

Abstract
Defensins, antimicrobial peptides of the innate immune system, protect human mucosal epithelia and skin against microbial infections and are produced in large amounts by neutrophils. The bacterial pathogen Staphylococcus aureus is insensitive to defensins by virtue of an unknown resistance mechanism. We describe a novel staphylococcal gene, mprF, which determines resistance to several host defense peptides such as defensins and protegrins. An mprF mutant strain was killed considerably faster by human neutrophils and exhibited attenuated virulence in mice, indicating a key role for defensin resistance in the pathogenicity of S. aureus. Analysis of membrane lipids demonstrated that the mprF mutant no longer modifies phosphatidylglycerol with l-lysine. As this unusual modification leads to a reduced negative charge of the membrane surface, MprF-mediated peptide resistance is most likely based on repulsion of the cationic peptides. Accordingly, inactivation of mprF led to increased binding of antimicrobial peptides by the bacteria. MprF has no similarity with genes of known function, but related genes were identified in the genomes of several pathogens including Mycobacterium tuberculosis, Pseudomonas aeruginosa, and Enterococcus faecalis. MprF thus constitutes a novel virulence factor, which may be of general relevance for bacterial pathogens and represents a new target for attacking multidrug resistant bacteria.
Keywords: host defense peptides, oxygen-independent killing, Staphylococcus aureus virulence, phospholipids, innate immunity
Introduction
The human pathogen Staphylococcus aureus is a major cause of community- and hospital-acquired skin, respiratory, endovascular, soft tissue, bone, and joint infections 1. The increasing prevalence of multidrug resistant strains and the recent appearance of strains with reduced susceptibility to vancomycin, the antibiotic of last resort, raises the specter of untreatable staphylococcal infections and adds urgency to the search for new antiinfective strategies 2.
S. aureus has evolved the means to resist antimicrobial host components such as lysozyme, the α-defensins human neutrophil peptide (HNP)1–3 3, and the β-defensin hBD2 4. Defensin peptides constitute a shield against microbial infections on skin, on epithelia of the respiratory, gastrointestinal, and genitourinary tracts (β-defensins), and are found in large amounts in the granules of phagocytes and intestinal Paneth cells (α-defensins; reference 5). Although defensins account for 50% of the neutrophil granule proteins, they fail to kill S. aureus effectively. Accordingly, patients with inherited oxidative burst deficiency (chronic granulomatous disease [CGD]) are particularly susceptible to S. aureus infections 6. When keratinocytes from human skin come into contact with bacterial pathogens, they upregulate the expression of the defensin hBD-2 gene 4 and there is evidence that airway epithelial cells respond in a similar manner 7. Diminished defensin activity, caused by increased salt concentration in airway fluids, is thought to contribute to the susceptibility of cystic fibrosis patients to life-threatening S. aureus and Pseudomonas aeruginosa infections 8. The ability of bacterial pathogens to resist host defense peptides has a profound influence on their virulence as demonstrated for Salmonella typhimurium 9. However, the molecular basis for defensin resistance has remained elusive.
Defensins form pores in the bacterial cytoplasmic membrane 5; peptides with similar properties and activity have been found in several vertebrates and invertebrates as well as in plants 10 and bacteria 11. They include molecules with β-sheet structure such as porcine protegrins 12, α-helical peptides such as the amphibian magainins 13, and the bacterial lantibiotics bearing thioether bridges 11. Both S. aureus and the coagulase-negative Staphylococcus xylosus show high level innate tolerances to host defense peptides and lantibiotics.
Materials and Methods
Transposon Mutagenesis and DNA Sequence Analyses.
All bacterial strains were grown in BM broth (1% tryptone, 0.5% yeast extract, 0.5% NaCl, 0.1% K2HPO4, and 0.1% glucose) unless otherwise noted. A mutant library of S. xylosus C2a was constructed using transposon Tn917 and the delivery vector pTV1ts as described previously 3. Mutant clones (4,000) were transferred to BM agar plates containing 3 μg gallidermin/ml and monitored for impaired growth. Tn917-specific primers were used to sequence genomic DNA upstream and downstream from the transposon insertion site by cycle sequencing 14 and the sequence was completed by primer walking. With one of the primers used for sequencing of the S. xylosus mprF, part of the corresponding gene in S. aureus Sa113 was obtained and the sequence was completed by primer walking. The program tblastn with the Microbial Genomes Blast Databases at http://www.ncbi.nlm. nih.gov was used to perform sequence similarity searches. Prediction of the MprF transmembrane topology was accomplished using the program TMAP at http://www.mbb.ki.se/cgi-bin/tmap1.pl. It is based on a multiple alignment of the closely related MprF homologues from S. aureus, S. xylosus, Bacillus subtilis, and Enterococcus faecalis.
Construction of Plasmids and Homologous Recombination.
For in vitro recombination of DNA, standard methods and vectors were used 14. To delete the mprF gene of S. aureus Sa113, DNA fragments of 885 bp and 1,042 bp flanking mprF were amplified by PCR and cloned together with the ermB gene from Tn551 into the polylinker of the temperature-sensitive shuttle plasmid pBT2 using the restriction sites indicated in Fig. 1 A. After construction in Escherichia coli DH5a 14, the resulting plasmid pBTΔmprF was transferred to S. aureus Sa113 by electroporation 15 and mprF deletion mutants were enriched by incubation at 42°C in the presence of 2.5 μg/ml erythromycin; the recombination procedure has recently been described in detail 16. The proper integration of ermB was verified by sequencing of the genomic DNA at the borders of the PCR-derived regions. A 2,345-bp fragment comprising 93% of the mprF gene was deleted. Plasmid pRBmprF was constructed in E. coli DH5α by ligation of a 3,281-bp PCR fragment bearing the mprF gene of S. xylosus C2a, together with 359 bp upstream containing the putative promoter region and 400 bp downstream containing the terminator structure into the SmaI site of the shuttle vector pRB473 17. To construct plasmid pTXmprF, a 2,927-bp PCR fragment encoding the S. xylosus mprF gene was cloned in the expression vector pTX15 via BamHI and MluI to produce a transcriptional fusion with the xylA promoter 18. PCR primers were modified to introduce a BamHI site 34 bp upstream of the mprF start codon and a MluI site 370 bp downstream of the stop codon. The cloning host Staphylococcus carnosus TM300 was transformed with the pTXmprF ligation mixture by protoplast transformation 19. Plasmids pRBmprF and pTXmprF were subsequently transferred to S. aureus Sa113 or S. xylosus C2a by electroporation 15. The xylA promoter of pTXmprF is repressed by the plasmid-encoded repressor XylR and derepression was achieved by the addition of 0.5% xylose to the culture medium 18.
Figure 1.
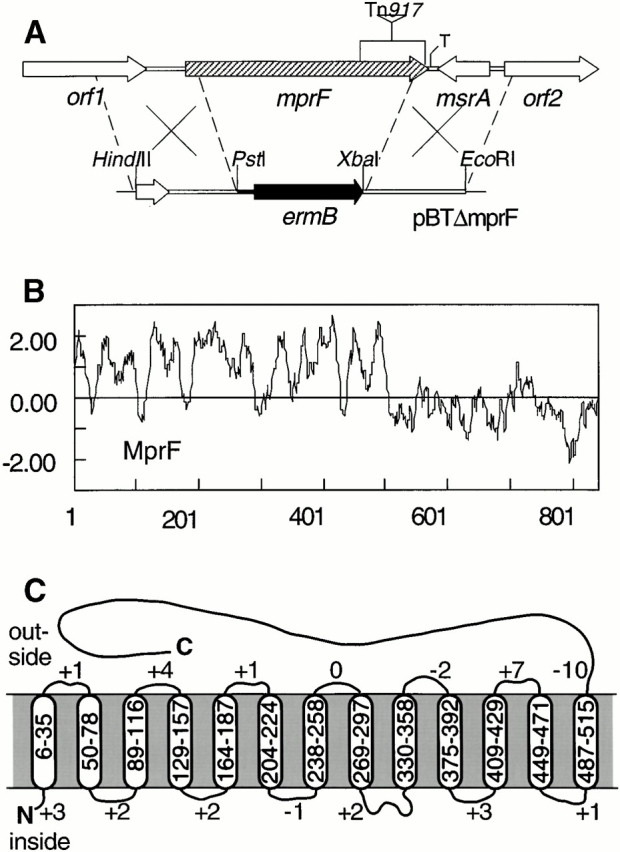
Inactivation of mprF (A), Kyte and Doolittle hydrophobicity profile (B), and putative transmembrane topology (C) of the S. aureus MprF. (A) The deletion caused by Tn917 integration in the S. xylosus mutant XG2 is indicated by the triangle. The mprF gene of S. aureus was disrupted by replacing the mprF gene with the erythromycin resistance gene ermB. T, transcriptional terminator. (C) The positions in the MprF sequence of putative transmembrane segments and the numbers of positive residues in the putative loops are indicated.
Antimicrobial Peptides and Antibacterial Assay.
Defensin HNP1-3 was isolated from human peripheral blood neutrophils by extraction of the neutrophil granules with 5% acetic acid and subsequent reversed phase HPLC (RP-HPLC) separation according to established methods 3 20. The resulting product comprised the three defensin variants HNP1, HNP2, and HNP3, differing only in the first amino acid position. Protegrins 3 and 5 and tachyplesin 1 were synthesized as peptide amides, oxidized to obtain the correct disulfide bridges, and purified by RP-HPLC (purities >95%), as recently described 3. The fluorescent-labeled derivative Lys(εDns)-tachyplesin 1 was synthesized, folded, and purified as above with the exception that the NH2 terminus was extended by a further lysine residue, the ε-amino group of which was dansylated. The purity and quality of the peptide preparations was confirmed by HPLC and electrospray ionization mass spectrometry (ESI-MS). Gallidermin was provided by Dr. K. Thomae (GmbH, Biberach, Germany). Nisin, synthetic (A8,13,18)-magainin II amide, melittin, gramicidin S, and gramicidin D were purchased from Sigma-Aldrich. Minimal inhibitory concentration (MIC) values of the various peptides were determined in serial dilution tests using Luria-Bertani (LB) broth (1% tryptone, 0.5% yeast extract, 0.5% NaCl) as described recently 21. As the activity of human defensins is very sensitive to the salt concentration 20, half-strength LB broth without NaCl was used for them; the tested strains all grew well in this medium.
Phagocytosis and Killing of S. aureus by Human Neutrophils.
Blood was drawn from healthy human volunteers and heparinized. Neutrophils were isolated from peripheral blood as described previously 22 and resuspended in HBSS-HSA (HBSS containing 0.05% human serum albumin). To prepare bacteria for killing experiments, Mueller Hinton Broth was inoculated with 1/100 volumes of overnight cultures and vigorously shaken at 37°C until mid-logarithmic phase was reached. The bacteria were washed twice in HBSS-HSA and adjusted to a density of 8.5 × 107 CFU/ml. Normal human serum was added to a final concentration of 4% and bacteria were opsonized for 10 min at 37°C. Prewarmed bacterial and neutrophil suspensions were mixed to final concentrations of 8.5 × 104 CFU/ml and 5 × 106 neutrophils/ml. 50-μl samples were shaken at 37°C and incubation was stopped by the addition of 2 ml ice-cold distilled water to disrupt the neutrophils. Appropriate sample volumes were spread on LB agar plates and colonies were counted after 24 h incubation at 37°C. When bacteria were incubated for 4 h under the same conditions without neutrophils, no significant changes in the colony numbers compared with the initial counts were observed.
In phagocytosis studies, bacteria were grown as described above, washed, resuspended in PBS, and inactivated by heating for 25 min at 70°C. Subsequently, 0.1 mg/ml FITC was added and the bacteria were labeled at 37°C for 5 h. After washing with PBS, the bacteria were resuspended in HBSS-HSA, adjusted to the same density, and opsonized as described above. 50-μl aliquots of the prewarmed bacterial and neutrophil suspensions were mixed and shaken at 37°C. The final concentrations of bacteria and neutrophils were 8.5 × 107/ml and 2.5 × 106/ml, respectively. Incubation was stopped by addition of 100 μl ice-cold 1% paraformaldehyde. The percentage of neutrophils bearing FITC-labeled bacteria was determined by flow cytometric analysis of 5,000 cells using a FACScan™ (Becton Dickinson).
Killing by Myeloperoxidase.
Bacteria were grown as described for neutrophil-mediated killing and washed twice with PBS containing 0.0005% HSA. 3 × 105 CFU/ml were incubated with 0.05 U myeloperoxidase (MPO)/ml (Calbiochem) and 10 μM H2O2 in the same buffer after preheating all solutions to 37°C. 154 mM NaCl from the PBS buffer was present in the samples to permit generation of toxic chlorinating compounds 23. Samples of 10 μl each were shaken at 37°C and killing was stopped after 45 min by 25-fold dilution in ice-cold PBS. CFUs were counted 24 h after plating appropriate amounts on LB agar.
Animal Studies.
Female NMRI mice aged 5–7 wk were injected in the tail vein with a bacterial suspension containing 1.8 × 107 CFU of either S. aureus Newman wild type or the isogenic mprF mutant and evaluated for weight loss, arthritis, and sepsis over a 7-d period as described previously 24. Mice were subsequently killed and kidneys were removed in order to assay bacterial loads. The differences between the means of the values in all groups were tested for significance with the two-tailed Student's t test. The between-group differences in the mortality rate, frequency of arthritis, body weight losses, and bacterial counts in kidneys were analyzed using the chi-square test. A P value ≤ 0.05 was considered statistically significant. All mice were treated in accordance with institutional guidelines.
Isolation and Detection of Membrane Lipids.
Bacteria were grown overnight in BM broth containing 0.25% glucose, washed once in sodium acetate buffer (20 mM, pH 4.5), and disrupted in the same buffer using glass beads and a Disintegrator S (Biomatic GmbH) as described elsewhere 18. Polar lipids were extracted by the Bligh-Dyer procedure 25, vacuum dried, and dissolved in chloroform/methanol (2:1, by volume). Two-dimensional TLC (2d-TLC) was carried out as described previously 26. In brief, equal amounts of lipid extracts were spotted onto silica 60 F254 HPTLC plates (Merck) and developed with chloroform/methanol/water (65:25:4, by volume) in the first direction and chloroform/acetic acid/methanol/water (80:15:12:4, by volume) in the second direction. All lipids were visualized with molybdatophosporic acid spray (Merck) followed by charring at 120°C and treatment with ammonia vapor to improve the contrast. Phospholipids or amino group–containing lipids were selectively stained with Molybdenum Blue (Sigma-Aldrich) or ninhydrin spray (Merck), respectively. For further analyses, lipid spots were stained with iodine vapor, scraped from the glass plates, and extracted with dichloromethane/methanol (2:1, by volume). Phosphatidylglycerol (PG) and diphosphatidylglycerol (DPG) were purchased from Sigma-Aldrich and used as standards to determine the positions of the PG and DPG spots in 2d-TLC.
Lipid Analysis by Mass Spectrometry and Gas Chromatography.
The masses of lipids from the TLC spot were determined by Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometry (MS) on an Apex II FT-ICR mass spectrometer (Bruker) in both the positive and negative ESI modes. To analyze the composition of the lipid, the samples were spiked with n-tridecanoic acid and d-lysine as internal standards and incubated at 110°C for 30 min in 200 μl 1.5 M ethanolic HCl. After addition of 200 μl distilled water, the hydrophobic compounds were extracted into pentane and subsequently analyzed by gas chromatography (GC) and GC-MS on an Agilent 6890/5973 (Agilent Technologies) and an HP-5 MS capillary (30 m × 0.25 mm). The ethanolic phase was evaporated to dryness, reesterified with ethanolic HCl to ensure complete esterification, and then trifluoroacetylated (trifluoroacetic anhydride/dichloromethane 4:1 at 110°C for 10 min). The derivatized amino acids were detected by chiral GC-MS on a Lipodex E capillary and determined by enantiomer labeling 27. Finally, the ethanolic layer was trimethylsilylated (bis[trimethylsilyl]trifluoroacetamide in pyridine [1:1]) at 60°C for 30 min and derivatives were analyzed by GC-MS on the HP-5 MS capillary. A sector from a TLC plate without sample but identically developed and worked up (blank) was used to correct the fatty acid amounts determined in corresponding spots from sample-containing plates.
Interaction of Tachyplesin 1 and Gallidermin with Bacterial Cells.
Strains were grown for 5 h in BM medium, harvested, washed three times, and resuspended in ice-cold 50 mM sodium phosphate buffer, pH 7.0. Bacteria (2 × 107/ml) were incubated with 2 μM Lys(εDns)-tachyplesin 1 for 20 min on ice, and pelleted by centrifugation. The relative fluorescence of the supernatant was determined spectrofluorometrically using excitation and emission wavelengths of 340 and 522 nm, respectively. Binding of gallidermin was analyzed in a similar way with the following modifications: bacteria were grown in LB broth with 0.5% xylose, incubated with 2.3 μM (S. aureus) or 11.5 μM (S. xylosus) gallidermin for 20 min at 37°C, and the bacterial density was 1.6 × 108/ml. The amount of gallidermin in the supernatant was determined by RP-HPLC as described previously 3.
Results
Identification of the mprF Gene and Its Influence on Susceptibility to Antimicrobial Peptides.
Transposon Tn917 insertion mutants of S. xylosus C2a were analyzed for sensitivity to the lantibiotic gallidermin. Mutant XG2 showed severely impaired growth on gallidermin agar plates, whereas growth was normal in the absence of the peptide. The transposon insertion had inactivated an open reading frame of 2,525 bp (Fig. 1 A). The corresponding gene from S. aureus Sa113 was sequenced and found to encode a predicted protein of 841 amino acids without similarity to any proteins of known function. The S. aureus and S. xylosus proteins share 80% similarity and were named MprF (“multiple peptide resistance factor”). The highly hydrophobic NH2-terminal regions are predicted to contain 13 transmembrane segments, whereas the COOH-terminal domains are hydrophilic and probably located at the outside of the cytoplasmic membrane (Fig. 1B and Fig. C). Upstream of mprF, orf1 encodes a membrane protein with 57 and 54% identity to proteins of unknown function from the genomes of Streptococcus mutans and Streptococcus pneumoniae, respectively (Fig. 1 A). The reading frame downstream of the S. aureus mprF shares up to 67% similarity with the methionine sulfoxide reductase genes (msrA) of various bacteria. msrA has recently been shown to play an important role in the bacterial defense against respiratory burst components in S. pneumoniae, Neisseria gonorrhoeae, and E. coli 28. It is followed by a putative transcriptional regulator gene (orf2) which has significant similarities to the enterococcal repressor gene psr involved in β-lactam resistance 29. In terms of growth and microscopic appearance, the mprF mutant was indistinguishable from the wild type (data not shown).
The S. aureus mprF gene was replaced by an erythromycin resistance gene (ermB) via homologous recombination (Fig. 1 A). In the absence of an intact mprF gene, the susceptibility of S. aureus Sa113 to defensin HNP1-3 from human neutrophils, protegrins 3 and 5 from porcine leukocytes, and tachyplesin 1 from horseshoe crab haemocytes was 8- to at least 30-fold higher (Fig. 2). The sensitivity to the lanthionine-containing peptides (lantibiotics) gallidermin from Staphylococcus gallinarum and to nisin from Lactobacillus lactis 11 was increased by factors of 8 to 38. The linear peptides magainin II from clawed frog skin and melittin from honeybee venom 13 were only slightly more active against the mprF mutant (three- to fourfold). The increased sensitivity of the mutant seemed to be restricted to extended cationic peptides, as the small, circular gramicidin S and the linear, neutral gramicidin D (both peptides from Bacillus brevis; reference 30) were almost equally active against wild-type and mutant strains. Interestingly, even the wild-type strain was particularly susceptible to these peptides. Complementation of the mprF mutant with plasmid pRBmprF resulted in normal or considerably higher tolerances to cationic antimicrobial peptides (Fig. 2).
Figure 2.

MIC values of the indicated antimicrobial peptides against S. aureus wild type and mprF mutant bearing the empty control plasmid pRB473 or the complementation vector pRBmprF. Positively or negatively charged peptide positions are highlighted in black or gray, respectively. The charges of amino acid side chains and the terminal amino and carboxyl groups were considered. *Although human defensin HNP-1 is shown, a mixture of HNP1, HNP2, and HNP3 which differ in the first amino acid was used in the antibacterial assay. ‡We used and show the synthetic variant A8,13,18-magainin II amide. §MICs of the fluorescent derivative Lys(εDns)-tachyplesin 1. Unusual amino acids: Lan, lanthionine; Mla, methyllanthionine; Dhb, dehydrobutyrine; Avc, S-aminovinyl-d-cysteine; Dha, dehydroalanine; O, ornithine.
The mprF Mutant Is Killed Faster by Human Neutrophils than the Wild-Type Strain.
The kinetics of killing by human neutrophils for S. aureus Sa113 wild-type and mprF mutant strains were compared. Log-phase bacteria were opsonized with normal human serum and incubated with neutrophils for various time intervals. Subsequently, the numbers of resistant bacteria were determined. The mprF mutant was killed considerably faster than the wild type (Fig. 3 A). A 50% reduction in the number of applied wild-type or mutant bacteria was noted after 100 and 22 min, respectively. The uptake kinetics of the two strains revealed no differences (Fig. 3 B), suggesting that the faster killing of mutant bacteria results from an increased susceptibility to neutrophil antimicrobial activities rather than increased phagocytosis.
Figure 3.

Kinetics of killing and phagocytosis of wild-type (•) and mprF mutant bacteria (○) by neutrophils. (A) The numbers of viable bacteria (CFU) after incubation with neutrophils are expressed as percentage of the initial counts (means and SD of three counts from a representative experiment). (B) The percentage of neutrophils bearing FITC-labeled S. aureus cells are given (means and SD of four independent experiments).
As normal neutrophils use oxygen-dependent killing mechanisms in addition to defensins, we compared, using human neutrophil MPO, the rates of inactivation of S. aureus Sa113 wild type and mprF mutant. Bacteria were incubated with MPO in the presence of H2O2 and chloride ions to permit generation of toxic oxidizing and halogenizing products 23. The kinetics of killing by MPO revealed no significant differences: 38.3 ± 4.8% and 36.0 ± 16% (means and SD of five counts from a representative experiment) of wild-type and mutant bacteria, respectively, were inactivated after 45 min incubation indicating that improved killing of the mprF mutant by neutrophils results from increased susceptibility to oxygen-independent mechanisms rather than to respiratory burst.
Disruption of mprF Results in Attenuated Virulence.
The virulence of the mprF mutant was analyzed in a mouse model of sepsis and septic arthritis 24 using S. aureus Newman, which is more virulent than the laboratory strain Sa113. The mprF mutation caused a similar sensitivity to antimicrobial peptides in strain Newman as in strain Sa113 (data not shown). Significant differences in the mortality of NMRI mice injected intravenously with 1.8 × 107 CFU bacteria were noted, with 33% mortality in the wild-type infected group and no deaths in the mutant infected group at day 7 after infection (Table ). The incidence of arthritis in the two groups of animals was also different. At day 7 after infection, the frequencies of joint swelling were 21% for the mutant and 94% for the wild type. In addition, the wild-type infected animals lost significantly more weight during the course of the experiment than the mutant group, and the numbers of bacteria recovered from the kidneys of mice infected with the mprF mutant were significantly lower than those from wild-type infected animals (Table ), suggesting that the mprF mutant causes less systemic effects of disease.
Table 1.
Comparative Virulence in NMRI Mice of S. aureus Newman Wild Type and mprF Mutant
| Strain | Days after infection | Mortality | Arthritis | Weight loss | Bacterial loads in kidneys |
|---|---|---|---|---|---|
| no. dead mice/total no. | no. positive mice/total no. | g | ×108 CFU | ||
| Wild type | 3 | 2/24 | 12/22 | 5.1 ± 1.0 | ND |
| mprF mutant | 3 | 0/24 | 5/24 | 1.9 ± 1.8 | ND |
| Wild type | 7 | 8/24 | 15/16 | 8.4 ± 1.7 | 5.9 ± 4.9 |
| mprF mutant | 7 | 0/24 | 5/24 | 4.1 ± 2.7 | 1.1 ± 1.3 |
mprF Is Involved in Esterification of Membrane PG with l-Lysine.
To analyze whether mprF-dependent peptide resistance is based on modification of the target site, the cytoplasmic membrane, we compared the membrane lipid patterns of the wild-type and mutant strains by 2d-TLC analysis. One of the prominent and one minor lipid spot were lacking in the mprF mutant but reappeared upon complementation with plasmid pRBmprF (Fig. 4 A). The lipids in question stained positive with Molybdenum Blue and ninhydrin reagents (data not shown) indicating that they represent amino group–containing phospholipids. Previous analyses have revealed that PG and derivatives thereof are the major phospholipids of S. aureus 31; unmodified PG and the dimer (DPG) constitute 38–76% and 5–30% of the total phospholipids, respectively, and between 14 and 38% represent PG esterified with l-lysine (lysylphosphatidylglycerol [L-PG]; references 31 and 32).
Figure 4.



Detection of membrane lipids (A), structure (B), and mass spectrometry analysis (C) of L-PG. (A) The polar lipids from S. aureus Sa113 wild type, mprF mutant, and mutant complemented with pRBmprF were analyzed by 2d-TLC. The first panel shows the position of the lipids DPG, PG, L-PG, and the putative 2′ L-PG isomer (L-PG2). Two unidentified lipids were negative (P−N−) or positive (P+N+) with both phosphate- or amino group–specific reagents, respectively. (B) Structure of L-PG. (C) Part of the FT-ICR mass spectrogram of the TLC spot lacking in the mprF mutant. The molecular masses of singly protonated ions (m/z) are given above the peaks. The major peaks represent L-PG species with two saturated fatty acids (indicated with a dot; compare Table ). The minor peaks represent species with one unsaturated fatty acid (mass difference of −2) or molecules containing one or two 13C atoms (mass differences of +1 or +2).
Upon hydrolysis, the lipid from the larger TLC spot was analyzed by GC-MS and found to contain all expected components of L-PG. Moreover, l-lysine was the sole amino acid detected. The total amounts of l-lysine and of fatty acids in the TLC spot were 1.9 and 3.4 × 10−5 mmol, respectively, corresponding reasonably well with the theoretical molar ratio of 1:2 for L-PG (Fig. 4 B). S. aureus has previously been shown to produce a variety of fatty acids, some of which are unsaturated and/or branched and ranging from 14 to 20 carbon atoms 33, randomly incorporated into the membrane lipids. Accordingly, we detected several fatty acids, with branched C15 and C17 members predominating (data not shown). Moreover, both positional isomers of glycerol monophosphate along with minor amounts of free glycerol and phosphate were identified (data not shown), indicating an incomplete hydrolysis of the phosphodiester bonds. FT-ICR-MS analysis of the nonhydrolyzed sample revealed a series of masses with differences of 14 (Fig. 4 C), which fit well with the calculated masses of L-PG esterified with fatty acids with total carbon atom numbers between 29 and 35 (Table ). Taken together, these results demonstrate that the lipid lacking in the mprF mutant is L-PG and that mprF is essential for L-PG biosynthesis. FT-ICR-MS of the minor TLC spot revealed the same masses as found in the larger spot. We assume that it represents an L-PG isomer bearing the lysyl group at the 2′ rather than the 3′ position (Fig. 4 B). This isomer has been shown to arise spontaneously from the 3′ species 34 and to migrate slightly differently in 2d-TLC 35.
Table 2.
Experimentally Determined and Calculated Masses of L-PG in Relation to the Fatty Acid Composition
| Total carbon atom number of both fatty acids | Calculated masses of L-PG (MH+) | Experimental masses (MH+) |
|---|---|---|
| 29 | 809.56507 | 809.5654 |
| 30 | 823.58072 | 823.5795 |
| 31 | 837.59637 | 837.5943 |
| 32 | 851.61202 | 851.6075 |
| 33 | 865.62767 | 865.6233 |
| 34 | 879.64332 | 879.6408 |
| 35 | 893.65897 | 893.6546 |
MprF-deficient Cells Bind More Antimicrobial Peptides.
Whereas PG and DPG are negatively charged and attract cationic antimicrobial peptides to the cytoplasmic membrane surface, the lysinylated PG bears a net positive charge reducing attractive electrostatic interaction (Fig. 5 B). To analyze whether L-PG–containing bacterial cells accumulate less cationic peptide, the capacity of wild-type and mprF mutant cells to bind Lys(εDns)-tachyplesin 1, a synthetic fluorescent-labeled derivative of tachyplesin 1, was determined. The modification of tachyplesin 1 caused a 2.5-fold increase in the MIC, but the mprF mutant was still 33-fold more sensitive to the peptide than the wild type (Fig. 2). Whole cells were incubated with Lys(εDns)-tachyplesin 1 and the amount remaining in the supernatant after centrifugation was determined spectrofluorometrically. In the supernatants of wild-type cells, a considerably higher amount of tachyplesin 1 was detected compared with that found in supernatants of the mutant strain (Fig. 5 A), indicating that in the presence of L-PG, antimicrobial peptides bind less efficiently to the cells. The wild-type and mutant bacteria were not killed by Lys(εDns)-tachyplesin 1 at the concentration used. Similar results were obtained when bacteria were incubated with gallidermin and the concentration of unbound lantibiotic was determined by RP-HPLC. Moreover, complementation of the mprF-deficient mutant with plasmid pTXmprF restored bacterial repulsion of gallidermin (Fig. 5 A).
Figure 5.

Increased binding of antimicrobial peptides in the absence of L-PG (A) and putative mode of action of the resistance system (B). (A) S. aureus or S. xylosus wild-type strains (black bars) and mprF mutants (white bars) were incubated with a fluorescent variant of tachyplesin 1 or gallidermin. The amount of unbound tachyplesin 1 or gallidermin in the supernatants was determined. In binding studies with gallidermin, mutant strains bearing the mprF gene on the plasmid pTXmprF (shaded bars) were also tested, whereas the wild-type and mutant strains contained the empty control plasmid pTX16. The means and SD of three independent experiments are shown. (B) The staphylococcal cell wall and membrane are shown. The hydrophobic and positively charged portions of antimicrobial peptides are indicated in gray and white, respectively. A major amount of membrane lipids is esterified with l-lysine residues in wild-type bacteria. The positively charged l-lysyl groups cause a reduced binding capacity of the cell envelope for cationic antimicrobial peptides, whereas mutant cells lacking L-PG accumulate the harmful molecules in the membrane composed mainly of negatively charged lipids.
mprF-related Genes Are Present in Several Pathogens.
The two mprF genes share similarity with open reading frames of unknown function from several other bacteria comprising the human pathogens P. aeruginosa, Mycobacterium tuberculosis, Mycobacterium leprae, E. faecalis, (Fig. 6), Bacillus anthracis, and Corynebacterium diphtheriae, and the plant pathogens Agrobacterium tumefaciens and Xylella fastidiosa (data not shown). The deduced amino acid sequences exhibit very similar hydrophobicity profiles but the mycobacterial mprF homologues appear to lack some of the putative transmembrane segments. Whereas the hydrophobic NH2-terminal parts of the proteins reveal only limited sequence similarity, the hydrophilic COOH-terminal portions are 40–83% similar to MprF of S. aureus (Fig. 6). In most organisms bearing mprF-related genes, the genetic context is different and the genes are not encoded within operons.
Figure 6.

Alignment of the COOH-terminal 310 amino acids of the S. aureus MprF protein (Sa) with the corresponding parts of hypothetical proteins from S. xylosus (Sx), B. subtilis (Bs), E. faecalis (Ef), P. aeruginosa (Pa), A. tumefaciens (At), S. coelicolor (Sc), M. tuberculosis (Mt), and M. leprae (Ml). Identical or similar amino acids are highlighted in black or gray, respectively. The COOH-terminal ends are indicated by asterisks. The amino acid positions, calculated from the first possible start points, are given, except for the E. faecalis and A. tumefaciens proteins whose entire sequences are not yet available. The mprF sequences from S. aureus and S. xylosus are available from GenBank/EMBL/DDBJ under accession nos. AF145699 and AF145698, respectively.
Discussion
MprF is a novel virulence factor involved in the S. aureus escape from innate host defenses. The attenuated virulence in mice of the mprF mutant was reflected in increased susceptibility to neutrophil killing and in vitro sensitivity to defensins, whereas MPO-dependent killing was not affected. Our data thus demonstrate the important role of defensin resistance in S. aureus pathogenicity.
The mprF mutant no longer produces the unusual L-PG lipid that arises from esterification of PG with l-lysine 36. It remains to be determined whether mprF encodes the as yet unidentified L-PG synthase or whether it is involved in some other essential aspect of L-PG biosynthesis. The putative membrane location of MprF is consistent with a role in lipid metabolism. Positively charged L-PG accounts for up to 38% of the S. aureus membrane lipids, whereas the other phospholipids (PG and DPG) are negatively charged. Therefore, L-PG synthesis most probably has a significant impact on membrane surface charge and interactions with cationic antimicrobial peptides, which have to bind to the charged head groups of the membrane lipids before integration into the hydrophobic core of the cytoplasmic membrane (Fig. 5 B). Accordingly, the mprF mutant was much more susceptible to a broad variety of cationic antimicrobial peptides whereas the neutral gramicidin D was equally active toward wild-type and mprF mutant. The increased binding capacity of mutant cells for a tachyplesin 1 derivative and gallidermin demonstrates that the presence of L-PG leads to reduced attraction and binding of cationic antimicrobial peptides by the bacteria. We have recently demonstrated that reducing the negative charge of the staphylococcal cell wall by modification of the teichoic acid polymers with d-alanine affects the binding of cationic peptides 3, supporting the notion that S. aureus cells protect themselves against host defense peptides by modulating the electrostatic properties of their cell envelope. The location of mprF proximal to an S. aureus homologue of msrA, a locus involved in repair of bacterial proteins damaged by reactive oxygen species 28, indicates a clustering of genes involved in the escape from phagocyte functions.
Up to 60% of healthy individuals are permanently or intermittently colonized by S. aureus, and carriage is an important risk factor for life-threatening infections in patients undergoing surgery, bearing intravascular devices, or those with HIV infection and AIDS 37. The main ecological niches for S. aureus are the nasal epithelia, although these sites are protected by defensins produced by epithelial cells and submucosal glands 38. Defensin resistance thus may play a key role in the capacity of S. aureus to infect host tissues, in particular those of cystic fibrosis or chronic granulomatous disease patients 6 8. It should be noted that S. aureus produces L-PG in comparatively high amounts, whereas coagulase-negative staphylococci contain only traces (Staphylococcus epidermidis, S. xylosus) or no detectable amounts (Staphylococcus haemolyticus, Staphylococcus saprophyticus) of this unusual lipid 26. It is tempting to speculate that high L-PG content is a prerequisite for S. aureus survival on nasal epithelia and invasiveness.
Lysinylation of phospholipids may play a similar role in other bacteria. L-PG has been described in several bacterial species bearing mprF-related genes including E. faecalis, B. subtilis 39, and P. aeruginosa 40. In the remaining bacteria with mprF homologues, the membrane lipids seem to be only incompletely characterized 41. Defensin-like peptides also play a role in the defense of plants against microbial infections 10, perhaps explaining the presence of a mprF homologue adjacent to other virulence genes in the plant pathogen A. tumefaciens 42. MprF thus represents an interesting new target for novel antimicrobial drugs which block L-PG synthesis and thereby render the bacteria susceptible to antimicrobial host defenses.
Acknowledgments
We thank Robert I. Lehrer and Friedrich Götz for helpful discussions, Reinhold Brückner for providing transposon mutants, Gerold Schwarz and Manuela Braun for isolation of defensins, and Erik Heezius, Vera Augsburger, Lena Svensson, and Shahin Hajizadeh for technical assistance.
This work was supported by a fellowship from the European Molecular Biology Organization to A. Peschel (ASTF 9521) and by grants from the German Research Council to A. Peschel (Priority Program No. 1047), H. Kalbacher (Ka 767/4-2), and G. Jung (SFB 323). L.V. Collins acknowledges the support of the EU Biotechnology program (contract no. BIO4CT975130).
Footnotes
Abbreviations used in this paper: DPG, diphosphatidylglycerol; FT-ICR, Fourier transform ion cyclotron resonance; GC, gas chromatography; HNP, human neutrophil peptide; HSA, human serum albumin; L-PG, lysylphosphatidylglycerol; MIC, minimal inhibitory concentration; MPO, myeloperoxidase; MS, mass spectrometry; PG, phosphatidylglycerol; RP-HPLC, reversed phase HPLC; 2d-TLC, two-dimensional TLC.
R.W. Jack's present address is Department of Biology and Chemistry, City University of Hong Kong, 83 Tat Chee Ave., Kowloon Tong, Kowloon, Hong Kong.
M. Otto's present address is Laboratory of Human Bacterial Pathogenesis, National Institute of Allergy and Infectious Diseases, National Institutes of Health, 903 South 4th St., Hamilton, MT 59840.
References
- Lowy F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- Sieradzki K., Roberts R.B., Haber S.W., Tomasz A. The development of vancomycin resistance in a patient with methicillin-resistant Staphylococcus aureus infection. N. Engl. J. Med. 1999;340:517–523. doi: 10.1056/NEJM199902183400704. [DOI] [PubMed] [Google Scholar]
- Peschel A., Otto M., Jack R.W., Kalbacher H., Jung G., Götz F. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins and other antimicrobial peptides. J. Biol. Chem. 1999;274:8405–8410. doi: 10.1074/jbc.274.13.8405. [DOI] [PubMed] [Google Scholar]
- Harder J., Bartels J., Christophers E., Schröder J.-M. A peptide antibiotic from human skin. Nature. 1997;387:861. doi: 10.1038/43088. [DOI] [PubMed] [Google Scholar]
- Lehrer R.I., Ganz T. Antimicrobial peptides in mammalian and insect host defence. Curr. Opin. Immunol. 1999;11:23–27. doi: 10.1016/s0952-7915(99)80005-3. [DOI] [PubMed] [Google Scholar]
- Liese J.G., Jendrossek V., Jansson A., Petropoulou T., Kloos S., Gahr M., Belohradsky B.H. Chronic granulomatous disease in adults. Lancet. 1996;347:220–223. doi: 10.1016/s0140-6736(96)90403-1. [DOI] [PubMed] [Google Scholar]
- Diamond G., Kaiser V., Rhodes J., Russel J.P., Bevins C.L. Transcriptional regulation of beta-defensin gene expression in tracheal epithelial cells. Infect. Immun. 2000;68:113–119. doi: 10.1128/iai.68.1.113-119.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman M., Anderson G., Stolzenberg E.D., Kari U.P., Zasloff M., Wilson J.M. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell. 1997;88:553–560. doi: 10.1016/s0092-8674(00)81895-4. [DOI] [PubMed] [Google Scholar]
- Groisman E.A. How bacteria resist killing by host defense peptides. Trends Microbiol. Sci. 1994;2:444–448. doi: 10.1016/0966-842x(94)90802-8. [DOI] [PubMed] [Google Scholar]
- Hoffmann J.A., Kafatos F.C., Janeway C.A., Ezekowitz R.A. Phylogenetic perspectives in innate immunity. Science. 1999;284:1313–1318. doi: 10.1126/science.284.5418.1313. [DOI] [PubMed] [Google Scholar]
- Jack R.W., Bierbaum G., Sahl H.-G. Lantibiotics and Related Peptides 1998. Springer-Verlag, Berlin; Germany: pp. 224 pp [Google Scholar]
- Kokryakov V.N., Harwig S.S.L., Panyutich E.A., Shevchenko A.A., Aleshina G.M., Shamova O.V., Korneva H.A., Lehrer R.I. Protegrinsleukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 1993;327:231–236. doi: 10.1016/0014-5793(93)80175-t. [DOI] [PubMed] [Google Scholar]
- Bechinger B. Structure and functions of channel-forming peptidesmagainins, cecropins, melittin and alamethicin. J. Membr. Biol. 1997;156:197–211. doi: 10.1007/s002329900201. [DOI] [PubMed] [Google Scholar]
- Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Seidman J.G., Smith J.A., Struhl K. Current Protocols in Molecular Biology. John Wiley and Sons, Inc; New York: 1990. [Google Scholar]
- Augustin J., Götz F. Transformation of Staphylococcus epidermidis and other staphylococcal species with plasmid DNA by electroporation. FEMS (Fed. Eur. Microbiol. Soc.) Microbiol. Lett. 1990;66:203–208. doi: 10.1016/0378-1097(90)90283-v. [DOI] [PubMed] [Google Scholar]
- Brückner R. Gene replacement in Staphylococcus carnosus and Staphylococcus xylosus . FEMS (Fed. Eur. Microbiol. Soc.) Microbiol. Lett. 1997;151:1–8. doi: 10.1111/j.1574-6968.1997.tb10387.x. [DOI] [PubMed] [Google Scholar]
- Brückner R. A series of shuttle vectors for Bacillus subtilis and Escherichia coli . Gene. 1992;122:187–192. doi: 10.1016/0378-1119(92)90048-t. [DOI] [PubMed] [Google Scholar]
- Peschel A., Ottenwälder B., Götz F. Inducible production and cellular location of the epidermin biosynthetic enzyme EpiB using an improved staphylococcal expression system. FEMS (Fed. Eur. Microbiol. Soc.) Microbiol. Lett. 1996;137:279–284. doi: 10.1111/j.1574-6968.1996.tb08119.x. [DOI] [PubMed] [Google Scholar]
- Götz F., Schumacher B. Improvements of protoplast transformation in Staphylococcus carnosus . FEMS (Fed. Eur. Microbiol. Soc.) Microbiol. Lett. 1987;40:285–288. [Google Scholar]
- Harwig S.S.L., Ganz L., Lehrer R.I. Neutrophil defensinspurification, characterization, and antimicrobial testing. Methods Enzymol. 1994;236:160–172. doi: 10.1016/0076-6879(94)36015-4. [DOI] [PubMed] [Google Scholar]
- Peschel A., Vuong C., Otto M., Götz F. The d-alanine residues of Staphylococcus aureus teichoic acids alter the susceptibility to vancomycin and the activity of autolysins. Antimicrob. Agents Chemother. 2000;44:2845–2847. doi: 10.1128/aac.44.10.2845-2847.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz F.J., Veldkamp K.E., Van Kessel K.P., Verhoef J., Van Strijp J.A. Delta-toxin from Staphylococcus aureus as a costimulator of human neutrophil oxidative burst. J. Infect. Dis. 1997;176:1531–1537. doi: 10.1086/514152. [DOI] [PubMed] [Google Scholar]
- Klebanoff S.J., Waltersdorph A.M., Rosen H. Antimicrobial activity of myeloperoxidase. Methods Enzymol. 1984;105:399–403. doi: 10.1016/s0076-6879(84)05055-2. [DOI] [PubMed] [Google Scholar]
- Bremell T., Lange S., Yacoub A., Ryden C., Tarkowski A. Experimental Staphylococcus aureus arthritis in mice. Infect. Immun. 1991;59:2615–2623. doi: 10.1128/iai.59.8.2615-2623.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh E.G., Dyer W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Nahaie M.R., Goodfellow M., Minnikin D.E., Hajek V. Polar lipid and isoprenoid quinone composition in the classification of Staphylococcus . J. Gen. Microbiol. 1984;130:2427–2437. doi: 10.1099/00221287-130-9-2427. [DOI] [PubMed] [Google Scholar]
- Frank H., Nicholson G., Bayer E. Enantiomer labelling, a method for the quantitative analysis of amino acids. J. Chromatogr. 1978;167:187–196. doi: 10.1016/s0021-9673(00)91157-9. [DOI] [PubMed] [Google Scholar]
- Wizemann T.M., Moskovitz J., Pearce B.J., Cundell D., Arvidson C.G., So M., Weissbach H., Brot N., Masure H.R. Peptide methionine sulfoxide reductase contributes to the maintenance of adhesins in three major pathogens. Proc. Natl. Acad. Sci. USA. 1996;93:7985–7990. doi: 10.1073/pnas.93.15.7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massidda O., Dardenne O., Wahlen M.B., Zorzi W., Coyette J., Shockman G.D., Daneo-Moore L. The PBP 5 synthesis repressor (psr) gene in Enterococcus hirae ATCC 9790 is substantially longer than previously reported. FEMS (Fed. Eur. Microbiol. Soc.) Microbiol. Lett. 1998;166:355–360. doi: 10.1111/j.1574-6968.1998.tb13912.x. [DOI] [PubMed] [Google Scholar]
- Zuber P., Nakano M.M., Marahiel M.A. Peptide antibiotics. In: Sonenshein A.L., editor. Bacillus subtilis and Other Gram-positive Bacteria: Physiology and Molecular Genetics. American Society for Microbiology; Washington, DC: 1993. pp. 897–916. [Google Scholar]
- Haest C.W.M., de Gier J., op den Kamp J.A.F., Bartels P., van Deenen L.L.M. Changes in permeability of Staphylococcus aureus and derived liposomes with varying lipid composition. Biochim. Biophys. Acta. 1972;255:720–733. doi: 10.1016/0005-2736(72)90385-9. [DOI] [PubMed] [Google Scholar]
- Short S.A., White D.C. Metabolism of phosphatidylglycerol, lysylphosphatidylglycerol, and cardiolipin of Staphylococcus aureus . J. Bacteriol. 1971;108:219–226. doi: 10.1128/jb.108.1.219-226.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutberlet T., Dietrich U., Bradaczek H., Pohlentz G., Leopold K., Fischer W. Cardiolipin, alpha-d-glucopyranosyl, and lysylcardiolipin from Gram-positive bacteriaFAB MS, monofilm and X-ray powder diffraction studies. Biochim. Biophys. Acta. 2000;1463:307–322. doi: 10.1016/s0005-2736(99)00214-x. [DOI] [PubMed] [Google Scholar]
- Tocanne J.F., Verheij H.M., op de Kamp J.A.F., van Deenen L.L.M. Chemical and physicochemical studies of lysylphosphatidylglycerol derivatives. Occurrence of a 2′-3′ lysyl migration. Chem. Phys. Lipids. 1974;13:389–403. doi: 10.1016/0009-3084(74)90012-7. [DOI] [PubMed] [Google Scholar]
- Fischer W., Arneth-Seifert D. d-Alanylcardiolipin, a major component of the unique lipid pattern of Vagococcus fluvialis . J. Bacteriol. 1998;180:2950–2957. doi: 10.1128/jb.180.11.2950-2957.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennarz W.J., Bonsen P.P.M., van Deenen L.L.M. Substrate specificity of O-l-lysylphosphatidylglycerol synthase. Enzymatic studies on the structure of O-l-lysylphosphatidylglycerol. Biochemistry. 1967;6:2307–2312. doi: 10.1021/bi00860a005. [DOI] [PubMed] [Google Scholar]
- Kluytmans J., van Belkum A., Verbrugh H. Nasal carriage of Staphylococcus aureusepidemiology, underlying mechanism, and associated risk. Clin. Microbiol. Rev. 1997;10:505–520. doi: 10.1128/cmr.10.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H., Lim H.H., Lee H.M., Choi J.O. Expression of human beta-defensin 1 mRNA in human nasal mucosa. Acta Otolaryngol. 2000;120:58–61. doi: 10.1080/000164800760370846. [DOI] [PubMed] [Google Scholar]
- Fischer W., Nakano M., Laine R.A., Bohrer W. On the relationship between glycerophosphoglycolipids and lipoteichoic acids in Gram-positive bacteria. I. The occurrence of phosphoglycolipids. Biochim. Biophys. Acta. 1978;528:288–297. doi: 10.1016/0005-2760(78)90018-8. [DOI] [PubMed] [Google Scholar]
- Kenward M.A., Brown M.R., Fryer J.J. The influence of calcium or manganese on the resistance to EDTA, polymyxin B or cold shock, and the composition of Pseudomonas aeruginosa grown in glucose- or magnesium-depleted batch cultures. J. Appl. Bacteriol. 1979;47:489–503. doi: 10.1111/j.1365-2672.1979.tb01210.x. [DOI] [PubMed] [Google Scholar]
- Ratledge C., Wilkinson S.G. Microbial Lipids 1988. Academic Press; London: pp. 963 pp [Google Scholar]
- Wirawan I.G., Kang H.W., Kojima M. Isolation and characterization of a new chromosomal virulence gene of Agrobacterium tumefaciens . J. Bacteriol. 1993;175:3208–3212. doi: 10.1128/jb.175.10.3208-3212.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]