

Abstract
Monocyte chemoattractant protein (MCP)-1 plays a critical role in innate immunity by directing the migration of monocytes into inflammatory sites. Recent data indicated a function for this chemokine in adaptive immunity as a regulator of T cell commitment to T helper cell type 2 (Th2) effector function. Studies in a Th1-dependent animal model, experimental autoimmune encephalomyelitis (EAE), showed that MCP-1 was highly expressed in the central nervous system (CNS) of affected rodents, and MCP-1 antibodies could block relapses of the disease. Mice deficient for the major MCP-1 receptor, CC chemokine receptor (CCR)2, did not develop EAE after active immunization but generated effector cells that could transfer the disease to naive wild-type recipients. We analyzed EAE in mice deficient for MCP-1 to define the relevant ligand for CCR2, which responds to murine MCP-1, MCP-2, MCP-3, and MCP-5. We found that C57BL/6 MCP-1–null mice were markedly resistant to EAE after active immunization, with drastically impaired recruitment of macrophages to the CNS, yet able to generate effector T cells that transferred severe disease to naive wild-type recipients. By contrast, adoptive transfer of primed T cells from wild-type mice into naive MCP-1–null recipients did not mediate clinical EAE. On the SJL background, disruption of the MCP-1 gene produced a milder EAE phenotype with diminished relapses that mimicked previous findings using anti–MCP-1 antibodies. There was no compensatory upregulation of MCP-2, MCP-3, or MCP-5 in MCP-1–null mice with EAE. These results indicated that MCP-1 is the major CCR2 ligand in mice with EAE, and provided an opportunity to define the role of MCP-1 in EAE. Compared with wild-type littermates, MCP-1−/− mice exhibited reduced expression of interferon γ in draining lymph node and CNS and increased antigen-specific immunoglobulin G1 antibody production. Taken together, these data demonstrate that MCP-1 is crucial for Th1 immune responses in EAE induction and that macrophage recruitment to the inflamed CNS target organ is required for primed T cells to execute a Th1 effector program in EAE.
Keywords: autoimmune disease, chemokine, chemokine receptor, macrophage, T helper cell type 1/T helper cell type 2
Introduction
Chemokines are small proteins (8–12 kD) divided into four subfamilies (CXC, CC, C, and CX3C) according to the organization of positionally conserved cysteine residues 1. Monocyte chemoattractant protein 1 (MCP-1) is a prototype CC chemokine, active towards monocytes, dendritic cells, and NK cells, thereby playing an important role in innate immunity 2 3 4 5 6 7. However, MCP-1 is also a crucial factor for the development of adaptive Th2 responses. In this regard, MCP-1 directs the differentiation of Th0 cells to Th2 in vitro 8 by a mechanism dependent on IL-4. Administration of anti–MCP-1 Abs 9 or disruption of the MCP-1 gene 2 significantly reduced the size of schistosome egg antigen (SEA) secondary granulomata, a Th2-dominant disease model. Conversely, local overexpression of MCP-1 increased the size of SEA secondary granulomata 10. Immunization with trinitrophenol-derivatized ovalbumin plus IFA elicited a reduced Th2 and unaltered Th1 response in MCP-1–deficient (MCP-1−/−) mice. MCP-1−/− mice of Balb/c strain were relatively resistant to Leishmania major infection, indicating that lack of MCP-1 led to reduced Th2 immunity 11.
Experimental autoimmune encephalomyelitis (EAE), a model for autoimmune demyelination of the central nervous system (CNS), has been widely employed to explore pathogenic mechanisms underlying the human disease multiple sclerosis (MS [12, 13]). The generation of myelin protein–reactive T cells is an immunological hallmark of both EAE and MS and is required for disease expression in EAE. These autoreactive T cells traffic to the CNS, and initiate inflammation and destruction of CNS myelin with consequent neurological impairment 14 15.
Th1-type T cells, producing IFN-γ, IL-2, and TNF-β, are associated with cellular immune responses, delayed-type hypersensitivity, and macrophage activation, whereas Th2-type T cells, producing IL-4, IL-5, and IL-10, are important for humoral immune responses 16 17. The dynamic interplay and reciprocal inhibition between Th1 and Th2 cytokines has been demonstrated in numerous research reports. IL-4 is a major factor that governs Th2 differentiation and inhibits the development of IFN-γ–secreting cells 18. The activation of macrophages and the production of Th1 cytokines such as IFN-γ can also be inhibited by IL-10 19. Most encephalitogenic T cell clones examined are Th1 polarized 20 21 22, although exceptions have been reported 23. Th1 cytokines are markedly elevated in the CNS of animals during EAE attacks whereas Th2 cytokines are associated with disease recovery 24. IL-4–induced immune deviation is beneficial for recovery from EAE 25; EAE can be prevented and/or reversed by myelin antigen–specific T cells that are genetically transduced with either IL-4 or IL-10 genes 26 27; anti–IL-4 treatment reverses the tolerance induced by an altered peptide ligand 28, and absence of IL-4 in gene-targeted mice increases the severity of EAE 29. In summary, Th1 immune responses are pathogenic and Th2 responses are protective in the initiation and evolution of EAE.
However, antigen-specific T cells constitute only a small proportion of infiltrating leukocytes in EAE or MS lesions 30. Secondarily recruited inflammatory cells account for the vast majority of infiltrating cells and play a pivotal role in CNS tissue damage 31. Although the detailed mechanisms by which inflammatory cells influx into the CNS compartment are not completely understood, increasing evidence suggests that chemokines, in concert with adhesion molecules, are essential for this process 32. In EAE, elevated expression of MCP-1 by CNS parenchymal cells, tightly linked to clinical disease, has been demonstrated repeatedly 33 34 35. Further, anti–MCP-1 Abs blocked relapses of adoptive transfer EAE in SJL mice 36. Additionally, mice that lacked CC chemokine receptor 2 (CCR2), the major receptor on monocytes for MCP-1, failed to develop EAE after active immunization and were resistant to induction of EAE by the adoptive transfer of primed T cells from syngeneic wild-type mice 37 38. It was uncertain whether MCP-1 was the relevant ligand for CCR2 in these experiments, as this receptor also responds to MCP-2, MCP-3, and MCP-5. However, there is also support for the possibility that regulation of Th2 responses by MCP-1 could be important for the pathogenesis of EAE; in particular, MCP-1 was critical for the development of tolerance after oral administration of a proteolipid protein (PLP) peptide containing residues 139–151 39
Therefore, the phenotype of EAE in MCP-1–deficient mice could not readily be predicted. On one hand, defective MCP-1–dependent monocyte recruitment might lead to attenuated disease. Alternatively, if functional replacement of MCP-1 by another MCP mediated monocyte accumulation in the CNS of these mice, defective Th2 responses might lead to very severe, nonremitting disease. Finally, in view of redundancy in the immune/inflammatory system, it remained possible that MCP-1–null mice would manifest EAE identically to wild-type animals. Given these considerations, the role of MCP-1 in the pathogenesis of EAE merited further characterization.
These concerns are also likely pertinent for the human disorder MS. Patients with active disease, manifest by clinical attacks, showed significantly decreased MCP-1 in the cerebrospinal fluid, as compared with controls. Of eight chemokines measured, only MCP-1 was reduced in the cerebrospinal fluid of patients with active MS. However, abundant MCP-1 has been readily detected by immunohistochemistry in autopsy brain sections containing MS lesions 40 41 42. Therefore, the role of MCP-1 in the pathogenesis of MS remains to be clarified.
In this report, we describe the phenotype of EAE in MCP-1–null mice. These mice exhibited markedly reduced clinical and histological EAE after active immunization and did not develop clinical disease after receiving encephalitogenic T cells from wild-type animals. Expressions of MCP-2, MCP-3, and MCP-5 in the CNS of both wild-type and MCP-1–null mice with EAE were virtually identical. These findings indicated that MCP-1 was the major ligand for CCR2 in murine EAE. In this EAE model, we found that disruption of the MCP-1 gene led to an attenuated Th1 autoimmune response and complimentarily increased Th2 response. These results indicated a crucial role for MCP-1 in generating CNS inflammatory reactions that mediate the effector phase of myelin-specific Th1 autoimmune responses. Therefore, the data suggested that primed encephalitogenic Th1 cells cannot manifest effector functions in the CNS without recruiting hematogenous macrophages.
Materials and Methods
Mice.
The disruption of the MCP-1 gene has been described previously 2. MCP-1−/− mice were backcrossed onto the C57BL/6 (B6) strain for eight generations. One F8 MCP-1−/− mouse was further backcrossed to a B6 mouse (obtained from The Jackson Laboratory). The heterozygous offspring were intercrossed to produce F9 wild-type (+/+), heterozygous (−/+), and MCP-1−/− mice. F10 mice were generated in a similar manner. MCP-1+/+, MCP-1−/+, and MCP-1−/− F9 and F10 mice on the B6 background were used in this study.
MCP-1–deficient mice on the B6/129 background were also backcrossed onto SJL for seven generations. MCP-1+/+ and MCP-1−/− F7 mice on SJL background were used in this study.
Mice were genotyped using a PCR-based analysis of genomic DNA extracted from tail clips. Primers MCP-1F, 5′-GGA GCA TCC ACG TGT TGG C-3′ and MCP-1R, 5′-ACA GCT TCT TTG GGA CAC C-3′ amplified a DNA fragment within the MCP-1 gene. Primers NeoF, 5′-CGC TTC CTT TTT GTC AAG AC-3′ and NeoR, 5′-ATC CTC GCC GTC GGG CAT GC-3′ amplified a fragment in the neomycin resistance gene insert. PCR reactions were performed in a PerkinElmer 9700 cycler (annealing temperature, 50°C) and products were visualized by electrophoresis on ethidium bromide–stained NuSeive GTG® agarose gel.
Rat Myelin Oligodendroglial Glycoprotein and Mouse Proteolipid Protein Peptides.
Rat myelin oligodendroglial glycoprotein (MOG)35–55 and mouse PLP139–151 peptides were obtained from (BIO·SYNTHESIS) and purified by HPLC with a purity of 98%. The sequence of MOG35–55 was MEVGWYRSPFSRVVHLYRNGK and that of PLP139–151 was HSLGKWLGHPDKF.
Active Induction of EAE with MOG and PLP Peptides and Clinical Evaluation.
Mice of 8–9 wk of age were subcutaneously injected with 300 μg MOG35–55 emulsified in CFA (Difco) containing 400 μg Mycobacterium tuberculosis. Mice were intravenously injected with pertussis toxin (Sigma-Aldrich) as indicated in the figure legends on day 0 and 2 postimmunization (pi). The immunization in SJL mice was carried out as described previously 43. All mice were weighed, examined, and graded daily for neurological signs in a double blind manner by one of us (J. Wang) as follows: 0, no disease; 1, decreased tail tone or slightly clumsy gait; 2, tail atony and moderately clumsy gait and/or poor righting ability; 3, limb weakness; 4, limb paralysis; and 5, moribund state. Disease relapse was determined when an increase of one EAE score unit was observed. Signs of neurological impairment were typically accompanied by an abrupt, substantial weight loss (>7%). The average day of EAE onset was calculated by adding the first day of clinical signs for individual mice and divided by the number of mice in the group. Day of EAE onset in mice that showed no clinical EAE was deliberately regarded as 1 d after the experiment was terminated 44. The EAE index was calculated by adding all the daily EAE scores to obtain cumulative score and dividing by day of EAE onset. Active immunization with MOG35–55 induced monphasic EAE in B6 mice and was followed for 65 d. Chronic relapsing EAE induced by PLP139–151 was monitored for 90 d.
T Cell Proliferation Assay.
Mice were killed and draining lymph nodes (popliteal and inguinal lymph nodes [PILNs]) were dissected on day 10 pi. Single cell suspensions (5 × 106/ml) were prepared and cultured in triplicate in 96-well flat-bottomed plates (Falcon; Becton Dickinson) in 200 μl/well in the presence or absence of MOG35–55, PLP139–151, LPS, or anti-CD3 (R&D Systems) in RPMI 1640 (GIBCO BRL) supplemented with 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ ml streptomycin, 10% FCS, and 5.5 × 10−5 M 2-mercaptoethanol. Cells were pulsed with [3H]thymidine (Amersham Pharmacia Biotech), 0.5 μCi/well 72 h after culture initiation, and incubated 10 h further. Plates were harvested using a harvester (INOTECH). Incorporated radioactivity was measured in a MicroBeta PLUS liquid scintillation counter with software v3.3 (Wallac Co.).
Cell Cultures and Cytokine Assay.
Mononuclear cell (MNC) suspensions (5 × 106/ml) were prepared from PILNs of mice that had been immunized with MOG35–55 plus CFA for 10 d. Cells were cultured at 2 × 106/ml in RPMI 1640 supplemented as above in cell culture tubes (Falcon; Becton Dickinson) in the presence or absence of MOG35–55 and anti-CD3ε Ab at 5% CO2 and 95% humidity. The supernatants were collected after 24, 48, and 72 h of in vitro restimulation and kept at −80°C until assay. Levels of IFN-γ, IL-4, and IL-10 in sera and cell culture supernatants were determined using ELISA kits commercially obtained from R&D Systems. The standard curves were made on the same occasion and the sensitivities for the methods were 2.0, 2.0, and 4.0 pg/ml for IFN-γ, IL-4, and IL-10, respectively. All samples were measured in duplicate and diluted if necessary.
Analysis of Chemokine and Chemokine Receptor mRNA Levels by RNase Protection Assay.
Mice were anesthetized with sodium pentobarbital and intracardially perfused through the left ventricle with ice-cold PBS. Spinal cords were extruded by flushing the vertebral canal with PBS, rinsed in PBS, and kept at −80°C. Total cellular RNA was prepared from spinal cord tissue by TRIzol (Life Technologies). Quantification of CC chemokines and chemokine receptors in CNS tissue was done by RNase protection assay (RPA) with Template Sets and In vitro Transcription Kit (BD PharMingen) according to the manufacturer's instructions. Protected fragments were visualized and quantified by autoradiography with PhosphorImager (Molecular Dynamics).
Reverse Transcription PCR Detection for Levels of MCPs, Cytokine, CD3ε, CD8, and Mb-1 mRNA.
Total RNA was extracted from the PBS-perfused, snap-frozen spinal cords using TRIzol. cDNA was synthesized using a RNA PCR Core kit (GeneAmp®; PerkinElmer). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression was used as an unregulated control and amplified using primers: GAPDHf, 5′-GGT GGA GGT CGG AGT CAA CG-3′ and GAPDHr, 5′-CAA AGT TGT CAT GGA TGA CC-3′. MCP-2, MCP-3, and MCP-5 cDNA were amplified by specific primer pairs, mcp-2f, 5′-ACA TCA CCT GCT TGG TCT GGA AAA C-3′ and mcp-2r, 5′-ACT AAA GCT GAA GAT CCC CCT TCG-3′; mcp-3f, 5′-CAC ATT CCT ACA GAC AGC TC-3′ and mcp-3r, 5′-AGC TAC AGA AGG ATC ACC AG-3′; and mcp-5f, 5′-CTC CTT ATC CAG TAT GGT CC-3′ and mcp-5r, 5′-TCT CCC TCC ACC ATG CAG AG-3′. IFN-γ, IL-4, and IL-10 were amplified with primer pairs: IFNgf, 5′-AGC GGC TGA CTG AAC TCA GAT TGT-3′ and IFNgr, 5′-GTC ACA GTT TTC AGC TGT ATA GGG-3′; il4f, 5′-TCG GCA TTT TGA ACG AGG TC-3′ and il4r, 5′-GAA AAG CCC GAA AGA GTC TC-3′; and il10f, 5′-CAT CAT GTA TGC TTC TAT GC-3′ and il10r, 5′-TAC CTG GTA GAA GTG ATG CC-3′. CD3ε was detected with primer pair: CD3ef, 5′-ATG GAG CAG AGG AAG GGT CTG-3′ and CD3er, 5′-TCA CTT CTT CCT CAG TTG GTT-3′. Levels of CD8 cDNA were detected using primer pair: CD8f, 5′-TCT GTC GTG CCA GTC CTT C-3′ and CD8r, 5′-CCT TCC TGT CTG ACT AGC GG-3′, while that of Mb-1 (the gene encoding IgGα, expressed by B cells; reference 45): Mb-1f, 5′-GCC AGG GGG TCT AGA AGC-3′ and Mb-1r, 5′-TCA CTT GGC ACC CAG TAC AA-3′. RNA Amplification Kit SYBR Green I was used in all PCR reactions that were performed in a real-time LightCycler system (Roche Molecular Biochemicals). The level of specific mRNA was quantified and expressed as the cycle number at which the LightCycler System detected the upstroke of the exponential phase of PCR product accumulation, and normalized by the level of GAPDH expression in each individual sample. Therefore, levels of input mRNAs correlate negatively with numbers of cycles.
Determination of Serum Anti–MOG35–55 IgG, IgG1, and IgG2a.
Detection of anti–MOG35–55 Abs was performed as described previously 46. In brief, rat MOG35–55 peptide (0.1 ml/well, 3.0 μg/ml) was added to 96-well microtiter plate (Nunc) in coating buffer and incubated at +4°C overnight. After being washed three times with PBS-Tween (0.1%), the plates were blocked with 5% FCS in PBS for 2 h at room temperature. Diluted sera (0.1 ml/well, 1:5 for IgG2a, 1:50 for total IgG and IgG1) were added to and incubated at room temperature for 2 h, followed by washing with PBS-Tween three times. A series of serum dilutions were examined in the preliminary experiments. The OD values obtained negatively correlated with the dilutions of the samples. For the detection of IgG2a, alkaline phosphatase–conjugated anti–mouse IgG2a (1:2,000 dilution; BD PharMingen) was added and incubated for 2 h at room temperature, followed by five washes. Color was developed by adding p-nitrophenyl phosphate substrate (Sigma-Aldrich) and measurements were performed at 450 nm in an ELISA reader (Wallac Co.). For the detection of total IgG and IgG1, biotinylated anti–mouse IgG (1:3,000; Sigma-Aldrich) and IgG1 (1:500; BD PharMingen) Abs were added and incubated at room temperature for 2 h. After incubation with ABC Vectastain (Vector Laboratories), the reaction was developed with 2,2′-azino-bis(3-ethylbenz-thiazoline-6-sulfonic acid) (ABTS) peroxidase substrate (Vector Laboratories) and read at 405 nm in the ELISA reader. In each assay for IgG, IgG1, or IgG2a, all the samples were measured in duplicate on a single plate.
Flow Cytometry.
Cells from the CNS were isolated as described previously 37. Samples were washed in FACS® buffer (1% FCS and 0.1% sodium azide in PBS). After blocking with CD16/CD32 Fc Block (BD PharMingen), cells were stained for surface markers with directly conjugated Abs in FACS® buffer. Abs used were CD4-FITC, CD11b-PE, CD45–Cy-Chrome, and CD8–Cy-Chrome, and were titrated using mouse peripheral blood. All Abs were obtained from BD PharMingen.
Adoptive Transfer of EAE.
Mice were immunized with 200 μg MOG35–55 peptide in CFA. Lymph nodes were harvested and single MNC suspensions were prepared 10 d pi and cultured in the presence of 30 μg/ml MOG35–55 and 20 ng/ml murine rIL-12 (R&D Systems). After 4 d incubation, cells were collected and washed in PBS. 2 × 107 living cells were intravenously injected into wild-type or MCP-1–deficient mice. Recipients were given 200 ng pertussis toxin intravenously on the day of cell transfer and 48 h after transfer 37 47. Mice were weighed and EAE scored daily by an investigator (J. Wang) who was blinded to mouse genotype or source of transferred cells. Mice were monitored for 40 d after transfer.
Statistical Analyses.
The Instat 2.02 software was used for the analyses of the difference between the MCP-1+/+, MCP-1−/+, and MCP-1−/– mice. The Mann-Whitney U-test was used for the comparisons of disease severity and cytokine and chemokine gene expressions. A chi-square test was used for the comparisons of disease incidence in groups of MCP-1+/+, MCP-1−/+, and MCP-1−/− mice. A P value <0.05 was considered as significant.
Results
C57BL/6 MCP-1–deficient Mice Are Relatively Resistant to Active EAE Induction with MOG35–55.
MCP-1+/+, MCP-1−/+, and MCP-1−/− F9 mice (n = 9 in each group) were immunized with 300 μg MOG35–55 plus 500 ng pertussis toxin intravenous injection on day 0 and 2 pi. One MCP-1−/− mouse died of immunization on day 4 pi. The remainder of the mice, regardless of genotype, developed clinical EAE. MCP-1+/+ mice showed EAE signs around day 10 pi, consistent with previously reported results in C57BL/6 mice 44 48. MCP-1−/− manifested significantly delayed EAE with an average onset on day 21 pi (Fig. 1). Three out of nine wild-type mice died of EAE, and another three had to be killed because of moribund state. None of the MCP-1−/− mice died of EAE or had to be killed throughout the experiment and they recovered from the disease significantly faster and more completely than wild-type littermate controls. Heterozygote MCP-1−/+ mice developed EAE with an intermediate kinetics and severity. None of the MCP-1−/+ mice died of EAE, whereas three were killed in a moribund state due to severe EAE. Analyses of CNS tissue histology revealed massive inflammatory infiltrates in wild-type control mice (+++ to ++++ in regions of lumbar and sacral spinal cord, n = 4; Fig. 2 A) but markedly reduced inflammatory reaction in MCP-1–null mice (+ to ++ in regions of corresponding levels of affected spinal cord, n = 4; Fig. 2 B; reference 49). Examination of demyelination using Luxol Fast Blue staining revealed significant reduction in MCP-1−/− mice compared with their littermates controls 65 d after immunization (data not shown).
Figure 1.

Effect of MCP-1 gene disruption on MOG35-55–induced EAE. F9 MCP-1+/+, MCP-1−/+, and MCP-1−/− mice were immunized with MOG35–55 emulsified in CFA and intravenously injected with pertussis toxin (PT) on the day of immunization and 48 h later (500 ng/injection). Three wild-type mice died of EAE and another three had to be killed due to severe EAE attack. None of the MCP-1−/+ mice died of EAE, but three were killed. None of the MCP-1−/− mice died of EAE or had to be killed. Shown are EAE score (mean ± SD) in each group of mice that had been followed throughout the experiment (n = 3, 6, and 8 for MCP-1+/+, MCP-1−/+, and MCP-1−/− group, respectively). This graph is representative of three experiments with similar results.
Figure 2.
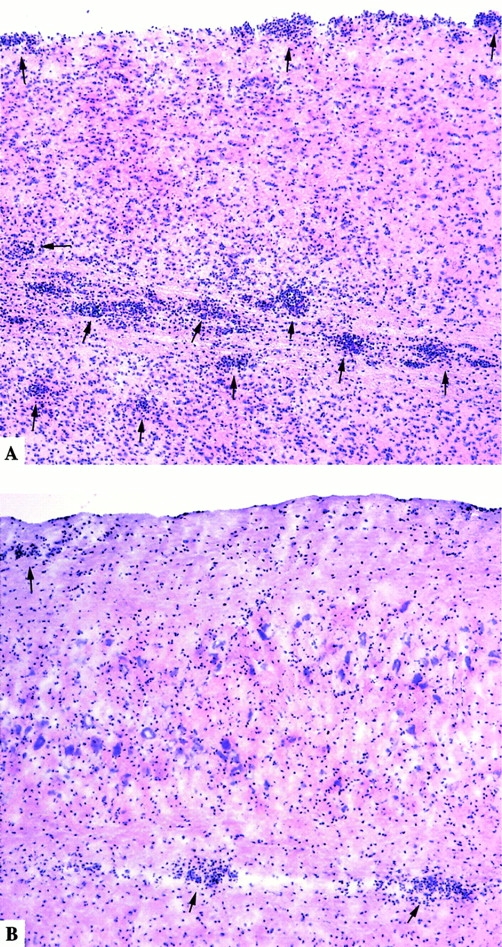
Spinal cord histology of MCP-1+/+ and MCP-1−/− mice with EAE score 4.0. Hematoxylin and eosin staining of longitudinal cryosections. Note the numerous perivascular cuffs and subpial infiltrates (arrows) as well as leukocytes disseminated in the white matter of MCP-1+/+ mice (A), whereas merely fewer inflammatory infiltrates were found in MCP-1–deficient mice (B).
F10 MCP-1–deficient mice and wild-type littermate controls were analyzed in a subsequent experiment. As pertussis toxin has been shown to increase the permeability of the blood-brain barrier (BBB [50, 51]), enhance delayed type hypersensitivity (DTH) responses and the production of IFN-γ 52 53 54, augment expression of CD80, CD86 on antigen-presenting cells and CD28 on T cells 55, and T cell immune responses 55 56, we reduced the amount of pertussis toxin in the immunization to 200 ng per injection in an attempt to reduce the high death rate observed in wild-type mice in the F9 experiments. In this experiment, all MCP-1+/+ and MCP-1−/− mice showed signs of clinical EAE; none died of EAE or required killing. In contrast to the MCP-1+/+ mice that developed full-blown EAE, the disease was largely suppressed with significantly delayed onset and milder neurological impairment (Fig. 3) and significantly less weight loss (data not shown) in MCP-1–deficient mice. These data demonstrated that MCP-1–null mice and CCR2-deficient mice exhibited strikingly similar EAE phenotypes 37 38 and suggested that MCP-1 may be the relevant ligand for CCR2 in this model.
Figure 3.

Attenuated MOG35–55–induced EAE in MCP-1–null mice. F10 MCP-1−/− mice (n = 6) and their littermate wild-type controls (n = 6) were immunized with MOG35–55 in CFA and intravenously injected with pertussis toxin (PT; 200 ng/injection). Shown are EAE score (mean ± SD) of individuals in each group.
Disruption of MCP-1 Gene Attenuates the Severity of PLP-induced EAE and Reduces the Number of Relapses in SJL Mice.
11 MCP-1+/+ and 10 MCP-1−/− mice on SJL background were immunized with PLP139–151 peptide emulsified in CFA. One MCP-1+/+ and two MCP-1−/− mice died of immunization within the first week after immunization. The reminder of MCP-1+/+ mice (10/10) and 7 out of 8 MCP-1−/− mice showed clinical EAE. Compared with the MCP-1−/− group, a significantly higher percentage of MCP-1+/+ mice died of EAE during the first attack and relapses thereafter. Among the mice that survived the first attack, six MCP-1+/+ mice had eight relapses (four mice had one attack each and two had two attacks each), whereas three out of seven MCP-1−/− mice had single attacks. At day 90 pi, the average EAE index was significantly higher in MCP-1+/+ group (n = 4) than that in MCP-1−/− group (n = 7) (Fig. 4). These results implicated a role for MCP-1 in eliciting relapses of EAE in this model. These findings were consistent with previous reports showing reduction of relapses using anti–MCP-1 Abs in a passive EAE model 36 and in mice receiving vaccine containing naked DNA encoding for MCP-1 57.
Figure 4.

Milder disease and reduced number of relapses in PLP-induced EAE MCP-1−/− SJL mice. MCP-1−/− SJL mice and their littermate wild-type controls were immunized with PLP139–151 in CFA plus intravenous injection of pertussis toxin as described in Materials and Methods. Mice were monitored for 90 d after immunization. MCP-1−/− SJL mice showed significantly decreased EAE index, reduced number of relapses, and nonsignificantly delayed disease onset.
There Is No Compensatory Upregulation of MCP-2, MCP-3, or MCP-5 in the CNS of MCP-1−/− Mice with EAE.
Because CCR2 is shared in common among all MCPs 58 59 60, we analyzed MCP-2, MCP-3, and MCP-5 mRNA expression in the CNS of C57BL/6 mice with MOG-induced EAE, using quantitative real-time reverse transcription (RT)-PCR. Levels of MCP-2 and MCP-3 but not MCP-5 were elevated in CNS tissue from EAE mice. There was no significant difference in MCP-2 expression in CNS tissue between MCP-1+/+ and MCP-1−/− group (23.8 ± 0.4 vs. 23.7 ± 0.5, mean ± SD; n = 5 in each group). No significant difference of MCP-3 expression in CNS tissue from MCP-1+/+ (31.0 ± 0.6, n = 5) and MCP-1−/− (33.1 ± 2.6, n = 5, P = 0.4) mice was found, whereas CNS MCP-5 expression in mice with EAE was essentially undetectable (data not shown). The unaltered levels of MCP-2 and MCP-5 and a trend towards decreased expression of MCP-3 in MCP-1−/− mice (higher PCR cycle number) uncovered no compensatory expression of other MCPs in this system, consistent with a previous report in autoimmune kidney disease model in MCP-1−/− MRL-Faslpr mice 61. These findings supported the hypothesis that the similarity of EAE phenotype in MCP-1– and CCR2-deficient mice is caused by absence of the ligand, i.e., MCP-1 signaling pathway through its major receptor CCR2.
T Cell Proliferation to MOG35–55 Peptide.
To examine the afferent limb of the immune response to MOG peptide in wild-type and MCP-1–null mice, MNC suspensions were prepared from PILNs primed with 200 μg MOG35–55 peptide in CFA and rechallenged with MOG35–55 in vitro. MCP-1–deficient mice showed a nonsignificantly higher recall T cell response than wild-type controls. No T cell recall response was induced by stimulation with PLP139–151 in vitro, a specificity control. No difference in anti-CD3ε–induced T cell proliferation was found between wild-type and MCP-1–deficient mice (data not shown). These data indicated that the CD3 pathway was intact in MCP-1−/− mice, and that MOG35–55–specific T cells can be generated in MCP-1–deficient mice.
MCP-1−/− Mice Do Not Develop Clinical EAE in Passive Transfer Model.
An adoptive transfer EAE model was used to further address whether MCP-1–null mice could develop pathogenic autoimmune responses to MOG35–55 peptide. MCP-1+/+ and MCP-1−/− B6 mice were immunized with MOG35–55 peptide in CFA, and MNCs isolated from draining lymph nodes were cultured in the presence of MOG35–55 peptide and IL-12 before transfer into MCP-1−/− mice or littermate controls. As expected, MCP-1+/+ mice receiving MCP-1+/+ T cells developed clinical EAE (Fig. 5). MCP-1−/− T cells showed approximately the same encephalitogenic capacity in this adoptive transfer model, resulting in a comparable incidence, severity, and clinical course of EAE in MCP-1+/+ recipients. In contrast, MCP-1−/− mice that received MCP-1+/+ T cells failed to develop clinical EAE (Fig. 5). This result indicated that the attenuated EAE in MCP-1−/− mice was not caused by impaired generation of encephalitogenic T cells. The data also demonstrated that absence of MCP-1 expression in the recipient rendered the mice unable to respond to encephalitogenic signals produced by wild-type T cells.
Figure 5.

Clinical course of EAE in MCP-1−/− and littermate control recipient mice that received MOG35–55–reactive T cells generated from MCP-1+/+ or MCP-1−/− littermates. Data are presented as the EAE score (mean ± SD) in each group. Each group consists of four recipient mice.
To dissect the mechanisms underlying the relatively resistance to EAE induction in MCP-1–deficient mice, the MOG35–55–induced EAE model was used in the following mechanistic studies. To obtain samples from MCP-1−/− mice with full-blown EAE, F10 mice were immunized with MOG35-55 in CFA plus 500 ng pertussis toxin per injection.
Reduced CD11b+CD4−/CD4+ Ratio but Unchanged Levels of CD3ε and CD8 Transcripts in CNS Tissue from MCP-1−/− Mice with EAE.
Results described above suggested that MCP-1–null mice were deficient in recruiting monocytes to the CNS during EAE. To address this issue, leukocytes were isolated from CNS tissue of wild-type and MCP-1–null mice with comparable severity of EAE, and analyzed with flow cytometry. Cell numbers in preparations isolated from MCP-1−/− mice during EAE attacks (score 4) were ∼1/3 of those from MCP-1+/+ littermate controls with comparable EAE severity. Compared with MCP-1+/+ littermate controls, MCP-1−/− mice showed a sharply reduced percentage of CD11b+CD4− cells in the CNS during EAE attacks (71.8 ± 4.6% vs. 44.0 ± 4.2%, mean ± SD, n = 5 and 4, respectively; P < 0.0001). In contrast, percentages of CD4+ cells were relatively increased in MCP-1−/− mice (20.2 ± 5.9% vs. 39.8 ± 2.7%, mean ± SD, n = 6 and 4, respectively; P < 0.001; Fig. 6). The CD4+ infiltrating T cells expressed high levels of CD11b (αMβ2 integrin) both in MCP-1+/+ (79.0 ± 5.6, n = 5) and MCP-1−/− (78.1 ± 6.8, n = 5) mice, indicating that most infiltrating CD4+ T cells are activated.
Figure 6.
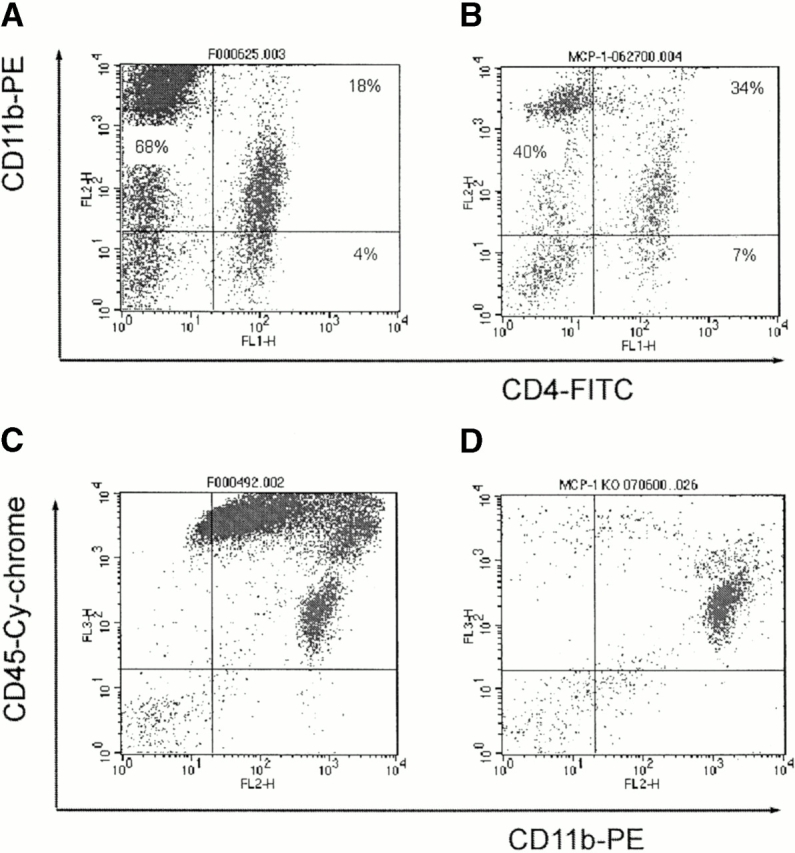
Altered pattern of CNS cell infiltrates in MCP-1–null mice. MCP-1−/− and MCP-1+/+ littermate controls were immunized with MOG35–55 and pertussis toxin (500 ng/injection) and killed at the peak of EAE (score 4). Cells were isolated from the CNS and stained with anti-CD4–FITC, anti-CD11b–PE, and anti-CD45–Cy mAbs. Compared with wild-type controls (A), the percentages of CD11b+CD4− cells were significantly decreased whereas the percentages of CD4+ T cells increased in MCP-1−/− mice (B). In contrast to MCP-1+/+ mice that developed full-blown EAE with numerous CNS CD11b+CD45high macrophages/activated microglia and T cells (C), the majority of cells isolated from CNS tissues of EAE symptom-free MCP-1−/− mice on day 14 pi were CD11b+CD45low microglia (D).
To normalize percentage of CD4+ T cells in the CNS infiltrates of wild-type and MCP-1–null mice with EAE, total CNS T cells were analyzed by determining levels of CD3ε in the CNS. We found no significant difference in CNS CD3ε mRNA levels between wild-type (0.789, n = 5; CD3ε/GAPDH) and MCP-1−/− (0.791, n = 5; CD3ε/GAPDH) mice with clinical EAE scores of 3.5–4.0, suggesting that total T cell numbers in the CNS of wild-type and MCP-1–null mice with EAE were equivalent. Therefore, the increased proportion of CD4+ cells in the CNS leukocyte infiltrates of MCP-1–null mice was caused by a marked reduction in the number of CD11b+CD4− cells in the CNS of MCP-1–deficient mice with EAE.
CD8+ T cells play an important downregulatory role in the pathogenesis of EAE 62. The possibility that the milder clinical EAE phenotype observed in MCP-1−/− mice might be due to an increased number of CD8+ T cells in the CNS infiltrates was unlikely based on the fact that levels of CD8-specific mRNA were virtually identical in MCP-1−/− (24.08 ± 0.3, n = 5) and MCP-1+/+ (24.21 ± 0.2, n = 5) CNS tissue during EAE attack. The passive transfer EAE model was used to examine further if CD8+ T cells can be preferentially recruited into CNS in the absence of MCP-1. MOG35–55–reactive MCP-1+/+ T cells were incubated in the presence of MOG35–55 and IL-12, and were injected intravenously into MCP-1+/+ and MCP-1−/− mice. To reduce the influence of secondarily recruited macrophages in the CNS, recipient mice were killed at day 3 and 4 after T cell transfer, before the onset of clinical EAE. CNS-infiltrating T cells were recovered and analyzed using flow cytometry. No difference was found between MCP-1+/+ and MCP-1−/− mice (data not shown). These results demonstrated that disruption of MCP-1 gene exerts no significant impact on the recruitment of adoptively transferred T cells into inflammatory CNS tissue. No significant difference in Mb-1 mRNA levels was found in EAE-affected CNS tissue from MCP-1+/+ and MCP-1−/− mice (data not shown).
We also examined the leukocyte infiltrates in MCP-1−/− mice at day 14 pi when the MCP-1+/+ controls were undergoing EAE attacks while the MCP-1−/− mice were still free of EAE signs. Shown in Fig. 6 C are numerous CD45highCD11b+ (mainly containing macrophages/activated microglia and activated T cells) isolated from MCP-1+/+ mice during EAE attack on day 14 pi. In contrast, the majority of cells isolated from MCP-1−/− mice were CD45lowCD11b+ microglia, and the components of the infiltrates (Fig. 6 D) were virtually the same as those from healthy unimmunized mice (data not shown).
Diminished MOG35–55–specific Th1 Cytokine Responses in MCP-1–null Mice.
Significant changes in cytokine production have been described in the Th1 immune response that typifies MOG35–55 peptide-induced EAE in B6 mice 35. Serum concentrations of IFN-γ, IL-4, and IL-10 were determined by ELISA from mice immunized with MOG35–55 on days 8 and 10 pi and at the peak of EAE (day 14 pi in wild-type controls and 25 pi in MCP-1–deficient mice). At day 8 pi, concentrations of IFN-γ were slightly but significantly higher in wild-type than in MCP-1–deficient mice. This difference between wild-type and MCP-1–deficient mice became strikingly evident on day 10 pi near the onset of EAE in wild-type controls. The onset of EAE was also associated with increased serum levels of IFN-γ in MCP-1–deficient mice, but the magnitude of increase was significantly less than in MCP-1+/+ mice (Fig. 7 A). Circulating IL-10 was detected at low levels in both MCP-1+/+ and MCP-1−/− mice before and after the onset of EAE. Before EAE onset at day 8 pi (Fig. 7 B), serum IL-10 was slightly but significantly higher in MCP-1−/− mice than in wild-type controls. Serum IL-4 remained below the limits of detection in both MCP-1+/+ and MCP-1−/− mice at all time points.
Figure 7.

Quantitation of circulating cytokine levels in MCP-1−/− and MCP-1+/+ mice immunized with MOG35–55 in CFA and pertussis toxin (500 ng/injection). Sera were collected at different times after immunization and measured for levels of IFN-γ (A), IL-10 (B), and IL-4 (undetectable, data not shown). Data are mean ± SD of five individual mice in each group. This histogram is representative of two experiments with similar results. WT, wild-type; KO, knockout.
These results were supported by data from in vitro restimulation experiments, using draining lymph node (PILN) cells from mice immunized with MOG35–55 in CFA. Upon rechallenge with MOG35–55 peptide in vitro, at all examined occasions during the cell culture, PILN cells from MCP-1−/− mice secreted ∼50% less IFN-γ than cells from MCP-1+/+ mice (Fig. 8, left). IL-4 and IL-10, signature Th2 cytokines in EAE 26 29 63 64, were also measured. Although low levels of IL-10 were found in the culture supernatants of restimulated PILN cells from both MCP-1+/+ and MCP-1−/− mice, significantly higher levels of IL-10 were detected in cultures of cells from MCP-1−/− mice (Fig. 8, right). IL-4 was undetectable in all cell culture supernatants.
Figure 8.

Cytokine levels in cell culture supernatants of lymphocytes from MOG35–55–immunized MCP-1–deficient mice and their littermate controls. Cells from draining lymph nodes were restimulated with MOG35–55 peptide in vitro. Supernatants were collected at different times after immunization as indicated and measured for IFN-γ, IL-10, and IL-4 (undetectable, data not shown). Data represent means ± SD and are representative of two experiments with similar results. WT, wild-type; KO, knockout.
CNS Cytokine mRNA Accumulation in MCP-1−/− and Wild-type Mice with EAE.
Local expression of IFN-γ in the CNS was analyzed by real-time RT-PCR. Significantly higher geometric mean levels of IFN-γ were found in spinal cord tissue from MCP-1+/+ mice at the peak of EAE attacks (MCP-1+/+: 27.7 ± 0.3, mean ± SD, n = 4; MCP-1−/−: 29.9 ± 0.8, n = 4, P < 0.05). This result indicated approximately a fourfold difference in the CNS expression of IFN-γ between mice with intact and disrupted MCP-1 genes, despite equal numbers of CNS-infiltrating T cells (see above). There was no difference in IL-10 expression in CNS tissue between MCP-1+/+ and MCP-1−/− mice with full-blown EAE. IL-4 gene expression was undetectable both in MCP-1+/+ and MCP-1−/− mice (data not shown).
Decreased Expression of IFN-γ–inducible 10-kD Protein, Macrophage Inflammatory Protein 1α, and RANTES in CNS Tissue from MCP-1−/− Mice with EAE.
CNS chemokine expression was quantified using RPA, in tissues from MCP-1+/+ and MCP-1−/− mice equally affected by EAE (score 3.5–4.0). MCP-1−/− mice had significantly lower levels of IFN-γ–inducible 10-kD protein (IP-10), macrophage inflammatory protein (MIP)-1α, and regulated upon activation, normal T cell expressed and secreted (RANTES) transcripts compared with wild-type littermate controls (Fig. 9). Expression of MCP-3 was low in MCP-1+/+ and MCP-1−/− mice at the peak of EAE attack without significant differences between MCP-1–deficient mice and littermate controls, supporting the results obtained using real-time RT-PCR. T cell activation gene (TCA)-3 expression in the CNS of MCP-1+/+ and MCP-1−/− mice with EAE was near the lower limits of detection. No significant difference was found in CCR gene expression (data not shown). However, there was a nonsignificant trend towards decreased levels of CCR2 expression in MCP-1−/− EAE CNS tissue compared with wild-type controls. In the absence of MCP-1, the presence of CCR2 in the affected tissue might indicate the action of other CCR2 ligand(s). Alternatively, the migration of CCR2-bearing cells into CNS might be a bystander phenomenon.
Figure 9.

Chemokine expression in spinal cords from MCP-1−/− and MCP-1+/+ mice immunized with MOG35–55 and pertussis toxin (500 ng/injection). Spinal cords were collected from mice that were equally affected by EAE (score 4) and levels of chemokine mRNA were measured using RPA. Data are presented as mean chemokine products ± SD. WT, wild-type; KO, knockout.
Compelling evidence has shown that IP-10, MIP-1α, and RANTES are potent factors that attract Th1 T cells into sites of inflammation 65 66 67 68 69 70. Further, Th1/Th2 T cells have been recently reported to differentially secrete RANTES, lymphotactin, and TCA-3, respectively 71. Enhanced expression of IP-10, MIP-1α, and RANTES in MCP-1+/+ EAE CNS tissue and undetectable TCA-3 expression in either MCP-1+/+ or MCP-1−/− support the notion that immune reactions within the CNS during EAE attacks are Th1 biased and such responses are more pronounced in mice with an intact MCP-1 gene.
Anti-MOG Ig Isotypes in MCP-1−/− and Wild-type Mice.
Sera from MCP-1+/+ and MCP-1−/− mice with EAE were analyzed for total IgG, IgG1, and IgG2a Abs against the immunizing MOG35–55 peptide. Wild-type and MCP-1–deficient mice produced similar amounts of total MOG-specific IgG (Fig. 10). Despite the disparity in clinical severity between wild-type and MCP-1–null mice, this finding was not unexpected, given the results of experiments using B cell–deficient mice, which showed that Ig does not play an important pathogenic role in MOG35–55 peptide-induced EAE in B6 mice 48.
Figure 10.

Serum concentrations of anti-MOG35–55 IgG, IgG1, and IgG2a Abs in MCP-1−/− and MCP-1+/+ littermate controls after immunization with MOG35–55 plus pertussis toxin (500 ng/injection). Indicated are days pi when sera were collected. Data are expressed as mean ± SD, n = 6, in each group. *P = 0.08, # P < 0.01. WT, wild-type; KO, knockout.
However, Ig isotype analyses differentiated the wild-type and MCP-1–deficient mice. Levels of anti-MOG35–55 IgG1 Abs in wild-type controls remained low from day 14 pi through day 60 pi (Fig. 10) and were not elevated at intermediate time points (data not shown). In contrast, significantly higher levels of anti-MOG35–55 IgG1 Abs were evident in MCP-1−/− mice (Fig. 10). Levels of anti-MOG35–55 IgG2a Abs showed a trend towards elevated levels in wild-type controls on day 14 pi, whereas no difference between wild-type and MCP-1–null mice was found on day 60 pi (Fig. 10).
Taken together, these results suggest that a polarized MOG35–55–induced Th1 immune response in wild-type mice leads to a suppressed Th2 response, characterized by undetectable IL-4, lower levels of IL-10, lower levels of anti-MOG35–55 IgG1, and higher levels of IgG2a. In this model, the absence of MCP-1 results in a shift towards a Th2-biased response, with reduced production of IFN-γ, enhanced secretion of IL-10, and higher levels of IgG1.
Discussion
We and others have previously shown that MCP-1 was markedly elevated in the CNS of SJL and B6 mice with EAE 33 35 and levels of MCP-1 expression correlated with the severity of relapsing EAE 72. Anti–MCP-1 Abs blocked relapses of EAE 36. CNS MCP-1 is largely produced by parenchymal astrocytes 34. MCP-1 expression by astrocytes in MS brain lesions has also been convincingly documented 40 41 42.
However, these studies did not establish a primary role for MCP-1 in disease pathogenesis. In recent definitive studies of EAE using CCR2-deficient mice 37 38, the relevant ligand for the deleted receptor was not defined. Moreover, MCP-1 exhibits attributes that argue for a role in restraining autoimmune demyelination. In particular, MCP-1 exerts a direct or indirect (via IL-4) impact on Th2 T cell development 11. Further, the presence of MCP-1 in in vitro cell culture systems decreased the encephalitogenic potential of T cells directed to PLP139–151 36. NK cells that inhibited the encephalitogenic potential of autoaggressive T cells in DA rats produced high levels of MCP-1 in vitro 73. The role(s) of MCP-1 in the pathogenesis and development of EAE (MS) has therefore been uncertain. Using gene-targeted mice, we demonstrate that lack of MCP-1 delays the onset of EAE and ameliorates its severity, by reducing the accumulation of inflammatory leukocytes within CNS. This phenotype was associated with impaired MOG35–55–specific Th1 immune responses.
Impaired macrophage recruitment into the CNS, as indicated by reduced total number of cells and percentage of CD11b+CD45++CD4− cells recovered from CNS in MCP-1–deficient mice in our study, is consistent with the reduction of macrophages in MCP-1−/− mice in contact hypersensitivity responses 2, in kidney and lung lesions of MCP-1−/− MRL-Faslpr mice 74, in aortic walls of MCP-1 and low density lipoprotein receptor double-deficient mice 75, and in atherosclerosis plaques from MCP-1−/− mice that overexpress apolipoprotein B 76.
Compelling evidence suggests that macrophages and their products can be detrimental in EAE and human MS. Expression of MHC class II is markedly elevated in EAE and MS 77, costimulatory molecules such as CD80, CD86 expressed mainly by macrophages have been demonstrated in MS lesions 78, and blockade of CD28/CD80, CD86 pathway prevents epitope spreading and clinical relapses of EAE 79. B7-1/B7-2−/− mice are resistant to EAE induction 47. Similarly, blocking of interactions between CD40 on macrophages and CD40L on T cells has been shown to effectively prevent EAE 80. Products of macrophages like TNF-α, IFN-γ, and nitric oxide have also been demonstrated to be critical in the effector phase of EAE 81 82. Macrophage depletion inhibits the induction of EAE 83. The absence of clinical EAE in MCP-1−/− recipients of wild-type encephalitogenic T cells further indicates the importance of CNS MCP-1 expression in recruiting macrophages to the CNS. We propose that it is the failure to recruit significant number of macrophages into CNS that constitutes the principal mechanism for resistance to EAE induction in MCP-1–deficient mice. Based on these studies, we cannot exclude the possibility that MCP-1 may directly alter trafficking pattern of dendritic cells in periphery or CNS, expression of costimulators, inflammatory cytokines, and adhesion molecules. Further studies are underway to address these issues.
As several studies have shown that MCP-1 is a critical factor for T cell commitment to the Th2 phenotype, we did not anticipate that MCP-1 gene disruption would result in reduced MOG35–55–specific Th1 immune response in these EAE experiments. Our results show that MOG35–55–specific MCP-1−/− T cells secreted a large amount of IFN-γ, although less than MCP-1+/+ T cells, but undetectable levels of IL-4. In view of the reciprocal regulation between IFN-γ and IL-10, the enhanced in vitro secretion of IL-10 by MCP-1−/− T cells might be secondary to reduced levels of IFN-γ. However, increased expression of IL-10 was not observed in vivo as demonstrated by the equal amount of IL-10 transcripts in MCP-1+/+ and MCP-1−/− EAE CNS tissue. The fact that MOG35–55–reactive MCP-1−/− T cells mediated severe EAE in wild-type recipient mice in the passive transfer EAE model further suggests that they were Th1 polarized. An explanation for the equal encephalitogenic capacity of MCP-1−/− T cells compared with MCP-1+/+ T cells could be that the defective IFN-γ production by MCP-1−/− T cells might be corrected by the presence of IL-12 in the cell culture system 84. Alternatively, IFN-γ concentrations beyond a threshold may be dispensable for encephalitogenic potential. T cells from MCP-1–null mice were shown to produce lower levels of IFN-γ in vitro upon MOG35–55 restimulation in our studies, and MCP-1−/− splenocytes secreted ∼50% less IFN-γ when restimulated with Schistosoma mansoni eggs in vitro 2, implying a role for MCP-1 in maximal expression of this cytokine under some circumstances. On the premise that MCP-1 is the major ligand for CCR2 in this model, our results are consistent with what has been recently reported in CCR2-deficient mice 38. Thus, a dual function is suggested for MCP-1 in regulating T cell immune responses: promoting Th2 immune responses in certain circumstance while facilitating Th1 responses in others. Such difference is not uncommon when molecules were tested in different animal strains, disease models, and using different immunogens.
We propose that the role of MCP-1 in EAE became manifest because of the extreme Th1 polarization implicated in this model. The impaired ability to mount Th2 responses was not relevant in these experiments because the disease was severely attenuated by the reduction of macrophage recruitment to the CNS. Such reduction of macrophage reaction might subsequently result in reduced Th1 immune responses. Remarkably, in the absence of recruited macrophages, highly polarized Th1 cells became unable to express the Th1 effector program, most clearly demonstrated by decreased circulating and CNS IFN-γ and failure to elicit EAE in MCP-1–deficient mice by MOG-primed MCP-1+/+ encephalitogenic T cells.
Taken in the context of recent reports 37 38, our results indicate that the MCP-1/CCR2 ligand/receptor pair is critical for the expression of EAE in mice. In turn, these findings motivate a continuing effort to characterize the function of this multipotential chemokine in human disease.
Acknowledgments
We thank Dr. W.J. Karpus (Northwestern University) for helpful discussion and sharing data before publication.
This work was supported by the National Institutes of Health (2RO1 NS32151 and 1PO1 NS38667 to R.M. Ransohoff; 2RO1 CA53091 to B.J. Rollins), and The National Multiple Sclerosis Society with a Pilot Project award to B.J. Rollins. We gratefully acknowledge the Williams Family Foundation for MS Research. D. Huang is a scholar of the Morgenthaler Family Foundation.
Footnotes
Abbreviations used in this paper: CCR, CC chemokine receptor; CNS, central nervous system; EAE, experimental autoimmune encephalomyelitis; IP-10, IFN-γ–inducible 10-kD protein; MCP, monocyte chemoattractant protein; MIP, macrophage inflammatory protein; MNC, mononuclear cell; MOG, myelin oligodendroglial glycoprotein; MS, multiple sclerosis; pi, postimmunization; PILN, popliteal and inguinal lymph node; PLP, proteolipid protein; RANTES, regulated upon activation, normal T cell expressed and secreted; RPA, RNase protection assay; RT, reverse transcription; TCA, T cell activation gene.
References
- Rollins B.J. Chemokines. Blood. 1997;90:909–928. [PubMed] [Google Scholar]
- Lu B., Rutledge B.J., Gu L., Fiorillo J., Lukacs N.W., Kunkel S.L., North R., Gerard C., Rollins B.J. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1–deficient mice. J. Exp. Med. 1998;187:601–608. doi: 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foti M., Granucci F., Aggujaro D., Liboi E., Luini W., Minardi S., Mantovani A., Sozzani S., Ricciardi-Castagnoli P. Upon dendritic cell (DC) activation chemokines and chemokine receptor expression are rapidly regulated for recruitment and maintenance of DC at the inflammatory site. Int. Immunol. 1999;11:979–986. doi: 10.1093/intimm/11.6.979. [DOI] [PubMed] [Google Scholar]
- Sallusto F., Palermo B., Lenig D., Miettinen M., Matikainen S., Julkunen I., Forster R., Burgstahler R., Lipp M., Lanzavecchia A. Distinct patterns and kinetics of chemokine production regulate dendritic cell function. Eur. J. Immunol. 1999;29:1617–1625. doi: 10.1002/(SICI)1521-4141(199905)29:05<1617::AID-IMMU1617>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Maghazachi A.A., Al-Aoukaty A., Schall T.J. C-C chemokines induce the chemotaxis of NK and IL-2-activated NK cells. Role for G proteins. J. Immunol. 1994;153:4969–4977. [PubMed] [Google Scholar]
- Maghazachi A.A., Al-Aoukaty A., Schall T.J. CC chemokines induce the generation of killer cells from CD56+ cells. Eur. J. Immunol. 1996;26:315–319. doi: 10.1002/eji.1830260207. [DOI] [PubMed] [Google Scholar]
- Taub D.D., Sayers T.J., Carter C.R., Ortaldo J.R. Alpha and beta chemokines induce NK cell migration and enhance NK-mediated cytolysis. J. Immunol. 1995;155:3877–3888. [PubMed] [Google Scholar]
- Karpus W.J., Lukacs N.W., Kennedy K.J., Smith W.S., Hurst S.D., Barrett T.A. Differential CC chemokine-induced enhancement of T helper cell cytokine production. J. Immunol. 1997;158:4129–4136. [PubMed] [Google Scholar]
- Chensue S.W., Warmington K.S., Ruth J.H., Sanghi P.S., Lincoln P., Kunkel S.L. Role of monocyte chemoattractant protein-1 (MCP-1) in Th1 (mycobacterial) and Th2 (schistosomal) antigen-induced granuloma formationrelationship to local inflammation, Th cell expression, and IL-12 production. J. Immunol. 1996;157:4602–4608. [PubMed] [Google Scholar]
- Matsukawa A., Lukacs N.W., Standiford T.J., Chensue S.W., Kunkel S.L. Adenoviral-mediated overexpression of monocyte chemoattractant protein-1 differentially alters the development of Th1 and Th2 type responses in vivo. J. Immunol. 2000;164:1699–1704. doi: 10.4049/jimmunol.164.4.1699. [DOI] [PubMed] [Google Scholar]
- Gu L., Tseng S., Horner R.M., Tam C., Loda M., Rollins B.J. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature. 2000;404:407–411. doi: 10.1038/35006097. [DOI] [PubMed] [Google Scholar]
- Martin R., McFarland H.F. Immunological aspects of experimental allergic encephalomyelitis and multiple sclerosis. Crit. Rev. Clin. Lab. Sci. 1995;32:121–182. doi: 10.3109/10408369509084683. [DOI] [PubMed] [Google Scholar]
- Steinman L. Multiple sclerosisa coordinated immunological attack against myelin in the central nervous system. Cell. 1996;85:299–302. doi: 10.1016/s0092-8674(00)81107-1. [DOI] [PubMed] [Google Scholar]
- Zamvil S.S., Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Annu. Rev. Immunol. 1990;8:579–621. doi: 10.1146/annurev.iy.08.040190.003051. [DOI] [PubMed] [Google Scholar]
- Rudick R.A., Cohen J.A., Weinstock-Guttman B., Kinkel R.P., Ransohoff R.M. Management of multiple sclerosis. N. Engl. J. Med. 1997;337:1604–1611. doi: 10.1056/NEJM199711273372207. [DOI] [PubMed] [Google Scholar]
- Finkelman F.D., Holmes J., Katona I.M., Urban J.F., Jr., Beckmann M.P., Park L.S., Schooley K.A., Coffman R.L., Mosmann T.R., Paul W.E. Lymphokine control of in vivo immunoglobulin isotype selection. Annu. Rev. Immunol. 1990;8:303–333. doi: 10.1146/annurev.iy.08.040190.001511. [DOI] [PubMed] [Google Scholar]
- Fiorentino D.F., Bond M.W., Mosmann T.R. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J. Exp. Med. 1989;170:2081–2095. doi: 10.1084/jem.170.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M.A., Hural J. Functions of IL-4 and control of its expression. Crit. Rev. Immunol. 1997;17:1–32. doi: 10.1615/critrevimmunol.v17.i1.10. [DOI] [PubMed] [Google Scholar]
- Davidson N.J., Fort M.M., Muller W., Leach M.W., Rennick D.M. Chronic colitis in IL-10−/− miceinsufficient counter regulation of a Th1 response. Int. Rev. Immunol. 2000;19:91–121. doi: 10.3109/08830180009048392. [DOI] [PubMed] [Google Scholar]
- Kuchroo V.K., Martin C.A., Greer J.M., Ju S.T., Sobel R.A., Dorf M.E. Cytokines and adhesion molecules contribute to the ability of myelin proteolipid protein-specific T cell clones to mediate experimental allergic encephalomyelitis. J. Immunol. 1993;151:4371–4382. [PubMed] [Google Scholar]
- Baron J.L., Madri J.A., Ruddle N.H., Hashim G., Janeway C.A., Jr. Surface expression of alpha 4 integrin by CD4 T cells is required for their entry into brain parenchyma. J. Exp. Med. 1993;177:57–68. doi: 10.1084/jem.177.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson T. Critical influences of the cytokine orchestration on the outcome of myelin antigen-specific T-cell autoimmunity in experimental autoimmune encephalomyelitis and multiple sclerosis. Immunol. Rev. 1995;144:245–268. doi: 10.1111/j.1600-065x.1995.tb00072.x. [DOI] [PubMed] [Google Scholar]
- Lafaille J.J., Keere F.V., Hsu A.L., Baron J.L., Haas W., Raine C.S., Tonegawa S. Myelin basic protein-specific T helper 2 (Th2) cells cause experimental autoimmune encephalomyelitis in immunodeficient hosts rather than protect them from the disease. J. Exp. Med. 1997;186:307–312. doi: 10.1084/jem.186.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Kuchroo V.K., Inobe J., Hafler D.A., Weiner H.L. Regulatory T cell clones induced by oral tolerancesuppression of autoimmune encephalomyelitis. Science. 1994;265:1237–1240. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- Racke M.K., Bonomo A., Scott D.E., Cannella B., Levine A., Raine C.S., Shevach E.M., Rocken M. Cytokine-induced immune deviation as a therapy for inflammatory autoimmune disease. J. Exp. Med. 1994;180:1961–1966. doi: 10.1084/jem.180.5.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw M.K., Lorens J.B., Dhawan A., DalCanto R., Tse H.Y., Tran A.B., Bonpane C., Eswaran S.L., Brocke S., Sarvetnick N. Local delivery of interleukin 4 by retrovirus-transduced T lymphocytes ameliorates experimental autoimmune encephalomyelitis. J. Exp. Med. 1997;185:1711–1714. doi: 10.1084/jem.185.9.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathisen P.M., Yu M., Johnson J.M., Drazba J.A., Tuohy V.K. Treatment of experimental autoimmune encephalomyelitis with genetically modified memory T cells. J. Exp. Med. 1997;186:159–164. doi: 10.1084/jem.186.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocke S., Gijbels K., Allegretta M., Ferber I., Piercy C., Blankenstein T., Martin R., Utz U., Karin N., Mitchell D. Treatment of experimental encephalomyelitis with a peptide analogue of myelin basic protein. Nature. 1996;379:343–346. doi: 10.1038/379343a0. [DOI] [PubMed] [Google Scholar]
- Falcone M., Rajan A.J., Bloom B.R., Brosnan C.F. A critical role for IL-4 in regulating disease severity in experimental allergic encephalomyelitis as demonstrated in IL-4-deficient C57BL/6 mice and BALB/c mice. J. Immunol. 1998;160:4822–4830. [PubMed] [Google Scholar]
- Cross A.H., Cannella B., Brosnan C.F., Raine C.S. Homing to central nervous system vasculature by antigen-specific lymphocytes. I. Localization of 14C-labeled cells during acute, chronic, and relapsing experimental allergic encephalomyelitis. Lab. Invest. 1990;63:162–170. [PubMed] [Google Scholar]
- Ransohoff R.M. Mechanisms of inflammation in MS tissueadhesion molecules and chemokines. J. Neuroimmunol. 1999;98:57–68. doi: 10.1016/s0165-5728(99)00082-x. [DOI] [PubMed] [Google Scholar]
- Karpus W.J., Ransohoff R.M. Chemokine regulation of experimental autoimmune encephalomyelitistemporal and spatial expression patterns govern disease pathogenesis. J. Immunol. 1998;161:2667–2671. [PubMed] [Google Scholar]
- Ransohoff R.M., Hamilton T.A., Tani M., Stoler M.H., Shick H.E., Major J.A., Estes M.L., Thomas D.M., Tuohy V.K. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB (Fed. Am. Soc. Exp. Biol.) J. 1993;7:592–600. doi: 10.1096/fasebj.7.6.8472896. [DOI] [PubMed] [Google Scholar]
- Glabinski A.R., Tani M., Strieter R.M., Tuohy V.K., Ransohoff R.M. Synchronous synthesis of alpha- and beta-chemokines by cells of diverse lineage in the central nervous system of mice with relapses of chronic experimental autoimmune encephalomyelitis. Am. J. Pathol. 1997;150:617–630. [PMC free article] [PubMed] [Google Scholar]
- Juedes A.E., Hjelmstrom P., Bergman C.M., Neild A.L., Ruddle N.H. Kinetics and cellular origin of cytokines in the central nervous systeminsight into mechanisms of myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. J. Immunol. 2000;164:419–426. doi: 10.4049/jimmunol.164.1.419. [DOI] [PubMed] [Google Scholar]
- Karpus W.J., Kennedy K.J. MIP-1alpha and MCP-1 differentially regulate acute and relapsing autoimmune encephalomyelitis as well as Th1/Th2 lymphocyte differentiation. J. Leukoc. Biol. 1997;62:681–687. [PubMed] [Google Scholar]
- Fife B.T., Huffnagle G.B., Kuziel W.A., Karpus W.J. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J. Exp. Med. 2000;192:899–906. doi: 10.1084/jem.192.6.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izikson L., Klein R.S., Charo I.F., Weiner H.L., Luster A.D. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J. Exp. Med. 2000;192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpus W.J., Kennedy K.J., Kunkel S.L., Lukacs N.W. Monocyte chemotactic protein 1 regulates oral tolerance induction by inhibition of T helper cell 1–related cytokines. J. Exp. Med. 1998;187:733–741. doi: 10.1084/jem.187.5.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Voorn P., Tekstra J., Beelen R.H., Tensen C.P., Van Der Valk P., De Groot C.J. Expression of MCP-1 by reactive astrocytes in demyelinating multiple sclerosis lesions. Am. J. Pathol. 1999;154:45–51. doi: 10.1016/S0002-9440(10)65249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus C., Berman J.W., Brett F.M., Staunton H., Farrell M., Brosnan C.F. MCP-1, MCP-2 and MCP-3 expression in multiple sclerosis lesionsan immunohistochemical and in situ hybridization study. J. Neuroimmunol. 1998;86:20–29. doi: 10.1016/s0165-5728(98)00002-2. [DOI] [PubMed] [Google Scholar]
- Simpson J.E., Newcombe J., Cuzner M.L., Woodroofe M.N. Expression of monocyte chemoattractant protein-1 and other beta-chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J. Neuroimmunol. 1998;84:238–249. doi: 10.1016/s0165-5728(97)00208-7. [DOI] [PubMed] [Google Scholar]
- Tuohy V.K., Lu Z., Sobel R.A., Laursen R.A., Lees M.B. Identification of an encephalitogenic determinant of myelin proteolipid protein for SJL mice. J. Immunol. 1989;142:1523–1527. [PubMed] [Google Scholar]
- Suen W.E., Bergman C.M., Hjelmstrom P., Ruddle N.H. A critical role for lymphotoxin in experimental allergic encephalomyelitis. J. Exp. Med. 1997;186:1233–1240. doi: 10.1084/jem.186.8.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.S., Hayakawa K., Hardy R.R. The regulated expression of B lineage associated genes during B cell differentiation in bone marrow and fetal liver. J. Exp. Med. 1993;178:951–960. doi: 10.1084/jem.178.3.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa M., Koh C.S., Inaba Y., Seki C., Inoue A., Itoh M., Ishihara Y., Bernard C.C., Komiyama A. IgG subclass switching is associated with the severity of experimental autoimmune encephalomyelitis induced with myelin oligodendrocyte glycoprotein peptide in NOD mice. Cell. Immunol. 1999;191:97–104. doi: 10.1006/cimm.1998.1414. [DOI] [PubMed] [Google Scholar]
- Chang T.T., Jabs C., Sobel R.A., Kuchroo V.K., Sharpe A.H. Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. J. Exp. Med. 1999;190:733–740. doi: 10.1084/jem.190.5.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmstrom P., Juedes A.E., Fjell J., Ruddle N.H. B-cell-deficient mice develop experimental allergic encephalomyelitis with demyelination after myelin oligodendrocyte glycoprotein sensitization. J. Immunol. 1998;161:4480–4483. [PubMed] [Google Scholar]
- Moore G.R., Traugott U., Farooq M., Norton W.T., Raine C.S. Experimental autoimmune encephalomyelitis. Augmentation of demyelination by different myelin lipids. Lab. Invest. 1984;51:416–424. [PubMed] [Google Scholar]
- Linthicum D.S., Munoz J.J., Blaskett A. Acute experimental autoimmune encephalomyelitis in mice. I. Adjuvant action of Bordetella pertussis is due to vasoactive amine sensitization and increased vascular permeability of the central nervous system. Cell. Immunol. 1982;73:299–310. doi: 10.1016/0008-8749(82)90457-9. [DOI] [PubMed] [Google Scholar]
- Levine S., Wenk E.J., Devlin H.B., Pieroni R.E., Levine L. Hyperacute allergic encephalomyelitisadjuvant effect of pertussis vaccines and extracts. J. Immunol. 1966;97:363–368. [PubMed] [Google Scholar]
- Sewell W.A., Munoz J.J., Vadas M.A. Enhancement of the intensity, persistence, and passive transfer of delayed-type hypersensitivity lesions by pertussigen in mice. J. Exp. Med. 1983;157:2087–2096. doi: 10.1084/jem.157.6.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewell W.A., Munoz J.J., Scollay R., Vadas M.A. Studies on the mechanism of the enhancement of delayed-type hypersensitivity by pertussigen. J. Immunol. 1984;133:1716–1722. [PubMed] [Google Scholar]
- Sewell W.A., de Moerloose P.A., McKimm-Breschkin J.L., Vadas M.A. Pertussigen enhances antigen-driven interferon-gamma production by sensitized lymphoid cells. Cell. Immunol. 1986;97:238–247. doi: 10.1016/0008-8749(86)90394-1. [DOI] [PubMed] [Google Scholar]
- Ryan M., McCarthy L., Rappuoli R., Mahon B.P., Mills K.H. Pertussis toxin potentiates Th1 and Th2 responses to co-injected antigenadjuvant action is associated with enhanced regulatory cytokine production and expression of the co-stimulatory molecules B7-1, B7-2 and CD28. Int. Immunol. 1998;10:651–662. doi: 10.1093/intimm/10.5.651. [DOI] [PubMed] [Google Scholar]
- Vistica B.P., McAllister C.G., Sekura R.D., Ihle J.N., Gery I. Dual effects of pertussis toxin on lymphoid cells in culture. Cell. Immunol. 1986;101:232–241. doi: 10.1016/0008-8749(86)90200-5. [DOI] [PubMed] [Google Scholar]
- Youssef S., Wildbaum G., Maor G., Lanir N., Gour-Lavie A., Grabie N., Karin N. Long-lasting protective immunity to experimental autoimmune encephalomyelitis following vaccination with naked DNA encoding C-C chemokines. J. Immunol. 1998;161:3870–3879. [PubMed] [Google Scholar]
- Franci C., Wong L.M., Van Damme J., Proost P., Charo I.F. Monocyte chemoattractant protein-3, but not monocyte chemoattractant protein-2, is a functional ligand of the human monocyte chemoattractant protein-1 receptor. J. Immunol. 1995;154:6511–6517. [PubMed] [Google Scholar]
- Combadiere C., Ahuja S.K., Van Damme J., Tiffany H.L., Gao J.L., Murphy P.M. Monocyte chemoattractant protein-3 is a functional ligand for CC chemokine receptors 1 and 2B. J. Biol. Chem. 1995;270:29671–29675. doi: 10.1074/jbc.270.50.29671. [DOI] [PubMed] [Google Scholar]
- Boring L., Gosling J., Chensue S.W., Kunkel S.L., Farese R.V., Jr., Broxmeyer H.E., Charo I.F. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J. Clin. Invest. 1997;100:2552–2561. doi: 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesch G.H., Maifert S., Schwarting A., Rollins B.J., Kelley V.R. Monocyte chemoattractant protein 1-dependent leukocytic infiltrates are responsible for autoimmune disease in MRL-Fas(lpr) mice. J. Exp. Med. 1999;190:1813–1824. doi: 10.1084/jem.190.12.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Zhang S.I., Pernis B. Role of CD8+ T cells in murine experimental allergic encephalomyelitis. Science. 1992;256:1213–1215. doi: 10.1126/science.256.5060.1213. [DOI] [PubMed] [Google Scholar]
- Bettelli E., Das M.P., Howard E.D., Weiner H.L., Sobel R.A., Kuchroo V.K. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J. Immunol. 1998;161:3299–3306. [PubMed] [Google Scholar]
- Samoilova E.B., Horton J.L., Chen Y. Acceleration of experimental autoimmune encephalomyelitis in interleukin-10-deficient miceroles of interleukin-10 in disease progression and recovery. Cell. Immunol. 1998;188:118–124. doi: 10.1006/cimm.1998.1365. [DOI] [PubMed] [Google Scholar]
- Sallusto F., Mackay C.R., Lanzavecchia A. The role of chemokine receptors in primary, effector, and memory immune responses. Annu. Rev. Immunol. 2000;18:593–620. doi: 10.1146/annurev.immunol.18.1.593. [DOI] [PubMed] [Google Scholar]
- Sallusto F., Mackay C.R., Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science. 1997;277:2005–2007. doi: 10.1126/science.277.5334.2005. [DOI] [PubMed] [Google Scholar]
- Sallusto F., Lanzavecchia A., Mackay C.R. Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol. Today. 1998;19:568–574. doi: 10.1016/s0167-5699(98)01346-2. [DOI] [PubMed] [Google Scholar]
- Sallusto F., Lenig D., Mackay C.R., Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J. Exp. Med. 1998;187:875–883. doi: 10.1084/jem.187.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonecchi R., Bianchi G., Bordignon P.P., D'Ambrosio D., Lang R., Borsatti A., Sozzani S., Allavena P., Gray P.A., Mantovani A., Sinigaglia F. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J. Exp. Med. 1998;187:129–134. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siveke J.T., Hamann A. T helper 1 and T helper 2 cells respond differentially to chemokines. J. Immunol. 1998;160:550–554. [PubMed] [Google Scholar]
- Zhang S., Lukacs N.W., Lawless V.A., Kunkel S.L., Kaplan M.H. Cutting edgedifferential expression of chemokines in Th1 and Th2 cells is dependent on Stat6 but not Stat4. J. Immunol. 2000;165:10–14. doi: 10.4049/jimmunol.165.1.10. [DOI] [PubMed] [Google Scholar]
- Kennedy K.J., Strieter R.M., Kunkel S.L., Lukacs N.W., Karpus W.J. Acute and relapsing experimental autoimmune encephalomyelitis are regulated by differential expression of the CC chemokines macrophage inflammatory protein-1alpha and monocyte chemotactic protein-1. J. Neuroimmunol. 1998;92:98–108. doi: 10.1016/s0165-5728(98)00187-8. [DOI] [PubMed] [Google Scholar]
- Smeltz R.B., Wolf N.A., Swanborg R.H. Inhibition of autoimmune T cell responses in the DA rat by bone marrow-derived NK cells in vitroimplications for autoimmunity. J. Immunol. 1999;163:1390–1397. [PubMed] [Google Scholar]
- Tesch G.H., Schwarting A., Kinoshita K., Lan H.Y., Rollins B.J., Kelley V.R. Monocyte chemoattractant protein-1 promotes macrophage-mediated tubular injury, but not glomerular injury, in nephrotoxic serum nephritis. J. Clin. Invest. 1999;103:73–80. doi: 10.1172/JCI4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L., Okada Y., Clinton S.K., Gerard C., Sukhova G.K., Libby P., Rollins B.J. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell. 1998;2:275–281. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- Gosling J., Slaymaker S., Gu L., Tseng S., Zlot C.H., Young S.G., Rollins B.J., Charo I.F. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J. Clin. Invest. 1999;103:773–778. doi: 10.1172/JCI5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R., McFarland H.F., McFarlin D.E. Immunological aspects of demyelinating diseases. Annu. Rev. Immunol. 1992;10:153–187. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- Windhagen A., Newcombe J., Dangond F., Strand C., Woodroofe M.N., Cuzner M.L., Hafler D.A. Expression of costimulatory molecules B7-1 (CD80), B7-2 (CD86), and interleukin 12 cytokine in multiple sclerosis lesions. J. Exp. Med. 1995;182:1985–1996. doi: 10.1084/jem.182.6.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S.D., Vanderlugt C.L., Lenschow D.J., Pope J.G., Karandikar N.J., Dal Canto M.C., Bluestone J.A. Blockade of CD28/B7-1 interaction prevents epitope spreading and clinical relapses of murine EAE. Immunity. 1995;3:739–745. doi: 10.1016/1074-7613(95)90063-2. [DOI] [PubMed] [Google Scholar]
- Gerritse K., Laman J.D., Noelle R.J., Aruffo A., Ledbetter J.A., Boersma W.J., Claassen E. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. USA. 1996;93:2499–2504. doi: 10.1073/pnas.93.6.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misko T.P., Trotter J.L., Cross A.H. Mediation of inflammation by encephalitogenic cellsinterferon gamma induction of nitric oxide synthase and cyclooxygenase 2. J. Neuroimmunol. 1995;61:195–204. doi: 10.1016/0165-5728(95)00091-f. [DOI] [PubMed] [Google Scholar]
- Lin R.F., Lin T.S., Tilton R.G., Cross A.H. Nitric oxide localized to spinal cords of mice with experimental allergic encephalomyelitisan electron paramagnetic resonance study. J. Exp. Med. 1993;178:643–648. doi: 10.1084/jem.178.2.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E.H., Hoekstra K., van Rooijen N., Dijkstra C.D., Owens T. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J. Immunol. 1998;161:3767–3775. [PubMed] [Google Scholar]
- Segal B.M., Shevach E.M. IL-12 unmasks latent autoimmune disease in resistant mice. J. Exp. Med. 1996;184:771–775. doi: 10.1084/jem.184.2.771. [DOI] [PMC free article] [PubMed] [Google Scholar]