

Abstract
NK T cells are a unique subset of T cells that recognize lipid antigens presented by CD1d. After activation, NK T cells promptly produce large amounts of cytokines, which may modulate the upcoming immune responses. Previous studies have documented an association between decreased numbers of NK T cells and the progression of some autoimmune diseases, suggesting that NK T cells may control the development of autoimmune diseases. To investigate the role of NK T cells in autoimmune diabetes, we crossed CD1 knockout (CD1KO) mutation onto the nonobese diabetic (NOD) genetic background. We found that male CD1KO NOD mice exhibited significantly higher incidence and earlier onset of diabetes compared with the heterozygous controls. The diabetic frequencies in female mice showed a similar pattern; however, the differences were less profound between female CD1KO and control mice. Early treatment of NOD mice with α-galactosylceramide, a potent NK T cell activator, reduced the severity of autoimmune diabetes in a CD1-dependent manner. Our results not only suggest a protective role of CD1-restricted NK T cells in autoimmune diabetes but also reveal a causative link between the deficiency of NK T cells and the induction of insulin-dependent diabetes mellitus.
Keywords: CD1, NK T cells, autoimmunity, diabetes, cytokines
Introduction
Nonobese diabetic (NOD) mice, which develop autoimmune (type I) diabetes spontaneously, have long been used as animal models to study the mechanisms involved in insulin-dependent diabetes mellitus (IDDM) in humans 1. In female mice, inflammation of the pancreatic islets (insulitis) usually begins at ∼3–5 wk of age, eventually leading to β cell destruction and overt diabetes by 4–6 mo of age. Both CD4+ T cells, especially the Th1 subset, which preferentially secretes IFN-γ and TNF-α, and CD8+ T cells have been implicated in the pathogenesis of IDDM 2 3 4 5. In parallel with these effector cells, the presence of regulatory T cells in prediabetic NOD mice that can block the development of IDDM has been suggested by adoptive transfer studies. These include CD4+CD62L+ thymocytes 6, CD4+CD25+ splenocytes 7, and NK T cells 8 9 10. Although the mechanisms of action of the regulatory T cells are not fully understood, it is believed that an imbalance between autoreactive effector T cells and regulatory T cells may trigger the development of destructive insulitis and IDDM.
NK T cells are a unique subset of T cells that coexpress receptors of the NK lineage and α/β TCR. Most NK T cells express an invariant TCR α chain encoded by the Vα14Jα281 gene segment, paired preferentially to Vβ8, Vβ7, and Vβ2 chain 11. Vα14+ NK T cells recognize lipid antigens presented by the MHC class I–like molecule CD1d 12. Similar subsets of T cells are also present in humans 13. CD1d-restricted NK T cells localize preferentially to thymus and liver and are mainly of CD4+ or CD4−CD8− phenotype 14. Upon TCR stimulation, NK T cells promptly produce large amounts of cytokines, including IL-4 and IFN-γ 11. In addition, NK T cells have cytotoxic effects to certain targets, such as tumor cells and immature thymocytes 15 16. Although the physiological ligand for CD1-restricted NK T cells is unknown, α-galactosylceramide (α-GalCer), a glycolipid isolated from marine sponges that specifically binds CD1d, has been shown to be a potent activator for Vα14+ NK T cells 17. After injection of α-GalCer into mice, the activation of NK T cells results in rapid production of cytokines, which leads to activation of cell types involved in both innate immunity (NK cells and dendritic cells) and adaptive immunity (T cells and B cells) 18 19 20 21. Recent studies indicated that NK T cells may be involved in various immune responses, including tumor rejection 22, early protection of infectious agents 23 24, and regulation of autoimmune diseases 25 26.
The potential role of NK T cells in autoimmune diabetes is suggested by the finding that the number and function of NK T cells is diminished in the thymus and peripheral lymphoid organs of young NOD mice 8 25 27. In particular, anti-CD3–induced IL-4 secretion is barely detectable in young NOD mice 27. Adoptive transfer of cell populations enriched for NK T cells prevents IDDM in NOD recipients 8 9 10. Thus, a relative lack of IL-4 production by NK T cells is associated with and might be causal to the onset of IDDM. This notion is further supported by the observation that the frequency of IL-4–producing NK T cells is decreased in the peripheral blood of patients with IDDM 28.
In this study, we directly addressed the question of whether functional deficiency in NK T cells has a causative effect on the onset of IDDM by comparing the cumulative incidence of diabetes and progression of insulitis in CD1-deficient NOD mice with CD1-heterozygous controls. We also examined the effect of α-GalCer treatment on the development of diabetes in NOD and CD1-deficient NOD mice. We found that lack of CD1-restricted NK T cells promotes the development of diabetes, whereas activation of Vα14+ NK T cells by α-GalCer suppresses disease in the NOD model. Our data suggests that NK T cells could be used as therapeutic targets to modulate autoimmune diabetes.
Materials and Methods
Generation and Genotyping of CD1-deficient NOD Mice.
Specific pathogen–free NOD/Lt and B6 mice were purchased from The Jackson Laboratory. In our NOD colony, the incidence of spontaneous diabetes at 24 wk is ∼70% in females and 20% in males. CD1-deficient mice were generated by homologous recombination in our laboratory as described previously 29 and were backcrossed up to nine generations onto NOD/Lt background and then intercrossed to generate CD1+/− and CD1 knockout (CD1KO) NOD mice. The CD1 genotype was determined with PCR using the following three primers: CD1-for, 5′-ATGTTTGATGGCTGCCTCTGCTCACTG-3′; CD1-rev, 5′-TGAGTACAGAGAAGCCAGTGGCCTGGT-3′; and neo-, 5′-CCAAGTGCCCAGCGGGGCTGCTAAAG-3′, which yields a 150-bp fragment for the mutated allele and 330-bp fragment for the wild-type allele. At the third backcross generation, three diabetes susceptibility loci, MHC I-A g7 (for Idd1), D3Mit5 (for Idd3), and D3Nds7 (for Idd10), were screened by PCR as described by Lenschow et al. 30. The specific primers used are as follows: for I-A g7, 5′-TGTCTTTTCTGTCACCCTAGAACA-3′ and 5′-CTCTTCAGGCTGGGATGCTCCAC-3′; for D3Mit5, 5′-AGCCCCTTCCAAGTGTCTCT-3′ and 5′-GGTTTCGGAATGAGATGAGC-3′; and for D3Nds7, 5′-TGCTCCTCACCAGTCATGC-3′ and 5′-TAGTGTTTGCCATGGCTCTC-3′. The PCR products were resolved on a 4% agarose gel. The CD1+/− mice homozygous for the NOD allele in I-A g7 and D3Mit5 loci were selected for further backcross.
Histopathology.
Pancreatic tissues were harvested at various time points, fixed with 10% buffered formalin, and embedded in paraffin. Four serial 5-μm sections were cut at intervals of 100 μm and stained with hematoxylin and eosin. A minimum of 30 islets from each animal was examined. The severity of insulitis was determined and graded using the following criteria according to the magnitude of mononuclear cell infiltration: grade 0, no mononuclear cell infiltration; grade 1, periinsulitis, mononuclear cells surrounding islets and ducts but no intraislet infiltration; grade 2, mononuclear cell infiltration in and around the islet, but intraislet infiltration in <20% of the islet area; grade 3, intraislet mononuclear cell infiltration in 20–50% of the islet area; grade 4, extensive intraislet infiltration occupying >50% of the islet area.
Assessment of Diabetes.
Starting at 10 wk of age, the urinary glucose analysis was performed every 2 wk with Diastix® reagent strips (Bayer Corporation). Hyperglycemia was confirmed by blood glucose determination with ONE-TOUCH® test strips (LifeScan, Inc.). Animals were considered diabetic after their urine glucose levels were >500 mg/dl for two consecutive days and their blood glucose levels were >250 mg/dl.
In Vivo α-GalCer Treatment.
A synthetic form of α-GalCer, KRN7000, was used to treat NOD and CD1KO NOD mice. At 3 wk of age, the mice in the experiment group were injected twice per week intraperitoneally with 2 μg per mouse of KRN7000 in 200 μl of 0.025% polysorbate-20 in PBS, and the control mice were injected with 0.025% polysorbate-20 in PBS.
In Vitro Activation of Lymphocytes and Analysis of Cytokine Production.
CD8+ T cells and B cells were depleted from the splenocytes of CD1KO NOD or CD1+/− NOD mice by incubating cells with CD8α–FITC and B220–FITC (PharMingen) and then incubated with avidin–magnetic beads and applied to magnetic separation (PerSeptive Biosystems). The CD4+-enriched cells (5 × 105 cells per well) were stimulated with either 100 ng/ml of α-GalCer or with an anti-CD3 (2C11) coated 96-well plate in a final volume of 200 μl of RPMI 1640 medium (supplemented with 10% FCS, 2 mM l-glutamine, 20 μM 2-ME, and 100 U/ml penicillin/streptomycin). After 48 h, the culture supernatants were harvested, and the levels of IL-4 and IFN-γ were quantitated by ELISA (PharMingen).
Statistical Analysis.
The time course of spontaneous diabetes onset was analyzed using the Kaplan Meier curve. The curves were tested for significance by log-rank test. Mean values for cytokine levels and degree of insulitis were compared using the unpaired Student's t test. Statistical significance was considered to be P < 0.05. All statistical analyses were performed with the Prism® program (GraphPad Software, Inc.).
Results
Increased Incidence of Diabetes in CD1-deficient NOD Mice.
The CD1KO mutation replaced a 12-kb DNA fragment encompassing exon 4–6 from both CD1d1 and CD1d2 with a neomycin-resistant cassette, which resulted in a complete loss of CD1 expression and impaired NK T cell development in homozygous mutant animals 29. We crossed the CD1KO mutation onto the NOD/Lt background to investigate the role of CD1-restricted NK T cells in the regulation of autoimmune diabetes. The development of diabetes in CD1KO NOD and control mice (from six to nine generations backcrossed) was monitored at biweekly intervals beginning after 10 wk of age. As seen in Fig. 1, >50% of the male CD1KO NOD mice were hyperglycemic by 40 wk of age, as compared with only 20% of the male CD1+/− NOD mice. Nearly 15% of the male CD1KO NOD mice developed diabetes by 20 wk of age, while all the CD1+/− controls were diabetes free within the same period. This data suggests that CD1 deficiency not only results in an increased incidence of diabetes but also accelerates the onset of diabetes in male NOD mice (P < 0.05; n = 51 and 43 for CD1KO and CD1+/−, respectively). Although the female CD1KO NOD mice also displayed a slightly increased incidence of diabetes over a period of 32 wk (P < 0.05; n = 43 and 41 for CD1KO and CD1+/−, respectively), we observed no significant differences in the onset of diabetes between the female CD1KO NOD and CD1+/− controls.
Figure 1.
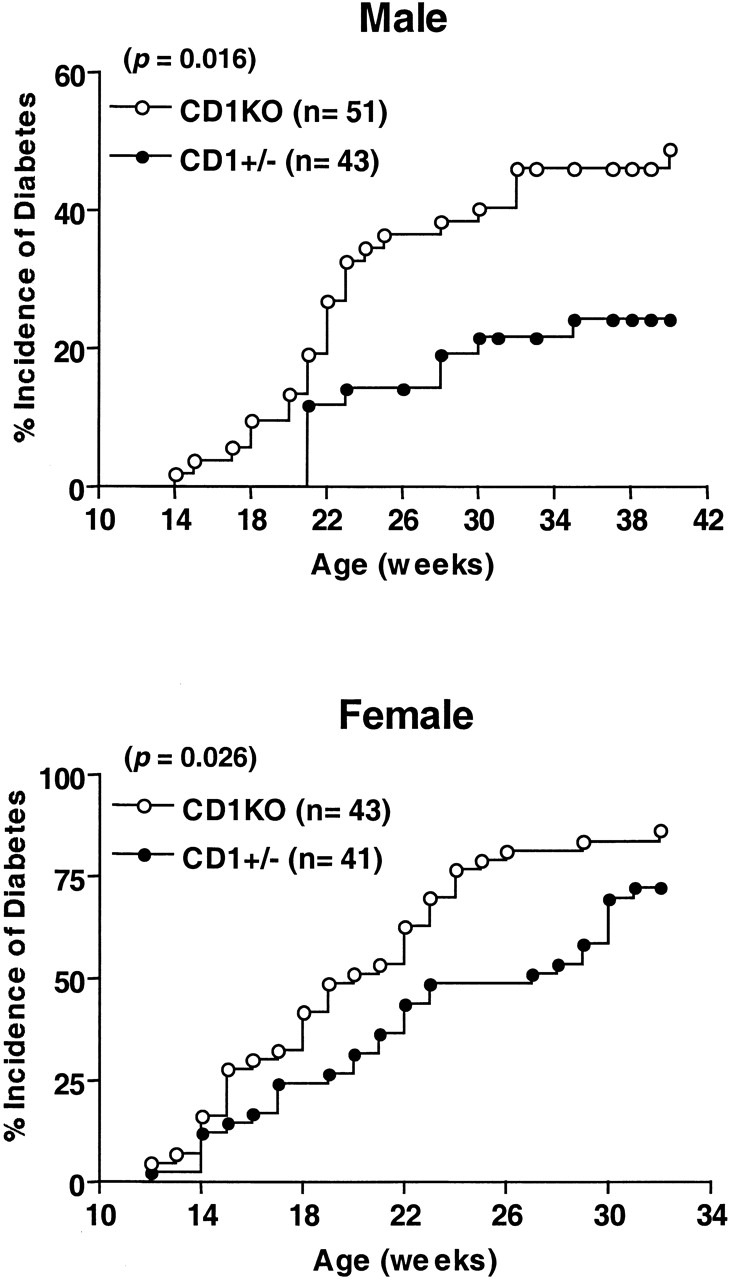
Increased onset and incidence of diabetes in CD1KO NOD mice. CD1KO mice were backcrossed six to nine generations onto NOD/Lt background and then intercrossed to generate CD1+/− and CD1KO NOD mice. Beginning at 10 wk of age, the urine glucose levels of NOD mice were monitored every 2 wk. Male (top) and female (bottom) mice were considered to have developed diabetes if two consecutive glucosuria levels were >500 mg/dl and blood glucose levels were >250 mg/dl. ○, homozygous CD1-deficient NOD mice; •, CD1 heterozygous NOD littermate mice.
A hallmark of the NOD mouse model for IDDM is the progressive infiltration of mononuclear cells into the pancreatic islets. In our wild-type NOD colony, insulitis becomes noticeable at 5–6 wk of age in females and 10–12 wk of age in males. To quantitate the development of insulitis, 30–40 islets from individual CD1KO NOD and heterozygous control mice (nine generations backcrossed) were assessed for the severity of insulitis (Fig. 2). In the male CD1+/− NOD mice, almost 70% of islets are free of infiltrating cells at 12 wk of age, at which time <40% of islets from CD1KO NOD mice are free of insulitis (n = 5, P < 0.05). At 16 wk of age, 35–45% of islets from CD1+/− controls still have no detectable insulitis, while only 10–15% of islets from CD1KO NOD male mice are free of infiltrating cells (n = 4, P < 0.05). In addition, the percentages of islets with heavy infiltrates (grades 3 and 4) were also significantly greater in CD1KO groups. In contrast, we found no significant difference in the degree of insulitis between female CD1KO NOD mice and their heterozygous littermate controls at 6, 9, and 12 wk of age (Fig. 2).
Figure 2.

Histopathologic examination of pancreatic insulitis in CD1KO NOD and CD1+/− NOD littermates. Pancreatic tissues were taken at serial time points. After hematoxylin and eosin staining, a minimum of 30 islets from each mouse was examined. The severity of insulitis was determined and graded semiquantitatively using the criteria as described in Materials and Methods. Data shown represent mean of four to five animals in each age group.
Reduced IL-4 and IFN-γ Secretion in CD1-deficient NOD Mice.
Cytokine imbalance has been attributed to be one of the mechanisms to trigger the development of autoimmune diabetes 31. To investigate whether lack of CD1-restricted NK T cells affects the balance between Th1 and Th2 cells in NOD mice, we analyzed the cytokine profile of CD4+ T cells from CD1KO and control littermates upon anti-CD3 stimulation in vitro. As shown in Fig. 3 A, splenic CD4+ T cells from both male and female CD1KO NOD mice produced significantly lower quantities of IL-4 compared with heterozygous control mice in all of the age groups (n = 5, P < 0.05). We found considerable variability in the levels of IFN-γ produced in individual mice with no statistical significance between CD1KO and heterozygous littermate controls (Fig. 3 A). Thus, lack of IL-4 secreted by NK T cells did not promote the Th1 response in CD1KO NOD mice. Nevertheless, the ratios of IFN-γ/IL-4 are consistently greater in CD1KO NOD mice than those in the control mice (Fig. 3 B), which might be an important factor in controlling the IDDM.
Figure 3.


IL-4 and IFN-γ production by splenic T cells from CD1+/− and CD1KO NOD mice. (A) The CD4+-enriched cells (5 × 105 cells per well) from the indicated mice were cultured with immobilized anti-CD3 (10 μg/ml). Supernatants were harvested 48 h later and assayed for IL-4 (top) and IFN-γ (bottom) by ELISA. (B) The ratio of IFN-γ/IL-4 produced by T cells from CD1KO NOD and littermate controls. Data shown represent mean ± SE of four to five mice in each group.
α-GalCer Treatment Delays the Onset and Reduces the Incidence of Diabetes in NOD Mice. α-GalCer has been shown to be a potent immunostimulator for CD1-restricted NK T cells both in vivo and in vitro 17. As NOD mice have diminished NK T cell activity compared with other strains of mice, we examined whether α-GalCer can activate NK T cells from NOD mice. Fig. 4 A shows that splenic T cells from NOD mice secrete both IL-4 and IFN-γ upon stimulation with α-GalCer, although the response is significantly weaker than that in B6 mice. To determine the effect of enhanced NK T cell activity on the development of spontaneous autoimmune diabetes, NOD mice were treated, starting at 3 wk of age, with α-GalCer or vehicle control. Treating NOD mice with α-GalCer significantly reduced the cumulative incidence of diabetes at the age of 32 wk (from 80 to 40%, n = 15, P < 0.05; Fig. 4 B). There was also a delayed onset of diabetes in α-GalCer–treated NOD mice. None of the α-GalCer–treated mice developed diabetes before 16 wk of age, whereas nearly 30% of vehicle-treated NOD mice had already developed diabetes. In contrast, treating CD1KO NOD mice with α-GalCer did not have any effect on the development of diabetes. Therefore, the effect of α-GalCer on the development of diabetes is strictly CD1 dependent, presumably mediated through CD1-restricted NK T cells.
Figure 4.

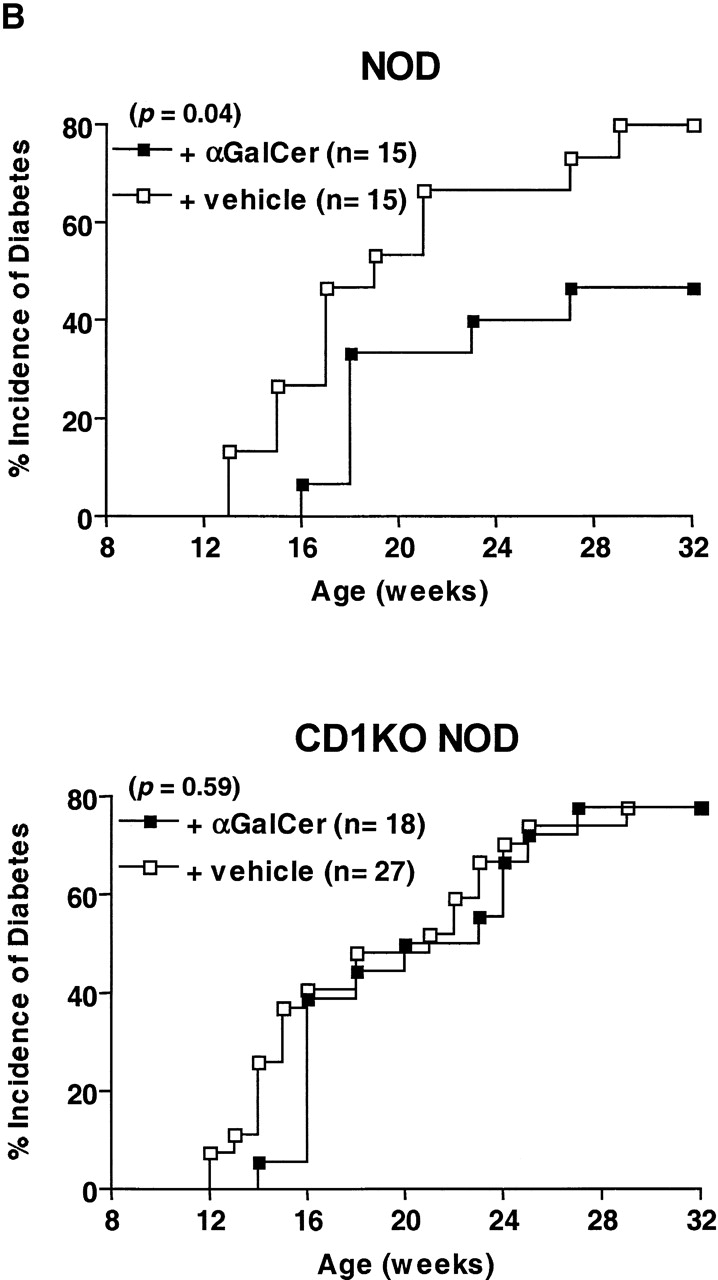

α-GalCer protects NOD mice from developing IDDM. (A) In vitro activation of spleen cells from NOD and B6 mice with α-GalCer. Spleen cells (5 × 105 cells per well) from the indicated mice were cultured with 100 ng/ml of α-GalCer. 2 d later, cytokine production in the culture supernatant was measured by ELISA. Spleen cells cultured with vehicle (0.1% DMSO) did not secrete detectable amounts of IFN-γ and IL-4 (<1 U/ml). (B) The effect of α-GalCer on the incidence of diabetes. Female NOD (top) and CD1KO NOD (bottom) mice were injected intraperitoneally twice per week with α-GalCer (▪) or 0.025% polysorbate-20 vehicle (□) as described in Materials and Methods. Starting at 10 wk of age, diabetes was monitored by measurements of urine and blood glucose levels. (C) The effect of α-GalCer on the insulitis of NOD mice. Female NOD mice were treated with α-GalCer or 0.025% polysorbate vehicle starting at 3 wk of age, as described in Materials and Methods. Pancreata were microscopically evaluated for the degree of insulitis as described above. Data shown represent the mean of five animals in each group.
To examine the effect of α-GalCer treatment on prediabetic islet infiltration, we analyzed hematoxylin and eosin–stained sections of pancreata from α-GalCer– and vehicle-treated mice at weeks 3, 6, and 9 after the start of treatment. As shown in Fig. 4 C, significantly greater percentages of islets were free of insulitis from α-GalCer–treated mice than those from the control mice at all three time points (n = 5, P < 0.05). Furthermore, the degree of insulitis in α-GalCer–treated mice is less severe than that in the vehicle-treated mice. Thus, activation of NK T cells by α-GalCer suppressed the inflammatory infiltration of the islets in NOD mice, which could subsequently delay the onset and reduce the incidence of diabetes.
Discussion
As in human IDDM, diabetes development in NOD mice is controlled by a number of disease susceptibility or resistance genes (Idds). The CD1 gene is close to the Idd3 and Idd10 loci in murine chromosome 3 32. Based on the PCR typing (data not shown), all of the mice (CD1 KO and CD1+/−) used in this study were homozygous for the NOD allele in Idd3 locus. The CD1 gene is tightly linked with Idd10. The CD1+/− mice used in this study are heterozygous for the Idd10 locus, while CD1KO mice retain the 129 allele. As CD1KO NOD mice contain one less Idd-susceptible allele than controls, it is unlikely that the increased susceptibility to diabetes is due to a linked gene carried along during the backcross. Our observation of the acceleration of autoimmune disease in CD1KO NOD mice suggested that CD1-restricted NK T cells play a role in resistance to diabetes in NOD mice even though NK T cell function is genetically diminished relative to that in other strains of mice 25. Activation of NK T cells with α-GalCer inhibited IDDM development in NOD but not CD1KO NOD mice, which further supports the protective role of CD1-restricted NK T cells in autoimmune diabetes.
In the NOD model, there exists a remarkable gender disparity in the cumulative incidence of diabetes, indicating that female NOD mice might have a greater deficit in various immunoregulatory mechanisms. Therefore, lack of functional activity from NK T cells, one out of many immunoregulatory mechanisms, appears to have less profound effect on the progression of diabetes in female NOD mice than in male NOD mice. However, we cannot eliminate the possibility that NK T cells may play a more dominant role in regulating the IDDM in male NOD mice.
There are several distinct mechanisms that may explain why lack of NK T cells in CD1KO NOD mice would render them more susceptible to autoimmune disease. First, lower levels of IL-4 detected in CD1KO NOD mice might not be sufficient to stimulate mice to downregulate Th1 diabetogenic cells. However, as IL-4KO NOD mice do not develop accelerated disease 33, lack of IL-4 secreted by NK T cells could not be solely responsible for the increased susceptibility to diabetes in CD1KO NOD mice. A recent study showed that IFN-γ secreted by NK T cells in the periphery might be critical for protection against diabetes 10. Although the majority of CD1KO NOD mice secreted lower levels of IFN-γ compared with controls, some CD1KO NOD mice produced more IFN-γ than their littermate controls, presumably from other CD4+ T cells. Thus, it is unlikely that lack of IFN-γ secreted by NK T cells would contribute significantly to the exacerbation of IDDM in CD1KO mice. Second, NK T cells have been shown to have a cytotoxic effect on immature thymocytes 15, indicating that NK T cells may regulate T cell selection in the thymus. Therefore, lack of NK T cells might affect the repertoire of autoreactive T cells. Third, NK T cells or the expression of CD1 might be involved in maintaining the survival and function of some regulatory T cells. We performed FACS® analysis and found that CD1KO NOD and heterozygous controls have comparable numbers of CD4+CD25+ T cells (data not shown), a subset of T cells that has been shown to be critical in controlling spontaneous autoimmune diabetes 7. However, it remains possible that other regulatory T cells might be affected by CD1 deficiency.
Treatment with α-GalCer has been shown to modulate several immune responses in vivo, such as enhancing the antitumor 34 and antiviral activity 35, mediating protection against murine malaria 36, and regulating intestinal inflammation 37. Here we showed that α-GalCer exerts a protective effect on autoimmune diabetes in NOD mice. The precise mechanism of how α-GalCer activates NK T cells to protect IDDM is not clear. Previous studies showed that in vivo administration of α-GalCer polarizes adaptive immune response for production of Th2 cytokines 20 21. However, we did not detect a polarized Th2 response in α-GalCer–treated NOD mice (data not shown). It is possible that activation of NK T cells by α-GalCer leads to bystander activation of cell types (i.e., regulatory T cells, NK cells, and dendritic cells) that are critical in regulating the autoreactive T cells in NOD mice. Similar to mouse NK T cells, α-GalCer can also activate human NK T cells in a CD1d-dependent manner 38 39. Thus, our results suggest that activation of NK T cells by α-GalCer may represent a novel approach for therapeutic intervention of IDDM in humans.
Acknowledgments
We thank Dr. Yasuhiko Koezuka (Kirin Brewery, Gunma, Japan) for providing α-GalCer (KRN7000); Hanh Nguyen and Angela Parsons for critical reading of the manuscript; and Candice Mulder for technical assistance.
This work was supported by National Institutes of Health grant AI43407 (to C.-R. Wang) and the Merrell Foundation.
Footnotes
Abbreviations used in this paper: IDDM, insulin-dependent diabetes mellitus; KO, knockout; NOD, nonobese diabetic.
References
- Kikutani H., Makino S. The murine autoimmune diabetes modelNOD and related strains. Adv. Immunol. 1992;51:285–322. doi: 10.1016/s0065-2776(08)60490-3. [DOI] [PubMed] [Google Scholar]
- Bendelac A., Carnaud C., Boitard C., Bach J.F. Syngeneic transfer of autoimmune diabetes from diabetic NOD mice to healthy neonates. Requirement for both L3T4+ and Lyt-2+ T cells. J. Exp. Med. 1987;166:823–832. doi: 10.1084/jem.166.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B.J., Appel M.C., O'Neil J.J., Wicker L.S. Both the Lyt-2+ and L3T4+ T cell subsets are required for the transfer of diabetes in nonobese diabetic mice. J. Immunol. 1988;140:52–58. [PubMed] [Google Scholar]
- Yagi H., Matsumoto M., Kunimoto K., Kawaguchi J., Makino S., Harada M. Analysis of the roles of CD4+ and CD8+ T cells in autoimmune diabetes of NOD mice using transfer to NOD athymic nude mice. Eur. J. Immunol. 1992;22:2387–2393. doi: 10.1002/eji.1830220931. [DOI] [PubMed] [Google Scholar]
- Katz J.D., Benoist C., Mathis D. T helper cell subsets in insulin-dependent diabetes. Science. 1995;268:1185–1188. doi: 10.1126/science.7761837. [DOI] [PubMed] [Google Scholar]
- Herbelin A., Gombert J.M., Lepault F., Bach J.F., Chatenoud L. Mature mainstream TCR αβ+CD4+ thymocytes expressing L-selectin mediate “active tolerance” in the nonobese diabetic mouse. J. Immunol. 1998;161:2620–2628. [PubMed] [Google Scholar]
- Salomon B., Lenschow D.J., Rhee L., Ashourian N., Singh B., Sharpe A., Bluestone J.A. B7/CD28 co-stimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- Baxter A.G., Kinder S.J., Hammond K.J., Scollay R., Godfrey D.I. Association between αβTCR+CD4−CD8− T-cell deficiency and IDDM in NOD/Lt mice. Diabetes. 1997;46:572–582. doi: 10.2337/diab.46.4.572. [DOI] [PubMed] [Google Scholar]
- Lehuen A., Lantz O., Beaudoin L., Laloux V., Carnaud C., Bendelac A., Bach J.F., Monteiro R.C. Over expression of natural killer T cells protects Vα14- Jα281 transgenic nonobese diabetic mice against diabetes. J. Exp. Med. 1998;188:1831–1839. doi: 10.1084/jem.188.10.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcone M., Yeung B., Tucker L., Rodriguez E., Sarvetnick N. A defect in interleukin 12-induced activation and interferon γ secretion of peripheral natural killer T cells in nonobese diabetic mice suggests new pathogenic mechanisms for insulin-dependent diabetes mellitus. J. Exp. Med. 1999;190:963–972. doi: 10.1084/jem.190.7.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendelac A. Mouse NK1+ T cells. Curr. Opin. Immunol. 1995;7:367–374. doi: 10.1016/0952-7915(95)80112-x. [DOI] [PubMed] [Google Scholar]
- Bendelac A., Rivera M.N., Park S.H., Roark J.H. Mouse CD1-specific NK1 T cellsdevelopment, specificity, and function. Annu. Rev. Immunol. 1997;15:535–562. doi: 10.1146/annurev.immunol.15.1.535. [DOI] [PubMed] [Google Scholar]
- Exley M., Garcia J., Balk S.P., Porcelli S. Requirements for CD1d recognition by human invariant Vα24+CD4−CD8− T cells. J. Exp. Med. 1997;186:109–120. doi: 10.1084/jem.186.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberl G., Lees R., Smiley S.T., Taniguchi M., Grusby M.J., MacDonald H.R. Tissue-specific segregation of CD1d-dependent and CD1d-independent NK T cells. J. Immunol. 1999;162:6410–6419. [PubMed] [Google Scholar]
- Arase H., Arase N., Kobayashi Y., Nishimura Y., Yonehara S., Onoé K. Cytotoxicity of fresh NK1.1+ T cell receptor α/β+ thymocytes against a CD4+8+ thymocyte population associated with intact Fas antigen expression on the target. J. Exp. Med. 1994;180:423–432. doi: 10.1084/jem.180.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth M.J., Thia K.Y., Street S.E., Cretney E., Trapani J.A., Taniguchi M., Kawano T., Pelikan S.B., Crowe N.Y., Godfrey D.I. Differential tumor surveillance by natural killer (NK) and NKT cells. J. Exp. Med. 2000;191:661–668. doi: 10.1084/jem.191.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano T., Cui J., Koezuka Y., Toura I., Kaneko Y., Motoki K., Ueno H., Nakagawa R., Sato H., Kondo E. CD1d-restricted and TCR-mediated activation of Vα14 NKT cells by glycosylceramides. Science. 1997;278:1626–1629. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- Carnaud C., Lee D., Donnars O., Park S.H., Beavis A., Koezuka Y., Bendelac A. Cutting edgecross talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J. Immunol. 1999;163:4647–4650. [PubMed] [Google Scholar]
- Kitamura H., Iwakabe K., Yahata T., Nishimura S., Ohta A., Ohmi Y., Sato M., Takeda K., Okumura K., Van Kaer L. The natural killer T (NKT) cell ligand α-galactosylceramide demonstrates its immunopotentiating effect by inducing interleukin IL-12 production by dendritic cells and IL-12 receptor expression on NKT cells. J. Exp. Med. 1999;189:1121–1128. doi: 10.1084/jem.189.7.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N., Hong S., Scherer D.C., Serizawa I., Burdin N., Kronenberg M., Koezuka Y., Van Kaer L. Cutting edgeactivation of NK T cells by CD1d and α-galactosylceramide directs conventional T cells to the acquisition of a Th2 phenotype. J. Immunol. 1999;163:2373–2377. [PubMed] [Google Scholar]
- Burdin N., Brossay L., Kronenberg M. Immunization with α-galactosylceramide polarizes CD1-reactive NK T cells towards Th2 cytokine synthesis. Eur. J. Immunol. 1999;29:2014–2025. doi: 10.1002/(SICI)1521-4141(199906)29:06<2014::AID-IMMU2014>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Cui J., Shin T., Kawano T., Sato H., Kondo E., Toura I., Kaneko Y., Koseki H., Kanno M., Taniguchi M. Requirement for Vα14 NKT cells in IL-12-mediated rejection of tumors. Science. 1997;278:1623–1626. doi: 10.1126/science.278.5343.1623. [DOI] [PubMed] [Google Scholar]
- Kumar H., Belperron A., Barthold S.W., Bockenstedt L.K. Cutting edgeCD1d deficiency impairs murine host defense against the spirochete, Borrelia burgdorferi . J. Immunol. 2000;165:4797–4801. doi: 10.4049/jimmunol.165.9.4797. [DOI] [PubMed] [Google Scholar]
- Ishikawa H., Hisaeda H., Taniguchi M., Nakayama T., Sakai T., Maekawa Y., Nakano Y., Zhang M., Zhang T., Nishitani M. CD4+Vα14 NKT cells play a crucial role in an early stage of protective immunity against infection with Leishmania major . Int. Immunol. 2000;12:1267–1274. doi: 10.1093/intimm/12.9.1267. [DOI] [PubMed] [Google Scholar]
- Gombert J.M., Herbelin A., Tancrede-Bohin E., Dy M., Carnaud C., Bach J.F. Early quantitative and functional deficiency of NK1+-like thymocytes in the NOD mouse. Eur. J. Immunol. 1996;26:2989–2998. doi: 10.1002/eji.1830261226. [DOI] [PubMed] [Google Scholar]
- Godfrey D.I., Hammond K.J., Poulton L.D., Smyth M.J., Baxter A.G. NKT cellsfacts, functions and fallacies. Immunol. Today. 2000;21:573–583. doi: 10.1016/s0167-5699(00)01735-7. [DOI] [PubMed] [Google Scholar]
- Godfrey D.I., Kinder S.J., Silvera P., Baxter A.G. Flow cytometric study of T cell development in NOD mice reveals a deficiency in αβTCR+CDR−CD8− thymocytes. J. Autoimmun. 1997;10:279–285. doi: 10.1006/jaut.1997.0129. [DOI] [PubMed] [Google Scholar]
- Wilson S.B., Kent S.C., Patton K.T., Orban T., Jackson R.A., Exley M., Porcelli S., Schatz D.A., Atkinson M.A., Balk S.P. Extreme Th1 bias of invariant Vα24JαQ T cells in type 1 diabetes. Nature. 1998;391:177–181. doi: 10.1038/34419. [DOI] [PubMed] [Google Scholar]
- Chen Y.H., Chiu N.M., Mandal M., Wang N., Wang C.-R. Impaired NK1+ T cell development and early IL-4 production in CD1- deficient mice. Immunity. 1997;6:459–467. doi: 10.1016/s1074-7613(00)80289-7. [DOI] [PubMed] [Google Scholar]
- Lenschow D.J., Herold K.C., Rhee L., Patel B., Koons A., Qin H.Y., Fuchs E., Singh B., Thompson C.B., Bluestone J.A. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity. 1996;5:285–293. doi: 10.1016/s1074-7613(00)80323-4. [DOI] [PubMed] [Google Scholar]
- Delovitch T.L., Singh B. The nonobese diabetic mouse as a model of autoimmune diabetesimmune dysregulation gets the NOD. Immunity. 1997;7:727–738. doi: 10.1016/s1074-7613(00)80392-1. [DOI] [PubMed] [Google Scholar]
- Wicker L.S., Todd J.A., Peterson L.B. Genetic control of autoimmune diabetes in the NOD mouse. Annu. Rev. Immunol. 1995;13:179–200. doi: 10.1146/annurev.iy.13.040195.001143. [DOI] [PubMed] [Google Scholar]
- Wang B., Gonzalez A., Hoglund P., Katz J.D., Benoist C., Mathis D. Interleukin-4 deficiency does not exacerbate disease in NOD mice. Diabetes. 1998;47:1207–1211. doi: 10.2337/diab.47.8.1207. [DOI] [PubMed] [Google Scholar]
- Kawano T., Cui J., Koezuka Y., Toura I., Kaneko Y., Sato H., Kondo E., Harada M., Koseki H., Nakayama T. Natural killer-like nonspecific tumor cell lysis mediated by specific ligand-activated Vα14 NKT cells. Proc. Natl. Acad. Sci. USA. 1998;95:5690–5693. doi: 10.1073/pnas.95.10.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakimi K., Guidotti L.G., Koezuka Y., Chisari F.V. Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J. Exp. Med. 2000;192:921–930. doi: 10.1084/jem.192.7.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Aseguinolaza G., de Oliveira C., Tomaska M., Hong S., Bruna-Romero O., Nakayama T., Taniguchi M., Bendelac A., Van Kaer L., Koezuka Y. α-galactosylceramide-activated Vα14 natural killer T cells mediate protection against murine malaria. Proc. Natl. Acad. Sci. USA. 2000;97:8461–8466. doi: 10.1073/pnas.97.15.8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saubermann L.J., Beck P., De Jong Y.P., Pitman R.S., Ryan M.S., Kim H.S., Exley M., Snapper S., Balk S.P., Hagen S.J. Activation of natural killer T cells by α-galactosylceramide in the presence of CD1d provides protection against colitis in mice. Gastroenterology. 2000;119:119–128. doi: 10.1053/gast.2000.9114. [DOI] [PubMed] [Google Scholar]
- Brossay L., Chioda M., Burdin N., Koezuka Y., Casorati G., Dellabona P., Kronenberg M. CD1d-mediated recognition of an α-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J. Exp. Med. 1998;188:1521–1528. doi: 10.1084/jem.188.8.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spada F.M., Koezuka Y., Porcelli S.A. CD1d-restricted recognition of synthetic glycolipid antigens by human natural killer T cells. J. Exp. Med. 1998;188:1529–1534. doi: 10.1084/jem.188.8.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]