

Abstract
Tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) is expressed by in vitro activated natural killer (NK) cells, but the relevance of this observation to the biological function of NK cells has been unclear. Herein, we have demonstrated the in vivo induction of mouse TRAIL expression on various tissue NK cells and correlated NK cell activation with TRAIL-mediated antimetastatic function in vivo. Expression of TRAIL was only constitutive on a subset of liver NK cells, and innate NK cell control of Renca carcinoma hepatic metastases in the liver was partially TRAIL dependent. Administration of therapeutic doses of interleukin (IL)-12, a powerful inducer of interferon (IFN)-γ production by NK cells and NKT cells, upregulated TRAIL expression on liver, spleen, and lung NK cells, and IL-12 suppressed metastases in both liver and lung in a TRAIL-dependent fashion. By contrast, α-galactosylceramide (α-GalCer), a powerful inducer of NKT cell IFN-γ and IL-4 secretion, suppressed both liver and lung metastases but only stimulated NK cell TRAIL-mediated function in the liver. TRAIL expression was not detected on NK cells from IFN-γ–deficient mice and TRAIL-mediated antimetastatic effects of IL-12 and α-GalCer were strictly IFN-γ dependent. These results indicated that TRAIL induction on NK cells plays a critical role in IFN-γ–mediated antimetastatic effects of IL-12 and α-GalCer.
Keywords: rodent, tumor immunity, in vivo animal models, immunotherapy, interleukin 12
Introduction
Members of the TNF family of cytokines and receptors are critically involved in cellular activation, proliferation and death 1 2. Within this family, TNF, lymphotoxin-α (LT-α), Fas ligand (FasL; reference 1), Apo3L 3, and TNF-related apoptosis-inducing ligand (TRAIL 4) have been characterized as major mediators of apoptosis in vitro. TRAIL is a characteristic type II transmembrane protein, sharing homology with FasL in the extracellular receptor–binding motif 4. Like FasL, TRAIL induces apoptosis in tumor cells and virus-infected cells either in vitro or in vivo 4 5 6 7. Unlike other TNF family ligands such as TNF and FasL, TRAIL does not kill cells from most normal primary tissues 4 8. These properties have seen TRAIL pursued as an anticancer therapeutic itself or in combination with chemotherapeutic drugs 9 10 11. Despite current pursuit of TRAIL as a selective anticancer therapeutic, little is known of the natural physiological role of TRAIL.
Although TRAIL mRNA has been observed in various cell types 4 12, TRAIL protein expression in vivo has been comparatively poorly defined. TRAIL expression was characterized on IFN-α–stimulated peripheral blood T cells 13 and IFN-stimulated human monocytes and dendritic cells 14 15. TRAIL has also been demonstrated on CD3−NK1.1+ NK cells after stimulation with IL-2, IFNs, or IL-15 16 17. TRAIL-expressing CD4+ T cells and mouse NK cells were capable of mediating apoptosis in a TRAIL-specific fashion 17 18. These preliminary findings suggested that TRAIL may play a role in innate immune responses involving IFN and NK cells. We have established several mouse spontaneous and metastatic tumor models in which NK cells use the cytotoxic granule protein, perforin (pfp), to control tumor initiation and metastasis 19 20. More recently, we have shown that IFN-γ secretion independently contributes to the natural NK cell response 21 or the NK cell response activated by biological response modifiers such as IL-12 22. Therefore, we rationalized that TRAIL may contribute to NK cell–mediated antitumor responses that are IFN-γ dependent. In this study, we observed that potent inducers of IFN-γ and NK cell effector function partially mediate their antitumor activity by stimulating TRAIL expression on NK cells.
Materials and Methods
Mice.
Inbred BALB/c wild-type (WT) mice were purchased from The Walter and Eliza Hall Institute of Medical Research and Clear Japan Inc. The following gene-targeted mice were bred at the Peter MacCallum Cancer Institute: BALB/c pfp deficient (BALB/c pfp−/−; reference 23), BALB/c IFN-γ deficient (BALB/c IFN-γ−/−; reference 24), and BALB/c CD1d deficient (BALB/c CD1d−/−; provided by Dr. L. Van Kaer, Vanderbilt University School of Medicine, Nashville, TN). Mice of 6–10 wk of age were used in all experiments that were performed according to animal experimental ethics committee guidelines.
Tumor Cells and Reagents.
The BALB/c-derived renal adenocarcinoma cell line, Renca (H-2d), was provided by Dr. T. Sayers (National Cancer Institute-Frederick Cancer Research and Development Center) and maintained in RPMI 1640 containing 10% FCS and 2 mM l-glutamine. Recombinant mouse IL-12 and IL-2 was provided by Genetics Institute (Andover, MA) and Chiron Corp. (Emeryville, CA), respectively. Recombinant mouse IFN-γ was purchased from BD PharMingen. The preparations of IL-12 and IL-2 were diluted in PBS immediately before use. α-galactosylceramide (α-GalCer) was provided by the Pharmaceutical Research laboratories, Kirin Brewery (Gumna, Japan), and prepared as described 25. α-GalCer was suspended in saline supplemented with 0.5% polysorbate-20 (wt/vol) and the control vehicle was saline supplemented with 0.5% polysorbate-20 (wt/vol). Concanamycin A (CMA), which inhibits pfp-mediated cytotoxicity 17, was purchased from Wako Pure Chemicals.
Flow Cytometric Analysis.
Mononuclear cells (MNCs) were prepared from the spleen, liver, lung, and peripheral blood as described previously 26 27. To avoid the nonspecific binding of Abs to FcγR, the cells were preincubated with anti–mouse CD16/32 (2.4G2) mAb before staining. The cells were then incubated with a saturating amount (1 μg) of biotinylated anti–mouse TRAIL (N2B2) mAb 17 before incubation with a saturating amount of PE-conjugated streptavidin, FITC-conjugated anti–mouse NK cell (DX5) mAb, and Cy-Chrome–conjugated anti-CD3 (145-2C11) mAb. All these reagents, except anti–mouse TRAIL mAb, were obtained from BD PharMingen. After washing with PBS twice, the stained cells were analyzed on a FACScan™ (Becton Dickinson) and the data were processed by the CELLQuest™ program (Becton Dickinson).
Cytotoxicity and Serum IFN-γ Assays.
Cytotoxic activities of hepatic, spleen, and lung MNCs were tested against Renca cells by an 8 h 51Cr-release assay as described previously 13 17. In some experiments, the assay was performed in the presence of anti-TRAIL (N2B2) mAb (10 μg/ml), control Ig (rat IgG2a, R35-95; 10 μg/ml; BD PharMingen), and/or CMA (50 nM). Serum was collected from various treated mice as indicated and measured by a mouse IFN-γ–specific ELISA according to the manufacturer's protocol (BD PharMingen).
Experimental Metastasis Assay.
BALB/c WT and gene-targeted mice were injected intrasplenically or intravenously with Renca tumor cells as described previously 28. Mice were killed 14 d after tumor inoculation and liver (after intrasplenic) or lung (after intravenous) metastases were quantified with the aid of a dissecting microscope. Some mice received either anti–mouse TRAIL mAb (250 μg intraperitoneally) or control rat IgG2a (R35–95; 250 μg/ml intraperitoneally) on days 0, 1, and 7 after tumor inoculation. Some groups of mice were additionally depleted of NK cells in vivo by treatment with 20 μg polyclonal rabbit anti-asialoGM1 (anti-asGM1) Ab (Wako) on days −1, 0, and 7 relative to tumor inoculation. This depletion protocol has been shown to selectively deplete NK cells, but not other leukocyte subsets including NKT cells, in both C57BL/6 and BALB/c mouse strains 21. Anti-TRAIL mAb did not deplete NK cells in any organ examined (data not shown). Antimetastatic treatment protocols were as follows: IL-2, 100,000 U/200 μl PBS intraperitoneally daily on days 3 through 7 (where day 0 was the day of tumor inoculation); IL-12, 500 U/200 μl PBS intraperitoneally daily on days 3 through 7; and α-GalCer, 2 μg/200 μl PBS intraperitoneally on days 0, 4, and 8. These regimes were chosen based on previous efficacy studies of these immunotherapies in Renca and other tumor models (22, 27, 28, and data not shown).
Results
IL-12 and α-GalCer Specifically Induce TRAIL In Vivo.
IL-2 29, IL-12 30 31 32, and α-GalCer 25 33 34 have been shown to induce proliferation, IFN-γ production and cytotoxicity of NK cells in vivo, which are associated with their antitumor effects. Regimes of treatments using IL-2, IL-12, or α-GalCer alone were designed based on their efficacy against Renca tumor metastases. The MNCs in each of liver, spleen, lung, and peripheral blood (PBMC) were analyzed 24 h after the final treatment for DX5 and CD3 expression (Fig. 1 A). Two- to threefold more cells were present in the livers and spleens of treated mice compared with control treated mice (data not shown). TRAIL expression has not previously been characterized on lymphocyte populations stimulated with these biological response modifiers. A low level of constitutive TRAIL expression was found on freshly isolated liver CD3−DX-5+ NK cells, but not CD3+DX-5− T cells, CD3+DX-5+ T cells, or CD3−DX-5− cells including B cells and macrophages from the liver of untreated BALB/c mice (Fig. 1 B, control). NK cells freshly isolated from the spleen, lung, and peripheral blood (Fig. 1 B, control) did not express TRAIL. Administration of IL-12 (500 U intraperitoneally for 5 d) selectively upregulated TRAIL expression on NK cells in the liver and also induced TRAIL expression on spleen and lung NK cells (Fig. 1 B). A single injection of 500 U IL-12 was sufficient to induce expression of TRAIL on NK cells, but expression was comparatively low. Single or repeated IL-12 injections did not induce TRAIL on DX-5+ T cells (data not shown). Administration of α-GalCer (2 μg intraperitoneally for days 0, 4, and 8) also upregulated TRAIL on liver NK cells (on day 9) and induced TRAIL on spleen NK cells, but not on lung NK cells (Fig. 1 B). A single injection of 2 μg α-GalCer did not induce detectable TRAIL expression on NK cells until 48 h later, but TRAIL expression remained significant for at least 4 d. TRAIL expression was never detected on lung NK cells or any DX-5+ T cells (BALB/c) or NK1.1+ T cells (C57BL/6; 3, 6, 24, 72, or 96 h) after α-GalCer treatment (data not shown). Administration of IL-2 (100,000 U intraperitoneally for 5 d) induced NK cell proliferation (data not shown), but did not induce TRAIL expression on NK cells or T cells in any organs examined (Fig. 1 B). TRAIL was never induced on NK cells after IL-2 administration. These results indicated that IL-12 and α-GalCer selectively induce TRAIL expression on NK cells in vivo.
Figure 1.


IL-12 and α-GalCer induce TRAIL expression on NK cells. MNCs from the liver, spleen, lung, and peripheral blood (PBMC) were isolated from PBS or vehicle-treated (control) BALB/c mice or IL-12 (500 U intraperitoneally on days −5, −4, −3, −2, and −1), α-GalCer (α-GC; 2 μg intraperitoneally on days –8, −4, and 0), or IL-2 (100,000 U intraperitoneally on days −5, −4, −3, −2, and −1)–treated BALB/c mice at 24 h after the treatment. These cells were stained with biotin-conjugated anti-TRAIL mAb followed by PE-conjugated streptavidin, FITC-conjugated anti-DX5 mAb, and Cy-Chrome–conjugated anti-CD3 mAb. (A) DX5 (y-axis) vs. CD3 (x-axis) staining profiles of MNCs from each organ after the indicated treatments. Percentages of DX5+CD3− cells and DX5+CD3+ cells are indicated at the top left and top right, respectively. Two- to threefold more cells were present in the livers and spleens of IL-12–, α-Gal Cer–, or IL-2–treated mice. (B) Expression of TRAIL was analyzed on electronically gated CD3−DX-5+ (NK) cells, CD3+DX-5− (T) cells, CD3+DX-5+ T cells, and CD3−DX-5− (DN) cells by flow cytometry. Bold lines indicate staining with anti-TRAIL mAb and plain lines indicate staining with isotype-matched control IgG2a. These analyses (A and B) have been performed on more than three occasions and these profiles are representative.
IL-12 and α-GalCer Induce TRAIL-mediated Cytotoxicity.
To evaluate the contribution of TRAIL to the cytotoxicity of hepatic, splenic, and lung NK cells after the administration of IL-12, α-GalCer, or IL-2, the effect of neutralizing anti-TRAIL mAb was examined in a cytotoxicity assay using liver, spleen, or lung MNCs as the effector and TRAIL-sensitive Renca tumor cells as the target. The cytotoxicity of liver MNCs from untreated control mice was inhibited partially by anti-TRAIL mAb alone and completely by the combination with a pfp inhibitor CMA (Fig. 2, left). Administration of IL-12, α-GalCer, or IL-2 markedly augmented the cytotoxicity of liver MNCs, which was also inhibited partially by anti-TRAIL mAb alone and completely by combination with CMA. The cytotoxicity of spleen MNCs from untreated or IL-2–treated mice was completely abrogated by CMA, whereas that from IL-12– or α-GalCer–treated mice was inhibited partially by anti-TRAIL mAb alone and completely by the combination with CMA (Fig. 2, middle). The cytotoxicity of lung MNCs from untreated, α-GalCer–treated, or IL-2–treated mice was abrogated by CMA alone, whereas that from IL-12–treated mice was inhibited partially by anti-TRAIL mAb alone and completely by the combination with CMA (Fig. 3, right). All these partial contributions of TRAIL to the cytotoxicity were totally consistent with the TRAIL expression on liver NK cells, spleen NK cells after IL-12 or α-GalCer treatment, and lung NK cells after IL-12 treatment shown in Fig. 1.
Figure 2.

IL-12 and α-GalCer induce TRAIL-mediated cytotoxicity. Liver, spleen, and lung MNCs were isolated (day 0) from PBS or vehicle-treated (control) BALB/c mice or IL-12 (500 U intraperitoneally on days −5, −4, −3, −2, and −1), α-GalCer (α-GC; 2 μg intraperitoneally on days −8, −4, and 0), or IL-2 (100,000 U intraperitoneally on days −5, −4, −3, −2, and −1)–treated BALB/c mice at 24 h after the treatment. Their cytotoxic activities were tested against TRAIL-sensitive Renca tumor cells in the presence or absence of 50 nM CMA, 10 μg/ml of anti-TRAIL mAb, or 10 μg/ml of control rat IgG2a mAb (control IgG) by 8 h 51Cr-release assay at several effector/target ratios (100:1 spleen MNCs, 50:1 liver and lung MNCs, shown). Data are represented as the mean ± SD of triplicate samples. Similar results were obtained in three independent experiments. *P < 0.05, compared with control IgG by two sample t tests.
Figure 3.

The antimetastatic activity of IL-12– or α-GalCer–activated liver NK cells involves TRAIL effector function. Groups of 5 to 20 WT mice were inoculated intrasplenically with (A) between 1 × 104 and 3 × 105 Renca tumor cells or (B) 3 × 105 Renca tumor cells on day 0. As indicated, some groups of mice were treated with one or more of: 500 U IL-12 intraperitoneally on days 3 through 7; 100,000 U IL-2 intraperitoneally on days 3 through 7; 2 μg α-GalCer (α-GC) intraperitoneally on days 0, 4, and 8; 20 μg anti-asGM1 on days −1, 0, and 7; and 0.25 mg anti-TRAIL mAb or control IgG intraperitoneally on days 0, 1, and 7. The livers were removed from mice on day 14, and the metastatic nodules quantified. Data are recorded as the mean ± SE, with the significance of anti-TRAIL mAb-treated mice compared with untreated or control IgG-treated mice as defined by Mann Whitney U test, *P < 0.05, **P < 0.01. IL-12, α-GalCer, and IL-2 were also statistically effective alone compared with no treatment in the livers of WT mice (P < 0.01). There was no effect of rabbit IgG (control for anti-asGM1) on the efficacy of α-GalCer, IL-12, or IL-2 (data not shown).
Involvement of TRAIL in Antimetastatic Effects of IL-12 and α-GalCer.
IL-2, IL-12, and α-GalCer have been demonstrated to possess potent antimetastatic activity when administered to mice 22 26 27 28 35 36 37. In particular, IL-2 and IL-12 have previously been shown to substantially reduce Renca tumor cell metastasis in BALB/c mice 28. Varying doses of Renca cells were inoculated intrasplenically, and at the lower tumor doses administered, anti-TRAIL mAb significantly increased liver metastases, whereas isotype control IgG where tested was without effect (Fig. 3 A). This data was consistent with constitutive expression of cytotoxic TRAIL on liver NK cells (Fig. 1 B). A minimum inoculum of 3 × 105 Renca tumor cells that metastasized equivalently in untreated or anti-TRAIL–treated WT mice was used for subsequent immunotherapy experiments. After intrasplenic inoculation of 3 × 105 Renca cells, therapeutic administration of IL-2, IL-12, or α-GalCer significantly reduced numbers of Renca liver metastases (Fig. 3 B). These antimetastatic activities were completely abolished by anti-asGM1 Ab, indicating that all these modifiers mediated their activities via NK cell effector function. In concert with increased cytotoxicity by activated liver MNCs (Fig. 2), the neutralizing anti-TRAIL mAb significantly increased the number of Renca liver metastases in IL-12– or α-GalCer–treated mice (Fig. 3). By contrast, anti-TRAIL mAb only marginally reduced the antimetastatic activity of IL-2 (Fig. 3). These data indicated that IL-12– or α-GalCer–, but not IL-2–stimulated antitumor activity in the liver was partially effected by TRAIL.
As constitutive TRAIL expression was not observed on spleen or lung NK cells, we reasoned that TRAIL function may not be observed in Renca models of lung metastasis. Indeed at three Renca doses examined (1 × 104, 5 × 104, and 1 × 105), anti-TRAIL mAb did not significantly enhance Renca lung metastasis (Fig. 4 A). However, after treatment with IL-12, TRAIL expression was observed on lung NK cells (Fig. 1 B) and IL-12 significantly reduced the number of Renca lung metastases (Fig. 4 B). Furthermore, anti-TRAIL mAb significantly increased Renca lung metastases in IL-12–treated mice, indicating that part of the antimetastatic activity of IL-12 was mediated by TRAIL (Fig. 4 B). Lung NK cell TRAIL expression was not induced in α-GalCer–treated mice and no significant antimetastatic effect of TRAIL was detected in the lung (Fig. 4 B). NK cell TRAIL expression and function was not detected in the lung after an antimetastatic dose of IL-2 (Fig. 4 B).
Figure 4.

IL-12 stimulates TRAIL-mediated antimetastatic activity in the lung. Groups of 5 to 20 WT mice were inoculated intravenously with (A) between 104 and 105 Renca tumor cells or (B) 105 Renca tumor cells on day 0. As indicated, some groups of mice were treated with one or more of: 500 U IL-12 intraperitoneally on days 3 through 7; 100,000 U IL-2 intraperitoneally on days 3 through 7; 2 μg α-GalCer (α-GC) intraperitoneally on days 0, 4, and 8; 20 μg anti-asGM1 on days −1, 0, and 7; and 0.25 mg anti-TRAIL mAb or control IgG intraperitoneally on days 0, 1, and 7. The lungs were removed from mice on day 14, and the metastatic nodules quantified. Data are recorded as the mean ± SE, with the significance of anti-TRAIL mAb-treated mice compared with untreated or control IgG-treated mice as defined by Mann Whitney U test; *P < 0.05, **P < 0.01. IL-12, α-GalCer, and IL-2 were also statistically effective alone compared with no treatment in the livers of WT mice (P < 0.01). There was no effect of rabbit IgG (control for anti-asGM1) on the efficacy of α-GalCer, IL-12, or IL-2 (data not shown).
Key Role of IFN-γ in TRAIL-mediated Antimetastatic Effect of IL-12.
Using gene-targeted mice we next examined the relative role of pfp and IFN-γ in the TRAIL-mediated antimetastatic activity stimulated by IL-12. Consistent with our previous observations 21, both pfp and IFN-γ contributed to NK cell control of Renca tumor metastasis. IFN-γ played a greater relative role than pfp in IL-12–mediated antimetastatic activity of NK cells in the liver (Fig. 5 A). In the lung, pfp-mediated protection was more important (Fig. 5 B). In both liver (Fig. 5 A) and lung (Fig. 5 B) metastasis models, anti-TRAIL mAb enhanced metastasis in IL-12–treated pfp−/− but not IFN-γ−/− mice. These data indicated that TRAIL is a key effector molecule mediating the IFN-γ–dependent antimetastatic effect of IL-12.
Figure 5.
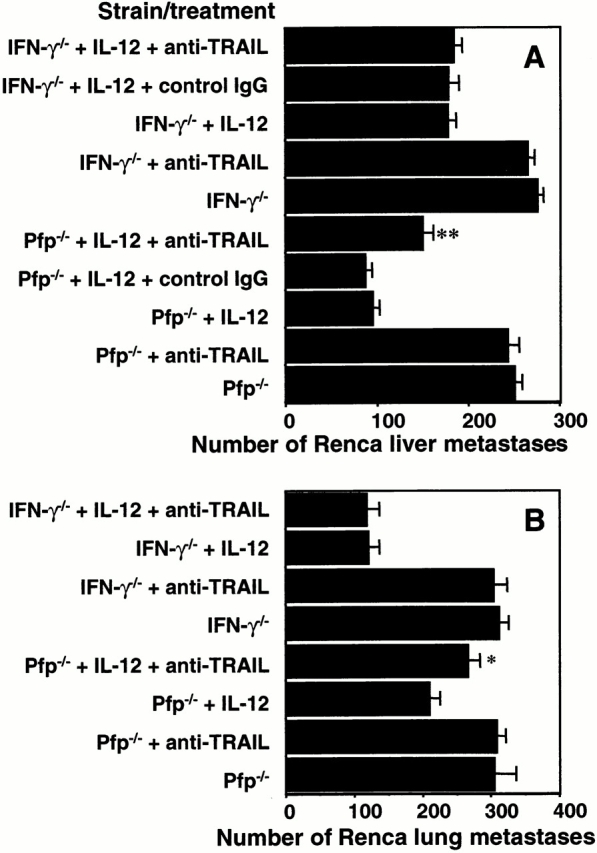
Key role of IFN-γ in TRAIL-mediated antimetastatic effect of IL-12. Groups of 5 to 15 IFN-γ−/− or pfp−/− mice were inoculated (A) intrasplenically with 3 × 105 Renca tumor cells or (B) intravenously with 1 × 105 Renca tumor cells on day 0. As indicated, some groups of mice were treated with: 500 U IL-12 intraperitoneally on days 3 through 7 and/or 0.25 mg anti-TRAIL mAb or control IgG intraperitoneally on days 0, 1, and 7. The (A) livers and (B) lungs were removed from mice on day 14, and the metastatic nodules quantified. Data are recorded as the mean ± SE, with the significance of anti-TRAIL mAb-treated mice compared with untreated or control IgG-treated mice as defined by *P < 0.05, **P < 0.01, Mann-Whitney U test. IL-12 was statistically effective alone compared with no treatment in the livers and lungs of both pfp−/− and IFN-γ−/− mice (P < 0.01). Control IgG was without effect in the experiment shown in B (data not shown).
Role of IFN-γ in Antimetastatic Effect of α-GalCer.
Using pfp−/− and IFN-γ−/− mice we next examined the involvement of IFN-γ and TRAIL in the antimetastatic activity stimulated by α-GalCer. In both liver (Fig. 6 A) and lung (Fig. 6 B) metastasis models, IFN-γ was essential for the α-GalCer activity. By contrast, α-GalCer significantly reduced Renca metastasis in pfp−/− mice, in both liver and lung. Anti-TRAIL mAb only enhanced liver metastasis in α-GalCer–treated pfp−/− mice and TRAIL antitumor function was not detected in the lung after α-GalCer administration. In summary, α-GalCer demonstrated IFN-γ–dependent antimetastatic activity in both liver and lung; however, TRAIL expression (Fig. 1 B) and function was only demonstrated in the liver.
Figure 6.

Role of IFN-γ and TRAIL in antimetastatic effect of α-GalCer. Groups of five IFN-γ−/− or pfp−/− mice were inoculated (A) intrasplenically with 3 × 105 Renca tumor cells or (B) intravenously with 1 × 105 Renca tumor cells on day 0. As indicated, some groups of mice were treated with: 2 μg α-GalCer (α-GC) intraperitoneally on days 0, 4, and 8 and/or 0.25 mg anti-TRAIL mAb on days 0, 1, and 7. The (A) livers and (B) lungs were removed from mice on day 14, and the metastatic nodules quantified. Data are recorded as the mean ± SE, with the signi-ficance of anti-TRAIL mAb-treated mice compared with mice treated with α-GalCer alone as defined by *P < 0.05, **P < 0.01, Mann-Whitney U test. Control IgG was without effect in pfp−/− or IFN-γ−/− mice (data not shown). α-GalCer was statistically effective alone compared with no treatment in the livers and lungs of pfp−/− mice (P < 0.01).
Correlation between IFN-γ Activity and NK Cell TRAIL Expression.
To further demonstrate the relationship between IFN-γ activity and TRAIL expression and function on NK cells, we characterized the kinetics of serum IFN-γ induction in mice that had received a single or multiple (regime) doses of IL-12, α-GalCer, or IL-2 (Fig. 7 A). After a single dose of IL-12 or α-GalCer a significant induction of IFN-γ was detected in the serum with levels peaking at between 6 to 10 ng/ml. IL-2 also induced serum IFN-γ; however, the levels were more moderate (<3 ng/ml). The rapid induction of serum IFN-γ in response to α-GalCer was also evident in anti-asGM1–treated mice, but did not occur in CD1d−/− mice, indicating the early and critical role for NKT cells in this response. IL-12–induced serum IFN-γ levels were clearly reduced in both anti-asGM1–treated WT mice or CD1d−/− mice, suggesting that both NK and NKT cells were contributing to IFN-γ production. IL-2–induced serum IFN-γ levels were completely dependent on NK cells. 24 h after the treatment regimes used for therapy of Renca metastases, serum IFN-γ levels were highest in IL-12–treated mice, then α-GalCer–treated mice and IL-2–treated mice (Fig. 7 B). These data indicated that appreciable serum IFN-γ levels were detected before TRAIL expression on NK cells, and that the reduced capacity of IL-2 to stimulate and maintain high IFN-γ levels correlated with its inability to induce TRAIL expression in vivo.
Figure 7.



Correlation between IFN-γ activity and NK cell TRAIL expression. (A) Serum was collected from untreated (▪) or anti-asGM1–treated BALB/c WT mice (•) or BALB/c CD1d−/− mice (▴) as indicated, and mouse IFN-γ measured by a specific ELISA. (B) Sera were also taken 24 h after treatment as per the Renca tumor therapy regime (above) from IL-12– (black bars), α-GalCer– (white bars), or IL-2 (striped bars)–treated mice. For A and B, data represent the mean ± SE (ng/ml) of duplicate samples from three different mice. The serum of all untreated mice contained <50 pg/ml of IFN-γ. (C) MNCs from the liver and spleen were isolated from PBS- or vehicle-treated (control) BALB/c IFN-γ−/− mice at 24 h after the treatment with IL-12 (500 U intraperitoneally on days −5, −4, −3, −2, and −1) or α-GalCer (α-GC; 2 μg intraperitoneally on days −8, −4, and 0), or at 3 h after the treatment with IFN-γ (50,000 U intraperitoneally). These cells were stained with biotin-conjugated anti-TRAIL mAb followed by PE-conjugated streptavidin, FITC-conjugated anti-DX5 mAb, and Cy-Chrome–conjugated anti-CD3 mAb. Expression of TRAIL was analyzed on electronically gated CD3−DX-5+ (NK) cells by flow cytometry. Bold lines indicate staining with anti-TRAIL mAb and plain lines indicate staining with isotype-matched control IgG2a.
We next examined the ability of IFN-γ, IL-12, and α-Gal Cer to induce NK cell TRAIL expression in BALB/c IFN-γ−/− mice (Fig. 7 C). Liver and spleen NK cells from IFN-γ−/− mice did not constitutively express TRAIL; however, treatment of these mice with IFN-γ induced both liver and spleen NK cell TRAIL expression. By contrast, treatment with either IL-12 or α-GalCer did not induce NK cell TRAIL. These data directly supported the conclusion that IFN-γ plays a key role in TRAIL expression and TRAIL-mediated antimetastatic function of NK cells.
Discussion
It has long been recognized that NK cell function can be activated by IFN-γ 38 and that IFN-γ can contribute to host antitumor immunity 39 40. Direct and macrophage-mediated antitumor activities of IFN-γ have been described 38, and IFN-γ has been shown to sensitize tumor cells to both pfp- 41 and FasL-mediated apoptosis 42. However, herein we provide the first evidence that IL-12 and α-GalCer immunotherapies that stimulate IFN-γ production can recruit the antitumor activity of NK cell TRAIL. At this stage, it is unknown what relative or general contribution NK cell TRAIL makes to the suppression of tumor metastasis, but under the appropriate activation conditions NK cell TRAIL can act in the liver and lung. NK cell TRAIL antitumor function appears to be limited to TRAIL-sensitive tumors (for example, Renca and 4T1 mammary carcinoma), as the anti-TRAIL mAb has no effect on immunity against TRAIL-resistant tumors (for example, B16 melanoma and 3LL lung carcinoma; our unpublished observations). Although Renca is controlled by NK cells in these models, it remains possible that other IFN-γ–regulated effector cells may mediate their antitumor activities via TRAIL. TRAIL with tumoricidal activity has also been shown to be induced on human monocytes and dendritic cells in vitro 14 15; however, we did not detect significant TRAIL expression on mouse non-NK leukocytes in the organs examined, nor did we observe monocyte/dendritic cell depletion after anti-asGM1 treatment. The documented involvement of IL-12/IFN-γ in the immune surveillance of methylcholanthrene-induced sarcoma 19 and IFN-γ in tumor development in p53 mutant mice 40 begs the question of whether TRAIL plays a broader role in tumor surveillance.
The constitutive expression of TRAIL on liver NK cells raises the issue of what normal physiological role TRAIL plays in the liver. It is possible that liver NK cells may express TRAIL in response to gut-derived endogenous endotoxin or in early responses to infections. Infections in the liver are most often controlled by NK cell secretion of antiviral cytokines, rather than by direct cytolysis; however, hepatocytes have almost undetectable MHC class I molecule expression and thus may represent significant targets for NK cell–mediated death. Although mouse hepatocytes are resistant to TRAIL under normal conditions, it is possible that after activation mouse hepatocytes may also be targets for TRAIL-mediated cell death. Human TRAIL has been demonstrated to cause apoptosis of human hepatocytes in vitro 43, but this finding remains unsubstantiated in vivo. Activated lymphocytes clearly cause liver injury in virus-infected or α-GalCer–treated mice 44 45, and it remains to be determined whether TRAIL participates in this damage. Alternatively, the liver is a site for lymphocyte cell death 46 47 and TRAIL may play a natural role in the surveillance of stressed cells in the liver 48.
Although T cells did not express TRAIL under any circumstances tested here, an implicit finding from our α-Gal Cer immunotherapy data is that NKT cells can initiate an IFN-γ–dependent induction of NK cell TRAIL function in the liver. Stimulation of serum IFN-γ by α-GalCer was rapid and dependent on NKT cells, consistent with NKT cells providing IFN-γ to secondarily activate NK cell function 49. NKT cells also played a role in IL-12 induction of IFN-γ, and we and others have previously shown in other tumor models that IL-12 preferentially stimulates NKT cell activation and IL-12 requires NKT cells for its antitumor activity 22 27 35. Induction of TRAIL on NK cells via autocrine NK cell IFN-γ secretion may also be important given that TRAIL is constitutively expressed on liver NK cells of WT, but not IFN-γ−/− mice. The rejection of TRAIL-insensitive tumors by α-GalCer also appeared to involve indirect activation of NK cells, as they too were inhibited by anti-asGM1 Ab (unpublished observations). Future NKT cell transfer experiments will be required to definitively show that NKT cells provide IFN-γ to activate NK cell TRAIL expression and function.
Although IFN-γ appears critical for effective TRAIL expression and function, not all inducers of IFN-γ activate NK cell TRAIL expression. Effective doses of IL-2 did not stimulate NK cell TRAIL expression or function in vivo, despite inducing moderate levels of serum IFN-γ. Previously, IL-18 did not induce TRAIL expression on murine NK cells in vitro 17, despite strongly inducing IFN-γ production by NK cells and augmenting pfp- and FasL-mediated NK cell cytotoxicity 50 51. None of the stimulators of serum IFN-γ–induced TRAIL expression on PBMC NK cells, suggesting that proximity between NKT cells and NK cells may be required. It was also curious that α-GalCer induced TRAIL expression on spleen NK cells, but not lung NK cells, despite the fact that IL-12 induced TRAIL on both populations of NK cells. There are fewer NKT cells in the lungs and the NKT cell/CD1d–α-GalCer interaction may not be efficient in the lung. In addition, IL-12 directly stimulates NK cell IFN-γ production and therefore NK cell TRAIL expression may not require indirect activation by lung NKT cells. Thus, although it is possible that continuous IFN-γ production in the environment of the NK cell is important for TRAIL induction, the complex issue of what other factors determine TRAIL expression in different tissues remains unresolved.
It has been recognized in mouse pathogen models that the immune effector response is controlled in a compartment-specific manner 52. Our data indicate that antitumor effector mechanisms also differ in their relative importance in the liver and lung. The two major effector mechanisms used by NK cells in their control of tumor metastasis are pfp and IFN-γ 21. Herein, we have shown that IFN-γ control of metastasis appears particularly important in the liver and TRAIL is at least one effector mechanism that acts downstream of IFN-γ during effective treatment with IL-12 or α-GalCer.
Acknowledgments
We thank Shayna Street and Christine Hall for genotyping the gene-targeted mice and Janice Kelly for technical assistance. We also thank Dr. Koezuka of Kirin Brewery for his kind provision of α-GalCer.
M.J. Smyth is supported by a National Health and Medical Research Council of Australia Research Fellowship. L.M. Sedger is currently supported by a U2000 University of Sydney Fellowship. The project was supported by grants in aid from the National Health and Medical Research Council of Australia, the Princess Takamatsu Cancer Research Fund (99-23110), and the Ministry of Education, Science and Culture, Japan.
Footnotes
Abbreviations used in this paper: α-GalCer, α-galactosylceramide; asGM1, asialo GM1; CMA, concanamycin A; MNC, mononuclear cell; pfp, perforin; TRAIL, TNF-related apoptosis-inducing ligand; WT, wild-type.
References
- Smith C.A., Farrah T., Goodwin R.G. The TNF receptor superfamily of cellular and viral proteinsactivation, costimulation, and death. Cell. 1994;76:959–962. doi: 10.1016/0092-8674(94)90372-7. [DOI] [PubMed] [Google Scholar]
- Van Parijs L., Abbas A.K. Role of Fas-mediated cell death in the regulation of immune responses. Curr. Opin. Immunol. 1996;8:355–361. doi: 10.1016/s0952-7915(96)80125-7. [DOI] [PubMed] [Google Scholar]
- Marsters S.A., Sheridan J.P., Pitti R.M., Brush J., Goddard A., Ashkenazi A. Identification of a ligand for the death-domain-containing receptor Apo3. Curr. Biol. 1998;8:525–528. doi: 10.1016/s0960-9822(98)70204-0. [DOI] [PubMed] [Google Scholar]
- Wiley S.R., Schooley K., Smolak P.J., Din W.S., Huang C.P., Nicholl J.K., Sutherland G.R., Smith T.D., Rauch C., Smith C.A. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–682. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- Snell V., Clodi K., Zhao S., Goodwin R., Thomas E.K., Morris S.W., Kadin M.E., Cabanillas F., Andreeff M., Younes A. Activity of TNF-related apoptosis-inducing ligand (TRAIL) in haematological malignancies. Br. J. Haematol. 1997;99:618–624. doi: 10.1046/j.1365-2141.1997.4393250.x. [DOI] [PubMed] [Google Scholar]
- Jeremias I., Herr I., Boehler T., Debatin K.M. TRAIL/Apo-2-ligand-induced apoptosis in human T cells. Eur. J. Immunol. 1998;28:143–152. doi: 10.1002/(SICI)1521-4141(199801)28:01<143::AID-IMMU143>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Sedger L.M., Shows D.M., Blanton R.A., Peschon J.J., Goodwin R.G., Cosman D., Wiley S.R. IFN-γ mediates a novel antiviral activity through dynamic modulation of TRAIL and TRAIL receptor expression. J. Immunol. 1999;163:920–926. [PubMed] [Google Scholar]
- Walczak H., Miller R.E., Ariail K., Gliniak B., Griffith T.S., Kubin M., Chin W., Jones J., Woodward A., Le T. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999;5:157–163. doi: 10.1038/5517. [DOI] [PubMed] [Google Scholar]
- Bonavida B., Ng C.P., Jazirehi A., Schiller G., Mizutani Y. Selectivity of TRAIL-mediated apoptosis of cancer cells and synergy with drugsthe trail to non-toxic cancer therapeutics (review) Int. J. Oncol. 1999;15:793–802. doi: 10.3892/ijo.15.4.793. [DOI] [PubMed] [Google Scholar]
- Gibson S.B., Oyer R., Spalding A.C., Anderson S.M., Johnson G.L. Increased expression of death receptors 4 and 5 synergizes the apoptosis response to combined treatment with etoposide and TRAIL. Mol. Cell. Biol. 2000;20:205–212. doi: 10.1128/mcb.20.1.205-212.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gliniak B., Le T. Tumor necrosis factor-related apoptosis-inducing ligand's antitumor activity in vivo is enhanced by the chemotherapeutic agent CPT-11. Cancer Res. 1999;59:6153–6158. [PubMed] [Google Scholar]
- Pitti R.M., Marsters S.A., Ruppert S., Donahue C.J., Moore A., Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 1996;271:12687–12690. doi: 10.1074/jbc.271.22.12687. [DOI] [PubMed] [Google Scholar]
- Kayagaki N., Yamaguchi N., Nakayama M., Eto H., Okumura K., Yagita H. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cellsa novel mechanism for the antitumor effects of type I IFNs. J. Exp. Med. 1999;189:1451–1460. doi: 10.1084/jem.189.9.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith T.S., Wiley S.R., Kubin M.Z., Sedger L.M., Maliszewski C.R., Fanger N.A. Monocyte-mediated tumoricidal activity via the tumor necrosis factor–related cytokine TRAIL. J. Exp. Med. 1999;189:1343–1354. doi: 10.1084/jem.189.8.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanger N.A., Maliszewski C.R., Schooley K., Griffith T.S. Human dendritic cells mediate cellular apoptosis via tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) J. Exp. Med. 1999;190:1155–1164. doi: 10.1084/jem.190.8.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamai L., Ahmad M., Bennett I.M., Azzoni L., Alnemri E.S., Perussia B. Natural killer (NK) cell-mediated cytotoxicitydifferential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J. Exp. Med. 1998;188:2375–2380. doi: 10.1084/jem.188.12.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N., Yamaguchi N., Nakayama M., Takeda K., Akiba H., Tsutsui H., Okamura H., Nakanishi K., Okumura K., Yagita H. Expression and function of TNF-related apoptosis-inducing ligand on murine activated NK cells. J. Immunol. 1999;163:1906–1913. [PubMed] [Google Scholar]
- Kayagaki N., Yamaguchi N., Nakayama M., Kawasaki A., Akiba H., Okumura K., Yagita H. Involvement of TNF-related apoptosis-inducing ligand in human CD4+ T cell-mediated cytotoxicity. J. Immunol. 1999;162:2639–2647. [PubMed] [Google Scholar]
- Smyth M.J., Thia K.Y.T., Street S.E.A., Kelly J.M., Trapani J.A., Taniguchi M., Kawano T., Pelikan S., Crowe N.Y., Godfrey D.I. Differential tumor surveillance by NK and NKT cells. J. Exp. Med. 2000;191:661–668. doi: 10.1084/jem.191.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth M.J., Thia K.Y.T., Street S.E.A., MacGregor D., Godfrey D.I., Trapani J.A. Cytotoxic lymphocyte perforin-mediated immune surveillance of spontaneous lymphoma. J. Exp. Med. 2000;192:755–760. doi: 10.1084/jem.192.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street S.E.A., Cretney E., Smyth M.J. Perforin and interferon-γ activities independently control tumor initiation, growth and metastasis. Blood. 2001;97:192–197. doi: 10.1182/blood.v97.1.192. [DOI] [PubMed] [Google Scholar]
- Smyth M.J., Taniguchi M., Street S.E.A. The anti-tumor activity of IL-12mechanisms of innate immunity that are model- and dose-dependent. J. Immunol. 2000;165:2665–2670. doi: 10.4049/jimmunol.165.5.2665. [DOI] [PubMed] [Google Scholar]
- Smyth M.J., Thia K.Y.T., Cretney E., Kelly J.M., Snook M.B., Forbes C.A., Scalzo A.A. Perforin is a major contributor to NK cell control of tumor metastasis. J. Immunol. 1999;162:6658–6662. [PubMed] [Google Scholar]
- Dalton D.K., Pitts-Meek S., Keshav S., Figari I.S., Bradley A., Stewart T.A. Multiple defects of immune cell function in mice with disrupted interferon-γ genes. Science. 1993;259:1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- Kitamura H., Iwakabe K., Yahata T., Nishimura S., Ohta A., Ohmi Y., Sato M., Takeda K., Okumura K., Van Kaer L. The natural killer T (NKT) cell ligand alpha-galactosylceramide demonstrates its immunopotentiating effect by inducing interleukin (IL)-12 production by dendritic cells and IL-12 receptor expression on NKT cells. J. Exp. Med. 1999;189:1121–1128. doi: 10.1084/jem.189.7.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama T., Takeda K., Shimozato O., Hayakawa Y., Atsuta M., Kobayashi K., Ito M., Yagita H., Okumura K. Perforin-dependent NK cell cytotoxicity is sufficient for anti-metastatic effect of IL-12. Eur. J. Immunol. 1999;29:1390–1396. doi: 10.1002/(SICI)1521-4141(199904)29:04<1390::AID-IMMU1390>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Takeda K., Hayakawa Y., Atsuta M., Hong S., Van Kaer L., Kobayashi K., Ito M., Yagita H., Okumura K. Relative contribution of NK and NKT cells to the anti-metastatic activities of IL-12. Int. Immunol. 2000;12:909–914. doi: 10.1093/intimm/12.6.909. [DOI] [PubMed] [Google Scholar]
- Wigginton J.M., Komschlies K.L., Back T.C., Franco J.L., Brunda M.J., Wiltrout R.H. Administration of interleukin 12 with pulse interleukin 2 and the rapid and complete eradication of murine renal carcinoma. J. Natl. Cancer Inst. 1996;88:38–43. doi: 10.1093/jnci/88.1.38. [DOI] [PubMed] [Google Scholar]
- Handa K., Suzuki R., Matsui H., Shimizu Y., Kumagai K. Natural killer (NK) cells as a responder to interleukin 2 (IL 2). II. IL 2-induced interferon gamma production. J. Immunol. 1983;130:988–992. [PubMed] [Google Scholar]
- Gately M.K., Warrier R.R., Honasoge S., Carvajal D.M., Faherty D.A., Connaughton S.E., Anderson T.D., Sarmiento U., Hubbard B.R., Murphy M. Administration of recombinant IL-12 to normal mice enhances cytolytic lymphocyte activity and induces production of IFN-γ in vivo. Int. Immunol. 1994;6:157–167. doi: 10.1093/intimm/6.1.157. [DOI] [PubMed] [Google Scholar]
- Kawamura T., Takeda K., Mendiratta S.K., Kawamura H., Van Kaer L., Yagita H., Abo T., Okumura K. Critical role of NK1+ T cells in IL-12-induced immune responses in vivo. J. Immunol. 1998;160:16–19. [PubMed] [Google Scholar]
- Trinchieri G. Proinflammatory and immunoregulatory functions of interleukin-12. Int. Rev. Immunol. 1998;16:365–396. doi: 10.3109/08830189809043002. [DOI] [PubMed] [Google Scholar]
- Burdin N., Brossay L., Koezuka Y., Smiley S.T., Grusby M.J., Gui M., Taniguchi M., Hayakawa K., Kronenberg M. Selective ability of mouse CD1 to present glycolipidsα-galactosylceramide specifically stimulates Vα14+ NK T lymphocytes. J. Immunol. 1998;161:3271–3281. [PubMed] [Google Scholar]
- Eberl G., MacDonald H.R. Selective induction of NK cell proliferation and cytotoxicity by activated NKT cells. Eur. J. Immunol. 2000;30:985–992. doi: 10.1002/(SICI)1521-4141(200004)30:4<985::AID-IMMU985>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Cui J., Shin T., Kawano T., Sato H., Kondo E., Toura I., Kaneko Y., Koseki H., Kanno M., Taniguchi M. Requirement for Vα14 NKT cells in IL-12-mediated rejection of tumors. Science. 1997;278:1623–1626. doi: 10.1126/science.278.5343.1623. [DOI] [PubMed] [Google Scholar]
- Nakagawa R., Motoki K., Ueno H., Iijima R., Nakamura H., Kobayashi E., Shimosaka A., Koezuka Y. Treatment of hepatic metastasis of the colon26 adenocarcinoma with an α-galactosylceramide, KRN7000. Cancer Res. 1998;58:1202–1207. [PubMed] [Google Scholar]
- Toura I., Kawano T., Akutsu Y., Nakayama T., Ochiai T., Taniguchi M. Cutting edgeinhibition of experimental tumor metastasis by dendritic cells pulsed with α-galactosylceramide. J. Immunol. 1999;163:2387–2391. [PubMed] [Google Scholar]
- Boehm U., Klamp T., Groot M., Howard J.C. Cellular responses to interferon-γ. Annu. Rev. Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- Dighe A.S., Richards E., Old L.J., Schreiber R.D. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity. 1994;1:447–456. doi: 10.1016/1074-7613(94)90087-6. [DOI] [PubMed] [Google Scholar]
- Kaplan D.H., Shankaran V., Dighe A.S., Stockert E., Aguet M., Old L.J., Schreiber R.D. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA. 1998;95:7556–7561. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthou C., Bourge J.F., Zhang Y., Soulie A., Geromin D., Denizot Y., Sigaux F., Sasportes M. Interferon-gamma-induced membrane PAF-receptor expression confers tumor cell susceptibility to NK perforin-dependent lysis. Blood. 2000;95:2329–2336. [PubMed] [Google Scholar]
- Xu X., Fu X.Y., Plate J., Chong A.S. IFN-γ induces cell growth inhibition by Fas-mediated apoptosisrequirement of STAT1 protein for up-regulation of Fas and FasL expression. Cancer Res. 1998;58:2832–2837. [PubMed] [Google Scholar]
- Jo M., Kim T.H., Seol D.W., Esplen J.E., Dorko K., Billiar T.R., Strom S.C. Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand. Nat. Med. 2000;6:564–567. doi: 10.1038/75045. [DOI] [PubMed] [Google Scholar]
- Liu Z.X., Govindarajan S., Okamoto S., Dennert G. NK cells cause liver injury and facilitate the induction of T cell-mediated immunity to a viral liver infection. J. Immunol. 2000;164:6480–6486. doi: 10.4049/jimmunol.164.12.6480. [DOI] [PubMed] [Google Scholar]
- Osman Y., Kawamura T., Naito T., Takeda K., Van Kaer L., Okumura K., Abo T. Activation of hepatic NKT cells and subsequent liver injury following administration of α-galactosylceramide. Eur. J. Immunol. 2000;30:1919–1928. doi: 10.1002/1521-4141(200007)30:7<1919::AID-IMMU1919>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Mehal W.Z., Juedes A.E., Crispe I.N. Selective retention of activated CD8+ T cells by the normal liver. J. Immunol. 1999;163:3202–3210. [PubMed] [Google Scholar]
- Eberl G., MacDonald H.R. Rapid death and regeneration of NKT cells in anti-CD3ε- or IL-12-treated micea major role for bone marrow in NKT cell homeostasis. Immunity. 1998;9:345–353. doi: 10.1016/s1074-7613(00)80617-2. [DOI] [PubMed] [Google Scholar]
- Rabinovich B.A., Shannon J., Su R.C., Miller R.G. Stress renders T cell blasts sensitive to killing by activated syngeneic NK cells. J. Immunol. 2000;165:2390–2397. doi: 10.4049/jimmunol.165.5.2390. [DOI] [PubMed] [Google Scholar]
- Carnaud C., Lee D., Donnars O., Park S.H., Beavis A., Koezuka Y., Bendelac A. Cutting edgecross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J. Immunol. 1999;163:4647–4650. [PubMed] [Google Scholar]
- Okamura H., Tsutsi H., Komatsu T., Yutsudo M., Hakura A., Tanimoto T., Torigoe K., Okura T., Nukada Y., Hattori K. Cloning of a new cytokine that induces IFN-γ production by T cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- Okamura H., Tsutsui H., Kashiwamura S., Yoshimoto T., Nakanishi K. Interleukin-18a novel cytokine that augments both innate and acquired immunity. Adv. Immunol. 1998;70:281–312. doi: 10.1016/s0065-2776(08)60389-2. [DOI] [PubMed] [Google Scholar]
- Tay C.H., Welsh R.M. Distinct organ-dependent mechanisms for the control of murine cytomegalovirus infection by natural killer cells. J. Virol. 1997;71:267–275. doi: 10.1128/jvi.71.1.267-275.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]