

Abstract
In latently infected B lymphocytes, the Epstein-Barr virus (EBV) suppresses signal transduction from the antigen receptor through expression of the integral latent membrane protein 2A (LMP2A). At the same time, LMP2A triggers B cell survival by a yet uncharacterized maintenance signal that is normally provided by the antigen receptor. The molecular mechanisms are unknown as LMP2A-regulated signaling cascades have not been described so far. Using a novel mouse model we have identified the intracellular adaptor protein Src homology 2 (SH2) domain–containing leukocyte protein (SLP)-65 as a critical downstream effector of LMP2A in vivo. Biochemical analysis of the underlying signaling pathways revealed that EBV infection causes constitutive tyrosine phosphorylation of one of the two SLP-65 isoforms and complex formation between SLP-65 and the protooncoprotein CrkL (CT10 regulator of kinase like). This leads to antigen receptor-independent phosphorylation of Cbl (Casitas B lineage lymphoma) and C3G. In contrast, phospholipase C-γ2 (PLC-γ2) activation is completely blocked. Our data show that in order to establish a latent EBV infection, LMP2A selectively activates or represses SLP-65–regulated signaling pathways.
Keywords: B lymphocytes, Epstein-Barr virus, antigen receptor, SLP-65, signal transduction
Introduction
EBV is an oncogenic herpes virus that infects human B lymphocytes through attachment to the CD21 surface marker (for a review, see references 1 and 2). EBV is the causative agent of infectious mononucleosis and is associated with numerous malignancies including Burkitt's lymphoma, Hodgkin's disease, and nasopharyngeal carcinoma. Like all herpes viruses, EBV is able to persist in infected cells in a latent form. Only a few virally encoded proteins are expressed in EBV-positive peripheral B cells, such as the EBV-associated nuclear antigen (EBNA) 1 and the integral latent membrane protein 2A (LMP2A). Infection of B cells in vitro results in expression of additional EBNAs together with LMP1, and an LMP2A splice variant, LMP2B 3. These B cells become immortalized and are called lymphoblastoid cell lines (LCLs). EBNAs are involved in maintaining the viral genome and, together with LMP1, are required for EBV-mediated B cell transformation. It is the expression of LMP2A that regulates EBV latency by remodeling signal transduction events from the B cell antigen receptor (BCR; references 4 5 6 7 8).
In the absence of antigen, a yet poorly understood BCR maintenance signal drives B cells through developmental checkpoints and is indispensable for the survival of mature, peripheral B cells 9 10 11. Antigen engagement triggers a BCR activation signal that allows proliferation and potentially differentiation of stimulated B cells into antibody-secreting plasma cells 12 13 14. Both BCR functions are dependent on the immunoreceptor tyrosine-based activation motif (ITAM) in the BCR signaling elements, Ig-α and Ig-β, and on cytoplasmic protein tyrosine kinases (PTKs) of the Src-, Syk-, and Tec families. An immediate early substrate of Syk is the Src homology 2 (SH2) domain-containing leukocyte adaptor protein (SLP)-65 15, (also called B cell linker protein [BLNK; reference 16] or B cell adaptor containing SH2 domain [BASH; reference 17]). SLP-65 contains at least five consensus tyrosine-phosphorylation sites at the N terminus, followed by several putative binding motifs for SH3 domains and a COOH-terminal SH2 domain. Phosphorylated SLP-65 couples Syk and the Tec family member Bruton's tyrosine kinase (Btk) to phosphorylation and hence activation of phospholipase C-γ2 (PLC-γ2; references 16 and 18 19 20 21). This triggers the mobilization of intracellular Ca2+ ions, which is a hallmark of the BCR activation signal 22. Mutations in BCR components or BCR effector proteins impair the development and activation of B cells in mouse and man 9 23 24 25 26. This is also true for loss of SLP-65 expression. SLP-65 knockout mice have been generated by multiple groups 27 28 29 30 and show a drastic reduction in the number of mature B cells due to a block at the transition from B220+CD43+ progenitor B (proB) to B220+CD43− precursor B (preB) cells. A small number of IgMhi B cells are seen in peripheral immune organs; however, mature IgMloIgDhi are not.
LMP2A is anchored in the plasma membrane by 12 hydrophobic membrane spanning regions and assembles into large aggregates 4 within glycolipid-enriched microdomains, so-called lipid-rafts or glycosphingolipid-enriched membrane microdomains (GEMs; reference 31). The termini are facing the cytosol and consist of 119 and 27 amino acids, respectively. LMP2A redirects BCR signaling in two ways. First, the constitutively phosphorylated ITAM tyrosines in the NH2-terminus of LMP2A recruit Syk via the PTK's Src homology (SH)2 domains 32 33. Similarly, Src family PTKs like Lyn and Fyn are sequestered by LMP2A 34 35 and therefore cannot be employed by the BCR to trigger an activation signal. As a consequence, LMP2A expression renders B cells largely unresponsive to BCR ligation by antigen in that they fail to induce PTK substrate phosphorylation and subsequent mobilization of Ca2+ ions 5 6 7 8. This signaling block prevents a BCR-induced lytic production of EBV particles and thereby contributes to a successful escape of the virus from immune recognition. Beside the dominant-negative effect on the BCR signaling machinery, a second function of LMP2A was initially suggested by the finding that its isolated ITAM-containing terminus can mimic BCR signaling events when expressed and ligated as a CD8 chimera 36 37. The in vivo role of this positive LMP2A signaling capacity has been recently demonstrated in the EμLMP2A transgenic mouse line E (TgE) with B cell lineage expression of LMP2A 38. TgE mice show a dramatic reduction in the number of BCR-positive B cells present in developing bone marrow and peripheral immune organs. Despite the absence of a cognate BCR, TgE B cells are capable of progressing out of the bone marrow and entering the peripheral immune system where they persist. This is in sharp contrast to wild-type (WT) mice, where these immunologically useless cells become rapidly eliminated by apoptosis 9. The lack of surface BCR expression on TgE B cells was found to be due to a block of Ig heavy chain gene rearrangement during B cell development 38. The results show that LMP2A provides B cells with a survival signal that can substitute the BCR maintenance signal to bypass normal developmental checkpoint controls. The molecular details underlying the BCR or LMP2A maintenance signal are not known to date. In this report we show that LMP2A signals B cell survival through the p70 isoform of human SLP-65 and the protooncogene product CT10 regulator of kinase like (CrkL).
Materials and Methods
Generation and Analysis of Transgenic Mice.
Construction and characterization of SLP-65/BLNK−/− mice (B6.129F1 background) and EμLMP2A transgenic animals have been described previously 27 38. EμLMP2A transgenic line E (TgE) mice have been bred for over 10 generations into the C57BL/6 background. TgE males were mated with SLP-65−/− females to obtain F1 progeny. F1 progeny were further mated to obtain transgenic LMP2A SLP-65−/− mice. All animals were housed at the Northwestern University Center for Experimental Animal Resources in accordance with university animal welfare guidelines. Isolation of tail DNA and polymerase chain reaction for the presence of the LMP2A transgene, recombination activating gene (RAG)-1, and the Neomycin cassette was done as described previously 38 39 40. Presence of the SLP-65/BLNK gene was determined by PCR using the primers BLNK sense (5′-GGACAAACAGGAAGTAGTG-3′) and BLNK antisense (5′-CTGGCA-GGGACAGTTATC-3′). Flow cytometry analysis of B cell populations was done as described previously 38 39 40 using antibodies purchased from BD PharMingen. Samples were run on a Becton Dickinson FACScan™ and data was analyzed using CELLQuest™ software.
Cell Culture.
LCLs were generated by infection of peripheral IgM- and IgG-positive B cells from healthy human donors with EBV strain B95-8 and maintained, like the EBV-negative Ramos B cell line 41 in RPMI 1640 supplemented with 10% fetal calf serum, 2 mM glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin at 37°C and 5% CO2. LCLs used in this study (BS, DK, NB, and OP) are heterogeneous for surface expression of IgM, IgD, and IgG. Surface IgM expression of LCL-BS was higher than that of Ramos B cells, while that of LCL-DK, LCL-DU, LCL-NB, and LCL-OP was reduced, and these cells became IgM-negative after a long time in culture. Stimulation of the cells with either F(ab′)2 fragments of anti-Ig antibodies or pervanadate/H2O2 has been described elsewhere 42 43 44.
Protein Isolation and Analysis.
After lysis of the unstimulated or stimulated B cells in 1% Nonidet P-40 buffer 44, immunoprecipitations were performed using agarose-coupled mouse monoclonal antibodies to phosphotyrosine (pTyr; 4G10; UBI), or with protein G-adsorbed mouse monoclonal antibodies to human SLP-65/BLNK (Babco), or with rabbit antibodies specific for the following BCR effector proteins: PLC-γ2 (Q20), CrkL, and C3G (Santa Cruz Biotechnology, Inc.); and Casitas B lineage lymphoma (Cbl; Transduction Laboratories/Becton Dickinson). Mouse monoclonal anti-CrkL (UBI) and anti-Syk (4D10; Santa Cruz Biotechnology, Inc.) were used for protein detection by immunoblot analysis. The fusion protein between the glutathione S-transferase (GST) and the tandem SH2 domains of Syk, and its application for affinity purification of phosphoprotein ligands are described in detail elsewhere 42 43 44.
Results
The Adaptor SLP-65/BLNK Is a Critical Downstream Effector of LMP2A.
The phenotype of TgE mice demonstrated that LMP2A is capable of altering normal B cell development and enhancing survival in vivo 38. Biochemical studies in LCLs revealed that this LMP2A function is dependent on its ITAM which becomes tyrosine phosphorylated and binds Syk 7 8 32 33. This suggested a role for the SLP-65 adaptor protein in initiating LMP2A-mediated signals, as SLP-65 is an early and specific Syk substrate 15 16. To test whether loss of SLP-65 affects LMP2A function in vivo, SLP-65/BLNK-deficient animals (described in reference 27) were bred to TgE mice. Spleen and bone marrow samples isolated from WT, TgE, SLP-65−/−, and TgE SLP-65−/− animals were analyzed by flow cytometry (Fig. 1). Cells were stained with antibodies to the pan-B cell marker CD19 and IgM and relative percentages of the different B cell populations were calculated based on a lymphocyte gate. As expected, WT animals showed the normal pattern of B cell development consisting of CD19+IgM− B cells (48 ± 7%) developing into CD19+IgM+ B cells (25 ± 7%) in the bone marrow. These WT CD19+IgM+ cells colonize peripheral immune organs, such as the spleen, where most B cells display a CD19+IgM+ phenotype (61 ± 7%) with variable levels of IgM surface expression. In contrast, TgE animals show the LMP2A phenotype with most developing bone marrow B cells displaying a CD19+IgM− phenotype (62 ± 9%) and a very low number of B cells being CD19+IgM+ (1 ± 1%). TgE CD19+IgM− B cells exit the bone marrow and colonize the spleen (14 ± 5%). SLP-65−/− animals display the expected phenotype of a block in B cell development from the pro-B to pre-B stage showing a small number of CD19+IgM+ B cells in the bone marrow (9 ± 1%) and the spleen (9 ± 2%). As observed previously, a small population of IgMhi B cells accumulates over time in SLP-65−/− mice 27. TgE SLP-65−/− animals were indistinguishable from SLP-65−/− animals with respects to B cell development. TgE SLP-65−/− bone marrow B cells were identified as being either CD19+IgM− B cells (43 ± 5%) or CD19+IgM+ B cells (6 ± 1%). Similarly to SLP-65−/− mice, TgE SLP-65−/− animals have a small number of CD19+IgM+ B cells in the spleen (6 ± 2%). Flow cytometric analysis of B220, CD19, and CD43 expression in bone marrow samples from TgE SLP-65−/− mice and SLP-65−/− littermate controls further confirmed that both animals display an almost identical block of B cell development, i.e., at the pre-BI (B220+CD19+CD43+) to pre-BII (B220+CD19+CD43−) stage (data not shown). Similar results were obtained from another LMP2A transgenic line displaying a weaker phenotype (Tg6) when bred into the SLP-65−/− background (data not shown). These results show that LMP2A is incapable of transmitting signals to alter B cell development and survival in vivo in the absence of SLP-65. The possibility that SLP-65 could be necessary for the differentiation of an EBV target population that will be subsequently driven by LMP2A signals can be excluded, as LMP2A expression in RAG-1–deficient animals (TgE RAG-1−/−) releases the block of pro-B cell development 38. Thus, early pro-B cells are already susceptible to LMP2A signaling, but this developmental stage is not affected by the loss of SLP-65 27 28 29 30.
Figure 1.

Loss of the LMP2A developmental and survival phenotype when bred into the SLP-65−/− background. (A) Mouse splenocytes (SP) and (B) bone marrow samples (BM) from WT, TgE SLP-65−/−, and TgE SLP-65−/− animals were subjected to flow cytometric analysis with at least 10,000 gated lymphocytes collected per animal using CD19-PE (CD19) and IgM-FITC (IgM) antibodies. Typical dot plot results of 10,000 gated lymphocytes from 5–7-wk-old animals (n = 5) are shown. The polygons represent distinct subpopulations of B cells with the average percentage of total gated lymphocytes indicated above each polygon. (A) The left polygon represents CD19+IgM− B cells typical in TgE animals; the right polygon represents CD19+IgM+ cells. (B) The left rectangle represents CD19+IgM− B cells; the right rectangle represents CD19+IgM+ B cells.
LMP2A Triggers Constitutive Tyrosine Phosphorylation of Syk and SLP-65.
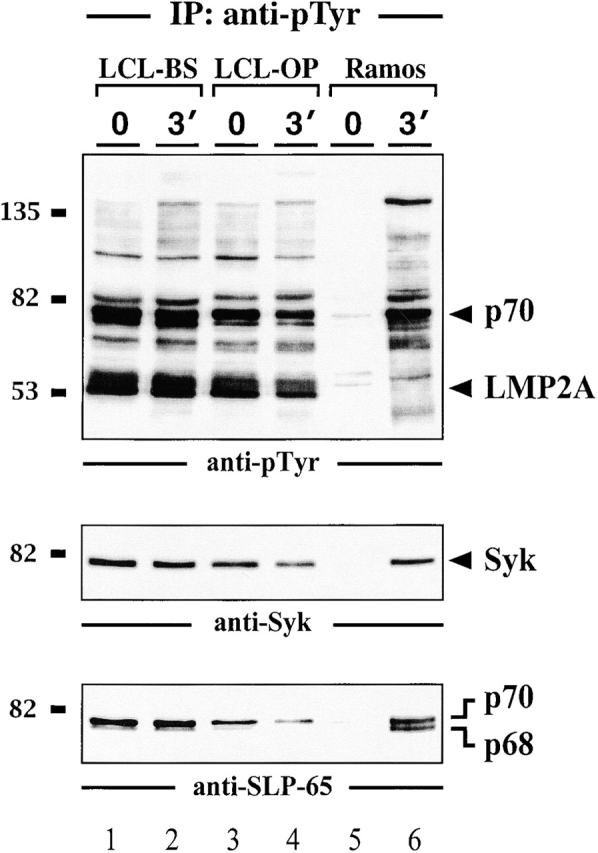
To biochemically analyze LMP2A signaling pathways and to address the role of SLP-65 in more detail several independent LCLs were established. As shown in Fig. 2 and in line with previous reports 7 32 33 34 35, our LCLs express the 53-kD LMP2A, which is constitutively phosphorylated on its ITAM tyrosines. The latter feature allows LMP2A to be specifically purified with a GST fusion protein encompassing the tandem SH2 domains of Syk and to be detected by anti-pTyr immunoblotting (Fig. 2 A, lanes 1–4). P53 is absent in the preparation from LMP2A-negative Ramos B cells (lanes 5 and 6). In this case, the Syk SH2 domains purify tyrosine-phosphorylated Ig-α after BCR activation of the cells. The results confirm that the previously published association between tyrosine-phosphorylated LMP2A and Syk 7 32 33 is mediated by the Syk SH2 domains, which possess an exquisite binding specificity for doubly phosphorylated ITAMs 42. The lack of tyrosine-phosphorylated Ig-α in stimulated LCLs (lanes 2 and 4) directly shows the LMP2A-mediated inhibition of BCR signal transduction 5 6 7 8. This is further demonstrated by anti-pTyr immunoblot analysis of LCLs (Fig. 2 B, top panel). In addition to LMP2A, a second major phosphoprotein of 70 kD (p70) can be detected (lanes 1–6). The intensity of all protein bands is independent of BCR engagement. Moreover and in marked contrast to EBV-negative Ramos B cells (lanes 7–9), BCR engagement on LCLs does not increase the complexity of the pTyr-containing protein pattern. In summary, the LCLs used in this study show the same features than those previously employed for the analysis of LMP2A function 5 6 7 8.
Figure 2.



Impaired BCR signaling but elevated levels of pTyr-containing proteins in EBV-infected B cells. Two independently obtained LCLs (lanes 1–4 in panel a and 1–6 in panel b) and EBV-negative Ramos B cells (lanes 5 and 6 in panel a and 7–9 in panel b) were left untreated or stimulated through their BCR with F(ab′)2 fragments of anti-IgM antibodies for the indicated time points (30 s or 3 min). (a) From the cleared cellular lysates, phosphoproteins were isolated by affinity purification (AP) with a GST fusion protein encompassing the tandem SH2 domains of Syk (GST-SYK[SH2]2) and detected by anti-pTyr immunoblotting. (b) Lysates were subjected to immunoprecipitation (IP) with anti-pTyr antibodies and the material was analyzed by subsequent immunoblotting with antibodies to pTyr (top panel), Syk (middle panel), and SLP-65 (bottom panel). (c) Cellular lysates of the different LCLs used in this study (lanes 1–4) and Ramos B cells (lane 5) were immunoblotted with anti–SLP-65 antibodies. Relative molecular masses of marker proteins are indicated on the left in kD.
As reported by Miller et al. 7, the p70 phosphoprotein band of LCLs is recognized by anti-Syk antibodies (Fig. 2 B, middle panel, lanes 1–6). However, repeated preclearing with anti-Syk antibodies failed to completely remove this band from anti-pTyr immunoprecipitates (data not shown). Probing of the same material with anti-SLP-65 antibodies revealed that anti-pTyr precipitates from unstimulated and stimulated LCLs also contain SLP-65 (Fig. 2 B, bottom panel, lanes 1–6). As for Syk, detection of SLP-65 in anti-pTyr precipitates from Ramos B cells required BCR ligation (lanes 7–9). However, whereas both SLP-65 isoforms, p68 and p70, are present in the preparation from activated Ramos B cells, predominantly the slower migrating form is detected in anti-pTyr precipitates from LCLs. Both isoforms are expressed by LCLs to similar amounts (Fig. 2 C). To directly test for tyrosine phosphorylation of SLP-65 in LCLs, anti–SLP-65 precipitates were analyzed by anti-pTyr immunoblotting. As shown in Fig. 3, the p70 isoform of purified SLP-65 from LCLs is constitutively tyrosine phosphorylated (lanes 1–4). In Ramos B cells, both SLP-65 isoforms become inducibly phosphorylated (lanes 5 and 6). Collectively, our data establish that EBV infection triggers constitutive tyrosine phosphorylation of the 70-kD form of human SLP-65.
Figure 3.

Constitutive tyrosine phosphorylation of the p70 isoform of SLP-65 in EBV-infected B cells. Two independent LCL clones (lanes 1–4) and Ramos B cells (lanes 5 and 6) were left untreated (lanes 1, 3, and 5) or stimulated through their BCR for 3 min (lanes 2, 4, and 6). From the cleared lysates of these cells, anti–SLP-65 immunoprecipitates (IP) were prepared and analyzed by anti-pTyr immunoblotting. Relative molecular mass of marker protein is indicated on the left in kD.
In LCLs, the Syk/SLP-65 Complex Is Uncoupled from PLC-γ2 Activation.
SLP-65 has been shown to be a specific substrate of activated Syk 16. Consistent with this, we find that Syk and SLP-65 coimmunoprecipitate after BCR ligation of Ramos B cells (Fig. 4, lanes 5 and 6). In LCLs, however, this association is stimulation independent, which explains why SLP-65 is constitutively phosphorylated in these cells (Fig. 4, lanes 1–4). A direct downstream effector protein of phosphorylated SLP-65 is PLC-γ2, which upon dual phosphorylation by Syk and Btk initiates mobilization of Ca2+ ions 16 18 19 20 21. In activated Ramos B cells, PLC-γ2 is tyrosine phosphorylated and coimmunoprecipitates with phosphorylated SLP-65 (Fig. 5, lanes 7 and 8). PLC-γ2 isolated from unstimulated or stimulated LCLs remains almost unphosphorylated (Fig. 5, top panel, lanes 1–6). In some LCLs, a weak association between PLC-γ2 and SLP-65 could be observed (Fig. 5, middle panel, lanes 4–6). The results demonstrate that EBV infection uncouples phosphorylation of Syk and SLP-65 from PLC-γ2 activation. The underlying mechanism for this phenomenon remains to be elucidated but the data provide an explanation for the described block of BCR-induced Ca2+ mobilization in LMP2A-expressing B cells 5. Moreover, our data suggest that in these cells, SLP-65 may be indirectly translocated to the plasma membrane by virtue of its association with phosphorylated Syk bound to the phospho-ITAM of LMP2A.
Figure 4.

Syk and SLP-65 associate in vivo. Anti–SLP-65 immunoprecipitates from unstimulated and BCR-stimulated LCLs (lanes 1–4) or Ramos B cells (lanes 5 and 6) were analyzed by anti-Syk immunoblotting. Relative molecular mass of marker protein is indicated on the left in kD. Note that a constitutive Syk/SLP-65 complex formation was observed in all LCLs that have been investigated (n = 4), but some variability in the amount of Syk present in SLP-65 immunoprecipitates was observed (compare lanes 1 and 4).
Figure 5.

Block of PLC-γ2 phosphorylation in LCLs. Anti–PLC-γ2 immunoprecipitates from unstimulated and BCR-stimulated LCLs (lanes 1–6) or Ramos B cells (lanes 7 and 8) were analyzed by immunoblotting with antibodies to pTyr (top panel), SLP-65 (middle panel), or PLC-γ2 (bottom panel). Relative molecular masses of marker proteins are indicated on the left in kD.
Constitutive SLP-65 Phosphorylation in LCLs Is Accompanied with a Constant Trigger of the CrkL-Cbl-C3G Pathway.
The 38-kD intracellular adaptor protein CrkL consists of one SH2 and two SH3 domains and has been implicated in positive and negative regulation of lymphocyte proliferation 45 46 47 48 49. The coimmunoprecipitation experiment shown in Fig. 6 A (top panel) identifies CrkL as a novel SLP-65–binding protein in vivo. The p68 and p70 isoforms of SLP-65 are detected in anti-CrkL precipitates from stimulated and weakly from unstimulated Ramos B cells (lanes 5 and 6). Also in anti-CrkL precipitates from LCLs, both SLP-65 isoforms are present but their detection is not regulated by BCR engagement (lanes 1–4). SLP-65 could not be detected in anti-Crk II precipitates (data not shown). These data show that EBV infection causes a constitutive complex formation between SLP-65 and the protooncogene product CrkL. The presence of the 68-kD isoform of SLP-65 in anti-CrkL precipitates from LCLs shows that the association is at least in part independent of SLP-65 phosphorylation and hence may involve the CrkL SH3 domain(s).
Figure 6.


Association of the protooncoprotein CrkL with SLP-65. (a) Anti-CrkL immunoprecipitates (IP) were prepared from cleared cellular lysates of untreated or BCR-stimulated LCLs (lanes 1–4) and Ramos B cells (lanes 5 and 6), and analyzed by subsequent immunoblotting with antibodies to SLP-65 (top panel), Cbl (middle panel), and C3G (bottom panel). (b) Anti-pTyr immunoprecipitates from the same cells were analyzed by anti-CrkL or anti-C3G immunoblotting (top and bottom panel, respectively). Relative molecular masses of marker proteins are indicated on the left in kD.
The SH2 and central SH3 domain of CrkL are known to bind the Fyn-associated ubiquitin ligase Cbl and the Rap1-specific guanine nucleotide exchange factor C3G 45 46 47 50 51 52 53. Consistent with previous reports we find an increased association of CrkL with Cbl after BCR ligation of Ramos B cells (Fig. 6a, middle panel, lanes 5 and 6). The amount of Cbl that co-immunoprecipitates with CrkL from two LCLs (lanes 1–4) is almost independent of BCR stimulation in that only a slight increase of complex formation is observed in only one of the two lines (lanes 1 and 2). Similarly, CrkL and C3G coimmunoprecipitate from LCL lysates independent of cellular stimulation (Fig. 6 A, bottom panel, lanes 1–4). After BCR ligation of Ramos B cells, a slight decrease of the anti-C3G signal was observed in some but not in all experiments (lanes 5 and 6). Fig. 6 B shows that CrkL and C3G are inducibly phosphorylated in Ramos B cells (lanes 5 and 6), but are constitutively phosphorylated in LCLs (lanes 1–4). In summary, this set of experiments revealed that LMP2A expression affects not only the phosphorylation status of the p70 SLP-65 isoform but also abolishes the requirement of BCR ligation for a phosphorylation-dependent complex formation to CrkL. The CrkL-Cbl-C3G signaling module seems to be permanently triggered in LCLs.
LCLs Are Unresponsive to Pervanadate/H2O2 Treatment.
To further compare the signaling behavior between EBV-positive and -negative B cells, LCLs and Ramos B cells were treated with the protein tyrosine phosphatase inhibitor pervanadate/H2O2. Subsequently, pTyr-containing proteins were purified with anti-pTyr antibodies. As reported for many cell types, pervanadate/H2O2 treatment of Ramos B cells changes the equilibrium between dephosphorylation and phosphorylation towards the latter and hence induces robust PTK substrate phosphorylation (Fig. 7, top panel, lanes 5 and 6). Unexpectedly, all LCLs tested in this assay were unresponsive in that no pervanadate-mediated increase of protein tyrosine phosphorylation could be observed (top panel, lanes 1–4). Probing of the filter with anti-Syk (middle panel) and anti–SLP-65 antibodies (bottom panel) showed that both signaling proteins could be purified to similar amounts from untreated and pervanadate-treated LCLs (lanes 1–4). In contrast, efficient anti-pTyr immunoprecipitation of Syk and SLP-65 from Ramos B cells required incubation of the cells with pervanadate/H2O2 (lanes 5 and 6). Pervanadate/H2O2 induces not only Syk- or Lyn-specific protein phosphorylation that can be affected by LMP2A. Hence, the data show for the first time that EBV infection renders B cells not only unresponsive to BCR activation but generally abrogates their ability to induce PTK substrate phosphorylation. Whether this is solely mediated by LMP2A remains to be elucidated.
Figure 7.
EBV-positive B cells are unresponsive to treatment with pervanadate/H2O2. Two independent LCL clones (lanes 1–4) and Ramos B cells (lanes 5 and 6) were left untreated (lanes 1, 3, and 5) or incubated with 20 μM pervanadate/H2O2 for 3 min at 37°C (lanes 2, 4, and 6). From the cleared cellular lysates, tyrosine-phosphorylated proteins were purified by immunoprecipitation (IP) and analyzed by subsequent immunoblotting with antibodies to pTyr (top panel), Syk (middle panel), and SLP-65 (bottom panel). Relative molecular masses of marker proteins are indicated on the left in kD.
Discussion
LMP2A reorganizes BCR-regulated survival and activation in EBV-infected B cells 5 6 7 8 32 33 38. Recent biochemical and genetic evidence identified the adaptor SLP-65 as a central BCR effector protein downstream of Syk 15 16 17 18 19 20 21 27 28 29 30 54. Here we have shown that loss of SLP-65 expression abrogates LMP2A-mediated signaling for B cell development and survival in vivo. Analysis of human LCLs provided insight into the molecular mechanism(s) of this phenotype. EBV infection of B cells causes constitutive tyrosine phosphorylation of human SLP-65, primarily of its p70 isoform. Other B cell signaling proteins, such as Ig-α or the SLP-65 effector protein PLC-γ2, remain unphosphorylated even after BCR ligation. Another novel finding is the association of SLP-65 with CrkL, which is regulated by the BCR in normal B cells but not in LCLs. Finally, we have demonstrated that after EBV infection, not only BCR-specific signal transduction but also pervanadate/H2O2-induced PTK substrate phosphorylation is rescinded. Hence, LMP2A expression seems to broadly inhibit the ability of cellular PTKs to respond to cellular stimulation. As a consequence, EBV infection may inhibit ligand-induced signaling from many surface receptors that transmit their activation signal through cellular PTKs. One mechanism to achieve this function might be related to the constitutive targeting of LMP2A into lipid-rafts 31. This may create a microenvironment, which prevents translocation of the activated BCR as well as of other stimulated receptors into the signaling-competent membrane compartment. Experiments are under way to test this directly.
After B cell activation, one of the earliest Syk substrates is the cytoplasmic adaptor protein SLP-65 15 16 43 55. In line with this, we found that Syk and SLP-65 coimmunoprecipitate from lysates of BCR-stimulated Ramos B cells. The stimulation-independent association between Syk and SLP-65 observed in LCLs explains the constitutive SLP-65 phosphorylation in these cells. As described previously, efficient coupling between Syk and SLP-65 is organized by the BCR and Y→F substitutions in the Ig-α ITAM reduce SLP-65 phosphorylation 43 55. The data presented here suggest that LMP2A can substitute this BCR function. Consistent with this, two of the eight tyrosine residues in the N terminus of LMP2A are organized as an ITAM 56, which is essential for LMP2A function in vitro and in vivo 33 40.
Lack of SLP-65 expression leads to a partial block of B cell development in mice and to a complete absence of mature B cells in a patient suffering from severe antibody deficiency 27 28 29 30 54. A nearly identical phenotype is caused by mutations in the btk gene, which lead to X-linked immunodeficiency (XID) in mice and X-linked agammaglobulinemia (XLA) in humans 25 57 58. These results suggest that SLP-65 and Btk function in a common BCR signaling pathway. In support of this, phosphorylated SLP-65 recruits both, PLC-γ 16 18 19 and Btk 20 21 through their SH2 domains. These are critical events for the phosphorylation and activation of PLC-γ allowing the subsequent generation of second messenger (for reviews, see references 14 and 22). In our LCLs, phosphorylation of PLC-γ2 is not detectable, despite the prominent and constitutive phosphorylation of Syk and SLP-65. At present we can only speculate about the mechanism that is responsible for this signaling block, but it may involve an LMP2A-regulated structural organization of proteins that prevents the interaction between PLC-γ2 and the SLP-65 signaling module. Possibly, LMP2A expression excludes PLC-γ2 from entry into lipid-rafts.
In human B cells, the primary slp-65 transcript is alternatively spliced generating the 68-kD and the 70-kD SLP-65 isoforms 16. Interestingly, p68 lacks amino acids 201–225 of p70, which encompass a putative binding motif for SH3 domains. Whether p68 and p70 perform different BCR signaling functions is not known. The selective and constitutive phosphorylation of p70 in LCLs together with the phenotype of TgE SLP-65−/− mice strongly suggest a dominant role of this SLP-65 isoform for the LMP2A-mediated B cell maintenance signal and probably also for the BCR maintenance signal. From our data, however, it is not completely clear whether the CrkL-Cbl-C3G pathway downstream of SLP-65 is involved in signaling cell survival or whether it is involved in establishing B cell anergy that renders EBV-infected B cells unresponsive to BCR ligation. Of the three known Crk isoforms (I, II, and L), particularly CrkL is known to transmit signals from lymphocyte antigen receptors 45 46 47. A positive signaling function of CrkL is suggested by its capacity to transform cells when mutated or overexpressed, and by the finding that CrkL is the dominant phosphoprotein in v-Abl–transformed pre-B cells 49 59. CrkL may also participate in negative regulation of lymphocytes through its SH2-mediated binding to the ubiquitin ligase Cbl 51 52. The constitutive phospho-Cbl/CrkL complex in LCLs could play a role in the ubiquitination-mediated degradation of signaling proteins. Indeed, three Nedd4-like ubiquitin ligases were recently found to directly associate with LMP2A 60 61. Moreover, the structurally unusual SH2 domain of Cbl competes with PLC-γ2 for binding to phosphorylated SLP-65 62, which could also contribute to the observed block of PLC-γ2 phosphorylation in our LCLs. In anergic T cells, which do not transcribe the Il-2 gene upon TCR activation, constitutive phosphorylation of Cbl leads to assembly of a ternary Cbl-CrkL-C3G complex that activates the Rap1 pathway independent of TCR ligation 48 53. Overexpression of activated Rap1 in Jurkat cells recapitulated T cell anergy. The constitutive association of Cbl with CrkL and C3G in LCLs suggests that a similar pathway could operate in EBV-infected B cells to block BCR-mediated activation of the cells. The BCR can activate Rap1 via a PLC-γ–dependent pathway, which may be independent of CrkL and C3G 63.
In EBV-negative B cells, the SLP-65/CrkL complex is regulated by the BCR, indicating that phosphorylated SLP-65 is bound by the CrkL SH2 domain. Consistent with this, the four YXXP phosphorylation motifs of SLP-65 are nearly identical to those found in known CrkL-binding proteins such as Cas (Crk-associated substrate) and Cbl. Similar amino acid sequences were selected by the CrkL SH2 domain from a phosphopeptide library 64. The finding that in LCLs, also the nonphosphorylated p68 isoform of SLP-65 coprecipitates with CrkL, suggests a function for the CrkL SH3 domain(s). At least seven PXXP consensus binding sites for SH3 domains are present in SLP-65 and a low level of SLP-65/CrkL complexes was repeatedly found in unstimulated Ramos B cells. We therefore conclude that the CrkL-derived SH2 and SH3 domains synergistically regulate the interaction with SLP-65 in resting and activated B cells.
In summary, our analysis revealed several novel aspects of LMP2A-mediated reorganization of B cell signaling elements. While a stimulation-dependent PTK substrate phosphorylation becomes generally suppressed, certain signaling pathways emanating from the SLP-65 module are selectively activated to keep EBV infected cells alive. Future studies on the assembly and function of the BCR or LMP2A signalosome will be required to better understand B lymphopoiesis and to finally interfere with it in pathological situations.
Acknowledgments
We are grateful to Ruth Draeger and Drs. Hans Hartmut Peter and Peter J. Nielsen for reagents and helpful comments.
The work was supported by the Deutsche Forschungsgemeinschaft through SFB 388, and the Ministry for Science, Research and Art of Baden-Württemberg through Forschungsschwerpunkt Immundefizienzen. R. Longnecker is supported by Public Health Service grants CA62234 and CA73507 from the National Cancer Institute and DE13127 from the National Institute of Dental and Craniofacial Research.
Footnotes
Abbreviations used in this paper: BCR, B cell antigen receptor; BLNK, B cell linker protein; Btk, Bruton's tyrosine kinase; Cbl, Casitas B lineage lymphoma; Crk, CT10 regulator of kinase; CrkL, Crk like; EBNA, EBV-associated nuclear antigen; GST, glutathione S-transferase; ITAM, immunoreceptor tyrosine-based activation motif; LCL, lymphoblastoid cell line; LMP2A, latent membrane protein 2A; PLC-γ2, phospholipase C-γ2; PTK, protein tyrosine kinase; pTyr, phosphotyrosine; RAG, recombination activating gene; SLP, SH2 domain-containing leukocyte protein; SH2, Src homology 2 domain; WT, wild-type.
References
- Rickinson A.B., Kieff E. Epstein-Barr viruses. In: Fields B.N., Knipe D.M., Howely P.M., editors. Fields Virology. Lippincott-Raven; Philadelphia, PA: 1996. pp. 2397–2446. [Google Scholar]
- Longnecker R. Molecular biology of Epstein-Barr virus. In: McCane D., editor. Human Tumor Viruses. American Society for Microbiology; Washington, D.C.: 1998. pp. 133–172. [Google Scholar]
- Kieff E. Epstein-Barr virus and its replication. In: Fields B.N., Knipe D.M., Howley P.M., editors. Fundamental Virology. Lippincott-Raven; Philadelphia, PA: 1996. pp. 1009–1162. [Google Scholar]
- Longnecker R., Kieff E. A second Epstein-Barr virus membrane protein (LMP2) is expressed in latent infection and colocalizes with LMP1. J. Virol. 1990;64:2319–2326. doi: 10.1128/jvi.64.5.2319-2326.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C.L., Lee J.H., Kieff E. Epstein-Barr virus latent membrane protein 2A blocks calcium mobilization in B lymphocytes. J. Virol. 1993;67:3087–3094. doi: 10.1128/jvi.67.6.3087-3094.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C.L., Lee J.H., Kieff E., Longnecker R. An integral membrane protein (LMP) blocks reactivation of Epstein-Barr virus from latency following surface immunoglobulin crosslinking. Proc. Natl. Acad. Sci. USA. 1994;91:772–776. doi: 10.1073/pnas.91.2.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C.L., Burkhardt A.L., Lee J.H., Stealey B., Longnecker R., Bolen J.B., Kieff E. Integral membrane protein 2 of Epstein-Barr-Virus regulates reactivation from latency through dominant negative effects on protein-tyrosine kinases. Immunity. 1995;2:155–166. doi: 10.1016/s1074-7613(95)80040-9. [DOI] [PubMed] [Google Scholar]
- Longnecker R., Miller C.L. Regulation of Epstein-Barr virus latency by latent membrane protein 2. Trends Microbiol. 1996;4:38–42. doi: 10.1016/0966-842x(96)81504-6. [DOI] [PubMed] [Google Scholar]
- Lam K.P., Kühn R., Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997;90:1073–1083. doi: 10.1016/s0092-8674(00)80373-6. [DOI] [PubMed] [Google Scholar]
- Neuberger M.S. Antigen receptor signaling gives lymphocytes a long life. Cell. 1997;90:971–973. doi: 10.1016/s0092-8674(00)80362-1. [DOI] [PubMed] [Google Scholar]
- Reth, M., and J. Wienands. 1999. The maintenance and the activation signal of the B-cell antigen receptor. Cold Spring Harbor Symposium on Quantitative Biology. Vol. LVIV. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 323–327. [DOI] [PubMed]
- Kurosaki T. Genetic analysis of B cell antigen receptor signaling. Annu. Rev. Immunol. 1999;17:555–592. doi: 10.1146/annurev.immunol.17.1.555. [DOI] [PubMed] [Google Scholar]
- Campbell K.S. Signal transduction from the B cell antigen-receptor. Curr. Opin. Immunol. 1999;11:256–264. doi: 10.1016/s0952-7915(99)80042-9. [DOI] [PubMed] [Google Scholar]
- Wienands J. The B cell antigen receptorformation of signaling complexes and the function of adaptor proteins Curr. Top. Microbiol. Immunol. 245 2000. 53 76a [DOI] [PubMed] [Google Scholar]
- Wienands J., Schweikert J., Wollscheid B., Jumaa H., Nielsen P.J., Reth M. SLP-65A new signaling component in B lymphocytes which requires expression of the antigen receptor for phosphorylation. J. Exp. Med. 1998;188:791–795. doi: 10.1084/jem.188.4.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu C., Turck C.W., Kurosaki T., Chan A.C. BLNKA central linker protein in B cell activation. Immunity. 1998;9:93–103. doi: 10.1016/s1074-7613(00)80591-9. [DOI] [PubMed] [Google Scholar]
- Goisuka R., Fujimura Y.-I., Mamada H., Umeda A., Morimura T., Uetsuka K., Doi K., Tsuji S., Kitamura D. BASH, a novel signaling molecule preferentially expressed in B cells of the Bursa of Fabricius. J. Immunol. 1998;161:5804–5808. [PubMed] [Google Scholar]
- Ishiai M., Kurosaki M., Pappu R., Okawa K., Ronko I., Fu C., Shibata M., Iwamatsu A., Chan A.C., Kurosaki T. BLNK required for coupling Syk to PLCγ2 and Rac1-JNK in B cells. Immunity. 1999;10:1–20. doi: 10.1016/s1074-7613(00)80012-6. [DOI] [PubMed] [Google Scholar]
- Ishiai M., Sugawara H., Kurosaki M., Kurosaki T. Association of phospholipase C-γ 2 Src homology 2 domain with BLNK is critical for B cell antigen receptor signaling. J. Immunol. 1999;163:1746–1749. [PubMed] [Google Scholar]
- Su Y.-W., Zhang Y., Schweikert J., Koretzky G.A., Reth M., Wienands J. Interaction of SLP adaptors with the SH2 domain of Tec family kinases. Eur. J. Immunol. 1999;29:3702–3711. doi: 10.1002/(SICI)1521-4141(199911)29:11<3702::AID-IMMU3702>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Hashimoto S., Iwamatsu A., Ishiai M., Okawa K., Yamadori T., Matsushita M., Baba Y., Kishimoto T., Kurosaki T., Tsukada S. Identification of the SH2 domain binding protein of Bruton's tyrosine kinase as BLNKFunctional significance of Btk-SH2 domain in B-cell antigen receptor-coupled calcium signaling. Blood. 1999;94:2357–2364. [PubMed] [Google Scholar]
- Kurosaki T., Tsukada S. BLNKConnecting Syk and Btk to calcium signals. Immunity. 2000;12:1–5. doi: 10.1016/s1074-7613(00)80153-3. [DOI] [PubMed] [Google Scholar]
- Torres R.M., Flaswinkel H., Reth M., Rajewsky K. Aberrant B cell development and immune response in mice with a compromised BCR complex. Science. 1996;272:1804–1808. doi: 10.1126/science.272.5269.1804. [DOI] [PubMed] [Google Scholar]
- Reichlin A., Hu Y., Meffre E., Nagaoka H., Gong S., Kraus M., Rajewsky K., Nussenzweig M.C. B cell development is arrested at the immature B cell stage in mice carrying a mutation in the cytoplasmic domain of immunoglobulin β. J. Exp. Med. 2001;193:13–23. doi: 10.1084/jem.193.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.I., Islam K.B., Vorechovsky I., Olerup O., Wallin E., Rabbani H., Baskin B., Hammarstrom L. X-linked agammaglobulinemia and other immunoglobulin deficiencies. Immunol. Rev. 1994;138:159–183. doi: 10.1111/j.1600-065x.1994.tb00851.x. [DOI] [PubMed] [Google Scholar]
- Wienands J. Signal transduction elements of the B cell antigen receptor and their role in immunodeficiencies Immunobiology. 202 2000. 120 133b [DOI] [PubMed] [Google Scholar]
- Pappu R., Cheng A.M., Li B., Gong Q., Chiu C., Griffin N., White M., Sleckman B.P., Chan A. Requirement for B cell linker protein (BLNK) in B cell development. Science. 1999;286:1949–1954. doi: 10.1126/science.286.5446.1949. [DOI] [PubMed] [Google Scholar]
- Jumaa H., Wollscheid B., Mitterer M., Wienands J., Reth M., Nielsen P.J. Abnormal development and function of B-lymphocytes in mice lacking the signaling adaptor protein SLP-65. Immunity. 1999;11:547–554. doi: 10.1016/s1074-7613(00)80130-2. [DOI] [PubMed] [Google Scholar]
- Xu S., Tan J.E., Wong E.P., Manickam A., Ponniah S., Lam K. B cell development and activation defects resulting in xid-like immunodeficiency in BLNK/SLP-65-deficient mice. Int. Immunol. 2000;12:397–404. doi: 10.1093/intimm/12.3.397. [DOI] [PubMed] [Google Scholar]
- Hayashi K., Nittono R., Okamoto N., Tsuji S., Hara Y., Goitsuka R., Kitamura D. The B cell-restricted adaptor BASH is required for normal development and antigen receptor-mediated activation of B cells. Proc. Natl. Acad. Sci. USA. 2000;97:2755–2760. doi: 10.1073/pnas.040575697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykstra M.L., Longnecker R., Pierce S. Epstein-Barr virus coopts lipid rafts to block the signaling and antigen transport functions of the BCR. Immunity. 2001;14:57–67. doi: 10.1016/s1074-7613(01)00089-9. [DOI] [PubMed] [Google Scholar]
- Fruehling S., Lee S., Herrold R., Frech B., Laux G., Kremmer E., Grässer F.A., Longnecker R. Identification of latent membrane protein 2A (LMP2A) domains essential for the LMP2A dominant-negative effect on B-lymphocyte surface immunoglobulin signal transduction. J. Virol. 1996;70:6216–6226. doi: 10.1128/jvi.70.9.6216-6226.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruehling S., Longnecker R. The immunoreceptor tyrosine-based activation motif of Epstein-Barr virus LMP2A is essential for blocking BCR-mediated signal transduction. Virology. 1997;235:241–251. doi: 10.1006/viro.1997.8690. [DOI] [PubMed] [Google Scholar]
- Longnecker R., Druker B., Roberts T.M., Kieff E. An Epstein-Barr virus protein associated with cell growth transformation interacts with a tyrosine kinase. J. Virol. 1991;65:3681–3692. doi: 10.1128/jvi.65.7.3681-3692.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt A.L., Bolen J.B., Kieff E., Longnecker R. An Epstein-Barr virus transformation-associated membrane protein interacts with Src family tyrosine kinases. J. Virol. 1992;66:5161–5167. doi: 10.1128/jvi.66.8.5161-5167.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alber G., Kim K.-M., Weiser P., Riesterer C., Carsetti R., Reth M. Molecular mimicry of the antigen receptor signalling motif by transmembrane proteins of the Epstein-Barr virus and the bovine leukaemia virus. Curr. Biol. 1993;3:333–339. doi: 10.1016/0960-9822(93)90196-u. [DOI] [PubMed] [Google Scholar]
- Beaufils P., Choquet D., Mamoun R.Z., Malissen B. The (YXXL/I)2 signalling motif found in the cytoplasmic segments of the bovine leukaemia virus envelope protein and Epstein-Barr virus latent membrane protein 2A can elicit early and late lymphocyte activation. EMBO J. 1993;12:5105–5112. doi: 10.1002/j.1460-2075.1993.tb06205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell R.G., Wilson J.B., Anderson S.J., Longnecker R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity. 1998;9:405–411. doi: 10.1016/s1074-7613(00)80623-8. [DOI] [PubMed] [Google Scholar]
- Caldwell R.G., Brown R.C., Longnecker R. Epstein-Barr virus LMP2A-induced B-cell survival in two unique classes of EμLMP2A transgenic mice. J. Virol. 2000;74:1101–1113. doi: 10.1128/jvi.74.3.1101-1113.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant M.C., Longnecker R. The LMP2A ITAM is essential for providing B cells with developmental and survival signals in vivo. J. Virol. 2000;74:9115–9124. doi: 10.1128/jvi.74.19.9115-9124.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein G., Giovanelli B., Westman A., Stehlin J.S., Mumford D. An EBV-genome-negative cell line established from an American Burkitt lymphoma; receptor characteristics. EBV infectibility and permanent conversion into EBV-positive sublines by in vitro infection. Intervirology. 1975;5:319–334. doi: 10.1159/000149930. [DOI] [PubMed] [Google Scholar]
- Wienands J., Freuler F., Baumann G. Tyrosine-phosphorylated forms of Ig-β, CD22, TCR-ζ and HOSS are major ligands for tandem SH2 domains of Syk. Int. Immunol. 1995;7:1701–1708. doi: 10.1093/intimm/7.11.1701. [DOI] [PubMed] [Google Scholar]
- Wienands J., Larbolette O., Reth M. Evidence for a preformed transducer complex organized by the B cell antigen receptor. Proc. Natl. Acad. Sci. USA. 1996;93:7865–7870. doi: 10.1073/pnas.93.15.7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann G., Maier D., Freuler F., Tschopp C., Baudisch K., Wienands J. In vitro characterization of major ligands for Src homology 2 domains derived from tyrosine kinases, from the adaptor protein SHC and from GTPase-activating protein in Ramos B cells. Eur. J. Immunol. 1994;24:1799–1807. doi: 10.1002/eji.1830240812. [DOI] [PubMed] [Google Scholar]
- Ingham R.J., Krebs D.L., Barbazuk S.M., Turck C.W., Hirai H., Matsuda M., Gold M.R. B cell antigen receptor signaling induces the formation of complexes containing the Crk adapter proteins. J. Biol. Chem. 1996;271:32306–32314. doi: 10.1074/jbc.271.50.32306. [DOI] [PubMed] [Google Scholar]
- Panchamoorthy G., Fukazawa T., Miyake S., Soltoff S., Reedquist K., Druker B., Shoelson S., Cantley L., Band H. p120cbl is a major substrate of tyrosine phosphorylation upon B cell antigen receptor stimulation and interacts in vivo with Fyn and Syk tyrosine kinases, Grb-2 and Shc adaptors, and the p85 subunit of phosphytidylinositol 3-kinase. J. Biol. Chem. 1996;271:3187–3194. doi: 10.1074/jbc.271.6.3187. [DOI] [PubMed] [Google Scholar]
- Smit L., van der Horst G., Borst J. Sos, Vav, and C3G participate in B cell receptor-induced signaling pathways and differentially associate with Shc-Grb2, and Crk-L adaptors. J. Biol. Chem. 1996;271:8564–8569. doi: 10.1074/jbc.271.15.8564. [DOI] [PubMed] [Google Scholar]
- Boussiotis V.A., Freeman G.J., Berezovskaya A., Barber D.L., Nadler L.M. Maintenance of human T cell anergyblocking of Il-2 gene transcription by activated Rap1. Science. 1997;278:124–127. doi: 10.1126/science.278.5335.124. [DOI] [PubMed] [Google Scholar]
- Sattler M., Salagia R. Role of the adapter protein CrkL in signal transduction of normal hematopoietic and BCR/Abl-transformed cells. Leukemia. 1998;12:637–644. doi: 10.1038/sj.leu.2401010. [DOI] [PubMed] [Google Scholar]
- Reedquist K.A., Fukazawa T., Panchamoorthy G., Langdon W.Y., Shoelson S.E., Druker B.J., Band H. Stimulation through the T cell receptor induces Cbl association with Crk proteins and the guanine nucleotide exchange protein C3G. J. Biol. Chem. 1996;271:8435–8442. doi: 10.1074/jbc.271.14.8435. [DOI] [PubMed] [Google Scholar]
- Luther M.L.J., Rao N., Eck M.J., Band H. The Cbl proto-oncoproteina negative regulator of immune receptor signal transduction. Immunol. Today. 1999;20:375–382. doi: 10.1016/s0167-5699(99)01484-x. [DOI] [PubMed] [Google Scholar]
- Rudd C.E., Schneider H. Cbl sets the threshold for autoimmunity. Curr. Biol. 2000;10:344–347. doi: 10.1016/s0960-9822(00)00463-2. [DOI] [PubMed] [Google Scholar]
- Henning S.W., Cantrell D.A. GTPases in antigen receptor signalling. Curr. Opin. Immunol. 1998;10:322–329. doi: 10.1016/s0952-7915(98)80171-4. [DOI] [PubMed] [Google Scholar]
- Minegishi Y., Rohrer J., Coustan-Smith E., Lederman H.M., Pappu R., Campana D., Chan A.C., Conley M.E. An essential role for BLNK in human B cell development. Science. 1999;286:1954–1957. doi: 10.1126/science.286.5446.1954. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Wienands J., Zurn C., Reth M. Induction of the antigen receptor expression on B lymphocytes results in rapid competence for signaling of SLP-65 and Syk. EMBO J. 1998;17:7304–7310. doi: 10.1093/emboj/17.24.7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reth M. Antigen receptor tail clue. Nature. 1989;338:383. doi: 10.1038/338383b0. [DOI] [PubMed] [Google Scholar]
- Khan W.N., Alt F.W., Gerstein R.M., Malynn B.A., Larsson I., Rathbun G., Davidson L., Müller S., Kantor A.B., Herzenberg L.A. Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3:283–299. doi: 10.1016/1074-7613(95)90114-0. [DOI] [PubMed] [Google Scholar]
- Kerner J.D., Appleby M.W., Mohr R.N., Chien S., Rawlings D.J., Maliszewski C.R., Witte O.N., Perlmutter R.M. Impaired expansion of mouse B cell progenitors lacking Btk. Immunity. 1995;3:301–312. doi: 10.1016/1074-7613(95)90115-9. [DOI] [PubMed] [Google Scholar]
- Feller S.M., Posern G., Voss J., Kardinal C., Sakkab D., Zheng J., Knudsen B.S. Physiological signals and oncogenesis mediated through Crk family adapter proteins. J. Cell. Physiol. 1998;177:535–552. doi: 10.1002/(SICI)1097-4652(199812)177:4<535::AID-JCP5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Winberg G., Matskova L., Chen F., Plant P., Rotin D., Gish G., Ingham R., Ernberg I., Pawson T. Latent membrane protein 2A of Epstein-Barr virus binds WW domain E3 protein-ubiquitin ligases that ubiquitinate B-cell tyrosine kinases. Mol. Cell. Biol. 2000;20:8526–8535. doi: 10.1128/mcb.20.22.8526-8535.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M., Ikeda A., Longan L.C., Longnecker R. The Epstein-Barr virus latent membrane protein 2A PY motif recruits WW domain-containing ubiquitin-protein ligases. Virology. 2000;268:178–191. doi: 10.1006/viro.1999.0166. [DOI] [PubMed] [Google Scholar]
- Yasuda T., Maeda A., Kurosaki M., Tezuka T., Hironaka K., Yamamoto T., Kurosaki T. Cbl suppresses B cell receptor-mediated phospholipase (PLC) γ activation by regulating B cell linker protein-PLC-γ2 binding. J. Exp. Med. 2000;191:641–650. doi: 10.1084/jem.191.4.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod S.J., Ingham R.J., Bos J.L., Kurosaki T., Gold M.R. Activation of the Rap1 GTPase by the B cell antigen receptor. J. Biol. Chem. 1998;273:29218–29223. doi: 10.1074/jbc.273.44.29218. [DOI] [PubMed] [Google Scholar]
- Songyang Z., Shoelson S.E., Chaudhuri M., Gish G., Pawson T., Haser W.G., King F., Roberts T., Ratnofsky S., Lechleider R.J. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]