

Abstract
Recent data from mice deficient for phosphatase and tensin homologue deleted from chromosome 10 or src homology 2 domain–containing 5′ inositol phosphatase, phosphatases that negatively regulate the phosphatidylinositol 3-kinase (PI3K) pathway, revealed an increased number of macrophages in these animals, suggesting an essential role for the PI3K pathway for macro-phage survival. Here, we focused on the role of the PI3K-regulated serine/threonine kinase Akt-1 in modulating macrophage survival. Akt-1 was constitutively activated in human macrophages and addition of the PI3K inhibitor, LY294002, suppressed the activation of Akt-1 and induced cell death. Furthermore, suppression of Akt-1 by inhibition of PI3K or a dominant negative (DN) Akt-1 resulted in loss of mitochondrial transmembrane potential, activation of caspases-9 and -3, and DNA fragmentation. The effects of PI3K inhibition were reversed by the ectopic expression of constitutively activated Akt-1 or Bcl-xL. Inhibition of PI3K/Akt-1 pathway either by LY294002 or DN Akt-1 had no effect on the constitutive or inducible activation of nuclear factor (NF)-κB in human macrophages. However, after inhibition of the PI3K/Akt-1 pathway, a marked decrease in the expression of the antiapoptotic molecule Mcl-1, but not other Bcl-2 family members was observed, and Mcl-1 rescued macrophages from LY294002-induced cell death. Further, inhibition of Mcl-1 by antisense oligonucleotides, also resulted in macrophage apoptosis. Thus, our findings demonstrate that the constitutive activation of Akt-1 regulates macrophage survival through Mcl-1, which is independent of caspases, NF-κB, or Bad.
Keywords: cell death, apoptosis, PI3K, dominant negative Akt-1, mitochondrial transmembrane potential
Introduction
Macrophages are critical in the pathogenesis of diseases such as rheumatoid arthritis 1, atherosclerosis and Crohn's disease 2. Unlike monocytes, macrophages are long-lived cells, resistant to many apoptotic stimuli including Fas and TNF-α death receptor ligation, ionizing radiation, and multiple antineoplastic or cytotoxic agents 3. We recently demonstrated that expression of Fas-associated death domain–like IL-1β–converting enzyme–inhibitory protein protected differentiated macrophages from Fas-mediated apoptosis 3 and that constitutively activated nuclear factor (NF)-κB is essential for the expression of A1/Bfl-1, mitochondrial homeostasis and macrophage survival 4. However, other mechanisms that may uniquely contribute to macrophage survival have not been fully elucidated.
Apoptosis may be initiated by ligation of specific death receptors, such as Fas or TNFR1. Death receptor signaling activates caspase-8 5, which is capable of cleaving caspase-3. In certain cell types, caspase-8 activation may directly induce loss of mitochondrial transmembrane potential (ΔΨm) through activation of Bid 6. Alternatively, apoptosis may be initiated at the mitochondria by disruption of ΔΨm, and release of cytochrome c into the cytosol 7. Mitochondria integrity is regulated by the Bcl-2 family of proteins, which includes both antiapoptotic (e.g., Bcl-2, Bcl-xL, and A1/Bfl-1; reference 8) and proapoptotic (e.g., Bax, Bik, Bak, Bad, and Bid; reference 7) family members.
Mice deficient in either src homology 2 domain–containing 5′ inositol phosphatase or phosphatase and tensin homologue deleted from chromosome 10 (PTEN) possess increased numbers of macrophages 9 10. Both of these phosphatases normally function to suppress the phosphatidylinositol 3-kinase (PI3K) pathway 11 12, suggesting that this pathway is critical for macrophage survival. The serine/threonine kinase Akt-1 is a major target of PI3K 13, which may be activated by a wide variety of stimuli including cytokines, chemokines, growth and survival factors, and integrin ligation 14 15. Activation of the PI3K/Akt-1 pathway protects against cell death induced by a variety of apoptotic stimuli. Although the mechanisms responsible for protection have not been fully elucidated, the phosphorylation Bad or caspase-9, suppression of FasL expression, and NF-κB activation may be induced through the PI3K/Akt-1 pathway 16 17 18 19 20 21 22. Although a prior study demonstrated that inhibition of the PI3K pathway resulted in apoptosis of murine peritoneal macrophages 15, the mechanism by which the PI3K/Akt-1 pathway promotes macrophage survival is not known.
In this study, we investigated the role of Akt-1 in the survival of primary human monocyte-differentiated macrophages. We observed that Akt-1 was constitutively expressed and activated in human macrophages. Inhibition of Akt-1 activation resulted in macrophage apoptosis mediated through the loss of ΔΨm and the activation of caspases-9 and -3. Activated Akt-1 did not contribute to activation of NF-κB in macrophages. Additionally, the inhibition of the Akt-1 had no effect on the expression of Fas or FasL, nor the expression or phosphorylation of Bad. In contrast, inhibition of PI3K/Akt-1 was associated with decreased Mcl-1 expression, while other anti- or proapoptotic Bcl-2 proteins were not altered. These observations demonstrate that constitutively activated PI3K resulted in activation of Akt-1 and the expression of Mcl-1, which were essential for macrophage survival.
Materials and Methods
Materials.
Trypan blue, LY294002, and polymyxin B sulfate were from Sigma-Aldrich. Lipofectamine was from GIBCO BRL. Propidium iodide (PI) was from Roche Molecular Biochemicals and rhodamine 123 (Rh123) was from Molecular Probes. The caspase inhibitors and substrate, zVAD.fmk, zLEHD.fmk, and Ac-LEVD-AFC, were obtained from Enzyme System Products.
Cell Isolation and Culture.
Human monocytes were isolated by countercurrent elutriation from commercially obtained buffy coats as described previously 4. Monocytes were differentiated in vitro for 7 d in RPMI 1640 containing 20% heat-inactivated FBS and 1 μg/ml polymyxin B sulfate. RAW 264.7 cells were obtained from American Type Culture Collection and cultured in DMEM with 10% heat-inactivated FBS.
Adenovirus Infection of Human Macrophages.
Differentiated macrophages were infected for 2 h in serum-free RPMI 1640 at the indicated multiplicity of infection (moi) with replication-defective adenoviruses (Ads) expressing either β-galactosidase (Adβgal), green fluorescence protein (AdGFP), dominant negative (DN) Akt-1 (AdDNAkt-1), activated Akt-1 (AdMyrAkt-1), or Bcl-xL (AdBcl-xL) as described previously 4.
Electrophoretic Mobility Shift Assay.
Nuclear extracts were prepared as described previously 23 from macrophages incubated in control medium or medium containing 50 μm LY294002 or infected with either AdGFP or AdDNAkt-1 for the times indicated in the figures. An oligonucleotide spanning the κB binding sites of HIV/Ig, previously shown to detect NF-κB binding, was employed for the electrophoretic mobility shift assay (EMSA) as described previously 23.
Promoter Activity Assay.
RAW 264.7 cells were transiently transfected using lipofectamine with 1 μg of the 3xκB promoter-reporter plasmid 4 plus 1 μg of a plasmid-expressing constitutively active Akt-1 or a control plasmid, with or without 1 μg of a plasmid-expressing NF-κB p65. Promoter activity is presented as relative light units (RLUs) normalized for protein concentration (RLU per μg protein).
Viability Assays.
PI exclusion and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) cleavage were employed to assess macrophage viability. PI was added just before analysis by flow cytometry and the data are presented as the percentage of cell death (PI+ cells) in each culture. MTT assays were performed as instructed by the supplier and values were calculated relative to control cultures. Additionally, transient cotransfections were employed to determine if expression of Mcl-1 could rescue RAW 264.7 cells from death induced by LY294002 treatment. RAW 264.7 cells were cotransfected with 0.5 μg of CMV-EGFP expression plasmid plus 2.5 μg of plasmids pCI-neo or pCI-Mcl-1. After transfection, cultures were incubated for 24 h and then were treated with 50 μM LY294002 for an additional 24 h. RAW 264.7 cells were collected and GFP-expressing cells were quantified by flow cytometry.
Mcl-1 Antisense.
Mcl-1 phosphorothioate antisense oligonucleotides to Mcl-1 (AS) and control inverted antisense oligonucleotides (INV) were synthesized by Intergrated DNA Technology, Inc. The sequences employed were described previously as Mcl-AS-8 and MclinvAS-8 24. Primary macrophages were incubated with either Mcl-1 AS or INV (100 μM) for 24 h, then harvested, and examined for Mcl-1 expression by Western blot analysis and apoptosis by determination of subdiploid DNA content.
Determination of Subdiploid DNA Content.
At the indicated time points, cultures were stained with PI as described previously 25. The subdiploid DNA peak (<2N DNA), immediately adjacent to the G0/G1 peak (2N DNA), represents apoptotic cells and was quantified by histogram analyses.
Determination of Mitochondrial Permeability Transition.
Mitochondrial dysfunction was assessed using the cationic lipophilic green fluorochrome rhodamine 123 as described previously 3 4. Mean fluorescence was recorded for each sample, and control cultures at each time point were designated as 100% fluorescence.
Western Blot Analysis.
Western blot analysis was conducted as described previously 4 using the indicated antibodies: rabbit anti–phospho-Akt-1 or total Akt-1 (New England Biolabs); rabbit anti–caspase-9 (Calbiochem); rabbit anti-PKCδ; rabbit anti–Mcl-1 (Santa Cruz Biotechnology, Inc.); mouse anti–caspase-3; mouse anti-Bcl-2; rabbit anti-Bcl-xL; mouse anti-Bax (Transduction Laboratories); rabbit anti-Bad; and phospho-Bad (Cell Signaling Technology), or mouse anti-tubulin (Calbiochem). For immunoprecipitation of Bad and phospho-Bad, 600 μg extracts were incubated with 2 μg rabbit anti-Bad and 50 μl of protein A Sepharose beads (Boehringer) overnight at 4°C. The immunoprecipitated complexes were electrophoresed on two separate gels and transferred to membranes which were used for Western blotting using rabbit anti-Bad and phospho-Bad, respectively. To examine cytochrome c, cells were suspended in lysis buffer and incubated for 3 min on ice, as described previously by us 26. Lysates were homogenized for 1 min and centrifuged for 15 min (750× g), and supernatants containing the cytosolic fraction were used to detect cytochrome c using a monoclonal anti–cytochrome c antibody (BD PharMingen).
Analysis of Caspase-9 Activity.
Macrophages were treated with 50 μM LY294002 and/or 100 μM zVAD.fmk, as indicated, for 48 h. Cells were harvested and suspended in 50 μl of lysis buffer for 10 min. The reaction buffer and the caspase-9 substrate Ac-LEVD-AFC were added and incubated at 37°C for 1 h. Fluorescence was measured using a fluorometer at 400-nm excitation and 505-nm emission as described previously 4.
Reverse Transcription PCR.
Total cellular RNA was extracted and was used for reverse transcription as described previously 4. The cDNA was amplified using specific glyceraldehyde 3-phosphate dehydrogeanse or Mcl-1, A1, and Bcl-xL primer pairs, respectively. We used the following sequences for the Mcl-1: 5′-CGGCAGTCGCTGGAGATTAT-3′ (526–545, sense) and 5′-GTGGTGGTGGTTGGTTA-3′ (1,062–1,081, antisense; reference 27). The sequences for A1 were: 5′-CAGCACATTGCCTCAACAGC-3′ and 5′-TGCAGATAGTCCTGAGCCAGC-3′ 28. The sequences for Bcl-xL were: 5′-CCAG-AAAGGATACAGCTGG-3′ and 5′-CTCCTGGATCCAAGGCTCTA-3′. The primers for GAPDH were purchased from CLONTECH Laboratories, Inc.
Statistical Analysis.
Results were expressed as the mean ± SE. Significance was determined by an unpaired two-tailed Student's t test.
Results
Akt-1 Is Constitutively Activated by PI3K in Human Monocyte-differentiated Macrophages.
Earlier studies demonstrated that PI3K activation and pp70s6k were important in survival of peritoneal thioglycollate elicited macrophages 15, although the role of Akt-1 was not examined. Therefore, unstimulated in vitro–differentiated human macrophages were examined to determine if Akt-1 was constitutively activated. Total and active phospho-Akt-1 were determined by Western blot analysis. Akt-1 was constitutively activated in monocyte-differentiated macrophages and the PI3K inhibitor LY294002 suppressed activation, but not the expression of Akt-1 at 24 and 36 h (Fig. 1). Therefore, Akt-1 was constitutively activated in normal macrophages and its activation was mediated through the PI3K pathway.
Figure 1.

Akt-1 is constitutively activated in human macrophages, which is PI3K dependent. The expression of total and activated Akt-1 was determined by immunoblot analysis. Activated Akt-1 was detected using an antibody specific to phospho-Akt-1 (P-Akt-1, the activated form of Akt-1). Whole cell lysates from macrophages treated with LY294002, or control medium (DMSO), for the indicated periods of time, were probed for Akt-1 and P-Akt-1 by rabbit anti-total Akt-1 or phospho-Akt-1 antibodies (New England Biolabs). The final concentration of LY294002 (LY) is 50 μΜ. The results shown are representative of four experiments.
Inhibition of PI3K or Akt-1 in Macrophages Induces Cell Death and Apoptosis of Human Monocyte-differentiated Macrophages.
To characterize the mechanism of Akt-1 activation, monocyte-differentiated macrophages were treated with varying concentrations of the PI3K inhibitor, LY294002 (from 12 μm to 100 μm), or control medium containing DMSO for 48 h. Cell death, determined by MTT, was observed at each concentration of LY2942002 employed (Fig. 2 A). At 50 μM, cell death was detected at 24 h, with ≥80% death by 48 h (Fig. 2 C). To determine if cell death was due to inhibition of Akt-1, macrophages were infected with an adenoviral vector expressing a DN version of Akt-1 (AdDNAkt-1) or the control vector (AdGFP) at concentrations ranging from 25 to 200 moi for 48 h 4 23. Macrophage cell death was observed in a dose-responsive fashion when cells were infected with AdDNAkt-1 compared with AdGFP (Fig. 2 B). Again, cell death was observed by 24 h of infection, increasing to ≥80% by 72 h (Fig. 2 D). These observations indicate that PI3K-activated Akt-1 is essential for macrophage survival.
Figure 2.

Inhibition of PI3K or Akt-1 activation results in macrophage cell death in a time and dose-dependent fashion. Macrophages were incubated with the PI3K inhibitor LY294002 at concentrations ranging from 12 to 100 μM (A) or at 50 μM (C). Control medium possessed DMSO alone. Adenoviral infection of macrophages was performed with a vector expressing a DN form of Akt-1 (AdDNAkt-1) or a control vector (AdGFP; B and D). The dosage of Ad used is presented on the abscissa in B and was 200 moi in D. The cells were harvested at time points indicated (C and D) or at 48 h (A and B). Survival was determined by relative MTT values (A and B), compared with control-treaded cells (DMSO or AdGFP), or as viable cell number determined by counting Trypan blue negative cells (C and D). *P < 0.05 determined by unpaired Student's t test compared with control. The results (mean ± 1 SE) of a single experiment, performed in triplicate, are presented, which was representative of three experiments.
The mechanism of macrophage death resulting from inhibition of Akt-1 was examined. DNA fragmentation was observed by 12 h, which increased over the next 24 to 48 h after the addition of LY294002 (Fig. 3 A) or infection with AdDNAkt-1 (Fig. 3 B). Supporting the results obtained with LY294002, another PI3K inhibitor, Wortmannin, also induced cell death and DNA fragmentation in human monocyte-differentiated macrophages (data not shown). Additionally, LY294002 and Wortmannin induced cell death and apoptosis in murine macrophage cell line RAW 264.7 (data not shown).
Figure 3.

Inhibition of PI3K (LY294002) or DN Akt-1 results in the loss of ΔΨm and DNA fragmentation. Macrophages were incubated with the PI3K inhibitor LY294002 (50 μM) or control DMSO (A and C) for 12–72 h, as indicated. Macrophages were infected with AdDNAkt-1 or the control vector (AdGFP or Adβgal; B and D) at 200 moi for 12–72 h. The effects on apoptosis were determined by DNA fragmentation by PI staining (<2N DNA; A and B), and on ΔΨm determined by Rh123 retention (mean fluorescence, percentage of control) (C and D). *P < 0.05, **P < 0.01 determined by unpaired Student's t test compared with control. The results (mean ± 1 SE) of a single experiment, performed in triplicate, are presented, which was representative of three experiments.
The role of the mitochondria in the induction of DNA fragmentation was also determined by examining the loss of ΔΨm, by Rh123 retention. The loss of ΔΨm was observed as early as 12 h after the addition of LY294002 (Fig. 3 C) or infection with the AdDNAkt-1 (Fig. 3 D). In each instance, the loss of ΔΨm was associated with the induction of apoptosis, determined by DNA fragmentation (Fig. 3A and Fig. B). The loss of ΔΨm and DNA fragmentation were detected by 12 h after infection with AdDNAkt-1, where as significant differences were first observed at 24 h after treatment with LY294002.
Constitutively Activated Akt-1 or Bcl-xL Protects Macrophages from LY294002-induced DNA Fragmentation.
To determine if the apoptotic cell death induced by inhibition of PI3K in macrophages was specifically due to inhibition of Akt-1 signaling, a constitutively activated form of Akt-1 (MyrAkt-1) was employed. Macrophages were infected with replication-defective Ad expressing constitutively activated Akt-1 (AdMyrAkt-1) or a control vector (Adβgal) for 48 h and then treated with LY294002 for an additional 48 h. Expression of MyrAkt-1 protected against loss of ΔΨm, DNA fragmentation and cell death (PI uptake) (Fig. 4). Since the loss of ΔΨm appeared temporally to coincide with the induction of DNA fragmentation after PI3K/Akt-1 inhibition, experiments were performed to determine if the expression of an antiapoptotic Bcl-2 family member was also protective. Infection of macrophages with an adenoviral vector expressing Bcl-xL, important in maintaining mitochondrial integrity 29, not only protected against the loss of ΔΨm induced by LY294002, but also against DNA fragmentation and PI uptake (Fig. 4). These observations indicate that in macrophages activated Akt-1 suppressed the induction of apoptosis by protecting mitochondrial integrity.
Figure 4.



Overexpression of activated Akt-1 (Myr-Akt-1) and Bcl-xL protects against the loss of ΔΨm, DNA fragmentation and PI uptake after the inhibition of PI3K. Macrophages were infected with Ad vectors expressing activated Akt-1 (MyrAkt-1), Bcl-xL, or control βgal at 200 moi for 48 h, then treated with LY294002 (50 μm) for an additional 48 h. The cells were collected and analyzed for mitochondrial integrity by Rh123 retention (mean flourescence, percentage of control; A), DNA fragmentation (% <2N DNA, B), and PI uptake (C). *P < 0.05 determined by unpaired Student's t test compared with control. The results (mean ± 1 SE) of a single experiment, performed in triplicate, are presented, which was representative of three experiments.
Inhibition of PI3K/Akt-1 Activation Results in Release of Cytochrome c, Activation of the Caspase Cascade, and Cleavage of PKCδ.
To characterize the mechanism of the apoptosis, the effects of PI3K/Akt-1 inhibition on cytochrome c release and caspase activation were examined. 7-d macrophages were treated with LY294002 or infected by AdDNAkt-1 for 24 h. Cytosolic cytochrome c was detected by Western blot assay after LY294002 treatment or Akt-1 inhibition (Fig. 5 A). No cytochrome c oxidase subunit IV was detected in the same cytosolic fractions (data not shown). To further characterize the mechanism of apoptosis induced by PI3K/Akt-1 inhibition, caspase activation was assessed. 7-d macrophages were infected with AdGFP or AdDNAkt-1 at 200 moi for 12 and 24 h. Immunoblot analyses revealed that the proforms of caspases-9 and -3 were decreased by 24, but not 12, h after the infection of AdDNAkt-1 (Fig. 5 B). Additionally full-length PKCδ, which is cleaved by active caspase-3, was reduced at 24, but not 12, h after infection with AdDNAkt-1. These observations suggest that after inhibition of the PI3K/Akt-1 pathway, the loss of ΔΨm, and cytochrome c release result in activation of caspases-9 and -3, cleavage of PKCδ, and DNA fragmentation.
Figure 5.


Inhibition of Akt-1 in macrophages results in the release of cytochrome c, activation of caspases-9 and -3, and cleavage of PKCδ. (A). Macrophages were treated with 50 μM LY294002 or infected with AdDNAkt-1 or the control AdGFP (200 moi) for 24 h. The cells were harvested and Western blots performed on the cytosolic fraction for cytochrome c or tubulin. (B). Macrophages were infected with adenoviral vectors expressing DN Akt-1, or the control GFP, at 200 moi for 12 and 24 h, as indicated. Cell lysates were collected and Western blot analysis performed as described in Materials and Methods using a rabbit anti–caspase-9, rabbit anti-PKCδ, mouse anti-caspase-3, and mouse anti-tubulin. The expression of the pro-form of caspases-9 and -3 and full-length PKCδ are shown. The results presented are representative of two experiments.
Caspase Inhibition Reduces DNA Fragmentation Induced by the Inhibition of Akt-1 Activation, but Does Not Protect Mitochondrial Integrity or Promote Cell Survival.
To further characterize the role of caspase activation during macrophage apoptosis induced by PI3K/Akt-1 inhibition, cells were cultured with either a general caspase inhibitor (z-VAD.fmk) or a specific inhibitor of caspase-9 (z-LEHD.fmk) in the presence of LY294002. Treatment with 100 μM of z-VAD.fmk or z-LEHD.fmk failed to prevent LY294002-induced mitochondrial dysfunction (Fig. 6 A) or cell death (PI-positive cells) (Fig. 6 C). In contrast, DNA fragmentation was significantly decreased by the addition of either z-VAD.fmk or z-LEHD-.fmk (Fig. 6 B). The role of caspase activation was further characterized by examining the effect of z-VAD.fmk, on the activation of caspases-9 and -3. Caspase 9 (Fig. 6 E) and caspase-3 activity (data not shown) induced by inhibition of PI3K were both suppressed by treatment with z-VAD.fmk. Consistent with this observation, the cleavage of caspase-3 and PKCδ (Fig. 6 E) induced by inhibition of PI3K/Akt-1 pathway was also suppressed. These observations suggest that the loss of ΔΨm induced by inhibition of the PI3K/Akt-1 pathway, resulted in cell death, independent of caspase activation.
Figure 6.





Caspase inhibitors do not prevent the loss of ΔΨm or macrophage PI uptake after PI3K/Akt-1 inhibition. Macrophages were treated with 100 μM zVAD.fmk, the caspase-9 inhibitor z-LEHD.fmk (100 μM) or control medium for 1 h, then 50 μM LY294002 was added for an additional 48 h. The cells were harvested and analyzed for mitochondrial retention of Rh123 (mean fluorescence, percentage of control; A), DNA fragmentation (% <2N DNA, B), PI uptake (percentage of PI-positive cells, C), caspase-9 activity (D), and the expression of proform of caspase-3 and full-length PKCδ (E). Each of the methods has been described previously. The results presented in each panel are representative of two or three experiments.
Our previous studies have shown that cell surface Fas–FasL interactions on human macrophages may contribute to the induction of apoptosis after the reduction of Fas-associated death domain–like IL-1ß–converting enzyme–inhibitory protein 3. Activated Akt-1 may suppress the expression of FasL, which may be mediated by phosphorylation of forkhead transcription factors 30. However, the expression of Fas and FasL on the surface of macrophages, determined by flow cytometry, was not affected by LY294002 treatment (data not shown). Additionally, in order to determine if Fas–FasL interactions may have contributed to the induction of apoptosis after the inhibition of the PI3K/Akt-1 pathway, macrophages were pretreated with antagonistic anti-FasL antibody for 1 h before the addition of LY294002. The antagonistic anti-FasL antibody had no effect on the loss of ΔΨm, DNA fragmentation, or cell death induced by LY294002 (data not shown). Additionally, TNF-α was not detected in the culture supernatants of macrophages, even after adenoviral infection (data not shown). These data, together with the lack of protection of mitochondrial integrity by zVAD.fmk, suggest that death receptor signaling was not responsible for initiating LY294002-induced ΔΨm loss and apoptosis.
There Is No Relationship between Constitutively Activated NF-κB and Akt-1 in Normal Macrophages.
We recently demonstrate that NF-κB was constitutively activated, and protected against apoptosis, in differentiated macrophages 4 23. Additionally, recent studies have demonstrated that Akt-1 mediates the activation of NF-κB in certain cell types 21 22 31 32. To characterize the potential connection between the Akt-1 and NF-κB activation, studies were performed to determine if inhibition of Akt-1 activation decreased the constitutive activation of NF-κB in normal human macrophages. Macrophages were treated with the PI3K inhibitor LY294002 for 24 and 48 h, and nuclear proteins were extracted for analysis by EMSA. Neither LY294002 nor DN Akt-1 inhibited the constitutive activation of NF-κB in human monocyte-differentiated macrophages (Fig. 7a Fig. b Fig. c). Additionally, LY294002 did not interfere with the activation of NF-κB by TNF-α in macrophages (Fig. 7 A). To further clarify the relationship between the activation of Akt-1 and NF-κB, macrophages were infected with an adenoviral vector expressing NF-κB–inducing kinase (NIK; reference 33). The ectopic expression of NIK resulted in a marked increase of NF-κB activation, determined by EMSA, but failed to protect against apoptosis induced by treatment with LY294002 (data not shown). Our results demonstrate that Akt-1 did not contribute to the constitutive activation of NF-κB in macrophages determined by the nuclear translocation of NF-κB and that increased NF-κB activation did not protect against the apoptosis induced after he inhibition of Akt-1.
Figure 7.




The activation of Akt-1 does not contribute to the constitutive or the induced activation of NF-κB in macrophages. (A) Macrophages were washed three times and cultured in serum-free medium for 18 h. The cells were then treated with 50 μM LY294002 or control DMSO for 2 h. Finally macro-phages were treated with 10 ng/ml TNF-α, 100 ng/ml LPS, or control medium as indicated for an additional 6 h. Nuclear extracts were harvested and EMSA was conducted. The composition of the NF-κB complex was determined by supershift analysis using antibodies specific for NF-κB p65 and p50, as indicated. (B) Macrophages were infected with AdDNAkt-1 or control AdGFP and cultured in serum containing medium for another for 24 and 48 h. (C) Treatment with LY294002 does not inhibit the constitutive activation of NF-κB. Macrophages were treated with 50 μM LY294002 or control DMSO for 24 or 48 h, before harvesting. (D) The 3xκB promoter-reporter plasmid (1 μg) was cotransfected with plasmid-expressing activated Akt-1 (1 μg MyrAkt) alone or together with a plasmid-expressing NF-κB p65 (1 μg) as described in Materials and Methods. The cells were harvested after 18 h, and cell lysates were prepared to measure luciferase activity, which was determined as RLU per microgram of protein. The results are presented as fold compared with cells transfected with the control plasmid (no). The results (mean ± 1 SE) of a single experiment, performed in triplicate, are presented, which was representative of three experiments.
Recent studies have suggested that activated Akt-1 may enhance the transcriptional activity of NF-κB 34. Therefore, the effect of activated Akt-1 on the transcriptional activity of NF-κB in macrophages was examined. In transient transfection assays, the ectopic expression of activated Akt-1 (Myr Akt-1) rescued RAW 264.7 cells from apoptosis induced by LY294002 (data not shown). In contrast, Myr Akt-1 failed to enhance NF-κB transcriptional activity, which we previously demonstrated in these cells 4, determined by cotransfection of a 3xκB promoter-reporter (Fig. 7 D). Furthermore, there was no enhancement of NF-κB activation by cotransfection of the plasmid-expressing Myr Akt-1 together with one expressing the transcriptionally active p65 subunit of NF-κB (Fig. 7 D). Further supporting the lack of effect on the transcriptional activity of NF-κB, inhibition of the PI3K/Akt-1 pathway in macrophages, did not reduce the expression of A1/Bfl-1 by reverse transcription PCR (Fig. 9 B), which is exquisitely sensitive to the constitutive activation of NF-κB in macrophages 4. Together, these observations demonstrate that in macrophages, the protection against apoptosis observed with activated Akt-1 is not mediated through the activation of NF-κB.
Figure 9.


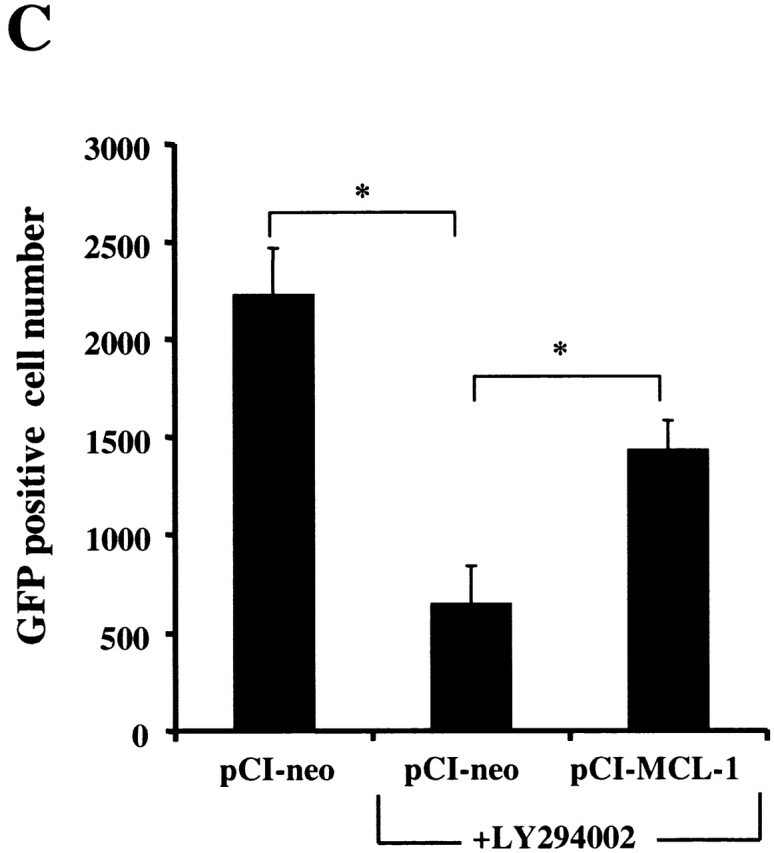


PI3K/Akt-1-regulated Mcl-1 is necessary for macrophage survival. Inhibition of PI3K/Akt-1 downregulates the expression of Mcl-1 at the protein (A) and transcriptional (B) levels. (A) Macrophages were treated with 50 μM LY294002 or infected with AdDNAkt-1 (200 moi) for 24 or 36 h as indicated in the figure. Cell lysates were collected and Western blots were performed with rabbit anti-Mcl-1 and mouse anti-tubulin antibodies as indicated. (B) Macrophages were treated with 50 μM LY294002 for 12, 24, and 36 h as indicated in the figure. Then the total RNA was extracted and used for the synthesis of cDNA. PCR was conducted using primers specific for Mcl-1, A1, Bcl-xL, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), as described in Materials and Methods. (C) The expression of Mcl-1 rescues RAW 264.7 macrophage-like cells from cell death after the inhibition of the PI3K/Akt-1 pathway. RAW cells were transfected with a plasmid-expressing enhanced GFP (0.5 μg) plus a control plasmid (pCl-neo) or one expressing Mcl-1 (pCl-MCL-1), each at 2.5 μg, as described in Materials and Methods. The transfected cells were then treated with 50 μM LY294002, where indicated, for 24 h. The cells were harvested and the number of enhanced GFP-positive cells was determined by flow cytometry. Macrophages were treated with Mcl-1 antisense oligonucleotides (AS) or control oligonucleotides (INV) for 24 h, cells were collected, and Mcl-1 expression detected by Western blot assay (D) and apoptosis determined by DNA fragmentation (<2N DNA; E). The results presented in each panel are representative of two or three experiments.
Inhibition of PI3K/Akt-1 Has No Effect on the Expression of Apoptosis-related Molecules Bcl-2, Bcl-xL, Bax or Bad, but Results in Inhibition of Mcl-1.
Since members of the Bcl-2 family control mitochondrial integrity, the effect PI3K/Akt-1 pathway inhibition on the expression of Bcl-2 family members was examined. Inhibition of PI3K/Akt-1 by LY294002 did not influence the expression of antiapoptotic molecules Bcl-2 or Bcl-xL or the proapoptotic molecule Bax, as determined by Western blot analysis (Fig. 8 A). Similar results were obtained after infection with AdDNAkt-1 (data not shown). Additionally, no change in the expression of phosphorylated Bad, which may contribute to protection of mitochondrial integrity, was observed in response to LY294002 treatment (Fig. 8 B). Furthermore, the expression of Bcl-xL and A1/Bfl-1 35 36 were not reduced after PI3K/Akt-1 inhibition by LY294002 (Fig. 9 B). In contrast, inhibition of PI3K by treatment with LY294002, or infection with AdDNAkt-1, suppressed Mcl-1 expression in macrophages by 24 h (Fig. 9a and Fig. b) before the activation of caspase-3 (Fig. 8 A). These observations suggest that Mcl-1 may be the downstream target of the PI3K/Akt-1 pathway responsible for protecting macrophage mitochondrial integrity.
Figure 8.


Inhibition of PI3K/Akt-1 has no effect on the expression of apoptosis-related molecules Bcl-2, Bcl-xL, Bax, or Bad. (A) Macrophages were treated with LY294002 (LY) at 50 μM for 24 or 48 h, as indicated. Cell lysates were collected and Western blot analyses were conducted using mouse anti–caspase-3, mouse anti-Bcl-2, rabbit anti-Bcl-xL, mouse anti-Bax, or mouse anti-tubulin antibodies as described in the Materials and Methods. (B) Macrophages were treated with 50 μM LY294002 for 48 h, then cell lysates was collected and immunoprecipitated using rabbit anti-Bad antibody. Western blot analysis was performed probing the immunoprecipitated complex with specific rabbit anti-Bad (Bad) or rabbit anti-phospho-Bad (P-Bad) antibodies. The result shown is one of three experiments with similar results.
Mcl-1 Is Essential for Macrophage Viability.
Since reduction of Mcl-1 was associated with cell death, experiments were performed to determine if Mcl-1 protected against cell death induced by inhibition of the PI3K/Akt-1 pathway. These experiments were performed with RAW 264.7 murine macrophages, since inhibition of the PI3K/Akt-1 pathway induced apoptosis, by a mechanism that was the same as observed in primary human macrophages (data not shown). A GFP-expressing plasmid was cotransfected with a control vector or one expressing Mcl-1. After 24 h, the cells were treated with LY294002 or vehicle control and cell viability was determined. Mcl-1 protected the RAW 264.7 macrophages from cell death induced by the inhibition of the PI3K/Akt-1 pathway (Fig. 9 C). These observations suggest that the constitutive activation of Akt-1 maintains the expression of Mcl-1, which is necessary for the protection of mitochondrial integrity in macrophages. To confirm that Mcl-1 was essential for macrophage viability, 7-d differentiated human macrophages were incubated with Mcl-1 antisense or control (INV) oligonucleotides. The reduction of Mcl-1 by the antisense resulted in macrophage apoptosis (Fig. 9d and Fig. e). These observations support an essential role for Mcl-1 in protecting macrophages from apoptosis induced by the loss of mitochondrial integrity.
Discussion
Prior studies using mice deficient in src homology 2 domain–containing 5′ inositol phosphatase or PTEN demonstrated an expansion of their macrophage populations 9 10. Both of these phosphatases normally function to suppress the PI3K pathway 11 12, suggesting that this pathway may be unique and critical for macrophage survival. The potential mechanisms by which this pathway may be important in macrophage survival have not been characterized. This study is the first to demonstrate that Akt-1 was constitutively activated in human monocyte-differentiated macrophages, and that specific inhibition of Akt-1 resulted in apoptotic cell death mediated by the loss of mitochondrial integrity. To date most studies have focused on the role of the PI3K/Akt-1 pathway in the regulation of apoptosis in proliferating cells rather than in terminally differentiated cells, such as macrophages.
Although the mechanism responsible for the constitutive activation of the PI3K/Akt-1 pathway in the differentiated human macrophages was not characterized in our manuscript, it is possible that factors present in serum may be important. Lysophosphatidic acid, M-CSF, and 1α, 25-dihydroxyvitamin D3, which may be present in normal serum, are capable of activating the PI3K pathway 15 37 38. It is not possible to withhold serum during monocyte to macrophage differentiation, since we have shown that most monocytes die by Fas–FasL–mediated apoptosis without serum 3. However, serum deprivation for 24 to 96 h after differentiation did not result in macrophage death (data not shown), suggesting that the continued presence of a factor in serum was not necessary for the survival of terminally differentiated human macrophages. In contrast, murine thioglycollate-elicited macrophages and bone marrow–derived macrophages, which proliferate in culture 39, died in the absence of serum or CSF-1 15 40. These observations suggest that the murine macrophages 15 40 may not be terminally differentiated or that the mechanism of protection against cell death was different, perhaps due to proliferation or activation that occurred during recruitment and isolation.
Akt-1 has been shown to promote cell survival, and protect against apoptosis, by a number of mechanisms. However, the mechanisms by which the PI3K/Akt-1 pathway contributes to the survival of terminally differentiated macrophages have not been described previously. Akt-1 activation, in certain cell types, may promote survival by protecting against Fas signaling 41. Additionally, PTEN+/− mice demonstrate impaired Fas-mediated apoptosis that is restored by PI3K inhibitors 10. However, our data do not support a role for death receptor signaling after the inhibition of the PI3K/Akt-1 pathway. The broad-based caspase inhibitor z-VAD.fmk protected against DNA fragmentation, a characteristic feature of the apoptotic phenotype, however, it failed to protect against the loss of ΔΨm or cell death, suggesting that activation of the initiator caspase-8 was not responsible. Additionally, we observed no change in the expression of Fas or FasL on the cell surface, and antagonistic anti-FasL antibody did not prevent the apoptosis observed (data not shown). These observations demonstrate that, after inhibition of the PI3K/Akt-1 pathway, death receptor ligation and caspase-8 activation did not initiate macrophage apoptosis and cell death.
Akt-1 may regulate cell survival at a postmitochondrial level, after the release of cytochrome c, by inhibiting the activation or activity of caspase-9 18 42. In a previous study, cytochrome c–induced activation of caspase-9 was inhibited by either z-VAD.fmk or activated Akt-1 42. In macrophages, inhibition of the PI3K/Akt-1 pathway with either LY294002 or the DN Akt-1 resulted in loss of ΔΨm and cytochrome c release. However, caspase inhibition by either z-VAD.fmk or a caspase-9–specific inhibitor partially protected against DNA fragmentation, but failed to protect against the loss of ΔΨm or cell death. These observations demonstrate that the protection provided by constitutively activated Akt-1, that was essential for macrophage survival, was upstream of the loss of ΔΨm and did not require caspase activation. Inhibition of caspase activation altered the apoptotic phenotype (i.e., DNA fragmentation and PKCδ cleavage), but did not protect against cell death. It is possible that mechanisms other than the loss of ΔΨm may have contributed directly to cell death. Apoptosis-inducing factor (AIF), when released from the mitochondria after the loss of ΔΨm, translocates to the nucleus, inducing chromatin condensation and DNA fragmentation, independent of caspase activation 43 44. Consistent with a role AIF in cell death, z-VAD.fmk only partially protected against DNA fragmentation, after inhibition of the PI3K/Akt-1 pathway. Therefore, the loss of ΔΨm and the subsequent release of AIF may both have directly contributed to macrophage death after inhibition of the PI3K/Akt-1 pathway.
Supporting the central role for the loss of ΔΨm in the induction of cell death after the inhibition of the PI3K/Akt-1 pathway, the ectopic expression of Bcl-xL protected not only against DNA fragmentation, but also prevented the loss of ΔΨm and cell death, even though no reduction of endogenous Bcl-xL or Bcl-2 was observed. Prior studies have demonstrated that activated Akt-1 phosphorylated Bad 17 45, which inhibited the binding of Bad with Bcl-xL, thereby promoting survival and protecting against apoptosis. Although this mechanism would result in apoptosis and cell death initiated by loss of ΔΨm, we observed no effect on the expression or phosphorylation of Bad in macrophages after inhibition of PI3K/Akt-1 activity. Similar to this observation, other studies have not observed an effect on the phosphorylation of Bad after the inhibition of Akt-1 in fibroblasts and a neuronal cell line 19 46. These data suggest that, although Bad may be involved in a cell-type and stimulus-specific fashion, it was not involved in macrophage apoptosis observed after the inhibition of the PI3K/Akt-1 pathway.
We recently demonstrated that the constitutive activation of NF-κB was essential for the survival of macrophages by maintaining mitochondrial integrity 4. Therefore, experiments were performed to determine if Akt-1 contributed to NF-κB activation in macrophages, as recently demonstrated in other cell types 31 32 34 47. In contrast to these observations, inhibition of the PI3K/Akt-1 pathway had no effect on the constitutive or TNF-α–stimulated activation of NF-κB in macrophages, as determined by EMSA. Consistent with this observation, TNF-α failed to induce activation of Akt-1 in monocytes 48. We also observed no effect of Akt-1 on the transcriptional activity of NF-κB in the RAW 264.7 macrophage cell line, in contrast to recent studies in other cell types 34 47. Our study directly examined the effects of Myr Akt-1 on the constitutive and the NF-κB p65 induced activation of a NF-κB promoter-reporter construct. The differences observed between the studies may be due to the experimental design or cell-type differences 34 47. Cell-type differences may be important since Myr Akt-1 activated a κB promoter in 3T3 cells 34, but not in RAW 264.7 macrophages, even though Myr Akt-1 protected the RAW 264.7 cells against cell death induced by inhibition of the PI3K pathway.
Further supporting a mechanism independent of NF-κB, the ectopic expression of NIK, which resulted in strong activation of NF-κB in macrophages, failed to protect against cell death induced after the inhibition of the PI3K pathway. Also, expression of the activated form of Akt-1 in macrophages, which protected against the apoptosis observed after treatment with LY294002, failed to activate NF-κB (data not shown). Additionally, we previously demonstrated that the antiapoptotic molecule A1 was an important downstream target of the constitutively activated NF-κB in macrophages 4. However, after PI3K/Akt-1 inhibition, no reduction in the expression of A1/Bfl-1 was observed. This observation is consistent with the lack of effect of PI3K/Akt-1 on the transcriptional activity of NF-κB, since A1/Bfl-1 expression in macrophages was exquisitely regulated by constitutively activated NF-κB 4. These observations demonstrate that the protection provided by the constitutively activated PI3K/Akt-1 pathway in macrophages was independent of the NF-κB activation. These observations support the independent role of A1, regulated by constitutively activated NF-κB and Mcl-1, regulated by the PI3K/Akt-1 pathway, in maintaining mitochondrial integrity, protecting against spontaneous apoptosis, in primary differentiated macrophages. The independence of the pathways is further supported by the observation that the apoptosis induced by inhibition of A1/NF-κB does not result in activation of caspase-3 4, while that which results from inhibition of PI3K/Akt-1/Mcl-1 does result in caspase-3 activation. This suggests that multiple pathways promote macrophage survival, even without specific stimulation or stress.
Since the loss of ΔΨm induced by PI3K/Akt-1 inhibition was protected by Bcl-xL, the expression of Bcl-2 family members was examined. Although Akt-1 regulated T lymphocyte survival, NF-κB activation and Bcl-xL expression 49, we did not detect an effect of PI3K/Akt-1 inhibition on the expression of Bcl-xL consistent with other studies 19 46. These differences may be due to cell-type and/or stimulus-specific differences. Also, no change in the expression of the antiapoptotic molecules Bcl-2 or A1 or the proapoptotic molecule Bax was observed. In contrast, the expression of Mcl-1 mRNA and protein was reduced after inhibition of the PI3K/Akt-1 pathway in macrophages. Supporting the importance of Mcl-1 in macrophages, Mcl-1 expression protected against the apoptotic cell death observed in the RAW 264.7 macrophage cell line after inhibition of the PI3K/Akt-1 pathway. Supporting the relevance of this observation, apoptosis induced in this cell line was associated with loss of ΔΨm, DNA fragmentation, and cell death that was prevented by the expression of activated Akt-1. The essential role of Mcl-1 in primary macrophages was further supported by the observation that reduction of Mcl-1 by antisense oligonucleotides resulted in macrophage apoptosis. An earlier study demonstrated that mcl-1 was regulated the PI3K/Akt-1 pathway through a transcription factor complex containing the cAMP response–element-binding protein 50. These observations support an essential role for Mcl-1, mediated by the PI3K/Akt-1 pathway, in promoting macrophage viability, by maintaining mitochondrial integrity.
Mcl-1, a Bcl-2 family member, is critical to embryonic development since deletion of this gene resulted in periimplantation embryonic lethality 51. Mcl-1 has been found to exhibit differentiation stage-specific expression in a variety of hematopoietic lineages 52. Overexpression of Mcl-1 delayed apoptosis induced by c-myc, growth factor withdrawal, and other cytotoxic agents, and reduced the release of cytochrome c from mitochondria 53 54 55. In contrast, our data suggest that Mcl-1 expression was essential in maintaining mitochondrial integrity in macrophages, in the absence of an exogenous death-inducing signal. Supporting this conclusion, mice transgenic for Mcl-1 demonstrated prolonged viability of both mature and immature myeloid cells 56. Additionally, antisense-mediated inhibition of Mcl-1 resulted in apoptosis of PMA-treated U937 monocytic cells 24. Overall, these observations support the importance of the PI3K/Akt-1 mediated–expression of Mcl-1 in maintaining macrophage viability.
Although the mechanism by which Mcl-1 protects macrophage mitochondrial integrity was not examined, in a yeast two-hybrid analysis, Mcl-1 bound avidly to Bax 57. Bax overexpression resulted in death by induction of mitochondrial permeability transition 58 59 , similar to the results observed after the inhibition of the PI3K/Akt-1 pathway. Supporting this mechanism, reduction of Bcl-xL, another Bcl-2 family member capable of binding to Bax, resulted in Bax-mediated cell death 60. These observations suggest that reduction of Mcl-1 in macrophages may lead to enhanced Bax activity, although the expression of Bax was not altered, which resulted in cell death mediated through the loss of mitochondrial integrity. These observations provide insights into potentially novel mechanisms to regulate macrophage survival in conditions such as atherosclerosis or rheumatoid arthritis.
Acknowledgments
We thank Kenneth Walsh (Tufts University, Boston, MA) for providing AdDNAkt-1, AdMyrAkt-1, and AdBcl-xL, and Dr. Stephen Nimer (Memorial Sloan-Kettering Cancer Center, New York, NY) for providing plasmid pCI-Mcl-1. We also thank Kathleen Carrigan for assistance in virus preparation and Mary Paniaqua for the flow cytometry conducted at the Robert H. Lurie Comprehensive Cancer Center, Flow Cytometry Core Facility of the Northwestern University Medical School, Chicago, IL.
This work was supported by National Institutes of Health (N01-AR62221 and P60-AR30692) and a grant from the Arthritis Foundation to R.M. Pope.
Footnotes
Abbreviations used in this paper: ΔΨm, mitochondrial transmembrane potential; Ad, adenovirus; AIF, apoptosis-inducing factor; DN, dominant negative; EMSA, electrophoretic mobility shift assay; GFP, green fluorescence protein; INV, inverted antisense oligonucleotides; moi, multiplicity of infection; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NF, nuclear factor; NIK, NF-κB–inducing kinase; PI, propidium iodide; PI3K, phosphatidylinositol 3-kinase; PTEN, phosphatase and tensin homologue deleted from chromosome 10.
References
- Feldmann M., Brennan F.M., Maini R.N. Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- Schwertschlag U.S., Trepicchio W.L., Dykstra K.H., Keith J.C., Turner K.J., Dorner A.J. Hematopoietic, immunomodulatory and epithelial effects of interleukin-11. Leukemia. 1999;13:1307–1315. doi: 10.1038/sj.leu.2401514. [DOI] [PubMed] [Google Scholar]
- Perlman H., Pagliari L.J., Georganas C., Mano T., Walsh K., Pope R.M. FLICE-inhibitory protein expression during macrophage differentiation confers resistance to Fas-mediated apoptosis. J. Exp. Med. 1999;190:1679–1688. doi: 10.1084/jem.190.11.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliari L.J., Perlman H., Liu H., Pope R.M. Macrophages require constitutive NF-κB activation to maintain A1 expression and mitochondrial homeostasis. Mol. Cell. Biol. 2000;20:8855–8865. doi: 10.1128/mcb.20.23.8855-8865.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.Y., Mayo M.W., Korneluk R.G., Goeddel D.V., Baldwin A.S., Jr. NF-κB antiapoptosisinduction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Bellacosa A., Testa J.R., Staal S.P., Tsichlis P.N. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254:274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- Cryns V., Yuan J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- Kluck R.M., Bossy-Wetzel E., Green D.R., Newmeyer D.D. The release of cytochrome c from mitochondriaa primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Helgason C.D., Damen J.E., Rosten P., Grewal R., Sorensen P., Chappel S.M., Borowski A., Jirik F., Krystal G., Humphries R.K. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12:1610–1620. doi: 10.1101/gad.12.11.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristofano A., Kotsi P., Peng Y.F., Cordon-Cardo C., Elkon K.B., Pandolfi P.P. Impaired Fas response and autoimmunity in Pten+/− mice. Science. 1999;285:2122–2125. doi: 10.1126/science.285.5436.2122. [DOI] [PubMed] [Google Scholar]
- Aman M.J., Lamkin T.D., Okada H., Kurosaki T., Ravichandran K.S. The inositol phosphatase SHIP inhibits Akt/PKB activation in B cells. J. Biol. Chem. 1998;273:33922–33928. doi: 10.1074/jbc.273.51.33922. [DOI] [PubMed] [Google Scholar]
- Wu X., Senechal K., Neshat M.S., Whang Y.E., Sawyers C.L. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. USA. 1998;95:15587–15591. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffer P.J., Jin J., Woodgett J.R. Protein kinase B (c-Akt)a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem. J. 1998;335:1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S.R., Brunet A., Greenberg M.E. Cellular survivala play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Koh J.S., Lieberthal W., Heydrick S., Levine J.S. Lysophosphatidic acid is a major serum noncytokine survival factor for murine macrophages which acts via the phosphatidylinositol 3-kinase signaling pathway. J. Clin. Invest. 1998;102:716–727. doi: 10.1172/JCI1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke T.F., Kaplan D.R., Cantley L.C. PI3Kdownstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- Datta S.R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y., Greenberg M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Cardone M.H., Roy N., Stennicke H.R., Salvesen G.S., Franke T.F., Stanbridge E., Frisch S., Reed J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- Kennedy S.G., Kandel E.S., Cross T.K., Hay N. Akt/protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol. Cell. Biol. 1999;19:5800–5810. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sizemore N., Leung S., Stark G.R. Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-κB p65/RelA subunit. Mol. Cell. Biol. 1999;19:4798–4805. doi: 10.1128/mcb.19.7.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraud C., Henzel W.J., Baeuerle P.A. Involvement of regulatory and catalytic subunits of phosphoinositide 3-kinase in NF-κB activation. Proc. Natl. Acad. Sci. USA. 1999;96:429–434. doi: 10.1073/pnas.96.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane L.P., Shapiro V.S., Stokoe D., Weiss A. Induction of NF-κB by the Akt/PKB kinase. Curr. Biol. 1999;9:601–604. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- Liu H., Sidiropoulos P., Song G., Pagliari L.J., Birrer M.J., Stein B., Anrather J., Pope R.M. TNF-α gene expression in macrophagesregulation by NF-κB is independent of c-Jun or C/EBPβ. J. Immunol. 2000;164:4277–4285. doi: 10.4049/jimmunol.164.8.4277. [DOI] [PubMed] [Google Scholar]
- Moulding D.A., Giles R.V., Spiller D.G., White M.R., Tidd D.M., Edwards S.W. Apoptosis is rapidly triggered by antisense depletion of Mcl-1 in differentiating U937 cells. Blood. 2000;96:1756–1763. [PubMed] [Google Scholar]
- Perlman H., Sata M., Le Roux A., Sedlak T., Branellec D., Walsh K. Bax-mediated cell death by the Gax homeoprotein requires mitogen-activation but is independent of cell cycle activity. EMBO J. 1998;17:3576–3586. doi: 10.1093/emboj/17.13.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman H., Georganas C., Pagliari L.J., Koch A.E., Haines K., III, Pope R.M. Bcl-2 expression in synovial fibroblasts is essential for maintaining mitochondrial homeostasis and cell viability. J. Immunol. 2000;164:5227–5235. doi: 10.4049/jimmunol.164.10.5227. [DOI] [PubMed] [Google Scholar]
- Ando T., Shibata H., Suzuki T., Kurihara I., Hayashi K., Hayashi M., Saito I., Kawabe H., Tsujioka M., Saruta T. The possible role of apoptosis-suppressing genes, bcl-2 and mcl-1/EAT in human adrenal tumors. Endocr. Res. 1998;24:955–960. doi: 10.3109/07435809809032715. [DOI] [PubMed] [Google Scholar]
- Gerber H.-P., Dixit V., Ferrara N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J. Biol. Chem. 1998;273:13313–13316. doi: 10.1074/jbc.273.21.13313. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Chandel N.S., Williamson E.K., Schumacker P.T., Thompson C.B. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- Biggs W.H., III, Meisenhelder J., Hunter T., Cavenee W.K., Arden K.C. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romashkova J.A., Makarov S.S. NF-κB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- Ozes O.N., Mayo L.D., Gustin J.A., Pfeffer S.R., Pfeffer L.M., Donner D.B. NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- Hu W.H., Johnson H., Shu H.B. Tumor necrosis factor-related apoptosis-inducing ligand receptors signal NF-κB and JNK activation and apoptosis through distinct pathways. J. Biol. Chem. 1999;274:30603–30610. doi: 10.1074/jbc.274.43.30603. [DOI] [PubMed] [Google Scholar]
- Madrid L.V., Wang C.Y., Guttridge D.C., Schottelius A.J., Baldwin A.S., Jr., Mayo M.W. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-κB. Mol. Cell. Biol. 2000;20:1626–1638. doi: 10.1128/mcb.20.5.1626-1638.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreb J.S., Schweder M. Human A1, a Bcl-2-related gene, is induced in leukemic cells by cytokines as well as differentiating factors. Leukemia. 1997;11:998–1004. doi: 10.1038/sj.leu.2400719. [DOI] [PubMed] [Google Scholar]
- Lin E.Y., Orlofsky A., Wang H.-G., Reed J.C., Prystowsky M.B. A1, a Bcl-2 family member, prolongs cell survival and permits myeloid differentiation. Blood. 1996;87:983–992. [PubMed] [Google Scholar]
- Kelley T.W., Graham M.M., Doseff A.I., Pomerantz R.W., Lau S.M., Ostrowski M.C., Franke T.F., Marsh C.B. Macrophage colony-stimulating factor promotes cell survival through Akt/protein kinase B. J. Biol. Chem. 1999;274:26393–26398. doi: 10.1074/jbc.274.37.26393. [DOI] [PubMed] [Google Scholar]
- Hmama Z., Nandan D., Sly L., Knutson K.L., Herrera-Velit P., Reiner N.E. 1α,25-dihydroxyvitamin D(3)–induced myeloid cell differentiation is regulated by a vitamin D receptor-phosphatidylinositol 3-kinase signaling complex. J. Exp. Med. 1999;190:1583–1594. doi: 10.1084/jem.190.11.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton J.A., Chan J., Byrne R.J., Bischof R.J., Jaworowski A., Kanagasundaram V. MRL/lpr and MRL+/+ macrophage DNA synthesis in the absence and the presence of colony-stimulating factor-1 and granulocyte-macrophage colony-stimulating factor. J. Immunol. 1998;161:6802–6811. [PubMed] [Google Scholar]
- Smith J.L., Schaffner A.E., Hofmeister J.K., Hartman M., Wei G., Forsthoefel D., Hume D.A., Ostrowski M.C. ets-2 is a target for an akt (protein kinase B)/Jun N-terminal kinase signaling pathway in macrophages of motheaten-viable mutant mice. Mol. Cell. Biol. 2000;20:8026–8034. doi: 10.1128/mcb.20.21.8026-8034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausler P., Papoff G., Eramo A., Reif K., Cantrell D.A., Ruberti G. Protection of CD95-mediated apoptosis by activation of phosphatidylinositide 3-kinase and protein kinase B. Eur. J. Immunol. 1998;28:57–69. doi: 10.1002/(SICI)1521-4141(199801)28:01<57::AID-IMMU57>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Zhou H., Li X.-M., Meinkoth J., Pittman R.N. Akt regulates cell survival and apoptosis at a postmitochondrial level. J. Cell Biol. 2000;151:483–494. doi: 10.1083/jcb.151.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Brenner C., Larochette N., Prevost M.C., Alzari P.M., Kroemer G. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J. Exp. Med. 1999;189:381–394. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin S.A., Daugas E., Ravagnan L., Samejima K., Zamzami N., Loeffler M., Costantini P., Ferri K.F., Irinopoulou T., Prevost M.C. Two distinct pathways leading to nuclear apoptosis. J. Exp. Med. 2000;192:571–580. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Peso L., Gonzalez-Garcia M., Page C., Herrera R., Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- Eves E.M., Xiong W., Bellacosa A., Kennedy S.G., Tsichlis P.N., Rosner M.R., Hay N. Akt, a target of phosphatidylinositol 3-kinase, inhibits apoptosis in a differentiating neuronal cell line. Mol. Cell. Biol. 1998;18:2143–2152. doi: 10.1128/mcb.18.4.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy S.A., Huang J.H., Liao W.S. Phosphatidylinositol 3-kinase as a mediator of TNF-induced NF-κB activation. J. Immunol. 2000;164:1355–1363. doi: 10.4049/jimmunol.164.3.1355. [DOI] [PubMed] [Google Scholar]
- Hayes A.L., Smith C., Foxwell B.M.J., Brennan F.M. CD-45-induced tumor necrosis factor α production in monocytes is phosphatidylinositol-kinase dependent and nuclear factor-κB-independent. J. Biol. Chem. 1999;274:33455–33461. doi: 10.1074/jbc.274.47.33455. [DOI] [PubMed] [Google Scholar]
- Jones R.G., Parsons M., Bonnard M., Chan V.S., Yeh W.C., Woodgett J.R., Ohashi P.S. Protein kinase B regulates T lymphocyte survival, nuclear factor κB activation, and Bcl-X(L) levels in vivo. J. Exp. Med. 2000;191:1721–1734. doi: 10.1084/jem.191.10.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.M., Chao J.R., Chen W., Kuo M.L., Yen J.J., Yang-Yen H.F. The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol. Cell. Biol. 1999;19:6195–6206. doi: 10.1128/mcb.19.9.6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkenberger J.L., Horning S., Klocke B., Roth K., Korsmeyer S.J. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 2000;14:23–27. [PMC free article] [PubMed] [Google Scholar]
- Krajewski S., Bodrug S., Krajewska M., Shabaik A., Gascoyne R., Berean K., Reed J.C. Immunohistochemical analysis of Mcl-1 protein in human tissues. Differential regulation of Mcl-1 and Bcl-2 protein production suggests a unique role for Mcl-1 in control of programmed cell death in vivo. Am. J. Pathol. 1995;146:1309–1319. [PMC free article] [PubMed] [Google Scholar]
- Chao J.R., Wang J.M., Lee S.F., Peng H.W., Lin Y.H., Chou C.H., Li J.C., Huang H.M., Chou C.K., Kuo M.L. mcl-1 is an immediate-early gene activated by the granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling pathway and is one component of the GM-CSF viability response. Mol. Cell. Biol. 1998;18:4883–4898. doi: 10.1128/mcb.18.8.4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Qian L., Kozopas K.M., Craig R.W. Mcl-1, a Bcl-2 family member, delays the death of hematopoietic cells under a variety of apoptosis-inducing conditions. Blood. 1997;89:630–643. [PubMed] [Google Scholar]
- Wang X., Studzinski G.P. Antiapoptotic action of 1,25-dihydroxyvitamin D3 is associated with increased mitochondrial MCL-1 and RAF-1 proteins and reduced release of cytochrome c. Exp. Cell. Res. 1997;235:210–217. doi: 10.1006/excr.1997.3667. [DOI] [PubMed] [Google Scholar]
- Zhou P., Qian L., Bieszczad C.K., Noelle R., Binder M., Levy N.B., Craig R.W. Mcl-1 in transgenic mice promotes survival in a spectrum of hematopoietic cell types and immortalization in the myeloid lineage. Blood. 1998;92:3226–3239. [PubMed] [Google Scholar]
- Sedlak T.W., Oltvai Z.N., Yang E., Wang K., Boise L.H., Thompson C.B., Korsmeyer S.J. Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc. Natl. Acad. Sci. USA. 1995;92:7834–7838. doi: 10.1073/pnas.92.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino J.G., Chen S.T., Tafani M., Snyder J.W., Farber J.L. The overexpression of Bax produces cell death upon induction of the mitochondrial permeability transition. J. Biol. Chem. 1998;273:7770–7775. doi: 10.1074/jbc.273.13.7770. [DOI] [PubMed] [Google Scholar]
- Pastorino J.G., Tafani M., Rothman R.J., Marcinkeviciute A., Hoek J.B., Farber J.L. Functional consequences of the sustained or transient activation by Bax of the mitochondrial permeability transition pore. J. Biol. Chem. 1999;274:31734–31739. doi: 10.1074/jbc.274.44.31734. [DOI] [PubMed] [Google Scholar]
- Zhang L., Yu J., Park B.H., Kinzler K.W., Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science. 2000;290:989–992. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]