 2 C) nor anti-CD154–treated mice (Fig. 2 D) show any sign of inflammation of the CNS compared with mice treated with H-Ig as established by histology (Fig. 2 B). On day 7 after disease onset, cells derived from spinal cord cell suspension were separated over a discontinuous Percoll gradient and stained with anti-CD45 and anti-CD11b mAbs. Cytofluorometric analysis of the CNS-infiltrating cells shows that infiltrating lymphocytes (CD11b−/CD45hi) and blood-derived phagocytes (CD11b+/CD45hi) are absent in immunized CD40−/− mice (Fig. 2 E) compared with CD40+/+ mice (Fig. 2 F). No significant changes were observed in the number of resident microglial cells (CD11b+/CD45lo). In summary, in the absence of CD40, naive MOG-specific T cells are not sufficiently stimulated to become encephalitogenic and fail to penetrate the blood–brain barrier and cause disease.
Figure 2.

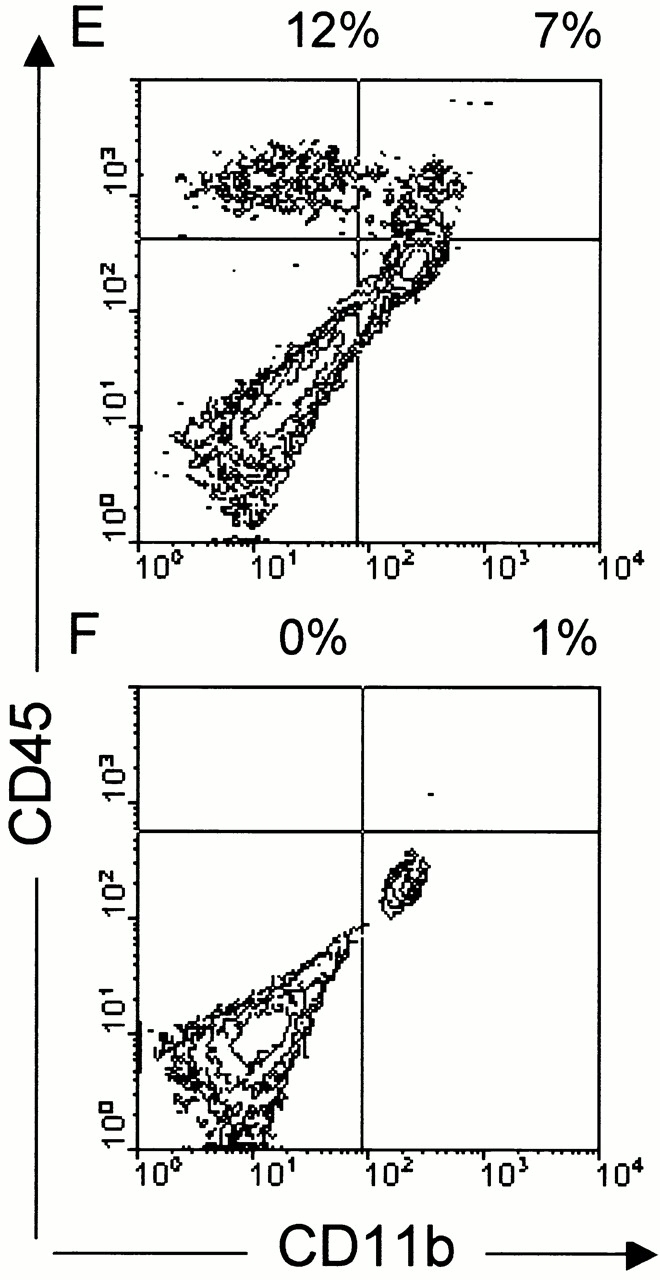
CD40–CD154 blockade inhibits extravasation of leukocytes into the CNS. (A–D) Genetic deficiency of CD40 or anti-CD154 treatment reduces leukocyte infiltration into the CNS as assessed by histochemistry. EAE was induced in either CD40+/+ or CD40−/− mice using MOG35–55 emulsified in CFA as described. CD40+/+ mice were either treated with anti-CD154 or irrelevant H-Ig at the time of immunization (three times per week). To quantify inflammatory infiltration into the CNS, 5–10 mice per group were killed 7 d after disease onset. Spinal cords and cerebella were collected and either fixed in 10% formaldehyde/PBS or homogenized. Spinal cords from: (A) CD40+/+ mice, nonimmune; (B) CD40+/+ mice, immune and treated with H-Ig; (C) CD40−/− mice, immune; and (D) CD40+/+ mice, immune and treated with anti-CD154 at the time of immunization, were sectioned and stained with hematoxylin and eosin. Displayed is a representative of at least three individual experiments. (E and F) Infiltration of leukocyte subsets into the CNS is reduced in CD40−/− mice. Cells derived from spinal cord cell suspension were separated over a discontinuous Percoll gradient and stained with anti-CD45 and anti-CD11b mAbs. Cells were analyzed by flow cytometry; (E) CD40+/+ mice; (F) CD40−/− mice.
Analysis of the Induction of Actively Induced EAE in Mice That Do Not Express CD40 within the CNS but Express CD40 in the Peripheral Hematopoietic Compartment.
In order determine if CD40 expression within the CNS contributes to the development and progression of EAE, BM chimeric mice were produced in which CD40 expression was limited to the radioresistant or radiosensitive compartment of the host. Fig. 3A–D, shows the staining of the CNS of BM chimeric mice 2 mo after reconstitution with CD45 (Ly5)-congenic BM. Although resident microglial cells are hematopoietically derived, they do not turnover after irradiation and BM reconstitution 20. Hence, the CD45low/Mac1+ CNS-resident microglia express the host congenic CD45.2 marker (Fig. 3 A). After induction of EAE, CNS-infiltrating CD45high cells predominantly expressed the donor CD45.1 marker, indicating that the host immune system is replaced by donor BM cells (Fig. 3 D). Only a minor population of CNS infiltrating lymphocytes are radioresistant and of host origin (Fig. 3 C). The extent of peripheral hematopoietic chimerism was confirmed through the analysis of the PBMCs, spleen, and LN. Although small numbers of host-derived cells are found in the blood (2%) and LNs/spleen (5%), the host-derived cells consisted entirely of CD3+ T cells. Neither myeloid cells nor B lymphocytes (CD40-expressing cells) were found to be of host origin (<0.1%) as assessed by B220 (B cells), CD11b (monocytes/macrophages), and GR-1 staining (granulocytes; data not shown).
Figure 3.

Analysis of the induction of actively induced EAE in irradiated BM chimeric mice. (A–D) In BM chimeric mice, the parenchymal microglial cells are host derived and the infiltrating leukocytes are predominantly donor derived. Mice were irradiated and reconstituted with CD45.1-congenic BM cells. Engraftment took place over 2 mo. CNS cells of nonimmune mice were stained with either the (A) host (CD45.2) or (B) donor-congenic CD45.1 marker. C and D show the degree of chimerism of CNS-infiltrating leukocytes in BM chimeric mice with EAE induced by immunization with MOG35–55 in CFA. (E and F) The lack of CD40 expression on radioresistant cells in the host does not influence peripheral T cell responses. After 2 mo of recovery, mice were immunized with MOG/CFA and LNs were removed 5 d later. Recall proliferation (E) and IFN-γ secretion (F) in response to either 100 μg MOG peptide or irrelevant PLP peptide was assessed in vitro. (G) The lack of CD40 in the CNS reduces EAE. CD40+/+→CD40+/+, CD40+/+→CD40−/−, CD40−/−→CD40+/+, and CD40−/−→CD40−/− mice were produced, allowed to recover for 2 mo, then immunized with MOG35–55 in CFA as described above and scored for clinical disease (n = 5/group; representative of four individual experiments). The clinical score of CD40+/+→CD40+/+ vs. CD40+/+→CD40−/− is statistically significantly different (P < 0.05). EAE was scored over 24 d. (H and I) The lack of CD40 expression in the CNS reduces the extent of leukocyte infiltration. 7 d after disease onset, leukocyte infiltration into the CNS of (H) CD40+/+→CD40+/+ and (I) CD40+/+→CD40−/− mice was analyzed as described.
Systemic and CNS-restricted immune responses were evaluated in CD40 BM chimeric mice. Data presented in Fig. 3E and Fig. F, show that CD40+/+→CD40−/− mice and CD40+/+→CD40+/+ mice had comparable systemic (peripheral) immune responses. Chimeric mice were immunized with MOG35–55 in CFA, and LN cells were isolated 5 d after immunization for the assessment of MOG recall responses in vitro. CD40−/−→CD40+/+ and CD40−/−→ CD40−/− mice do not generate strong T cell responses to MOG as assessed by proliferation (Fig. 3 E) and IFN-γ production (Fig. 3 F). In contrast, both CD40+/+→ CD40−/− and CD40+/+→CD40+/+ mice demonstrated equivalent capacities to prime T cells and for those T cells to proliferate and secrete IFN-γ when rechallenged with MOG in vitro. Thus, CD40 expression by the peripheral hematopoietic cells is essential for priming of encephalitogenic T cells. However, the lack of CD40 expression on nonhematopoietic cells in the CD40+/+→CD40−/− chimeras (endothelial cells, epithelial cells, etc.) appears to have little or no impact on the priming and early differentiation of antigen-reactive T cells in the periphery. Hence, the per-ipheral T cell response depends solely on the genotype of the BM donor and the expression of CD40 on nonhematopoietic, host-derived cells is not necessary for the peripheral T cell response against MOG35–55.
In parallel to the measurement of systemic immune responses in chimeric mice, CNS-restricted immune responses were also measured (Fig. 3 G). Mice in which CD40 is absent from the systemic immune compartment (either CD40−/−→CD40−/− or CD40−/−→CD40+/+) are resistant to develop EAE upon active immunization. Thus, CD40 expression by the peripheral immune system is essential for the development of EAE; CD40 expression solely by nonhematopoietic cells or radioresistant hematopoietic cells (mainly parenchymal microglia) is inadequate to support the development of EAE.
To address the function of CD40 within the CNS, active induction of EAE in CD40+/+→CD40+/+ and CD40+/+ →CD40−/− chimeras was compared (Fig. 3 G). The lack of CD40 expression within the CNS parenchyma (CD40+/+ →CD40−/−) causes a significant delay in disease onset and decreased severity compared with chimeric mice that express CD40 on resident CNS cells (CD40+/+→CD40+/+). Table summarized the clinical development in chimeric mice obtained from three individual experiments.
Table 1.
EAE in CD40 BM Chimeras
| Mice | Incidence | Mean day of disease onset | Mean maximal clinical score |
|---|---|---|---|
| CD40+/+→CD40+/+ | 16/17 | 15.8 | 3.2 |
| CD40+/+→CD40−/− | 14/17 | 18.5 | 2.5 |
| CD40−/−→CD40+/+ | 3/17 | 18.3 | 2.0 |
| CD40−/−→CD40−/− | 1/16 | 18.0 | 2.8 |
This table summarizes the results of three independent experiments where EAE was induced by active immunization in BM chimeric animals. The overall clinical score of CD40+/+→CD40+/+ vs. CD40+/+→CD40−/− mice is significantly higher (P < 0.001), whereas CD40−/−→CD40+/+ and CD40−/−→CD40−/− do not differ significantly (P > 0.05) as assessed using Student's t test.
The data do not specifically address the role of perivascular CD40 expression, as mice in which CD40 is absent from the systemic/perivascular compartment do not generate encephalitogenic cells as described above. However, the absence of parenchymal CD40 expression, when CD40 is restricted to the hematopoietic, systemic, and perivascular/meningeal compartment, results in significantly reduced infiltration of leukocytes (Fig. 3H and Fig. I) as well as diminished and delayed clinical disease.
The Role of Systemic and CNS-restricted CD40 Expression in the Effector Phase of Disease.
To more extensively evaluate the function of CD40 in the effector phase of EAE, CD40−/− mice and CD40 BM chimeric mice were used in an adoptive transfer model of disease. In this case, a population of activated encephalitogenic effector T cells is adoptively transferred into mice that do or do not express CD40 in specific compartments. Furthermore, adoptive transfer bypasses the need for T cell priming in secondary lymphoid tissues and in contrast to active immunization, the largest depot of available Ag is the CNS itself. Upon passive transfer of MOG-reactive lymphocytes, CD40−/− mice are significantly less susceptible to EAE induction than CD40+/+ controls (Fig. 4 A). Like CD40−/− mice, CD40+/+ →CD40−/− chimeric mice are also less susceptible passively transferred EAE and display a diminished disease severity and disease incidence (Fig. 4 D). Significantly fewer inflammatory infiltrates can be recovered from the CNS of CD40−/− (Fig. 4 C) mice or CD40+/+→CD40−/− (Fig. 4 F) recipients compared with CD40+/+ (Fig. 4 B) mice or CD40+/+→CD40+/+ chimeric mice (Fig. 4 E), respectively. The data indicate that CD40 engagement within the CNS is critical for the infiltration/retention of encephalitogenic cells into the CNS.
Figure 4.

Encephalitogenic T cells are less effective at inducing EAE when transferred into a CD40-deficient host. (A) Adoptive transfer of MOG-reactive T cells into a CD40−/− host compared with transfer into CD40+/+ host results in less disease. MOG-reactive cells (15 × 106) were adoptively transferred into either CD40+/+ (▵) or CD40−/− (□) recipients and the mice were scored for clinical disease (n = 6/group). (B and C) Adoptive transfer of MOG-reactive T cells can less effectively infiltrate the CNS in a CD40−/− host. 7 d after disease onset, mice were killed and CNS infiltrates into (B) CD40+/+ and (C) CD40−/− mice were analyzed as described. (D) Adoptive transfer of MOG-reactive T cells into CD40+/+→CD40−/− BM chimeras results in less disease compared with CD40+/+→CD40+/+ mice. MOG-reactive cells (20 × 106) were adoptively transferred into either CD40+/+→CD40+/+ (⋄) or CD40+/+→CD40−/− (□) recipients and the mice were scored for clinical disease (n = 6/group). (E and F) Adoptive transfer of MOG-reactive T cells can less effectively infiltrate when CD40 is absent from the CNS. 7 d after disease onset, mice were killed and CNS infiltrates into (E) CD40+/+→CD40+/+ and (F) CD40+/+→CD40−/− were analyzed as described.
Although the CD40 genotype of the host influenced the magnitude of the T cell infiltration, no phenotypic differences of the T cells were noted. In general, a two- to fourfold difference in the total number of infiltrating cells were noted between hosts that expressed CD40 in the CNS versus those that did not express CD40. No differences in the phenotypic profile of CNS infiltrates between CD40+/+ and CD40−/− recipients with regards to expression of CD44, CD45RB, CD62L, and CD25 on CD4+ T cells were observed (not shown). Furthermore, upon rechallenge with MOG peptide in vitro, CNS infiltrating cells collected from the CNS of BM chimeric mice revealed that they are able to secrete IFN-γ regardless of whether they infiltrate a CD40+/+ or CD40−/− CNS (not shown).
Reduced T cell infiltration/recruitment and disease suggested that the absence of CD40 in the CNS impeded the production of critical inflammatory mediators within the CNS needed to recruit encephalitogenic T cells and cause disease. To address the validity of this hypothesis, mRNA expression levels of several relevant chemokines in the CNS of BM chimeric mice immunized with MOG35–55 peptide was quantitated. Fig. 5 A shows an RNase protection assay revealing that the expression of chemokines is drastically reduced in BM chimeras that do not express CD40 within the CNS upon active immunization. The mice were killed 5 d after disease onset. Consistent with delayed disease onset, only one of the CD40+/+→CD40−/− mice displayed clinical symptoms, in contrast to four mice in the CD40+/+→CD40+/+ group (n = 6 each group). At this early time point, striking differences with regards to chemokine expression were observed. The data suggest that the absence of CD40 on CNS-resident APCs results in the reduction of chemokine expression leading to a decreased level of leukocyte infiltration. To confirm that CD40 is critical during the effector phase of EAE, we adoptively transferred MOG-reactive T cells into either CD40+/+ or CD40−/− recipients (Fig. 5 B). RNase protection assays of spinal cords of these mice after disease onset confirms the lack of chemokine mRNA induction when CD40 was absent from the CNS. Furthermore, the data suggest that the exaggerated reduction in chemokine expression is not simply due to the reduced number of T cells, which are observed in the CD40−/− mice.
Figure 5.

Chemokine expression within the CNS of CD40+/+, CD40−/−, and BM chimeric mice. (A) Chemokine expression is reduced in BM chimeras where CD40 is absent in the CNS. EAE was induced in BM chimeric mice by active immunization (+→+ = CD40+/+ → CD40+/+; +→− = CD40+/+ → CD40−/−; −→+ = CD40−/− →CD40+/+; −→− = CD40−/−→CD40−/−). 5 d after disease onset the animals were killed and total RNA was isolated from spinal cords and analyzed for chemokine expression by RNase protection assay. Either 10 or 20 mg of RNA was loaded onto the gel. (B) Chemokine expression is reduced in CD40−/− mice upon passive transfer of MOG-reactive cells. EAE was induced in either CD40+/+ or CD40−/− mice by adoptive transfer of either 12 × 106 or 5 × 106 MOG-reactive cells and RNase protection assay was performed as described. Mip, macrophage inflammatory protein; IP-10, IFN-γ–inducible protein 10; MCP-1, monocyte chemoattractant protein 1; TCA-3, T cell activation gene 3; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Taken together, CD40 expression by CNS-resident cells (most likely parenchymal microglia) is important for the infiltration/retention of inflammatory leukocytes into the CNS. Confirmation that CD40 function within the CNS significantly contributes to the pathogenesis of EAE provides a reasonable explanation for the therapeutic activity of anti-CD154 therapy. In the absence of CD40 function, reduced T cell infiltration, reduced production of inflammatory cytokines and chemokines within the CNS, and blockade of peripheral CD40 function may all contribute to the therapeutic activities of anti-CD154. Studies in murine models of EAE have guided the application of humanized anti–human CD154 for the treatment of multiple sclerosis. Currently, anti-CD154 is in phase II clinical trials in patients with multiple sclerosis, and studies in mice continue to refine and redefine the function of this ligand–receptor pair in CNS inflammatory diseases.
Acknowledgments
This work was supported by National Institutes of Health grant AI37075 and the Schram foundation. B. Becher holds a fellowship from the National MS-Society.
Footnotes
Abbreviations used in this paper: BM, bone marrow; BSS, balanced salt solution; CNS, central nervous system; EAE, experimental autoimmune encephalomyelitis; H, hamster; MOG, myelin oligodendrocyte glycoprotein; PLP, myelin proteolipid protein.
References
- Grewal I.S., Flavell R.A. CD40 and CD154 in cell-mediated immunity. Annu. Rev. Immunol. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- Mackey M.F., Barth R.J.J., Noelle R.J. The role of CD40/CD154 interactions in the priming, differentiation, and effector function of helper and cytotoxic T cells. J. Leukoc. Biol. 1998;63:418–428. doi: 10.1002/jlb.63.4.418. [DOI] [PubMed] [Google Scholar]
- Durie F.H., Fava R.A., Foy T.M., Aruffo A., Ledbetter J.A., Noelle R.J. Prevention of collagen-induced arthritis with an antibody to gp39, the ligand for CD40. Science. 1993;261:1328–1330. doi: 10.1126/science.7689748. [DOI] [PubMed] [Google Scholar]
- Sadlack B., Lohler J., Schorle H., Klebb G., Haber H., Sickel E., Noelle R.J., Horak I. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur. J. Immunol. 1995;25:3053–3059. doi: 10.1002/eji.1830251111. [DOI] [PubMed] [Google Scholar]
- Gerritse K., Laman J.D., Noelle R.J., Aruffo A., Ledbetter J.A., Boersma W.J., Claassen E. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. USA. 1996;93:2499–2504. doi: 10.1073/pnas.93.6.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carayanniotis G., Masters S.R., Noelle R.J. Suppression of murine thyroiditis via blockade of the CD40-CD40L interaction. Immunology. 1997;90:421–426. doi: 10.1111/j.1365-2567.1997.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard L.M., Miga A.J., Vanderlugt C.L., Dal Canto M.C., Laman J.D., Noelle R.J., Miller S.D. Mechanisms of immunotherapeutic intervention by anti-CD40L (CD154) antibody in an animal model of multiple sclerosis. J. Clin. Invest. 1999;103:281–290. doi: 10.1172/JCI5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasa B., Krahl T., Patstone G., Lee J., Tisch R., McDevitt H.O., Sarvetnick N. CD40 ligand-CD40 interactions are necessary for the initiation of insulitis and diabetes in nonobese diabetic mice. J. Immunol. 1997;159:4620–4627. [PubMed] [Google Scholar]
- Grewal I.S., Foellmer H.G., Grewal K.D., Xu J., Hardardottir F., Baron J.L., Janeway C.A., Jr., Flavell R.A. Requirement for CD40 ligand in costimulation induction, T cell activation, and experimental allergic encephalomyelitis. Science. 1996;273:1864–1867. doi: 10.1126/science.273.5283.1864. [DOI] [PubMed] [Google Scholar]
- De Jong Y.P., Comiskey M., Kalled S.L., Mizoguchi E., Flavell R.A., Bhan A.K., Terhorst C. Chronic murine colitis is dependent on the CD154/CD40 pathway and can be attenuated by anti-CD154 administration. Gastroenterology. 2000;119:715–723. doi: 10.1053/gast.2000.16485. [DOI] [PubMed] [Google Scholar]
- Becher B., Prat A., Antel J.P. Brain-immune connectionimmuno-regulatory properties of CNS-resident cells. Glia. 2000;29:293–304. [PubMed] [Google Scholar]
- Hickey W.F., Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239:290–292. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- Becher B., Antel J.P. Comparison of phenotypic and functional properties of immediately ex vivo and cultured human adult microglia. Glia. 1996;18:1–10. doi: 10.1002/(SICI)1098-1136(199609)18:1<1::AID-GLIA1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Ford A.L., Goodsall A.L., Hickey W.F., Sedgwick J.D. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J. Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- Becher B., Fedorowicz V., Antel J.P. Regulation of CD14 expression on human adult central nervous system-derived microglia. J. Neurosci. Res. 1996;45:375–381. doi: 10.1002/(SICI)1097-4547(19960815)45:4<375::AID-JNR6>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Becher B., Dodelet V., Fedorowicz V., Antel J.P. Soluble tumor necrosis factor receptor inhibits interleukin 12 production by stimulated human adult microglial cells in vitro. J. Clin. Invest. 1996;98:1539–1543. doi: 10.1172/JCI118946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B., Blain M., Antel J.P. CD40 engagement stimulates IL-12 p70 production by human microglial cellsbasis for Th1 polarization in the CNS. J. Neuroimmunol. 2000;102:44–50. doi: 10.1016/s0165-5728(99)00152-6. [DOI] [PubMed] [Google Scholar]
- Aloisi F., Penna G., Polazzi E., Minghetti L., Adorini L. CD40-CD154 interaction and IFN-gamma are required for IL-12 but not prostaglandin E2 secretion by microglia during antigen presentation to Th1 cells. J. Immunol. 1999;162:1384–1391. [PubMed] [Google Scholar]
- Rinner W.A., Bauer J., Schmidts M., Lassmann H., Hickey W.F. Resident microglia and hematogenous macrophages as phagocytes in adoptively transferred experimental autoimmune encephalomyelitisan investigation using rat radiation bone marrow chimeras. Glia. 1995;14:257–266. doi: 10.1002/glia.440140403. [DOI] [PubMed] [Google Scholar]
- Hickey W.F., Vass K., Lassmann H. Bone marrow-derived elements in the central nervous systeman immunohistochemical and ultrastructural survey of rat chimeras. J. Neuropathol. Exp. Neurol. 1992;51:246–256. doi: 10.1097/00005072-199205000-00002. [DOI] [PubMed] [Google Scholar]
- Sean R.D., Korner H., Strickland D.H., Lemckert F.A., Pollard J.D., Sedgwick J.D. Challenging cytokine redundancyinflammatory cell movement and clinical course of experimental autoimmune encephalomyelitis are normal in lymphotoxin-deficient, but not tumor necrosis factor–deficient, mice. J. Exp. Med. 1998;187:1517–1528. doi: 10.1084/jem.187.9.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers K.J., Dougherty J.P., Ron Y. In vivo antigen presentation by both brain parenchymal cells and hematopoietically derived cells during the induction of experimental autoimmune encephalomyelitis. J. Immunol. 1993;151:2252–2260. [PubMed] [Google Scholar]
- Noelle R.J., Roy M., Shepherd D.M., Stamenkovic I., Ledbetter J.A., Aruffo A. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc. Natl. Acad. Sci. USA. 1992;89:6550–6554. doi: 10.1073/pnas.89.14.6550. [DOI] [PMC free article] [PubMed] [Google Scholar]