

Abstract
Vascular cellular adhesion molecule (VCAM)-1 is a membrane-bound cellular adhesion molecule that mediates adhesive interactions between hematopoietic progenitor cells and stromal cells in the bone marrow (BM) and between leukocytes and endothelial as well as dendritic cells. Since VCAM-1–deficient mice die embryonically, conditional VCAM-1 mutant mice were generated to analyze the in vivo function of this adhesion molecule. Here we show that interferon-induced Cre-loxP–mediated deletion of the VCAM-1 gene after birth efficiently ablates expression of VCAM-1 in most tissues like, for example, BM, lymphoid organs, and lung, but not in brain. Induced VCAM-1 deficiency leads to a reduction of immature B cells in the BM and to an increase of these cells in peripheral blood but not in lymphoid organs. Mature recirculating B cells are reduced in the BM. In a migration assay, the number of mature B cells that appears in the BM after intravenous injection is decreased. In addition, the humoral immune response to a T cell–dependent antigen is impaired. VCAM-1 serves an important role for B cell localization and the T cell–dependent humoral immune response.
Keywords: conditional, VCAM-1 mutant mice, B cell development, lymphocyte migration, cre/loxP, bone marrow
Introduction
The vascular cell adhesion molecule (VCAM)-1 (CD106) was originally described as a cytokine-induced glycoprotein (INCAM-110) expressed on human vascular endothelial cells 1. In mice, two isoforms of VCAM-1 are produced by alternative splicing. One form contains seven Ig-like domains, a transmembrane region, and a short COOH-terminal cytoplasmic tail 2 3 4, while the second, cytokine and LPS-inducible form, contains the first three domains only and is attached to the cell membrane by a glycosylphosphatidylinositol anchor 5. VCAM-1 directs the recruitment of leukocytes to sites of inflammation by binding predominantly to α4β1 integrin (very late antigen [VLA]-4; CD49d/CD29) and with lower affinity to α4β7 integrin 6 7 8. Binding of the seven domain form of VCAM-1 to its ligands is mediated by the first and fourth Ig-like domain 9 10.
A contribution of the VCAM-1/VLA-4 adhesion pathway for leukocyte migration in rheumatoid arthritis, lupus nephritis, inflammatory bowel disease, allograft rejection, atheriosclerosis, contact hypersensitivity, and experimental autoimmune encephalomyelitis has been suggested 11 12 13 14 15 16 17 18. Blocking VCAM-1/VLA-4–mediated cellular adhesion by mAbs often results in a less severe course of these diseases in animal models.
Differentiation and proliferation of hematopoietic progenitor cells occur in intimate contact with the bone marrow (BM) microenvironment which is composed of stromal cells and extracellular matrix proteins. Since stromal cells secrete numerous factors that are necessary for the growth and differentiation of hematopoietic precursor cells, it may be critical for these cells to stay in the BM in order to be exposed to a cytokine milieu and to receive all signals necessary for maturation. VCAM-1 is constitutively expressed on BM stromal cells 19. Several reports demonstrate that VCAM-1 plays a crucial role in hematopoietic progenitor cell trafficking and lodgement of transplanted murine hematopoietic progenitor cells to BM 19 20 21 22. Other authors have shown the significance of cytokines for hematopoietic progenitor cell (HPC) mobilization in vivo by the treatment of mice with fms-like tyrosine kinase 3 (Flt3) ligand 23. This suggests that cytokines may alter the expression or the activation state of adhesion molecules. VCAM-1 is also constitutively expressed on follicular dendritic cells 24 25, which serve a critical role for antigen presentation. The VLA-4–VCAM-1 interaction is involved in the adhesion of human B cells to follicular dendritic cells of germinal centers in vitro, which is hypothesized to affect the immune response maturation in the germinal center 24 25 26 27. Furthermore, a link between VCAM-1 function and rescue of germinal center B cells 26 27 28 or thymocytes 29 from apoptosis has been reported. Finally, VCAM-1 may also function in maturation and costimulation of T cells 30 31 32.
Since mice homozygous for the VCAM-1 deletion die early during embryogenesis 33 34 35, most experiments regarding the function of VCAM-1 have so far been carried out either in vitro or using mAbs blocking the function of VCAM-1. A very small number of VCAM-1–deficient mice survive the detrimental effects of the VCAM-1 mutation on embryogenesis 34. These mice exhibit elevated numbers of circulating blood mononuclear leukocytes 34; however, no phenotype with regard to hematopoiesis has been reported 36. It remains questionable whether these mice represent a suitable tool for evaluation of postnatal VCAM-1 function. To circumvent the limitations of the above mentioned approaches for the evaluation of the in vivo function of VCAM-1 for postnatal life, we used inducible gene targeting 37. This approach leads to the absence of VCAM-1 protein in most organs of mice in which the VCAM-1 gene was deleted by IFN-induced Cre-loxP–mediated recombination. Hereby, the critical function of VCAM-1 for retention of B cells during maturation in the BM and for localization of mature B cells in the BM could be established. Moreover, VCAM-1 plays a critical role in the humoral immune response to a T cell–dependent antigen.
Materials and Methods
Mice and Conditional Gene Inactivation.
Mice homozygous for the loxP flanked (floxed) VCAM-1 gene 35 and Mx-cre transgenic mice 37 were bred and crossed to generate homozygous VCAM-1 floxed mice carrying the Mx-cre transgene. In each litter, mice with and without the Mx-cre transgene were generated. Neonatally, at days 1, 4, and 7 the mice were injected with 106 U of IFN-α1/α2 intraperitoneally to inactivate the VCAM-1 gene in mice bearing the Mx-cre transgene and to use the Mx-cre nontransgenic mice as controls. At the age of 5 wk, mice were typed for the Mx-cre transgene by Cre-specific PCR and Southern blot analysis. The different genotypes, Mx-cre;VCAMflox/flox mice (conditional VCAM-1 mutant mice) and VCAMflox/flox mice (control mice), were identified at the expected Mendelian ratio. The mice were kept in a conventional animal facility. Sentinel animals were checked regularly according to the FELASA health monitoring report 38 and mice were used for studies at the age of 6–8 wk to exclude any influence of IFN-α treatment on the B and T cell pool 39.
Southern Blot Analysis.
Tissue of different organs from conditional VCAM-1 mutant mice was washed with PBS and digested at 56°C with 100 μg/ml proteinase K (Boehringer) in lysis buffer (100 mM Tris-HCl, pH 8.5, 5 mM EDTA, 0.2% SDS) overnight. 1 vol of isopropanol was added to the lysate and the precipitate was recovered. Depending on the size of the precipitate the DNA was dispersed in 100–400 μl of 10 mM Tris-HCl, 0.1 mM EDTA, pH 7.5. The prepared DNA was digested with BamHI (New England Biolabs, Inc.) and probed with a mixture of two 650-bp VCAM-1–specific SpeI-BglII (New England Biolabs, Inc.) fragments of vector 2BAM 35 nonradioactive labeled with the Gene Images™ labeling kit (Amersham Pharmacia Biotech). In an agarose gel the deleted VCAM-1 gene migrated at 4.5 kb, whereas the floxed gene gave two signals of 6 and 3 kb in size.
To test mice for the presence of the Mx-cre transgene tail, DNA was digested with BamHI and probed with a 750-bp BamHI-XbaI (New England Biolabs, Inc.) fragment of the Cre-coding region, resulting in two bands at 5 and 3.5 kb.
Quantification of the Deletion.
To assess the degree of VCAM-1 gene deletion in various tissues, DNA from organs of three different conditional VCAM-1 mutant mice was prepared and analyzed by at least two independent Southern blot each. The amount of deletion was calculated by scanning the signals of the 3-kb floxed and the 4.5-kb deleted VCAM-1 gene fragment generated during different exposure times on X-OMAT film (Eastman Kodak Co.) in a FluorS MultiImager (Bio-Rad Laboratories).
PCR.
To test for the presence of the Mx-cre transgene, a 1-kb fragment of the coding region was amplified by standard PCR procedure using the two primers cre− (5′-CAA TTT ACT GAC CGT ACA C-3′) and cre+ (5′-CAT CGC CAT CTT CCA GCA-G). Tissue from tail biopsies was lysed overnight at 56°C with 100 μg/ml proteinase K (Boehringer) in lysis buffer (100 mM Tris-HCl, pH 8.5, 5 mM EDTA, 0.2% SDS), precipitated with isopropanol, and resuspended in 150 μl Tris-HCl, 0.1 mM EDTA, pH 7.5. 1 μl of this DNA solution was used for PCR analysis in 50 ml vol overlayed with mineral oil. 200 mM dNTPs (Boehringer), 20 pmol of each primer, and 5 U Taq polymerase (produced in our own laboratory) were added, and the PCR reaction was performed on a thermal cycler (TRIO-Thermoblock; Biometra) in 10 mM Tris-HCl, pH 8.3, at 25°C, 50 mM KCl, and 3 mM MgCl2 (35 cycles: 40 s at 94°C, 1 min at 60°C, and 1.5 min at 72°C). After the last cycle, samples were incubated for another 10 min at 72°C and subsequently analyzed by gel electrophoresis on a 1% agarose gel.
Immunoprecipitation.
Mice were killed and the prepared organs were washed several times in PBS. Tissue was then homogenized and cells were disrupted with glass beads (Braun-Melsungen) in TBS (0.5% SDS, 1% Triton, 0.025 mM EDTA, 0.1 M Tris, pH 8.0) containing protease inhibitors as described 40. After adding 0.5% NP-40, membrane proteins were extracted at 4°C overnight. Cell debris was removed by high speed centrifugation and 0.4 ml of the supernatant was subjected to immunoprecipitation. After incubation with biotinylated goat anti–mouse IgG1 Ab (Dianova) overnight at 4°C and precipitation of unspecific bound proteins through adding streptavidin-coupled agarose (Sigma-Aldrich) for another night at 4°C, preclearing was finished by centrifugation preceeding precipitation of VCAM-1 protein from the supernatant. For that purpose the supernatant was incubated again overnight at 4°C with biotinylated rat anti–mouse VCAM-1 Ab (Clone 429; BD PharMingen) after another 24 h at 4°C in the presence of streptavidin-coupled agarose and precipitation by centrifugation. The pellet was washed two times in PBS and separated on a 10% acrylamide gel. Proteins were blotted on a PVDF membrane (Immobilon-P; Millipore) and VCAM-1 protein was detected with polyclonal goat anti–VCAM-1 Ab (sc-1504; Santa Cruz Biotechnology, Inc.) and anti–goat IgG-horseradish peroxidase (Dako) using an ECL detection kit (Amersham Pharmacia Biotech).
Immunohistochemistry.
Frozen sections of spleen were fixed in acetone, incubated with 0.1% phenylhydrazine (Sigma-Aldrich) in PBS, and blocked with PBS containing 4% FCS. For immunostaining biotinylated rat anti–mouse VCAM-1 mAb (clone 429; BD PharMingen), rat anti–mouse VCAM-1 mAb (MK-2, 9.DB3, V.7H1, V.4B12.1, V.6C3; provided by Dr. D. Vestweber, University of Muenster, Muenster, Germany), or polyclonal goat anti–VCAM-1 Ab (sc-1504; Santa Cruz Biotechnology, Inc.) was used. Primary Abs were detected with peroxidase-conjugated streptavidin (Dianova) or peroxidase-conjugated anti–rat IgG (Jackson ImmunoResearch Laboratories) or anti–goat IgG (Santa Cruz Biotechnology, Inc.). The detection was performed using 3,3′-diaminobenzidine (DAB) substrate kit (Pierce Chemical Co.).
Hematologic Analysis.
Blood samples were obtained from mouse tail vein; EDTA served as an anticoagulant. Erythrocyte and leukocyte counts were determined by an automatic cell counter (Beckman Coulter). Differential counts were assessed based on cellular morphology of blood smears by Wright-Giemsa stain (Sigma-Aldrich) and in peripheral blood by four-color flow cytometry using the following mAbs: PE-conjugated CD3e (clone 145-2C11; BD PharMingen), allophycocyanin (APC)-conjugated RA3-6B2 (anti–murine B220; references 41, 42), biotinylated GR-1 (clone RB6-8C5; BD PharMingen), FITC-conjugated F4-80 (Serotec), and streptavidin-CyChrome (GIBCO BRL). Monocytes and neutrophils were further quantified by cytochemistry on blood smears. Naphthol-AS-d-chloroacetate esterase activity was used to identify neutrophils 43 while monocytes were characterized by their α-naphthyl-acetate esterase activity 44.
Flow Cytometry.
Single cell suspensions from BM (femur) were prepared by flushing bones with medium (DMEM; GIBCO BRL), and for preparation of blood lymphocytes heparinized blood was separated by a 7.5% Ficoll (GIBCO BRL) gradient. Single cell suspensions from other organs were prepared by gentle dissection of these tissues in medium. All cell suspensions were subsequently incubated for 2 min on ice in erythrocyte lysis buffer (140 mM NH4Cl, 17 mM Tris, pH 7.65) followed by washing and resuspension in staining buffer (PBS supplemented with 1% BSA and 0.1% NaN3). Cells present in the lymphocyte gate, as defined by light scatter 45, were analyzed by three- and four-color immunofluorescence on a FACScan™ (Becton Dickinson) or on a FACStar™ (Becton Dickinson) using the CELLQuest™ program. Staining was done with fluorochrome APC-, FITC-, PE-, or CyChrome- or biotin-conjugated mAbs. Biotin-conjugated mAbs were detected using streptavidin-PE, streptavidin-CyChrome, or Texas red–avidin (GIBCO BRL). APC-conjugated RA3-6B2 (anti-murine B220; references 41, 42), PE-conjugated R33-24-12 (anti–murine μ; reference 46), FITC-conjugated 1.3-5 (anti–murine IgD; reference 47), biotinylated 493 (anti–murine pB130-140) Abs 48, and FITC-conjugated annexin V (BD PharMingen) or biotinylated annexin V (Roche) were used for surface marker staining.
Short-term Migration Assay.
Splenocytes were prepared according to standard procedures from CB20 mice (H-2d). 1.8 × 109 splenocytes were coupled to S7 beads (Miltenyi Biotec). Depletion of S7+ (CD43+) cells by MACS® 49 using VS+ columns resulted in 1.2 × 108 mature B cells at 93% purity, which were collected and stained with PKH-26 (Sigma-Aldrich). 3 × 107 stained cells were injected into the tail vein of control and conditional VCAM-1 mutant mice, respectively. After 20 h, mice were killed and lymphocytes from BM were prepared and analyzed by flow cytometry.
Immunization.
Mice 8–10 wk of age were immunized intraperitoneally with 100 μg alum-precipitated (4-hydroxy-3-nitrophenyl)-acetyl-chicken globulin (NP-CG) or 10 μg NP-Ficoll. After 3 wk NP-CG–injected mice were boosted in the same manner with 50 μg NP-CG. At days 0, 7, 14, 21, 28, and 35 (NP-CG immunization) or at days 0, 7, and 14 (NP-Ficoll immunization) mice were bled. The sera were analyzed by enzyme-linked immunosorbent assays for NP-conjugated antigens 50. Affinity maturation of serum IgG1 Ab was assessed in sera of NP-CG–immunized mice by measuring the ratio of binding to NP4-BSA/NP14-BSA 50. Germinal centers were stained in splenic sections with biotinylated peanut agglutinin (Vector Laboratories), detected with streptavidin–horseradish peroxidase (Dianova), developed with diaminobenzidine, and counterstained with hematoxylin (Sigma-Aldrich) by standard procedure 51.
Quantification of Plasma Cells.
IgG-secreting plasma cells in 10-mo-old mice were stained with TRITC (rhodamine)-conjugated goat anti–mouse IgG mAb (Southern Biotechnology Associates, Inc.) in cytospins of spleen and BM 52. Each cytospin contained 2 × 105 cells. At least 14 cytospins of two control and two conditional VCAM-1 mutant mice were counted.
Bromodeoxyuridine Treatment.
Mice (two control and two conditional VCAM-1 mutant mice) were fed with bromodeoxyuridine (BrdU) at a concentration of 1 mg/ml with 1 M sucrose in drinking water for 3 d. During the labeling period the drinking water was protected from light. Mice were then killed and surface staining of single cell suspensions was performed as described previously 53. 5–10 × 106 cells were first surface stained with FITC-conjugated Abs against B220 (mAb RA33.A1.CL6) or IgM (mAb R33-24-12). After fixation in methanol and denaturation of the DNA, cells were stained with a biotinylated anti-BrdU Ab (Alexis Biochemicals), then incubated with streptavidin-CyChrome and analyzed by FACS®.
Statistical Analysis.
Data are presented as mean ± SEM. Statistical significance of the results was assessed using the Student's t test.
Results
Conditional Gene Targeting of VCAM-1.
To circumvent embryonic lethality in VCAM-1– deficient mice 33 34 we set out to delete the VCAM-1 gene in mice in a conditional manner using the Mx-cre system 37. In this approach the target gene is flanked by loxP sites and Cre is expressed after IFN injection. This leads to a Cre-loxP–mediated deletion of the target gene. We have crossed mice in which the proximal promoter sequences and the first two exons of the VCAM-1 gene are flanked by loxP sites (VCAM-1flox/flox mice; reference 35), with Mx-cre transgenic mice (37; Fig. 1 A). On days 1, 4, and 7 after birth, mice homozygous for the VCAM-1 mutation and transgenic for Mx-cre (Mx-cre;VCAMflox/flox mice) were treated with 106 U IFN-α1/α2 (IFN-α) 54 to induce Cre-mediated recombination. Mice were analyzed for deletion of the VCAM-1 gene at the age of 6–8 wk.
Figure 1.



Efficient conditional ablation of VCAM-1 in vivo. (A) Southern blotting strategy for detection of IFN-α–induced VCAM-1 gene deletion. (B) Southern blot analysis of splenic DNA of mice without (lanes 1 and 2) and with (lanes 3 and 4) IFN-α injection (probed for VCAM-1). Without either the cre transgene (lanes 1 and 3) or IFN-α induction (lane 2), deletion of the VCAM-1 gene did not occur, as indicated by the 3- and 6-kb bands. By contrast, deletion is almost complete after IFN-α induction in the presence of the cre transgene (lane 4), resulting in a 4.5-kb signal. (C) Southern blotting of various tissues from IFN-α–treated, conditional VCAM-1 mutant mice revealed a high degree of deletion in most organs, but almost no deletion in brain (LI, liver; LN, lymph node; LU, lung; SP, spleen; TH, thymus; BR, brain; HE, heart; IN, intestine; KI, kidney). (D) Western blot analysis of VCAM-1 (110 kD) in conditional VCAM-1 mutant and control mice. After IFN-α–induced Cre expression, VCAM-1 could not be detected in BM, liver, lymph node, spleen, and thymus of conditional VCAM-1 mutant mice (lanes 4, 6, 8, 10, and 11), while minor traces of VCAM-1 are detectable in heart, intestine, and lung (lanes 2, 5, and 7) and brain contains VCAM-1 levels similar to control mice (lane 1). (E) Immunohistochemical analysis of spleen reveals VCAM-1 expression in control mice (left) and by contrast, no VCAM-1+ cells in spleen of conditional VCAM-1 mutant mice (right).
VCAM-1flox/flox mice (control mice) and Mx-cre;VCAM-1flox/flox mice (conditional VCAM-1 mutant mice) did not exhibit any detectable deletion of the VCAM-1 gene in spleen without IFN-α treatment. After IFN-α application, VCAM-1 deletion was complete in the spleens of conditional VCAM-1 mutant mice, while there was no deletion detectable in control mice (Fig. 1 B). This suggests that conditional targeting of the VCAM-1 gene using the Mx-cre system is efficient and does not exhibit any obvious leakiness either in the absence of IFN-α application or in the absence of the transgene. After induced deletion, conditional VCAM-1 mutant mice did not exhibit any overt disease and could be kept for at least 12 mo. When different organs were assessed by Southern blotting for deletion of the VCAM-1 gene in conditional VCAM-1 mutant mice after IFN-α treatment, it became evident that the extent of the deletion varied. While the deletion was complete in, for example, BM, liver, lymph nodes, spleen, and thymus, only partial deletion was observed in heart (45%), intestine (85%), kidney (84%), and lung (69%), and only minimal deletion could be detected in brain (16%; Fig. 1 C).
To confirm that conditional deletion of the VCAM-1 gene led to a lack of protein expression, Western blot analysis for VCAM-1 protein and immunhistochemical detection using mAbs against VCAM-1 were performed. By Western blot analysis, there was no VCAM-1 protein detectable in BM, liver, lymph nodes, spleen, and thymus of conditional VCAM-1 mutant mice after IFN-α treatment, while a minor protein band was recognizable in the lung and heart (Fig. 1 D). By contrast, abundant VCAM-1 protein with a molecular mass of ∼110 kD was present in the brain of these mice, as expected from the results of the Southern blot analysis (Fig. 1 C).
The immunohistochemical analysis confirmed the absence of VCAM-1 expression after conditional targeting of the VCAM-1 gene. A set of anti–VCAM-1 mAbs binding to different epitopes was used for indirect immunohistochemical detection. Frozen tissue sections from spleens of control mice after IFN-α treatment readily exhibited staining for VCAM-1 (Fig. 1 E). By contrast, no VCAM-1 expression was detectable in spleens of conditional VCAM-1 mutant mice after IFN-α application (Fig. 1 E).
Thus, the level of genomic deletion as assessed by Southern blot analysis corresponds well with the lack of protein expression of VCAM-1 as measured by Western blot analysis or immunohistochemistry.
VCAM-1 Retains Immature B Cells in the BM.
Spleen, lymph nodes, thymus, and BM from IFN-treated conditional VCAM-1 mutant mice and from IFN-treated control mice were analyzed for cellularity and lymphocyte subsets by flow cytometry. While there was no major difference in overall cell number and expression of B220, IgM, IgD, CD3, CD4, and CD8 in spleens, lymph nodes, and thymus, a marked alteration of B cell subset distribution was detected in BM. There was a consistent, but statistically not significant, small reduction of nucleated cells in the BM of conditional VCAM-1 mutant mice (1.5 ± 0.64 × 107 nucleated cells per femur for control mice, 1.2 ± 0.41 × 107 nucleated cells per femur in conditional VCAM-1 mutant mice, P = 0.27). However, the percentages of cells within the lymphocyte gate were similar (32.5 ± 5.6% in control mice and 33.4 ± 6.3% in conditional VCAM-1 mutant mice), whereas the fraction of B220+ cells within these gates is slightly reduced in VCAM-1–deficient animals (14.7 ± 3.7% in control mice versus 10.6 ± 2.9% in conditional VCAM-1 mutant mice, P < 0.05). Taken together, this results in a reduced absolute number of B220+ cells per femur in BM of conditional VCAM-1 mutant mice (1.4 ± 0.6 × 106 B220+ cells per femur) compared with control mice (2 ± 0.5 × 106 B220+ cells per femur, P < 0.05). Hardy et al. have developed a nomenclature to distinguish the different stages of B cell development, with the most immature pro-B cells present in fraction A while mature B cells establish fraction F 42. In conditional VCAM-1 mutant mice fraction E, which comprises the immature B cells expressing IgM but no IgD, is reduced by a mean of 40% compared with fraction E in control mice (Fig. 2 A). This reduction of fraction E is even more pronounced when absolute cell numbers are calculated since the overall cell number and the number of B220+ cells are reduced in conditional VCAM mutant mice (see above; Fig. 2 B).
Figure 2.
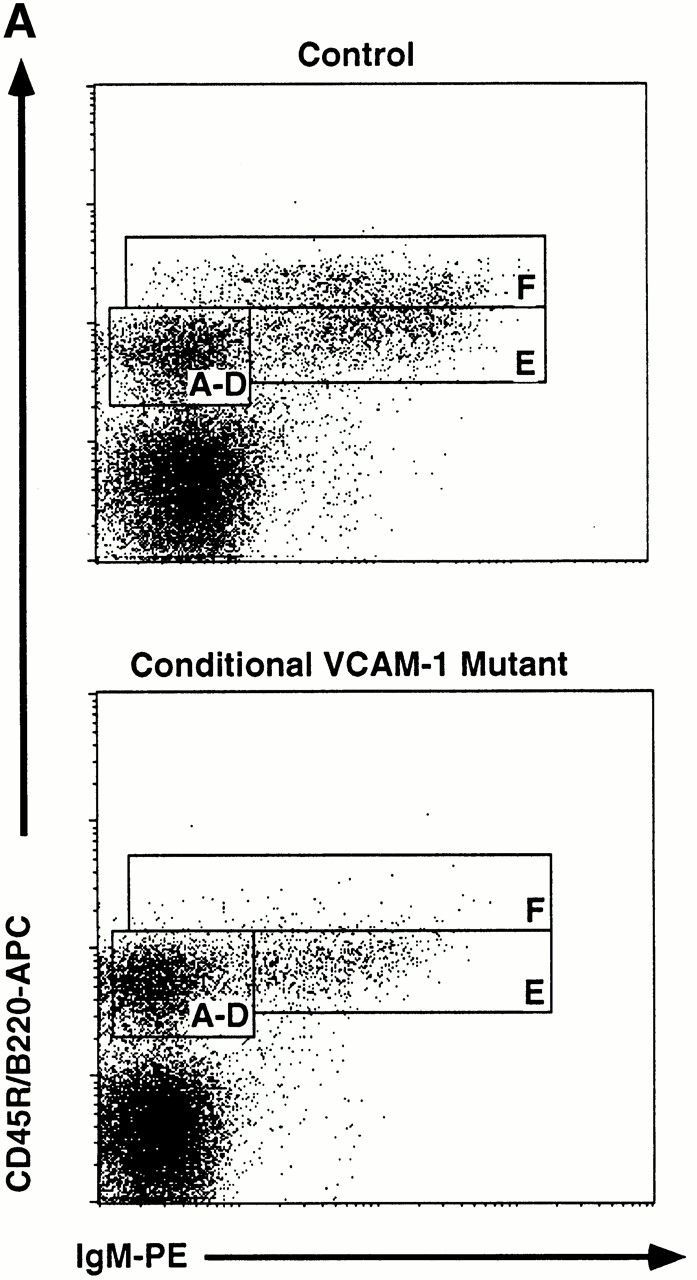
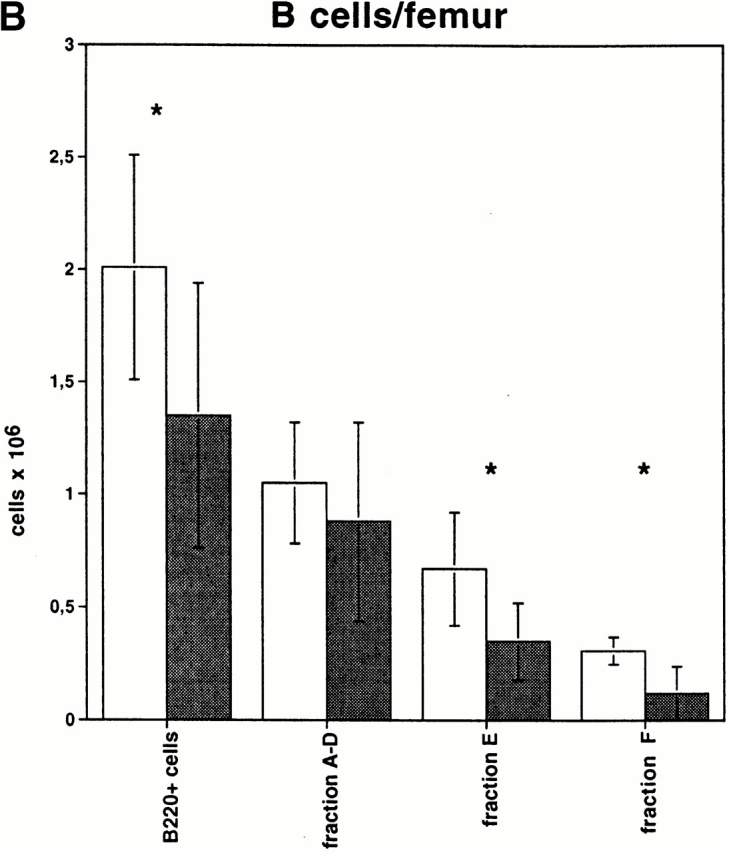
Altered B cell subsets in BM of conditional VCAM-1 mutant mice. (A) Flow cytometric analysis of BM cells within the lymphocyte gate (reference 45). B lineage cells are displayed by their CD45R/B220 and IgM expression. The letters in the rectangles indicate the different developmental stages according to Hardy et al. (reference 42). Conditional VCAM-1 mutant mice exhibit reduced numbers of B cells in fraction E and fraction F. 20,000 events were collected for both of the dot plots. More than 10 mice for both genotypes were analyzed, of which one representative experiment is depicted. (B) Comparison of absolute B cell numbers/femur (± SEM) between conditional VCAM-1 mutant mice (gray bars) and control mice (white bars). Values are calculated from four independent experiments including 8 control and 11 conditional VCAM-1 mutant mice. *Significantly different from value in control mice, P < 0.05.
Fraction E cells comprise immature B cells that will soon leave the BM to enter the blood. Therefore, we wondered whether this B cell subset, which was reduced in the BM of conditional VCAM-1 mutant mice, would be detectable outside of the BM. In peripheral blood from conditional VCAM-1 mutant mice an increased number of white blood cells (16.3 ± 3.9 × 103/mm3 blood versus 7.2 ± 1 × 103/mm3 blood in control mice) was detected compared with control mice. The ratio between lymphocytes, neutrophils, and monocytes, as well as the fraction of B220+ cells within the lymphocyte gate, was not significantly altered (Table ). These findings are in accordance with experiments of anti–VLA-4 treatment in primates leading to granulocytosis as well as lymphocytosis 20 21, whereas blocking of VLA-4 in rats caused a strong increase of mononuclear leukocytes and only a minor increase of polymorphonuclear leukocytes 55. However, in conditional VCAM-1 mutant mice there was a dramatic increase of IgM+IgD− B cells (Fig. 3), a fraction of cells that could possibly represent the fraction E cells found to be diminished in the BM (Fig. 2a and Fig. b). 30.5 ± 5.1% of all B220+ cells in conditional VCAM-1 mutant mice compared with 14.9 ± 1.8% of all B220+ cells in control mice belonged to the fraction of immature IgM+IgD− B cells in peripheral blood (Fig. 3). This translates into a 3.5-fold increase of this population with regard to absolute cell numbers (1.72 ± 0.41 × 103/mm3 blood in conditional VCAM-1 mutant mice versus 0.49 ± 0.04 × 103/mm3 blood in control mice). In addition, about threefold elevated levels of B220+IgM−IgD− B cells, which may comprise B cells earlier during development than fraction E, were detectable in peripheral blood of conditional VCAM-1 mutant mice (0.32 ± 0.13 × 103/mm3 blood in mutant mice versus 0.11 ± 0.04 × 103/mm3 blood in control mice).
Table 1.
Peripheral Blood Counts
| Blood cell types | Control | ConditionalVCAM-1 mutant | Foldincrement | P value |
|---|---|---|---|---|
| RBCs (106/mm3) | 8.7 ± 0.1 | 8.0 ± 0.6 | 0.9 | 0.13 |
| Hemoglobin (g/dl) | 14.2 ± 0.4 | 14.1 ± 0.7 | 1.0 | 0.92 |
| WBCs (103/mm3) | 7.2 ± 1.0 | 16.3 ± 3.9 | 2.3 | 0.008 |
| L (% of WBCs) | 71.9 ± 13.4 | 70.1 ± 11.3 | ||
| B cells (% of L) | 54.5 ± 0.5 | 51.2 ± 2.2 | ||
| T cells (% of L) | 37.3 ± 6.0 | 32.2 ± 4.1 | ||
| N (% of WBCs) | 19.9 ± 3.2 | 20.6 ± 4.8 | ||
| M (% of WBCs) | 3.5 ± 0.8 | 3.4 ± 0.4 | ||
| E (% of WBCs) | 1.6 ± 1.2 | 2.4 ± 1.2 | ||
| Ratio L/N/M/E | 45:12:2:1 | 44:13:2:1.5 |
Hemoglobin levels as well as numbers of erythrocytes and leukocytes were evaluated in three control and seven mutant mice. E, eosinophils; L, lymphocytes; M, monocytes; N, neutrophils; WBC, white blood cell.
Figure 3.
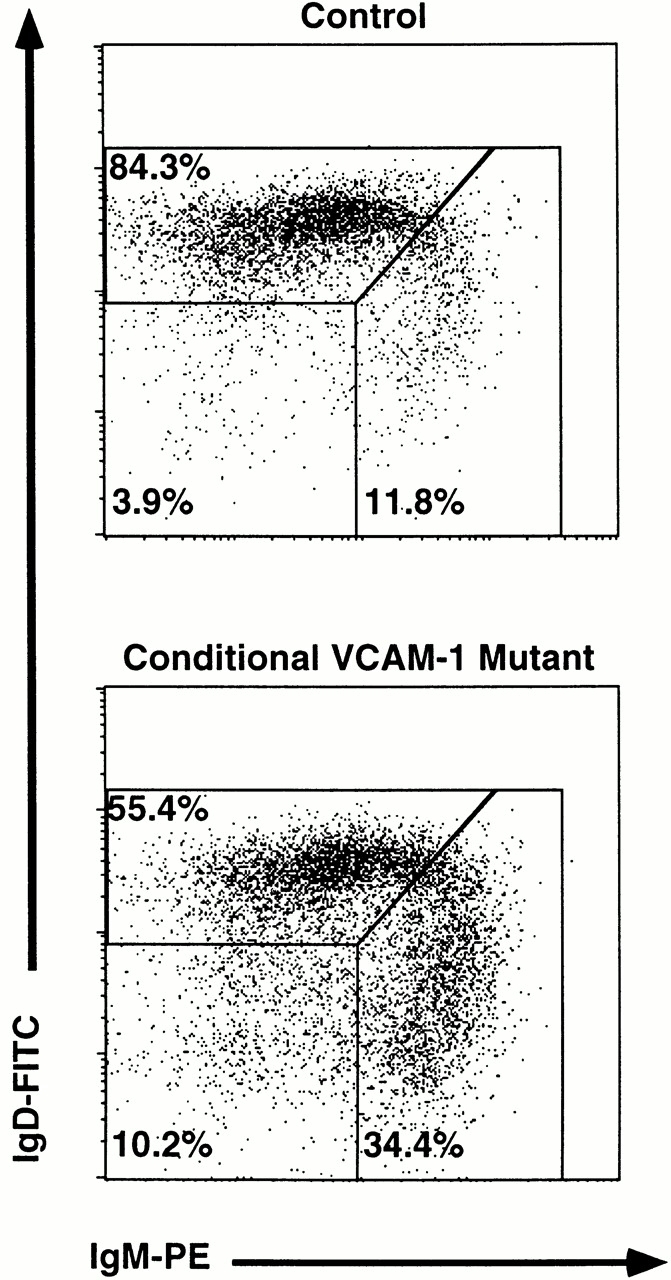
Increased number of immature B cells in peripheral blood of conditional VCAM-1 mutant mice. Flow cytometric analysis of peripheral blood B cells. In each experiment 20,000 events were acquired and subsequently gated for B cells (CD45R/B220+ cells). The dot blots represent the CD45R/B220+ cells within the lymphocyte gate in a control (top) and a conditional VCAM-1 mutant mouse (bottom) displayed by their IgM and IgD surface expression. In addition, the frequencies of mature (IgM+IgD+), immature (IgM+IgD2), and B220 only (IgM−IgD−) B cells are depicted as percentages of all B220+ cells. More than 15 mice for both genotypes were analyzed, of which one representative experiment is shown.
Taken together, there is a sizeable shift in the peripheral blood B cell pool of conditional VCAM-1 mutant mice towards the more immature cells. This shift goes along with the loss of fraction E cells in the BM, which led us to hypothesize that VCAM-1 may be important for retention of B lineage cells during development in BM until they reach a certain developmental stage.
In contrast, the distribution of T cells was not significantly altered in conditional VCAM-1 mutant mice. T cell subsets (CD3, CD4, CD8) in thymus, spleen, peripheral blood, and lymph nodes were similar in conditional VCAM-1 mutant mice and control mice (data not shown).
B Cells Accumulating in the Blood of Conditional VCAM-1 Mutant Mice Are Immature Emigrants from the BM.
Recently a novel mAb called 493 has been described which binds to a surface protein designated pB130-140 48. This Ab discriminates between long-lived recirculating 493− B cells and various stages of B cell development, including fraction E cells which are either inside the BM or have just left this site and still express pB130-140. Since the reduction of B cells in the BM is accompanied by an increase of a similar B cell fraction in blood, we examined the origin of these B cells in blood according to their pB130-140 expression. As predicted, the fraction of 493+ cells within the lymphocyte gate in peripheral blood of conditional VCAM-1 mutant mice was almost doubled compared with control mice (Fig. 4 A). A strong increase of the absolute number of 493+IgM+IgD− B cells and a minute increase of the 493+B220+IgM−IgD− B cells were noted (Fig. 4 B). We evaluated whether immature B cells would also be detectable at higher numbers in spleens of conditional VCAM-1 mutant mice. 10–20% of the immature B cells are supposed to reach the spleen as 493+ cells 48; however, the subsets of 493+ immature B cells have similar sizes in spleens of conditional VCAM-1 mutant mice and control mice. Compared with blood there is only a modest increase of 493+IgM+ cells detectable in spleens of VCAM-1 deficient mice (Fig. 4 C). There are several possible explanations: 493+ cells in peripheral blood of conditional VCAM-1 mutant mice have a reduced half life, 493 expression on these cells is rapidly downregulated, or, alternatively, these cells get trapped in sites other than the spleen.
Figure 4.
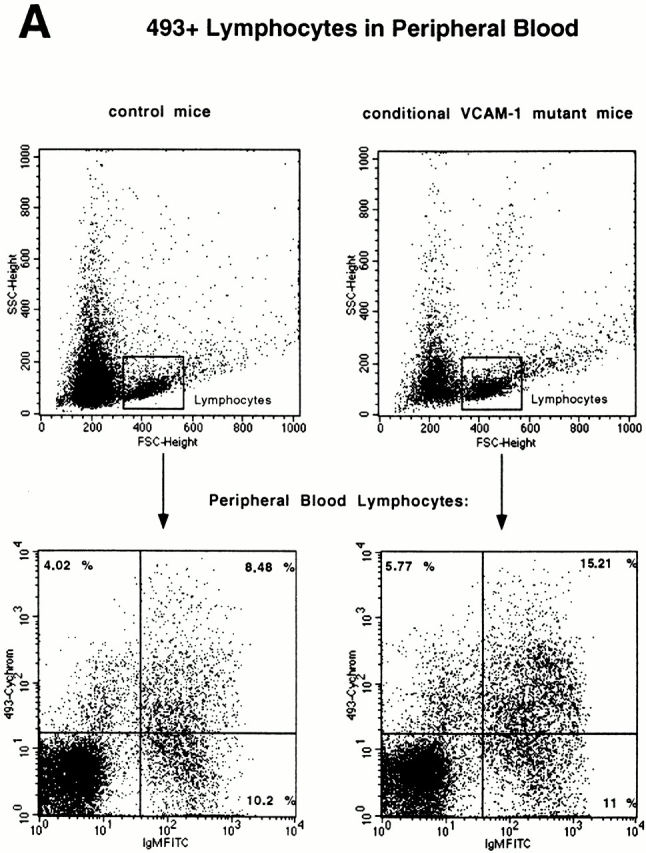
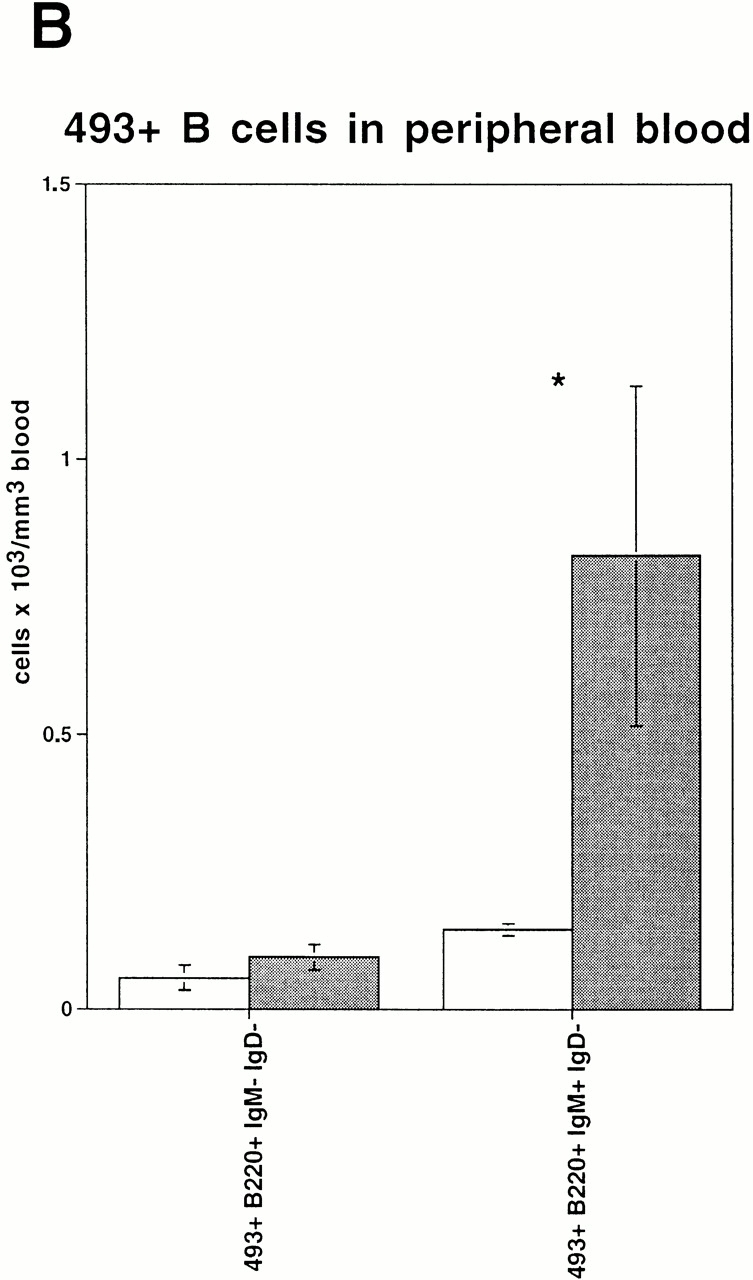
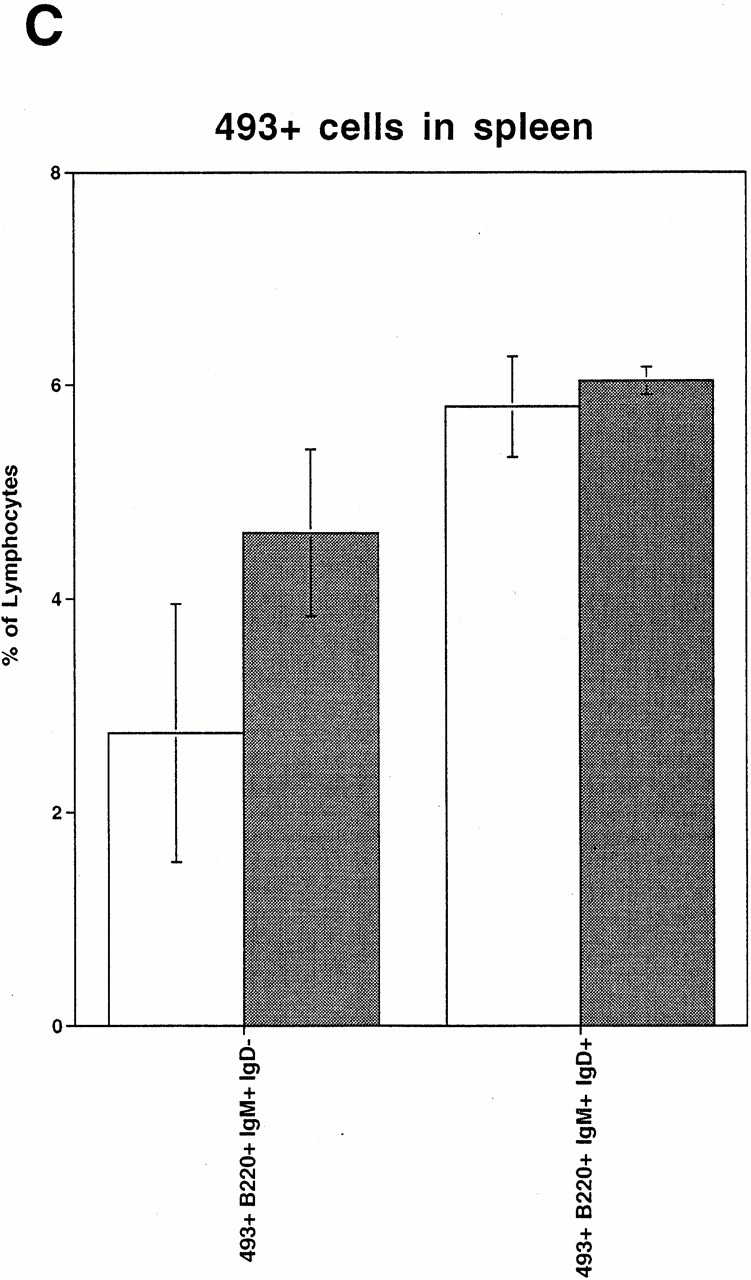
Expression of 493, a marker for immature B cells, on peripheral blood B cells. (A) Increased number of 493+IgM+ B cells in peripheral blood of conditional VCAM-1 mutant mice was detected. In each experiment, 50,000 events were acquired. The upper dot blots represent peripheral blood cells in a control (left) and a conditional VCAM-1 mutant mouse (right) to indicate gating for lymphocytes. The bottom displays the cells within the lymphocyte gate by their IgM and 493 surface expression. The frequencies of IgM−493+, IgM+493+, and IgM+493− cells are depicted as percentages of all lymphocytes. Out of three experiments, one representative flow cytometric study including three control and seven conditional VCAM-1 mutant mice is shown. (B) The graph illustrates the increase of 493+ B cells in peripheral blood of conditional VCAM-1 mutant mice compared with control mice. The mean absolute cell counts for leukocytes and lymphocytes determined in a separate experiment were used to calculate absolute numbers for the B cell subsets. White bars represent control mice; gray bars represent conditional VCAM-1 mutant mice. (C) Number of 493+ cells in spleen of two control mice (white bars) and two conditional VCAM-1 mutant mice (gray bars) are shown as a percentage of all lymphocytes. Immature IgD− B cells are identified by their IgM surface expression, and mature B cells according to their IgD+ staining. Values are depicted with the SEM from the flow cytometric analysis. *Significantly different from value in control mice, P < 0.05.
To determine the transit time of IgM+ cells from BM to blood and spleen, we performed BrdU labeling of dividing cells. After a labeling period of 3 d, we recovered 21 ± 7.9% IgM+BrdU+ cells in BM, 20.8 ± 1.2% in blood, and 26.8 ± 1.2% in spleen of control mice (as percentage of all B220+ cells). In contrast, conditional VCAM-1 mutant mice showed lower numbers of IgM+BrdU+ cells in BM (15.8 ± 4.2%) and increased amounts in blood (28.6 ± 2.1%) and spleen (33.3 ± 3.4%). If absolute cell numbers are considered, the increase of newly generated IgM+ cells in the blood of conditional VCAM-1 mutant mice is even higher. Taken together, these results suggest that the IgM+IgD− B cells in peripheral blood of conditional VCAM-1 mutant mice are recent emigrants from the BM.
Apoptosis of Immature B Cells in the Periphery.
Although the immature B cell compartment in blood is enlarged by premature transit of these cells from the BM (Fig. 4a and Fig. b), no significant increase in immature B cells was observed in spleen (Fig. 4 C) or lymph nodes of conditional VCAM-1 mutant mice. Since increased apoptosis of B cells in blood could account for this observation, we quantified the number of apoptotic cells in the peripheral blood. In both control mice and conditional VCAM-1 mutant mice, ∼12% of the IgM+ cells were stained with annexin V (data not shown). Therefore, the frequency of apoptotic B cells does not seem to differ in blood from mutant and control animals.
Recirculation of Mature IgM+IgD+ B Cells into the BM Is Influenced by VCAM-1 Expression.
In addition to the premature loss of immature B cells from the BM into the blood, we also observed a phenotype of the VCAM-1 deficiency for mature recirculating B cells. Fraction F cells in the BM were reduced by ∼63% compared with control mice. These cells are mature B cells expressing IgM and IgD, which are thought to recirculate from the periphery into the BM 56. The number of fraction F cells significantly decreased from 2.4% (0.31 ± 0.06 × 106 cells per femur) in control mice to 0.9% (0.12 ± 0.1 × 106 cells per femur) of all nucleated cells in conditional VCAM-1 mutant mice (Fig. 2a and Fig. b). We hypothesized that VCAM-1 may play a role in mediating migration of mature B cells into the BM. To address this we performed a short-term migration assay of mature B cells. Splenocytes of control mice were isolated and the immature CD43+ cells were depleted by MACS®. Enriched CD43− splenocytes, consisting mainly of IgM+IgD+ B cells, were labeled with PKH-26 and injected into the tail veins of conditional VCAM-1 mutant mice and control mice. First, in control mice we were readily able to detect PKH-26–labeled mature B cells in the BM after 20 h (rectangle FD in Fig. 5, left dot plot), which supports the idea that mature B cells enter the BM. More important, about half of the PKH-26–labeled cells, which were detectable in BM of control mice, were found in BM of conditional VCAM-1 mutant mice (Fig. 5). The reduced number of PKH-26–labeled mature B cells that were detected in the BM of conditional VCAM-1 mutant mice after cell transfer in size parallels the observed reduction of fraction F (which is depicted by the rectangle F in Fig. 5, right dot plot) in these animals. This could be explained by an impaired recirculation of mature B cells into the BM caused by VCAM-1 deficiency. However, we cannot rule out other possible explanations for this phenotype: for example, an increasing loss of mature IgD+ B cells from the BM or a rapid death of these cells in BM as a result of the VCAM-1 deficiency. In addition, the short-term migration assay in the mutant animals might be influenced by the leukocytosis present in these mice: competition of the PKH-26–labeled cells with leukocytes present in the blood stream might affect the efficiency of PKH-26–labeled cells to enter the BM. The latter explanation is unlikely, as short-term migration assays performed with total splenocytes did not show any differences with regard to lymphocyte homing to spleen and peripheral lymph nodes in mutant and control mice, while the impaired recirculation to BM was confirmed (preliminary data not shown).
Figure 5.

Short-term migration of mature B cells into the BM. Flow cytometric analysis of CD45R/B220+ gated lymphocytes in BM of control mice and conditional VCAM-1 mutant mice 20 h after injection of mature splenic PKH-26–labeled B cells. The BM B cells are displayed by their IgD surface and PKH-26 expression. Rectangle FD contains the donor-derived, PKH-26–labeled, recirculating mature B cells; rectangle F represents the fraction F of mature B cells present in the host. 0.06 ± 0.011% of all acquired events were recirculating B220+PKH-26+IgD+ B cells in control mice, while only 0.03 ± 0.01% of such cells were recovered from conditional VCAM-1 mutant BM. In total, 200,000 events were acquired for each dot plot; four control mice and four conditional VCAM-1 mutant mice were analyzed in two independent assays, of which one representative pair of mice is shown.
Conditional VCAM-1 Mutant Mice Exhibit an Impaired T Cell–dependent Humoral Immune Response.
To investigate whether the function of B cells is affected by the VCAM-1 deficiency, mice were immunized with NP-CG. The specific immune response in conditional VCAM-1–deficient mice and control mice was monitored by measuring the NP-specific Ig levels at different time points. Before immunization, slightly but not significantly reduced levels of IgA, IgM, IgG1, IgG2a, IgG2b, and IgG3 were detected in sera of conditional VCAM-1 mutant mice (data not shown). Upon NP-CG immunization, conditional VCAM-1 mutant mice exhibited impaired primary and greater than fivefold reduced secondary IgG1 responses compared with control mice (Fig. 6). Affinity maturation of the Ab response against NP and generation of germinal centers in conditional VCAM-1 mutant mice did not seem to be affected (data not shown; see Materials and Methods). In addition, the numbers of IgG secreting plasma cells in BM and spleen of control and VCAM-1 deficient mice did not show any significant difference. 10-mo-old control mice contained 230 ± 85 IgG-secreting plasma cells/2 × 105 cells in BM and 77 ± 17 IgG-secreting plasma cells/2 × 105 cells in spleen, versus 247 ± 90 in BM and 75 ± 21 in spleen of conditional VCAM-1 knockout mice. Although the generation of an efficient humoral immune response to a T cell–dependent antigen is dependent on VCAM-1, we could not distinguish whether this effect of the VCAM-1 deficiency is caused by an intrinsic B cell defect or is due to a defect in T and B cell interaction. Interestingly, when conditional VCAM-1 mutant mice were immunized with NP-Ficoll, which serves as a T cell–independent antigen, the immune responses were similar in mutant and control mice (anti-NP λ preimmune levels less than 0.08 μg/ml in mutant and control mice; 14 d after immunization 47 ± 6.2 μg/ml in control and 46.1 ± 22.8 μg/ml in conditional VCAM-1 mutant mice).
Figure 6.
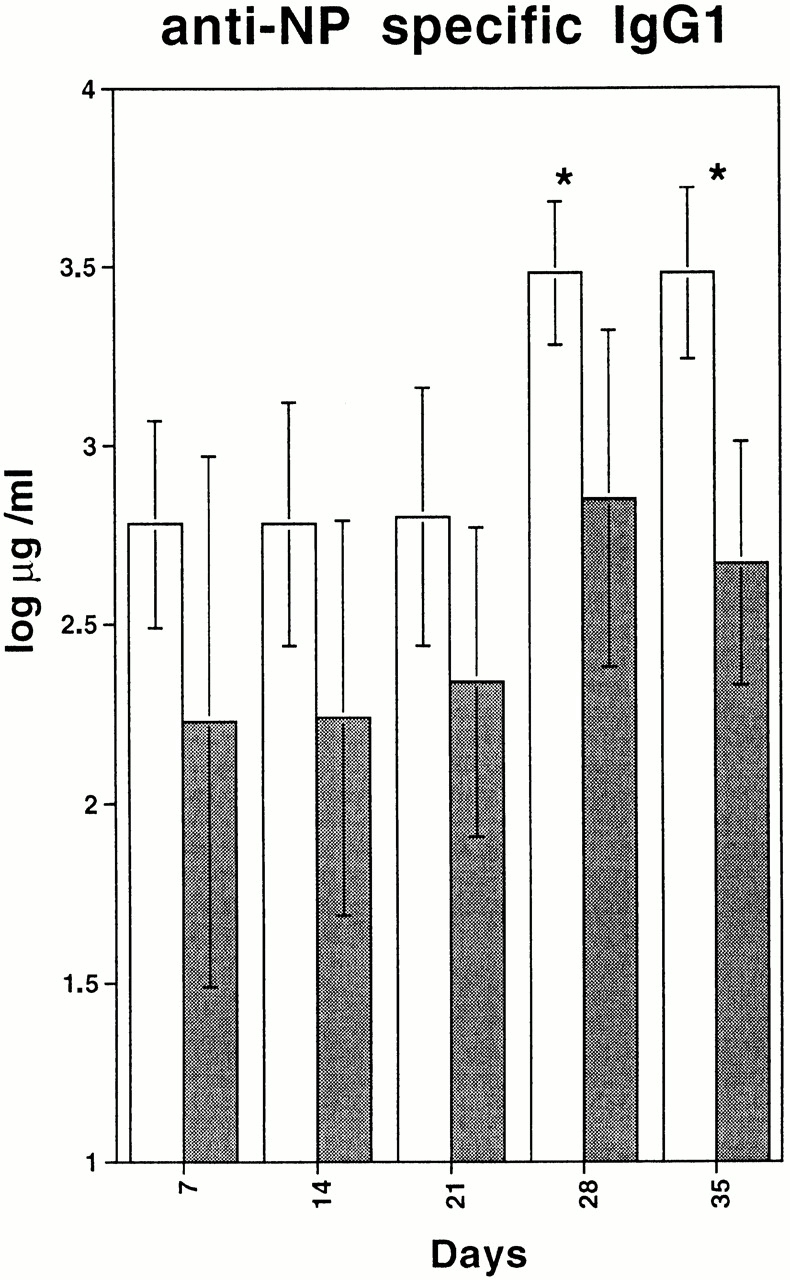
T cell–dependent immune response. Anti–NP-IgG1 levels in sera from control mice (white bars) and from conditional VCAM-1 mutant mice (gray bars) are depicted over 35 d after immunization. Two independent experiments were performed. The values shown were calculated from one experiment including three control mice and eight conditional VCAM-1 mutant mice. *Significantly different from value in control mice, P < 0.05.
Discussion
To study the in vivo function of the VCAM-1 gene, conditional VCAM-1 mutant mice were generated, since conventional gene targeting of VCAM-1 had resulted in embryonic lethality. VCAM-1flox/flox mice (here control mice) and conditional VCAM-1 mutant mice are viable, and VCAM-1flox/flox mice expressed similar levels of VCAM-1 compared with wild-type mice 35. After IFN-α induced Cre expression 37, the loxP-flanked VCAM-1 alleles were efficiently deleted in BM, liver, lymph nodes, spleen, and thymus. Southern blot analysis revealed almost complete deletion for VCAM-1 in these tissues and lower efficiency of deletion in heart, intestine, kidney, and lung (Fig. 1 C). In accordance with the marginal IFN-induced Cre expression in the brain of Mx-cre transgenic mice 37 there was only poor VCAM-1 deletion in the brain of conditional VCAM-1 mutant mice, probably caused by limited accessibility of this compartment for IFN-α 57. In addition, we confirmed the deficiency of VCAM-1 protein after induced deletion by Western blot analysis and by immunohistochemistry using various VCAM-1–specific Abs (Fig. 1D and Fig. e). With respect to the large amounts of immunoprecipitated VCAM-1 protein detectable in control mice (Fig. 1 D), we cannot exclude that VCAM-1 expression is upregulated by the neonatal treatment of these mice with IFN-α, which would be similar to the induction of VCAM-1 expression by IFN-α in humans 58.
We studied the role of VCAM-1 for hematopoiesis in conditional VCAM-1 mutant mice. In accordance with studies using mAbs that block VLA-4 function 20 a greater than twofold increase of the white blood cells was detected in the mutants. However, in contrast to the studies by Papayannopoulou et al. 20 and to the results obtained from mice with reduced VCAM-1 expression 36, we observed an altered distribution of immature and mature B cells in BM and peripheral blood of conditional VCAM-1 mutants. The strong increase of B220+IgM+ B cells in blood is paralleled by a loss of immature B cells (fraction E) from the BM. The expression of pB130-140 by these immature B cells as recognized by the 493 Ab suggests that they originate from the BM (Fig. 4a and Fig. b). This interpretation is further supported by the increase of BrdU+IgM+ B cells in the blood of conditional VCAM-1 mutant mice after a labeling period of 3 d, which is accompanied by a modest decrease of these cells in BM of VCAM-1–deficient mice. These results describe a novel function for VCAM-1: retention of B cells at late developmental stages (i.e., B220+IgM+IgD− immature B cells) in the BM. The loss of immature B cells from BM and their accumulation in peripheral blood resembles data obtained from mice reconstituted with CXC chemokine receptor (CXCR)4-deficient fetal liver cells 59. However, there are also differences in the phenotypes of conditional VCAM-1 mutants and mice reconstituted with CXCR4-deficient fetal liver cells. While conditional VCAM-1 mutant mice exhibit the most dramatic changes in the fraction of immature B cells in the BM, the B cell progenitors and the pre-B cells are mainly affected in mice reconstituted with CXCR4-deficient fetal liver cells as well as in mice with a deletion of the CXCR4 ligand, stromal cell–derived factor 1 (SDF-1; reference 60). A contribution of SDF-1 for migration of hematopoietic progenitor cells into the BM by activation of the VLA-4/VCAM-1 adhesion pathway has already been shown in vitro 61. Interestingly, the VCAM-1 and CXCR4 mutations have in common that they specifically affect B cells, while the T cell compartment so far has not been shown to be altered by either mutation.
CD24 heat stable antigen (HSA)–deficient mice also exhibit a phenotype that shares features of the VCAM-1 deficiency. Fraction E cells are significantly reduced in the BM of CD24-deficient mice; however, there are no data available on whether these cells appear in peripheral blood. Nielsen et al. have interpreted their findings as a leaky block in B cell development 62. Since another study has reported that CD24 HSA expression is a prerequisite for VLA-4/VCAM-1–mediated adhesion of pre-B cells to endothelioma cells 63, one may speculate that the mutation of VCAM-1 as well as of CD24 HSA both affect a common pathway which mediates retention of immature B cells in the BM.
Apart from the altered distribution of B cells between BM and peripheral blood in conditional VCAM-1 mutant mice, the development from pre-B cells to mature IgD+ B cells does not seem to be affected, given that we detected similar numbers of mature B cells in lymph nodes and spleen of conditional VCAM-1 mutant and control mice. Therefore, it may be that only a limited number of immature B cells, which are increased in blood of conditional VCAM-1 mutant mice, undergoes transition to mature B cells. We cannot rule out that these immature B cells are functionally not equivalent to the immature B cells in blood of control mice. Whether immature B cells in conditional VCAM-1 mutant mice possess the same ability to renew the mature B cell pool like immature B cells in control mice, which have spent a longer time in the BM, and whether they exhibit the same half-life and homing properties are still unknown. It has recently been suggested that the size of the peripheral mature B cell pool is autonomously regulated with the priority to maintain normal IgM serum levels and is not a direct function of the number of available B cell precursors 64. This mechanism may keep the mature B cell pool constant in the conditional VCAM-1 mutant mice as well, as these mice exhibit normal Ig levels. The immature B cells in blood, which are not used to replenish the mature B cell pool of conditional VCAM-1 mutant mice, may leave the blood and may either be trapped or die in other compartments. However, immature B cells do not accumulate in significant numbers in spleen or lymph nodes of mutant mice. In addition, we could not show that B cells undergo apoptosis in blood at significantly higher rates than in control animals. In general, similar statements apply also for T cells, e.g., enlarged amounts of T cells in peripheral blood, but no increased amounts of apoptotic T cells in blood or higher T cell numbers in secondary lymphoid tissues of young mice.
During fetal life β1 integrins are critical for hematopoiesis 65 and B and T cell development postnatally depends on α4 integrin 63 66 67. α4 integrin mediates efficient attachment and transmigration of pre-B cells beneath the BM stroma, which seems to be important for B cell development. In the absence of α4 integrin, B and T cell development is still possible, albeit at a very inefficient rate 67; B cell development in α4 integrin-deficient chimeric mice is impaired before the pro-B cell stage 66. By contrast, our data suggest that B and T cells can develop in the absence of the α4β1 integrin ligand, VCAM-1. We therefore hypothesize that, while VCAM-1 mediates retention of immature B cells in the BM, other ligands of α4 integrin serve additional functions during lymphopoiesis. The role of the α4 integrin ligand fibronectin for localization of HPCs is still controversial. In one study, treatment with reagents blocking binding to fibronectin (connecting sequence-1 [CS-1] inhibitor, mAbs against α5β1) did not lead either to an increase in the number of HPCs in peripheral blood or to a reduction of HPCs in BM 22, while a different study revealed increasing numbers of HPCs by intravenous injection of blocking peptides for all three primary fibronectin-binding sites 68. The expression of fms-like tyrosine kinase 3 (Flt3) ligand and IL-3 receptor on early B lineage cells and the in vitro finding that through these receptors binding of VLA-4 and VLA-5 to fibronectin is augmented 69 70 support the idea that particularly early developmental stages of B lineage cells may depend on interaction with BM stromal cells via fibronectin.
An additional important observation in the BM of conditional VCAM-1 mutant mice is the reduction of the mature B cells (fraction F) in this compartment (Fig. 2a and Fig. b). Short-term migration assays confirmed the hypothesis that in mice mature B cells recirculate from the peripheral blood to the BM, and showed that in the absence of VCAM-1 the lodgement of mature B cells to the BM is impaired (Fig. 5). Therefore, VCAM-1 not only plays a role in recruitment of HPCs to the BM, as recently shown 71, but is probably also involved in the recirculation of mature B cells from peripheral blood to BM. This is supported by recent findings of Koni et al., who have shown that migration of mature B cells into the BM is also impaired in TIE2Cre;VCAM-1flox/Δ mice 72. The functional significance of this recirculation of mature B cells to the BM is still unclear.
Taken together, our results show that in addition to the already described function of VCAM-1 for hematopoietic progenitor localization VCAM-1 plays a crucial role for the ordered trafficking of immature B cells and mature B cells between BM and periphery. Since, apart from the leukocytosis in peripheral blood (Table ), the observed consequences of the conditional VCAM-1 mutation exclusively affect B cells but not T cells (see Results), one can speculate that during lymphoid development VCAM-1 expression in BM becomes important for lymphocyte retention subsequent to the stage of a common lymphoid progenitor cell and after the division into T and B cell lineage commitment.
mAbs against either intercellular adhesion molecule (ICAM)-1, CD54, or VCAM-1 inhibit binding of human germinal center B cells to follicular dendritic cells in vitro 25. Moreover, ligation of the B cell receptor augments the adhesion of B cells to VCAM-1 and fibronectin in germinal centers by c-Met induction 73. The expression of VCAM-1 on follicular dendritic cells and the possible role of VCAM-1 for costimulation of T cells raised the question of whether deletion of VCAM-1 would have an impact on the immune response to a T cell–dependent antigen in vivo. The structurally related adhesion molecule ICAM-1, which in vitro had also been shown to participate in germinal center interactions and to function as a costimulatory molecule for T cells 25 74, does not seem to participate in the humoral immune response. ICAM-1–deficient mice generate normal Ab responses to immunization with OVA 75. By contrast, VCAM-1 is necessary for an efficient immune response against a T cell–dependent antigen, as shown after immunization of conditional VCAM-1 mutant mice (Fig. 6).
Blocking the VLA-4/VCAM-1 adhesion pathway has been demonstrated to increase apoptosis in human germinal center B cells 27. Therefore, VCAM-1 deficiency could also have affected affinity selection of B cells. However, we were unable to detect a difference in affinity maturation between conditional VCAM-1 mutant mice and control mice (data not shown). In addition, B cells in mutant animals were capable of mounting a robust T cell–independent humoral immune response (see Results). The impaired T cell–dependent humoral immune response could be explained by several mechanisms: B cell migration within the germinal center might be affected, and attachment of B cells to follicular dendritic cells might be impaired, leading to inefficient B cell proliferation. Alternatively, T cell help for mounting a humoral immune response might be impaired due to a lack of VCAM-1–mediated costimulation. Finally, recirculation of antigen-specific B cells to secondary lymphoid organs leading to a sustained immune response might depend on VCAM-1. We favor the idea that the reduced humoral immune response may be because of the absence of VCAM-1 expression on dendritic cells. At least IgG-secreting plasma cells are present in similar numbers in spleen and BM of control and mutant mice. Therefore, it is unlikely that an impaired migration of plasma cells to BM could explain the reduced humoral immune response in these mice.
In conclusion, the Mx-cre/loxP system can be used to inducibly delete genes which are expressed in endothelial cells. This approach was used to elucidate the in vivo function of VCAM-1 for B cell localization and humoral immune responses. We have detected a novel role for VCAM-1 in addition to its already known function for HPC migration, namely, its involvement in B cell homeostasis in BM and peripheral blood. VCAM-1 mediates retention of immature B cells in the BM and is probably involved in the recirculation of mature B cells from blood to the BM. Finally, VCAM-1 is critical for the T cell–dependent humoral immune response.
Acknowledgments
We thank B. Hampel, C. Uthoff-Hachenberg, and C. Göttlinger for their expert technical help. We are grateful to Dr. Klaus Rajewsky for his continuous support and critical reading of the manuscript. We thank Dr. Ralf Kühn for providing the Mx-cre transgenic mice. Dr. D. Vestweber generously donated anti–VCAM-1 mAbs. We thank P.A. Koni et al. for discussing with us their VCAM-1 mutant mice (TIE2Cre; VCAM-1flox/Δ mice) before submission for publication.
This study was supported by a grant from the Deutsche Forschungsgemeinschaft (WA1127/1-2, 1-3).
Footnotes
Abbreviations used in this paper: APC, allophycocyanin; BM, bone marrow; BrdU, bromodeoxyuridine; CXCR, CXC chemokine receptor; HPC, hematopoietic progenitor cell; HSA, heat stable antigen; ICAM, intercellular adhesion molecule; NP-CG, (4-hydroxy-3-nitrophenyl)-acetyl-chicken globulin; VCAM, vascular cell adhesion molecule; VLA, very late antigen.
References
- Rice G.E., Bevilacqua M.P. An inducible endothelial cell surface glycoprotein mediates melanoma adhesion. Science. 1989;246:1303–1306. doi: 10.1126/science.2588007. [DOI] [PubMed] [Google Scholar]
- Araki M., Araki K., Vassalli P. Cloning and sequencing of mouse VCAM-1 cDNA. Gene. 1993;126:261–264. doi: 10.1016/0378-1119(93)90377-f. [DOI] [PubMed] [Google Scholar]
- Cybulsky M.I., Allan-Motamed M., Collins T. Structure of the murine VCAM1 gene. Genomics. 1993;18:387–391. doi: 10.1006/geno.1993.1480. [DOI] [PubMed] [Google Scholar]
- Kumar A.G., Dai X.Y., Kozak C.A., Mims M.P., Gotto A.M., Ballantyne C.M. Murine VCAM-1. Molecular cloning, mapping, and analysis of a truncated form. J. Immunol. 1994;153:4088–4098. [PubMed] [Google Scholar]
- Terry R.W., Kwee L., Levine J.F., Labow M.A. Cytokine induction of an alternatively spliced murine vascular cell adhesion molecule (VCAM) mRNA encoding a glycosylphosphatidylinositol-anchored VCAM protein. Proc. Natl. Acad. Sci. USA. 1993;90:5919–5923. doi: 10.1073/pnas.90.13.5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elices M.J., Osborn L., Takada Y., Crouse C., Luhowskyj S., Hemler M.E., Lobb R.R. VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell. 1990;60:577–584. doi: 10.1016/0092-8674(90)90661-w. [DOI] [PubMed] [Google Scholar]
- Kamata T., Puzon W., Takada Y. Identification of putative ligand-binding sites of the integrin α4β1 (VLA-4, CD49d/CD29) Biochem. J. 1995;305:945–951. doi: 10.1042/bj3050945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegg C., Postigo A.A., Sikorski E.E., Butcher E.C., Pytela R., Erle D.J. Role of integrin α4β7/α4βP in lymphocyte adherence to fibronectin and VCAM-1 and in homotypic cell clustering. J. Cell Biol. 1992;117:179–189. doi: 10.1083/jcb.117.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn L., Vassallo C., Benjamin C.D. Activated endothelium binds lymphocytes through a novel binding site in the alternately spliced domain of vascular cell adhesion molecule 1. J. Exp. Med. 1992;176:99–107. doi: 10.1084/jem.176.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonderheide R.H., Tedder T.F., Springer T.A., Staunton D.E. Residues within a conserved amino acid motif of domains 1 and 4 of VCAM-1 are required for binding to VLA-4. J. Cell Biol. 1994;125:215–222. doi: 10.1083/jcb.125.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo A.A., Garcia-Vicuna R., Diaz-Gonzalez F., Arroyo A.G., De Landazuri M.O., Chi-Rosso G., Lobb R.R., Laffon A., Sanchez-Madrid F. Increased binding of synovial T lymphocytes from rheumatoid arthritis to endothelial-leukocyte adhesion molecule-1 (ELAM-1) and vascular cell adhesion molecule-1 (VCAM-1) J. Clin. Invest. 1992;89:1445–1452. doi: 10.1172/JCI115734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHale J.F., Harari O.A., Marshall D., Haskard D.O. Vascular endothelial cell expression of ICAM-1 and VCAM-1 at the onset of eliciting contact hypersensitivity in miceevidence for a dominant role of TNF-α. J. Immunol. 1999;162:1648–1655. [PubMed] [Google Scholar]
- Koizumi M., King N., Lobb R., Benjamin C., Podolsky D.K. Expression of vascular adhesion molecules in inflammatory bowel disease. Gastroenterology. 1992;103:840–847. doi: 10.1016/0016-5085(92)90015-q. [DOI] [PubMed] [Google Scholar]
- Wuthrich R.P. Vascular cell adhesion molecule-1 (VCAM-1) expression in murine lupus nephritis. Kidney Int. 1992;42:903–914. doi: 10.1038/ki.1992.367. [DOI] [PubMed] [Google Scholar]
- Sadahiro M., McDonald T.O., Allen M.D. Reduction in cellular and vascular rejection by blocking leukocyte adhesion molecule receptors. Am. J. Pathol. 1993;142:675–683. [PMC free article] [PubMed] [Google Scholar]
- Yednock T.A., Cannon C., Fritz L.C., Sanchez-Madrid F., Steinman L., Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4β1 integrin. Nature. 1992;356:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- Cybulsky M.I., Gimbrone M.A., Jr. Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991;251:788–791. doi: 10.1126/science.1990440. [DOI] [PubMed] [Google Scholar]
- Baron J.L., Madri J.A., Ruddle N.H., Hashim G., Janeway C.A., Jr. Surface expression of α4 integrin by CD4 T cells is required for their entry into brain parenchyma. J. Exp. Med. 1993;177:57–68. doi: 10.1084/jem.177.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake K., Medina K., Ishihara K., Kimoto M., Auerbach R., Kincade P.W. A VCAM-like adhesion molecule on murine bone marrow stromal cells mediates binding of lymphocyte precursors in culture. J. Cell Biol. 1991;114:557–565. doi: 10.1083/jcb.114.3.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papayannopoulou T., Nakamoto B. Peripheralization of hemopoietic progenitors in primates treated with anti-VLA4 integrin. Proc. Natl. Acad. Sci. USA. 1993;90:9374–9378. doi: 10.1073/pnas.90.20.9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papayannopoulou T., Craddock C., Nakamoto B., Priestley G.V., Wolf N.S. The VLA4/VCAM-1 adhesion pathway defines contrasting mechanisms of lodgement of transplanted murine hemopoietic progenitors between bone marrow and spleen. Proc. Natl. Acad. Sci. USA. 1995;92:9647–9651. doi: 10.1073/pnas.92.21.9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock C.F., Nakamoto B., Elices M., Papayannopoulou T. The role of CS1 moiety of fibronectin in VLA mediated haemopoietic progenitor trafficking. Br. J. Haematol. 1997;97:15–21. doi: 10.1046/j.1365-2141.1997.d01-2120.x. [DOI] [PubMed] [Google Scholar]
- Brasel K., McKenna H.J., Morrissey P.J., Charrier K., Morris A.E., Lee C.C., Williams D.E., Lyman S.D. Hematologic effects of flt3 ligand in vivo in mice. Blood. 1996;88:2004–2012. [PubMed] [Google Scholar]
- Freedman A.S., Munro J.M., Rice G.E., Bevilacqua M.P., Morimoto C., McIntyre B.W., Rhynhart K., Pober J.S., Nadler L.M. Adhesion of human B cells to germinal centers in vitro involves VLA-4 and INCAM-110. Science. 1990;249:1030–1033. doi: 10.1126/science.1697696. [DOI] [PubMed] [Google Scholar]
- Koopman G., Parmentier H.K., Schuurman H.J., Newman W., Meijer C.J., Pals S.T. Adhesion of human B cells to follicular dendritic cells involves both the lymphocyte function–associated antigen 1/intercellular adhesion molecule 1 and very late antigen 4/vascular cell adhesion molecule 1 pathways. J. Exp. Med. 1991;173:1297–1304. doi: 10.1084/jem.173.6.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman G., Keehnen R.M., Lindhout E., Zhou D.F., de Groot C., Pals S.T. Germinal center B cells rescued from apoptosis by CD40 ligation or attachment to follicular dendritic cells, but not by engagement of surface immunoglobulin or adhesion receptors, become resistant to CD95-induced apoptosis. Eur. J. Immunol. 1997;27:1–7. doi: 10.1002/eji.1830270102. [DOI] [PubMed] [Google Scholar]
- Koopman G., Keehnen R.M., Lindhout E., Newman W., Shimizu Y., van Seventer G.A., de Groot C., Pals S.T. Adhesion through the LFA-1 (CD11a/CD18)-ICAM-1 (CD54) and the VLA-4 (CD49d)-VCAM-1 (CD106) pathways prevents apoptosis of germinal center B cells. J. Immunol. 1994;152:3760–3767. [PubMed] [Google Scholar]
- Lindhout E., Mevissen M.L., Kwekkeboom J., Tager J.M., de Groot C. Direct evidence that human follicular dendritic cells (FDC) rescue germinal centre B cells from death by apoptosis. Clin. Exp. Immunol. 1993;91:330–336. doi: 10.1111/j.1365-2249.1993.tb05904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitseva M.B., Mojcik C.F., Salomon D.R., Shevach E.M., Golding H. Co-ligation of α4β1 integrin and TCR rescues human thymocytes from steroid-induced apoptosis. Int. Immunol. 1998;10:1551–1561. doi: 10.1093/intimm/10.10.1551. [DOI] [PubMed] [Google Scholar]
- Burkly L.C., Jakubowski A., Newman B.M., Rosa M.D., Chi-Rosso G., Lobb R.R. Signaling by vascular cell adhesion molecule-1 (VCAM-1) through VLA-4 promotes CD3-dependent T cell proliferation. Eur. J. Immunol. 1991;21:2871–2875. doi: 10.1002/eji.1830211132. [DOI] [PubMed] [Google Scholar]
- Damle N.K., Klussman K., Linsley P.S., Aruffo A. Differential costimulatory effects of adhesion molecules B7, ICAM-1, LFA-3, and VCAM-1 on resting and antigen-primed CD4+ T lymphocytes. J. Immunol. 1992;148:1985–1992. [PubMed] [Google Scholar]
- Schlegel P.G., Vaysburd M., Chen Y., Butcher E.C., Chao N.J. Inhibition of T cell costimulation by VCAM-1 prevents murine graft-versus-host disease across minor histocompatibility barriers. J. Immunol. 1995;155:3856–3865. [PubMed] [Google Scholar]
- Kwee L., Baldwin H.S., Shen H.M., Stewart C.L., Buck C., Buck C.A., Labow M.A. Defective development of the embryonic and extraembryonic circulatory systems in vascular cell adhesion molecule (VCAM-1) deficient mice. Development. 1995;121:489–503. doi: 10.1242/dev.121.2.489. [DOI] [PubMed] [Google Scholar]
- Gurtner G.C., Davis V., Li H., McCoy M.J., Sharpe A., Cybulsky M.I. Targeted disruption of the murine VCAM1 geneessential role of VCAM-1 in chorioallantoic fusion and placentation. Genes Dev. 1995;9:1–14. doi: 10.1101/gad.9.1.1. [DOI] [PubMed] [Google Scholar]
- Terry R.W., Kwee L., Baldwin H.S., Labow M.A. Cre-mediated generation of a VCAM-1 null allele in transgenic mice. Transgenic Res. 1997;6:349–356. doi: 10.1023/a:1018475031852. [DOI] [PubMed] [Google Scholar]
- Friedrich C., Cybulsky M.I., Gutierrez-Ramos J.C. Vascular cell adhesion molecule-1 expression by hematopoiesis-supporting stromal cells is not essential for lymphoid or myeloid differentiation in vivo or in vitro. Eur. J. Immunol. 1996;26:2773–2780. doi: 10.1002/eji.1830261133. [DOI] [PubMed] [Google Scholar]
- Kühn R., Schwenk F., Aguet M., Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- Rehbinder C., Baneaux P., Forbes D., van Herck H., Nicklas W., Rugaya Z., Winkler G. FELASA recomendations for the health monitoring of mouse, rat, hamster, gerbil, guinea pig and rabbit experimental units. Report of the Federation of European Laboratory Animal Science Associations (FELASA) Working Group on Animal Health accepted by the FELASA Board of Management, November 1995. Lab. Anim. 1996;30:193–208. doi: 10.1258/002367796780684881. [DOI] [PubMed] [Google Scholar]
- Lin Q., Dong C., Cooper M.D. Impairment of T and B cell development by treatment with a type I interferon. J. Exp. Med. 1998;187:79–87. doi: 10.1084/jem.187.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedder T.F., McIntyre G., Schlossman S.F. Heterogeneity in the B1 (CD20) cell surface molecule expressed by human B-lymphocytes. Mol. Immunol. 1988;25:1321–1330. doi: 10.1016/0161-5890(88)90047-8. [DOI] [PubMed] [Google Scholar]
- Coffman R.L. Surface antigen expression and immunoglobulin gene rearrangement during mouse pre-B cell development. Immunol. Rev. 1982;69:5–23. doi: 10.1111/j.1600-065x.1983.tb00446.x. [DOI] [PubMed] [Google Scholar]
- Hardy R.R., Carmack C.E., Shinton S.A., Kemp J.D., Hayakawa K. Resolution and characterization of pro-B and pre–pro-B cell stages in normal mouse bone marrow. J. Exp. Med. 1991;173:1213–1225. doi: 10.1084/jem.173.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloney W., McPherson K., Fliegelman L. Estrase activity in leukocytes demonstrated by the use of naphthol as-d chloroacetate substrate. J. Histochem. Cytochem. 1960;8:200–208. doi: 10.1177/8.3.200. [DOI] [PubMed] [Google Scholar]
- Grossi C., Cadoni A., Zicca A., Leprini A., Ferrarini M. Large granular lymphocytes in human peripheral bloodultrastructural and cytochemical characterization of the granules. Blood. 1982;59:277–283. [PubMed] [Google Scholar]
- Förster I., Vieira P., Rajewsky K. Flow cytometric analysis of cell proliferation dynamics in the B cell compartment of the mouse. Int. Immunol. 1989;1:321–331. doi: 10.1093/intimm/1.4.321. [DOI] [PubMed] [Google Scholar]
- Leptin M., Potash M.J., Grützmann R., Heusser C., Shulman M., Kohler G., Melchers F. Monoclonal antibodies specific for murine IgM I. Characterization of antigenic determinants on the four constant domains of the μ heavy chain. Eur. J. Immunol. 1984;14:534–542. doi: 10.1002/eji.1830140610. [DOI] [PubMed] [Google Scholar]
- Roes J., Müller W., Rajewsky K. Mouse anti-mouse IgD monoclonal antibodies generated in IgD-deficient mice. J. Immunol. Methods. 1995;183:231–237. doi: 10.1016/0022-1759(95)00059-j. [DOI] [PubMed] [Google Scholar]
- Rolink A.G., Andersson J., Melchers F. Characterization of immature B cells by a novel monoclonal antibody, by turnover and by mitogen reactivity. Eur. J. Immunol. 1998;28:3738–3748. doi: 10.1002/(SICI)1521-4141(199811)28:11<3738::AID-IMMU3738>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Miltenyi S., Müller W., Weichel W., Radbruch A. High gradient magnetic cell separation with MACS. Cytometry. 1990;11:231–238. doi: 10.1002/cyto.990110203. [DOI] [PubMed] [Google Scholar]
- Roes J., Rajewsky K. Immunoglobulin D (IgD)-deficient mice reveal an auxiliary receptor function for IgD in antigen-mediated recruitment of B cells. J. Exp. Med. 1993;177:45–55. doi: 10.1084/jem.177.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickert R.C., Rajewsky K., Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376:352–355. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- Kearny J., Lawton A. B lymphocyte differentiation induced by lipopolysaccharide. I. Generation of cells synthesizing four major immunoglobulin classes. J. Immunol. 1975;115:671–776. [PubMed] [Google Scholar]
- Müller W. Simultaneous flow cytometric detection of bromodeoxyuridine incorporation and cell surface marker expression. In: Radbruch A., editor. Flow Cytometry and Cell Sorting. 2nd ed. Springer-Verlag; New York: 2000. pp. 105–111. [Google Scholar]
- Weber H., Valenzuela D., Lujber G., Gubler M., Weissmann C. Single amino acid changes that render human IFN-α 2 biologically active on mouse cells. EMBO (Eur. Mol. Biol. Organ.) J. 1987;6:591–598. doi: 10.1002/j.1460-2075.1987.tb04795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issekutz T. Inhibition of in vivo lymphocytes migration to inflammation and homing to lymphoid tissues by the TA-2 monoclonal antibody. A likely role for VLA-4 in vivo. J. Immunol. 1991;147:4178–4184. [PubMed] [Google Scholar]
- Osmond D.G. Population dynamics of bone marrow B lymphocytes. Immunol. Rev. 1986;93:103–124. doi: 10.1111/j.1600-065x.1986.tb01504.x. [DOI] [PubMed] [Google Scholar]
- Smith R.A., Norris F., Palmer D., Bernhardt L., Wills R.J. Distribution of alpha interferon in serum and cerebrospinal fluid after systemic administration. Clin. Pharmacol. Ther. 1985;37:85–88. doi: 10.1038/clpt.1985.16. [DOI] [PubMed] [Google Scholar]
- Lechleitner S., Gille J., Johnson D.R., Petzelbauer P. Interferon enhances tumor necrosis factor–induced vascular cell adhesion molecule 1 (CD106) expression in human endothelial cells by an interferon-related factor 1–dependent pathway. J. Exp. Med. 1998;187:2023–2030. doi: 10.1084/jem.187.12.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q., Jones D., Springer T.A. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity. 1999;10:463–471. doi: 10.1016/s1074-7613(00)80046-1. [DOI] [PubMed] [Google Scholar]
- Ma Q., Jones D., Borghesani P.R., Segal R.A., Nagasawa T., Kishimoto T., Bronson R.T., Springer T.A. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl. Acad. Sci. USA. 1998;95:9448–9453. doi: 10.1073/pnas.95.16.9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K., Kobayashi M., Wang J., Ohiro Y., Hamada J., Cho Y., Imamura M., Musashi M., Kondo T., Hosokawa M., Asaka M. Selective transendothelial migration of hematopoietic progenitor cellsa role in homing of progenitor cells. Blood. 1999;93:149–156. [PubMed] [Google Scholar]
- Nielsen P.J., Lorenz B., Muller A.M., Wenger R.H., Brombacher F., Simon M., von der Weid T., Langhorne W.J., Mossmann H., Kohler G. Altered erythrocytes and a leaky block in B-cell development in CD24/HSA-deficient mice. Blood. 1997;89:1058–1067. [PubMed] [Google Scholar]
- Hahne M., Wenger R.H., Vestweber D., Nielsen P.J. The heat-stable antigen can alter very late antigen 4–mediated adhesion. J. Exp. Med. 1994;179:1391–1395. doi: 10.1084/jem.179.4.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agenes F., Rosado M.M., Freitas A.A. Independent homeostatic regulation of B cell compartments. Eur. J. Immunol. 1997;27:1801–1807. doi: 10.1002/eji.1830270731. [DOI] [PubMed] [Google Scholar]
- Hirsch E., Iglesias A., Potocnik A.J., Hartmann U., Fassler R. Impaired migration but not differentiation of haematopoietic stem cells in the absence of β1 integrins. Nature. 1996;380:171–175. doi: 10.1038/380171a0. [DOI] [PubMed] [Google Scholar]
- Arroyo A.G., Yang J.T., Rayburn H., Hynes R.O. Differential requirements for alpha4 integrins during fetal and adult heamtopoiesis. Cell. 1996;85:997–1008. doi: 10.1016/s0092-8674(00)81301-x. [DOI] [PubMed] [Google Scholar]
- Arroyo A.G., Yang J.T., Rayburn H., Hynes R.O. α4 integrins regulate the proliferation/differentiation balance of multilineage hematopoietic progenitors in vivo. Immunity. 1999;11:555–566. doi: 10.1016/s1074-7613(00)80131-4. [DOI] [PubMed] [Google Scholar]
- van der Loo J.C., Xiao X., McMillin D., Hashino K., Kato I., Williams D.A. VLA-5 is expressed by mouse and human long-term repopulating hematopoietic cells and mediates adhesion to extracellular matrix protein fibronectin. J. Clin. Invest. 1998;102:1051–1061. doi: 10.1172/JCI3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy C.L., Minguell J.J. Modulation of the adhesion of hemopoietic progenitor cells to the RGD site of fibronectin by interleukin 3. J. Cell. Physiol. 1995;164:315–323. doi: 10.1002/jcp.1041640212. [DOI] [PubMed] [Google Scholar]
- Shibayama H., Anzai N., Ritchie A., Zhang S., Mantel C., Broxmeyer H.E. Interleukin-3 and Flt3-ligand induce adhesion of Baf3/Flt3 precursor B-lymphoid cells to fibronectin via activation of VLA-4 and VLA-5. Cell. Immunol. 1998;187:27–33. doi: 10.1006/cimm.1998.1318. [DOI] [PubMed] [Google Scholar]
- Mazo I.B., Gutierrez-Ramos J.C., Frenette P.S., Hynes R.O., Wagner D.D., von Andrian U.H. Hematopoietic progenitor cell rolling in bone marrow microvesselsparallel contributions by endothelial selectins and vascular cell adhesion molecule 1 J. Exp. Med. 188 1998. 465 474[published erratum at 188:1001] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koni P.A., Joshi S.K., Temann U.-A., Olson D., Burkly L., Flavell R.A. Conditional vascular cell adhesion molecule 1 deletion in miceimpaired lymphocyte migration to bone marrow. J. Exp. Med. 2001;193:741–753. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pals S.T., Taher T.E., van der Voort R., Smit L., Keehnen R.M. Regulation of adhesion and migration in the germinal center microenvironment. Cell. Adhes. Commun. 1998;6:111–116. doi: 10.3109/15419069809004466. [DOI] [PubMed] [Google Scholar]
- Sligh J.E., Jr., Ballantyne C.M., Rich S.S., Hawkins H.K., Smith C.W., Bradley A., Beaudet A.L. Inflammatory and immune responses are impaired in mice deficient in intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. USA. 1993;90:8529–8533. doi: 10.1073/pnas.90.18.8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfield C.A., Brashler J.R., Winterrowd G.E., Bell F.P., Griffin R.L., Fidler S.F., Kolbasa K.P., Mobley J.L., Shull K.L., Richards I.M., Chin J.E. Intercellular adhesion molecule-1-deficient mice have antibody responses but impaired leukocyte recruitment. Am. J. Physiol. 1997;273:L513–L523. doi: 10.1152/ajplung.1997.273.3.L513. [DOI] [PubMed] [Google Scholar]