

Abstract
The stimulation of interferon (IFN)-γ by interleukin (IL)-12 has been shown to provide protection from intracellular pathogens such as Listeria monocytogenes. Tumor necrosis factor (TNF) is also a major player in the resolution of Listeria infections and is suggested to have more global effects than can be explained by the induction of IFN-γ alone. Since IL-18 synergizes with IL-12 to induce IFN-γ production by natural killer and T helper (Th)1 cells, we determined its role in responses to Listeria. IL-18 appeared to be even more potent than either IL-12 or IFN-γ for protection against this pathogen and IL-18 enhanced bacterial clearance in the complete absence of IFN-γ. Indeed IL-18 was comparable to TNF in its ability to resolve the infection and showed a lowered protective capacity in the absence of TNF. Moreover, IL-18 induced macrophages to secrete both TNF and nitric oxide after a Listeria infection. IL-18 was also essential for optimal IFN-γ production by antigen-specific T cells. Therefore, IL-18 operates via its effects on both the innate immune response, including macrophages, as well as on Th1 cells, to protect against Listeria.
Keywords: Listeria, IL-18, TNF, NO, Th1 cells
Introduction
Listeria monocytogenes (Listeria), a gram-positive facultative intracellular bacterium, is associated with severe infections in newborns, the elderly, and immunocompromised individuals 1 2 3 4 5. Murine models of listeriosis have been extensively studied to facilitate an understanding of the host immune response 6 7. Results of these studies demonstrated that there is coordinate activation of the innate immune response, including neutrophils, macrophages, and NK cells 8 9, together with the adaptive immune response of CD4+ Th1 and CD8+ T cells 6 10. Moreover, it has been clearly demonstrated that for resolution and immunity to develop to Listeria the induction of antigen-specific T cells is essential 2 10 11 12.
The release of proinflammatory cytokines from these various cell subsets was found to be pivotal in controlling primary immune responses to Listeria. IFN-γ, produced by NK and T cells, clears Listeria predominantly by enhancing the antimicrobial and antigen-presenting properties of macrophages 13 14 15. The key macrophage product, IL-12, provides protection from Listeria by inducing IFN-γ production 16. Likewise, TNF plays a role in the clearance of Listeria due in part to its induction of IFN-γ production 17 18. However, IFN-γ does not completely replace the ability of TNF to resolve Listeria infections 16, in keeping with observations that TNF appears to be more dominant than IL-12 in eliciting protective primary immune responses to Listeria 16 yet plays a lesser role than IL-12 in invoking IFN-γ production by Th1 cells 19 20. The role of these proinflammatory cytokines in the eradication of Listeria has also been confirmed in IFN-γ−/− 21, IFN-γ receptor−/− 22, IL-12−/− 23, and TNF receptor 1−/− 24 25 mice.
In contrast to a primary immune response, the secondary immune response to Listeria is more rapidly induced and is able to overcome a high, normally lethal, dose of bacteria 6. This acquired resistance is largely attributable to memory effector CD4+ and CD8+ T cells, in part by IFN-γ production 26 27 28. Although IL-12 is also produced in a secondary challenge with this organism, it offers less protection than in primary responses and cannot completely account for the effects of IFN-γ 28, indicating that there may be other players involved in IFN-γ production at this stage. TNF is clearly protective in secondary responses to Listeria, and this cytokine may complement the role of IFN-γ in the bacterial clearance 27 29. In addition to the role of CD4+ and CD8+ T cells in the memory response to Listeria, the innate immune response is also essential with neutrophils playing a critical role 30 31.
A potentially important player in the eradication of a Listeria infection is IL-18 32, which was originally shown to induce IFN-γ production in mice injected with heat-killed Propionibacterium acnes (P. acnes) and LPS 32 33. In vitro studies have since shown that IL-18 synergizes with IL-12 to induce IFN-γ from differentiating and committed CD4+ Th1, but not Th2, cells 34 as well as from NK cells 35. These two cytokines can also synergize in vivo since IL-12−/−IL-18−/− mice displayed more profound impairment of IFN-γ production after injection with Mycobacterium bovis than did either IL-12−/− or IL-18−/− mice individually 36. In addition to its IFN-γ–enhancing capacity, IL-18 can also augment the cytotoxic activity of both NK and T cells 36 37 38 39 40 and can enhance their production of other proinflammatory mediators 33 36 41 42 43. Moreover, studies of acute bacterial, fungal, parasitic, and viral infections have shown a role for IL-18 in the eradication of intracellular pathogens 44 45 46 47 48 49. However, a role for IL-18 in memory effector responses to such pathogens has not been investigated yet, nor has its ability to protect through mediators other than IFN-γ.
Responsiveness to IL-18 is conferred by binding to its cognate receptor, which consists of an IL-1R5/IL-1R7 heterodimer 50 51 52. Herein, using a neutralizing mAb to the IL-1R7 chain (anti–IL-18R; reference 53) we report an important and partly IFN-γ–independent role for IL-18 in both innate and adaptive immune responses to Listeria involving the production of TNF and nitric oxide (NO) as well as IFN-γ.
Materials and Methods
Mice.
Female Balb/c and C.B-17 severe combined immunodeficient (SCID) mice (both H-2d) were purchased from Taconic Farms. Female Balb/c IFN-γ–deficient mice (H-2d) were provided by Bob Coffman (DNAX Research Institute, Palo Alto, CA). Female mice transgenic for the DO11.10 αβ TCR 54 on a Balb/c genetic background were identified at age 4–6 wk by staining peripheral blood leukocytes with the anti-TCR clonotype-specific mAb KJ1-26 55; these mice were heterozygous for the TCR-α and -β transgenes. All mice were housed under specific pathogen-free conditions and were used at the age of 6–8 wk.
Bacteria and Bacterial Antigens.
Listeria (provided by H. Rogers, K. Murphy, and E. Unanue) has been maintained in a virulent state by repeated passage in mice. Bacteria were grown in BHI broth (Difco) to midlog phase as determined by OD560 measurements and aliquots were stored in 20% glycerol/PBS at –80°C.
Heat-killed Listeria monocytogenes (HKLM) was obtained by incubating live bacteria at 74°C for 120 min. Aliquots were stored at −80°C.
mAbs, Cytokines, and Reagents.
The mAbs anti-β galactosidase (isotype control: GL117), anti–IL-12 (C17.8.20), anti–IL-18R (TC30-28E3), and anti-TNF (XT22) were administered (1 mg) intraperitoneally to mice 1 h before infection with Listeria and at the additional times as indicated. Recombinant mouse cytokines were IL-2 (DNAX), TNF (Genzyme), IL-12 (BD PharMingen), and IL-18 (PeproTech). LPS was obtained from Sigma-Aldrich and azide-free, low endotoxin anti-CD3ε was obtained from BD PharMingen. The antigenic peptide from chicken OVA (OVA323–339) was synthesized on an Applied Biosystems model 430 peptide synthesizer.
Infection with Listeria.
An initial infection with Listeria was performed by intravenous injection of 1–2 × 103 CFU viable bacteria diluted in a volume of 0.2 ml PBS. For a second infection, mice were injected with 5 × 104 CFU viable bacteria on day 28 after the initial infection. Actual numbers of bacteria injected were determined by plating aliquots of relevant dilutions on BHI agar (Difco). Bacterial growth in the spleen was determined on the indicated days after infection by plating relevant dilutions of the spleen homogenates on BHI agar. Colonies were counted after 24 h of incubation at 37°C. LD50 were calculated as described previously 56.
Cell Culture Ex Vivo.
On the indicated day after infection, single cell spleen suspensions were prepared from individual mice in culture medium: RPMI 1640 medium (Bio-Whittaker) supplemented with 10% FCS (UT; Hyclone Laboratories), 1 mM l-glutamine, 10 mM Hepes, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 mM sodium pyruvate, and 0.05 mM β-2 mercaptoethanol (all GIBCO BRL). Cells were incubated in 96-well plates at a density of 106 cells per well with 3.9 × 104 CFU HKLM for 48 h at 37°C. Supernatants were harvested at the end of culture and stored at −80°C until analyzed for cytokine content.
T Cell Culture In Vitro.
CD4+ T cells were enriched from DO11.10 spleen cell preparations by negative selection using magnetic-activated cell sorting with a mixture of biotinylated anti-CD8α, anti–I-Ad, anti-B220, anti-GR1, and anti–Mac-1 mAbs (Miltenyi Biotec). The enriched cells were then purified further using a FACStarPlus™ flow cytometer (Becton Dickinson) to achieve >99% CD4+ T cells, demonstrated to be naive on the basis of bright L-selectin staining 57.
Primary stimulations of these sorted CD4+ T cells (2.5 × 105 cells per well) were conducted using 0.6 μM OVA323–339, 3.9 × 104 CFU HKLM, and RBC-lysed Balb/c spleen cells (5 × 106 cells per well; 3,000 rad) as APCs with or without the addition of 10 μg/ml of the anticytokine mAbs described above (all at 10 μg/ml). T cells were expanded threefold into fresh culture medium at 72 h and cells harvested on day 7 were washed three times, counted, and restimulated with fresh APCs, OVA plus or minus HKLM with or without the addition of anticytokine mAbs as detailed in the individual figure legends. All concentrations used for restimulation were the same as those described for priming. Supernatants were collected at 48 h and stored at −80°C until analyzed for cytokine content.
Macrophage Cultures In Vitro.
Macrophages from peritoneal exudates were selected by retaining adherent cells. After overnight culture in medium at 2 × 106 cells per milliliter in flat-bottomed 96-well plates, adherent or whole peritoneal cell populations were placed in the presence of IL-18 (100 ng/ml) or HKLM (3.9 × 104 CFUs) with or without anti–IL-18R (10 μg/ml) for 48 h, and supernatants were stored at −20°C until assayed for cytokine content. For intracellular cytokine staining, identical cultures were stimulated in the presence of 10 μg/ml Brefeldin A (Sigma-Aldrich) for 4 h.
Alternatively, whole peritoneal cell populations were incubated at 105 cells per milliliter in flat-bottomed 96-well plates with HKLM (3.9 × 104 CFUs) for 16 h and 50 μl of supernatant measured for nitrite content using 50 μl Greiss reagent (3% phosphoric acid, 1% p-amino-benzene-sulfonamide, and 1% n-1-nap-thylethylenediamide; Sigma-Aldrich) 58 and absorbance was read at 540 nm.
Cytokine Assays.
IFN-γ was detected using a two-site sandwich ELISA as described previously 59. The sensitivity was 125 pg/ml (1 U/ml = 0.1 ng/ml). TNF was detected using a Quantikine™ M ELISA (R&D Systems) according to the manufacturer's instructions. The sensitivity was <5 pg/ml. TNF-secreting cells were analyzed on a single cell basis by flow cytometric analysis of intracellular TNF as described previously 60.
Histology and Immunohistochemistry.
Tissue for immunohisto-chemistry was immersed in OCT compound (TissueTek) and frozen in the vapor phase of liquid N2. 8-μm frozen sections were thawed on gelatinized microscope slides, allowed to dry at room temperature, and fixed in acetone containing 0.04% (vol/vol) H2O2 (room temperature, 10 min). Primary and secondary mAbs were diluted in PBS containing 7.5% (wt/vol) BSA, 10% (vol/vol) normal mouse serum, and 10 mM NaN3. Primary mAbs were purified RM4-5 (anti–mouse CD4), purified 53–6.7 (anti–mouse CD8), purified or FITC-conjugated RB6-8C5 (anti-Ly6G, GR-1), purified or FITC-conjugated F4/80 (mouse macrophages), and the appropriate isotype controls (all BD PharMingen). Secondary mAbs were peroxidase-conjugated donkey-anti–rat (Jackson ImmunoResearch Laboratories) or peroxidase-conjugated sheep-antifluorescein (Roche). All mAbs were used at previously determined concentrations (10–50 μg/ml). Peroxidase activity was visualized using diaminobenzidine (DAB; Sigma-Aldrich). Section were lightly counterstained with hematoxylin solution (Sigma-Aldrich), dehydrated, and mounted in Hemode (Fisher Scientific).
Statistical Analysis.
The Dunnet procedure for all possible pairwise contrasts of means with a control mean was performed as indicated.
Results
IL-18 Is Critical for Resistance to Listeria.
To obtain quantitative information on the effect of IL-18 on survival to Listeria, mice were infected with graded doses of the organism in the presence of a neutralizing mAb to IL-18R 53. This was compared with an isotype control or anti–IL-12 mAb, since IL-12 is known to protect from Listeria 61. For those mice treated with an isotype control mAb (Fig. 1) or PBS (data not shown), the highest dose of 5 × 104 CFUs led to 100% mortality within 4 d, whereas 103 CFUs was sublethal for 100% of these animals. Administration of anti–IL-18R dramatically increased the susceptibility to Listeria with 100% of mice now succumbing to as little as 103 CFUs by day 4 (Fig. 1 and Table ). In contrast, anti–IL-12 had no effect on the survival of mice infected with 103 CFUs (Fig. 1). In fact, 100% mortality was not observed in anti–IL-12–treated mice until 5 d after they were infected with ≥104 CFUs (Fig. 1 and Table ). This dose was lethal for <50% of isotype control mice (Fig. 1) and their mean survival was extended to 6 d (Table ) suggesting that IL-12 was playing a protective role in the infection albeit to a lesser extent than IL-18. Impaired survival was seen to a similar extent whether mice were treated with anti–IL-18R alone or in conjunction with anti–IL-12 (Fig. 1 and Table ). Hence these findings indicate that IL-18 may play a more dominant role than IL-12 in survival from a Listeria infection.
Figure 1.

Anti–IL-18R treatment can increase sensitivity to Listeria to a greater extent than anti–IL-12. Balb/c mice were injected weekly with isotype control mAb (GL117), anti–IL-12, anti–IL-18R, or the combination of both anticytokine mAbs before infection with the indicated doses of Listeria as described in Materials and Methods. Survival was monitored over the next 8 d and plotted as a percentage of the total number of mice per group. 6 mice per group were assessed and the results shown were representative of three experiments.
Table 1.
Mean Survival Time
| Dose of Listeria (CFUs): | 103 | 2 × 103 | 5 × 103 | 104 | 5 × 104 |
|---|---|---|---|---|---|
| Group | |||||
| GL117 | ND | ND | ND | 6 | 4 |
| Anti–IL-18R | 4 | 4 | 3 | 3 | 3 |
| Anti–IL-12 | ND | ND | ND | 5 | 4 |
| Anti–IL-12 plus anti–IL-18R | 4 | 4 | 3 | 3 | 3 |
Displayed is the mean survival time (in days) of mice from the experiment described in Fig. 1.
IL-18–mediated Protection to Listeria Occurs even in the Absence of IFN-γ.
Since IL-18 synergizes with IL-12 for IFN-γ production by NK and Th1 cells 34 36 and the listericidal effects of IL-12 have been shown to be IFN-γ dependent 61, we determined how IL-18 compared with IL-12 and IFN-γ in its ability to resolve a Listeria infection. Clearance of Listeria from the spleen 3 d after infection was significantly impaired in those animals pretreated with anti–IL-18R mAb as compared with isotype control mAb, whether or not Listeria-infected mice were fully immunocompetent (Balb/c mice, Fig. 2 A) or lacked T and B cells (CB17.SCID mice, data not shown). This correlated with a significant reduction in the levels of IFN-γ produced upon antigen stimulation of spleen cells from the anti–IL-18R mAb treated group versus the isotype mAb treated controls (Fig. 2 B). Bacterial clearance was also reduced in Listeria-infected mice given anti–IL-12 or anti–IFN-γ mAbs as compared with isotype mAb treated control mice (Fig. 2 A) and as reported previously 16. However, the bacterial burden remained significantly lower in the anti–IL-12 or anti–IFN-γ mAb treated mice than in the anti–IL-18R mAb treated mice even though the antigen-specific IFN-γ levels of each group were impaired similarly (Fig. 2 B). There was no additive effect between anti–IL-18R and anti–IL-12 since the CFU counts when both mAbs were given together did not differ from those obtained using the individual mAbs (Fig. 2 A). Taken together, our observations reveal a critical role for IL-18 in the clearance of Listeria, which cannot be fully explained by its effects on IFN-γ production.
Figure 2.
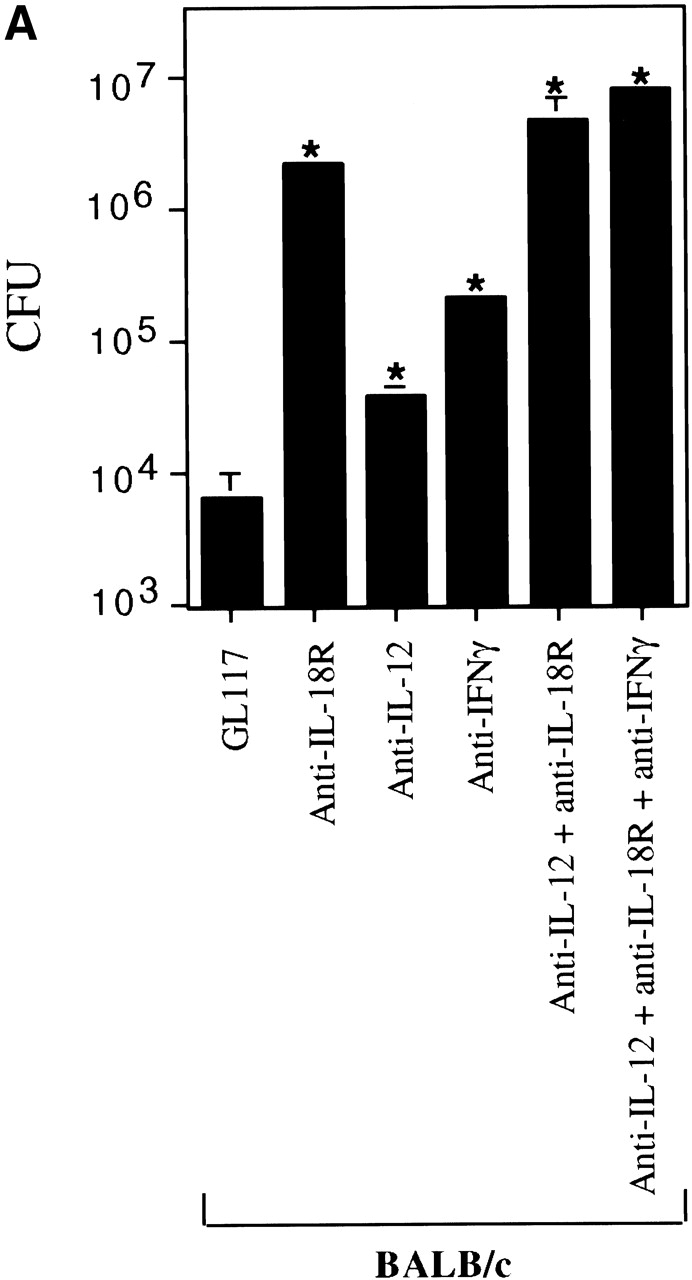
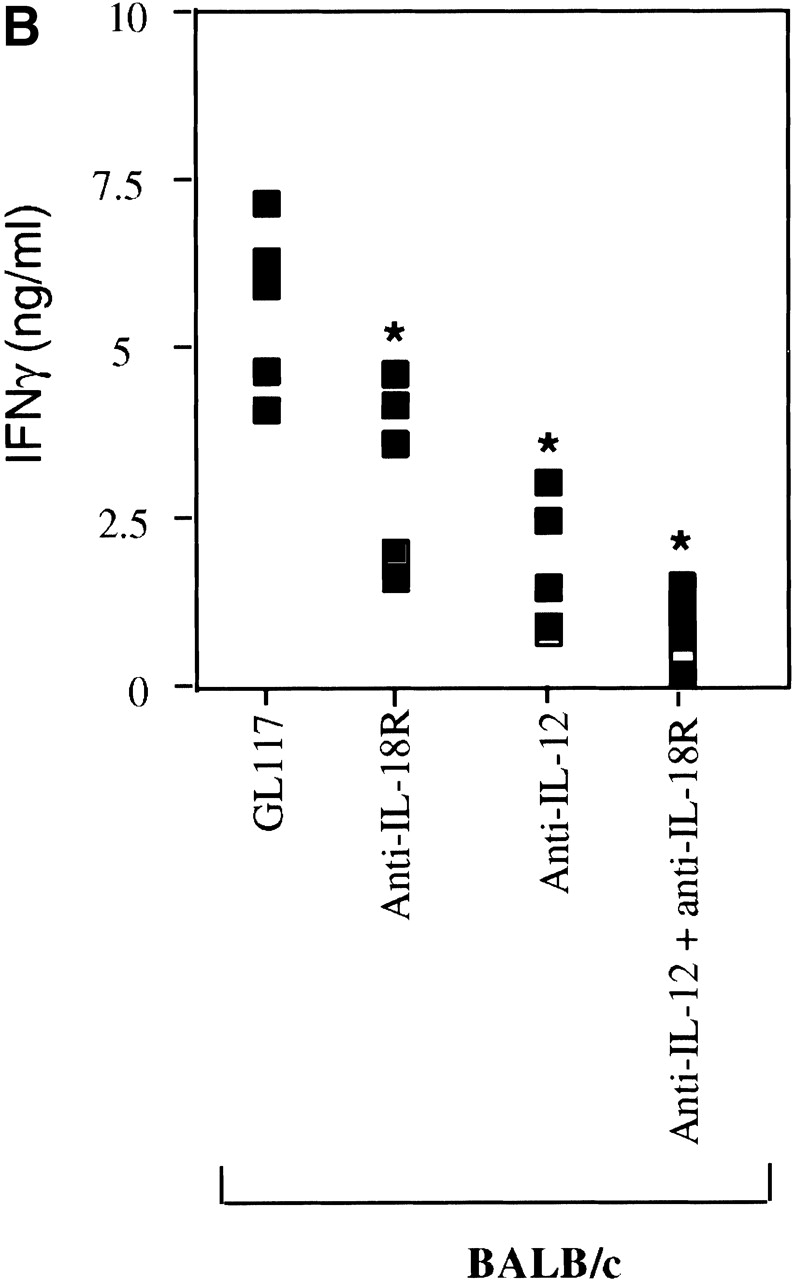
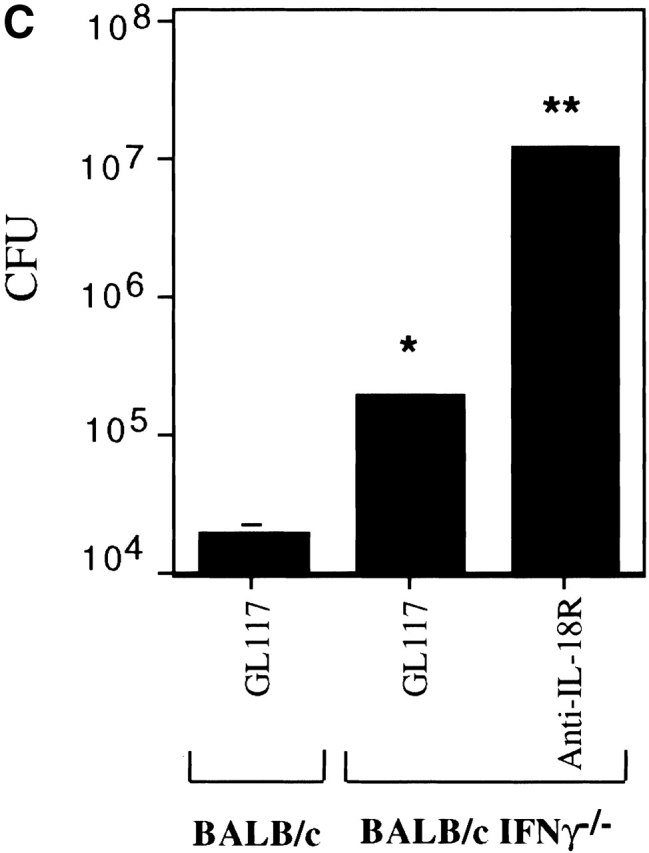
IFN-γ cannot completely account for the role of IL-18 in clearance of a Listeria infection. Mice were injected with isotype control mAb (GL117), anti–IL-18R, anti–IL-12, anti–IFN-γ, or the combination of anticytokine mAbs indicated, before infection with the indicated dose of Listeria as described in Materials and Methods. (A) The bacterial burden per spleen of Balb/c mice was evaluated 3 d after infecting with 2 × 103 CFUs. Each bar represents the mean CFU per group ± SD for ≥5 mice per group. (B) Spleen cells from the same Balb/c mice were also restimulated ex vivo with 3.9 × 104 CFU HKLM for 48 h and supernatants analyzed for IFN-γ. Data are shown for individual mice. (C) The bacterial burden per spleen of Balb/c and Balb/c.IFN-γ−/− mice was evaluated 3 d after infecting with 600 CFUs. Each bar represents the mean CFU per group ± SD for ≥5 mice per group. Results are representative of greater than three experiments. Statistical analysis was performed using Dunnett's. *P < 0.05 versus Balb/c isotype control. **P < 0.05 versus Balb/c.IFN-γ2/− isotype control.
To rule out the possibility that our findings that IL-18 was more dominant than IFN-γ in bacterial clearance was due to differing mAb efficacies, we also analyzed Balb/c.IFN-γ–deficient mice which are more susceptible to Listeria than regular Balb/c mice (Fig. 2 C; reference 21). Bacterial clearance was again increased significantly beyond isotype control levels by administering anti–IL-18R before infecting these mice (Fig. 2 C).
TNF Contributes to the Protective Effects of IL-18.
Since IL-18 can induce TNF in nonpathogen-driven systems 62 63 64 and TNF is critical in eradicating Listeria 17 18, we determined how these two cytokines compared in their ability to clear Listeria. Strikingly, anti–IL-18R had a similar capacity to anti-TNF in impairing bacterial clearance (Fig. 3 A). Those mice given either anti–IL-18R or anti-TNF mAb harbored colonies of Listeria in their spleen 2.5 logs higher than isotype mAb treated control mice by the third day of infection (Fig. 3 A). In addition, there was no additive effect between anti–IL-18R and anti-TNF since the CFU counts when both mAbs were given together did not differ from those obtained using the individual mAbs when assessed 2 d after infection and therefore, before the CFU plateau was reached (Fig. 3 A). Thus, we assessed bacterial clearance at this earlier time of infection in mice given mAbs to TNF and/or IFN-γ with or without a daily treatment of rIL-18 to determine whether or not IL-18 required TNF and/or IFN-γ for its protective effects.
Figure 3.


TNF contributes to the protective effects of IL-18. (A) Balb/c mice were injected with the mAbs indicated before infection with 2 × 103 CFU Listeria and the bacterial burden per spleen was evaluated after 2 or 3 d. (B) Balb/c mice treated with the mAbs indicated were given 2 × 103 CFU Listeria and treated daily with either 0.3 ml PBS or an equal volume containing 5 μg rIL-18 as indicated. The bacterial burden per spleen was evaluated after 2 d. Each bar represents the mean CFU per group ± SD for ≥5 mice per group. Results were reproducible in greater than three experiments. Statistical analysis was performed using Dunnett's. *P < 0.05 versus isotype control plus PBS. **P < 0.05 versus isotype control plus rIL-18.
The bacterial burden of isotype mAb treated control mice was indeed significantly reduced by administering rIL-18 (Fig. 3 B). When rIL-18 was given to mice depleted of IFN-γ the bacterial load was reduced to the level seen with the isotype controls given rIL-18 (Fig. 3 B) confirming that this treatment could prevent an otherwise exacerbated infection. In contrast, although rIL-18 treatment reduced the bacterial burden of mice given mAbs to TNF to a significant extent, it did not reduce it to the level seen with either the rIL-18–treated isotype controls or anti–IFN-γ–treated mice given rIL-18 (Fig. 3 B). The effects of rIL-18 on bacterial clearance were similar when mice had been treated with anti-TNF mAbs alone or in combination with anti–IFN-γ mAbs. Overall these findings indicate that the protective capacity of IL-18 largely required the production of TNF and did not appear to require IFN-γ in this early phase of bacterial clearance.
IL-18 Is Required for TNF and NO Production by Listeria-infected Macrophages.
Listeria-infected mice given anti–IL-18R were not impaired in their antigen-specific production of TNF upon restimulation with HKLM in vitro (data not shown). Therefore, we investigated the macrophage as an alternative source of TNF in response to IL-18, using adherent cells from the peritoneal cavity of Listeria-infected SCID mice. In these cultures the release of TNF was completely restricted to F4/80+ macrophages (data not shown) and was increased by stimulation with either IL-18 or HKLM (Fig. 4 A). Moreover, the TNF produced in response to HKLM was significantly reduced upon addition of an anti–IL-18R mAb (Fig. 4 A). Overall these findings show that IL-18 enhanced the production of TNF by macrophages from Listeria-infected mice. However, no effect on macrophage TNF regulation ex vivo was observed after Listeria infection in the presence of anti–IL-18R as compared with isotype control in vivo (data not shown) since the macrophage produces IL-18 upon stimulation in vitro thus masking the effects of anti–IL-18R treatment in vivo.
Figure 4.
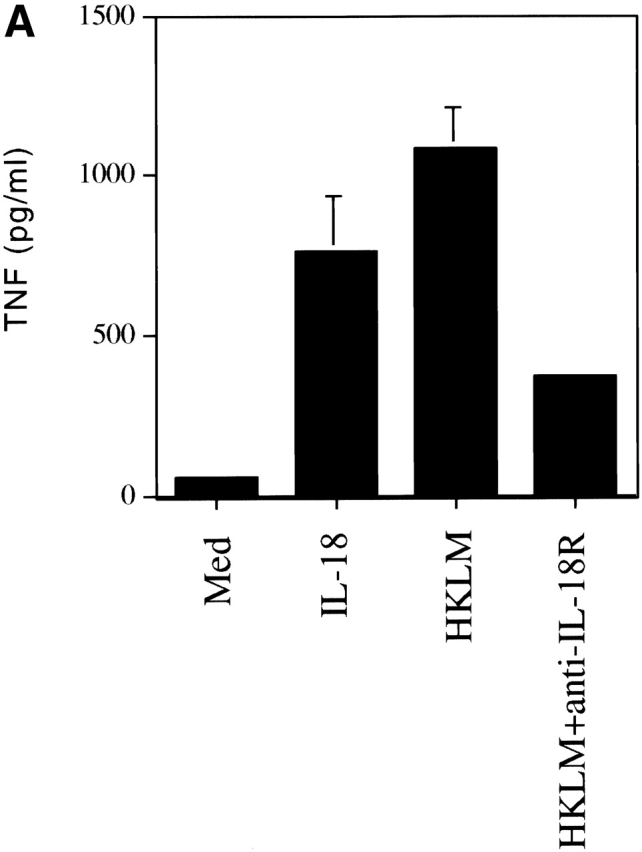
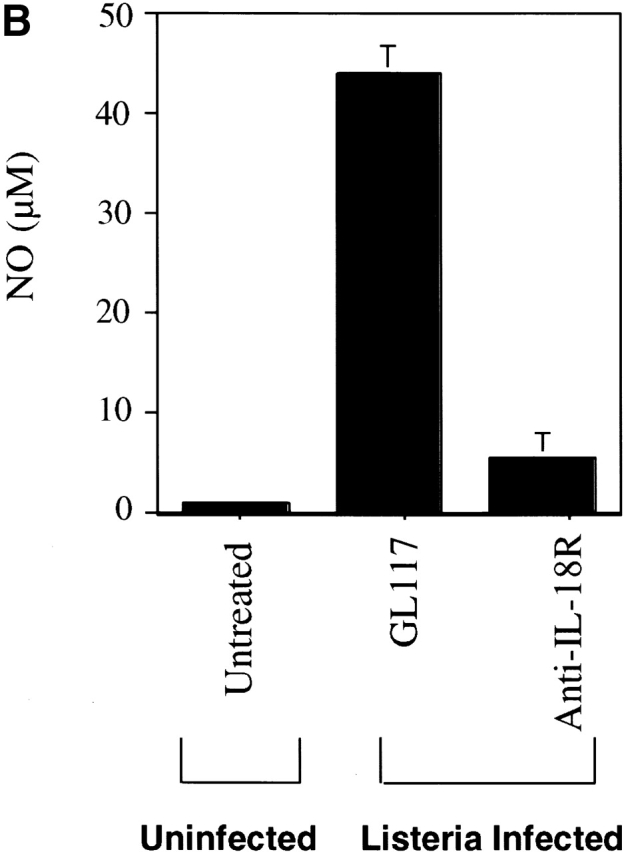
IL-18 induces TNF and NO production by macrophages from Listeria-infected mice. (A) Adherent peritoneal cells taken from SCID mice 3 d after infecting with 2 × 103 CFU Listeria were rested overnight and stimulated for 48 h with medium, IL-18, HKLM, or HKLM plus anti–IL-18R at concentrations detailed in Materials and Methods. The supernatants were assessed for TNF content by ELISA and data are represented as the mean ± SD of triplicate cultures. (B) Adherent peritoneal cells from SCID mice left untreated or taken 3 d after treating with the mAbs indicated ± infecting with 2 × 103 CFUs Listeria were stimulated for 16 h with HKLM and supernatants assessed for nitrite content as described in Materials and Methods. Data are represented as the mean ± SD of duplicate cultures.
NO forms part of a critical effector pathway involved in eliminating bacteria from the macrophage 65 66 67. We tested if this factor was also influenced by IL-18 using peritoneal (or splenic, data not shown) macrophages from SCID mice. In comparison to cells taken from noninfected mice, cells from Listeria-infected mice produced far higher levels of NO upon restimulation with HKLM (Fig. 4 B). Anti–IL-18R treatment before infection greatly impaired this response (Fig. 4 B) indicating that IL-18 was also required for this macrophage effector function. In addition, NO levels from all groups were lowered by addition of anti–IL-18R to the cultures themselves (data not shown).
Inhibition of IL-18 Action in Listeria Infection Greatly Enhances Neutrophilic Microfoci.
Livers from Listeria-infected mice treated with anti–IL-18R showed a dramatically increased GR1 staining for neutrophils, as compared with controls and this was restricted to microfoci within the tissue (Fig. 5). The frequency of neutrophil-rich microfoci in mice treated with mAbs to either IL-18R or TNF was similar and greater than seen using either anti–IFN-γ or anti–IL-12 mAbs, while the liver appeared normal by day 3 after infection in control animals (Fig. 5). Hence the frequency of these microfoci in the liver correlated completely with the capacity of each mAb to impair bacterial clearance in the spleen (Fig. 2 A and Fig. 3), but not with IFN-γ production (Fig. 2 B). F4/80 expression was only found on Kupffer cells distributed throughout the liver parenchyma, but not in the microfoci themselves, and was similar in all groups (Fig. 5), suggesting that additional macrophages had yet to influx the site at this time point. No CD4 and CD8 staining was found in the control group and only a few CD4+ or CD8+ cells were seen at the edge of the microfoci of those groups with pathology (Fig. 5).
Figure 5.

Histological analyses of Listeria-infected mouse livers confirms the dominant role of IL-18 and TNF in primary immune responses to Listeria infection. Displayed are frozen sections from the livers of Listeria-infected Balb/c mice treated with the mAb combinations indicated and stained with either GR1, F4/80, CD4, CD8, or isotype control (data not shown) mAbs as described in Materials and Methods.
IL-18 Has a Greater Effect on Th1 Effector Cells after their Differentiation.
Our studies suggest that IL-18 induced clearance of Listeria in part through its ability to increase IFN-γ production, which is in keeping with its reported ability to synergize in vitro with IL-12 for the induction of IFN-γ by NK and Th1 cells 34 68. We assessed the effect of anti–IL-18R on the development of Th1 cells in vitro by stimulating CD4+ Mel-14hi cells from DO11.10 mice with HKLM, OVA323–339, and APCs for 1 wk in the presence or absence of mAbs to IL-18R, IL-12, or TNF. The generation of Th1 cells was analyzed by measurement of IFN-γ production after restimulating with OVA and APCs. Results are shown in the absence of HKLM but were identical whether or not HKLM was present in these secondary recall cultures. IL-12 has been previously shown to play a dominant role in Th1 cell development in contrast to TNF 19 20. We now show that anti–IL-18R only slightly reduced the optimal development of Th1 cells seen in the absence of any blocking mAbs (Fig. 6 A). However, as suggested from our earlier studies 34, anti–IL-18R had a more pronounced effect on IFN-γ production during the recall responses of Th1 cells already developed in the absence of blocking mAbs for 7 d with HKLM, OVA, and APCs (Fig. 6 B). However, this was still less effective than anti–IL-12, which had a more prominent effect at this recall stage (Fig. 6 B). Anti-TNF had minimal effects not only in modulating the levels of IFN-γ detected from primary development but also in recall cultures (Fig. 6A and Fig. B) as shown previously 20 69. The role of IL-18 in enhancing IFN-γ production from already differentiated Th1 cells suggested that this cytokine might be critical for the optimal maintenance of Th1 responses necessary to generate potent effector memory cells, which are required for effective bacterial clearance of repeated Listeria infections.
Figure 6.

IL-18 has a greater effect on Th1 cells after their differentiation in vitro. Naive CD4+ spleen cells sorted from DO11.10 transgenic mice were set up in primary cultures with HKLM, OVA, and APCs (irradiated spleen cells) for 7 d. (A) Primary T cell cultures were performed in the presence of medium or mAbs to IL-12, IL-18R, or TNF as described in Materials and Methods. Cells were then restimulated with OVA plus APCs for 48 h and supernatants harvested to determine IFN-γ content. (B) Primary T cell cultures were stimulated with HKLM, OVA, and APCs in the absence of mAbs for 7 d and then restimulated with HKLM plus OVA plus APCs for 48 h in the presence of medium or mAbs to IL-12, IL-18R, or TNF when supernatants were harvested to determine IFN-γ content. All possible combinations of Ab were also analyzed in this experiment and their effects on lowering IFN-γ production were found to be additive (results not shown). Results were representative of three experiments and data are represented as the mean ± SD of triplicate cultures.
IL-18 Is a Dominant Factor in the Memory Effector Response Induced upon Reinfection with Listeria.
To test the role of IL-18 in memory effector responses to Listeria, mice given an initial sublethal Listeria infection of 2 × 103 CFUs were administered with anticytokine mAbs 27 d later, and then reinfected with a high dose of 5 × 104 CFU Listeria on day 28. The animals were assessed for bacterial burden and IFN-γ production after a further 3 d. Isotype-treated control mice were now able to completely clear this bacterial infection which is otherwise lethal in a primary response (Fig. 7 A) and this correlated with high levels of IFN-γ production upon antigenic stimulation (Fig. 7 B). In contrast to the controls, treatment with anti–IL-18R mAb led to a greatly enhanced bacterial burden, which was significantly greater than that observed after treatment with either anti–IL-12 or anti–IFN-γ mAbs (Fig. 7 A). This profound role of IL-18 in bacterial clearance was not completely attributable to reduction in the levels of IFN-γ. Specifically, treatment with anti–IL-18R mAb reduced the levels of IFN-γ to the same extent yet exacerbated the bacterial load significantly more than either anti–IL-12 or anti–IFN-γ mAbs (Fig. 7a and Fig. b). Indeed, the dominance of anti–IL-18R over anti–IL-12 was even more dramatic in impairing recall responses than had been observed in primary responses to Listeria. The effect of anti–IL-18R mAb on bacterial clearance was comparable to that of anti-TNF, although anti-TNF showed no effect on antigen-induced IFN-γ production (Fig. 7). At this challenge dose, there was no further enhancement in bacterial load using anti–IL-18R in combination with Abs to TNF, IFN-γ, or IL-12 (Fig. 7 A, and data not shown).
Figure 7.

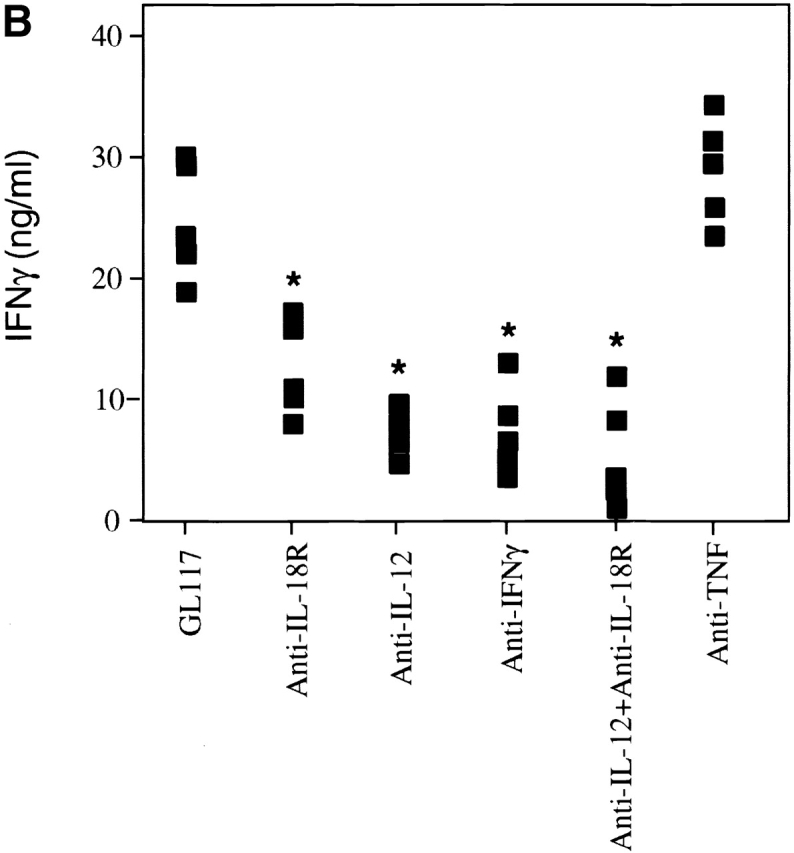
A critical role for IL-18 in memory recall responses and subsequent clearance of a repeat Listeria infection. Balb/c mice were infected with 2 × 103 CFU Listeria and 27 d later groups were treated with the indicated mAbs and then reinfected with 5 × 104 CFU Listeria on day 28. After a further 3 d the bacterial burden per spleen (A) and IFN-γ production by spleen cells upon restimulation with HKLM antigen for 48 h (B) were evaluated as described for Fig. 2. Results were representative of three experiments. Statistical analysis was performed using Dunnett's. *P < 0.05 versus isotype control.
In keeping with the similar effects of anti–IL-18R mAb in primary and secondary Listeria infections, the livers of mice treated with this mAb before a secondary infection also contained very high numbers of GR-1+ neutrophils within microfoci (data not shown). This is in keeping with an important role for IL-18 in the induction of recall responses.
Discussion
This study was performed to determine the role of IL-18 in protective immune responses to the intracellular pathogen Listeria. Our findings show that IL-18 provided significant protection in both primary and secondary responses to this pathogen and appeared more potent than either IL-12 or IFN-γ. Thus, IL-18 has a more extensive role in bacterial clearance than via its ability to induce IFN-γ production by Th1 and NK cells, since it can exacerbate the bacterial load even in IFN-γ–deficient mice. The main effects of IL-18 were observed only in the presence of TNF. Indeed, IL-18 induced macrophage production of TNF as well as NO, which is critical for its antimicrobial effects. Overall, our findings show that IL-18 plays a very important role in cell-mediated immunity via its effects on both the innate and adaptive immune response, which appear important for both primary and secondary challenge with this intracellular pathogen. Thus, IL-18 may prove to be beneficial when used as part of an antimicrobial treatment or when targeted as a therapeutic strategy for those disorders associated with Th1 inflammatory responses.
Our data show that IL-18 plays a dominant role in the clearance of Listeria in a manner not fully attributable to the induction of IFN-γ. Evidence for this came from the following findings. First, the bacterial load and mortality was higher in the absence of IL-18 than in the absence of IL-12, while the production of IFN-γ was reduced similarly in each case. Secondly, IL-18, but not IL-12, was dominant over IFN-γ in controlling the bacterial load after infection. Furthermore, the infection was greatly exacerbated by anti–IL-18R mAb even in IFN-γ−/− mice. Finally, IL-18 operated independently of IFN-γ to reduce the bacterial load early in infection. Although these findings are consistent with the IFN-γ–dependent role of IL-12 in protection to a primary challenge with Listeria 16 they suggest an additional role for IL-18. We had originally published findings describing the mechanism by which IL-18 induced IFN-γ production by Th1 cells in concert with IL-12 34. Moreover, many studies now suggest that the ability of IL-18 to enhance cell-mediated Th1-type responses is mainly via the upregulation of the IL-12 receptor 70 71. Our present findings, showing that IL-18 has effects over and above the induction of IFN-γ, demonstrate its more global role in immune responses than had originally been suggested. That IFN-γ levels remained detectable after anti–IL-18R mAb treatment also indicated that the responsive cells were not being depleted by this mAb.
We show that IL-18 and TNF display a similar capacity to protect mice from Listeria and that the protection mediated by IL-18 can only partially overcome the ability of anti-TNF to enhance the bacterial load, thereby demonstrating that IL-18 mediates protection to a large extent through TNF action in vivo. Since we were unable to demonstrate a requirement for IL-18 in the TNF produced by in vitro cultures designed to stimulate T and NK cells from Listeria-infected mice we investigated the macrophage as an alternative IL-18–dependent source of TNF. Indeed, we found that TNF production by F4/80+ peritoneal macrophages was induced by IL-18 after a Listeria infection, in keeping with previous findings in nonpathogen-driven systems 62 63 64. Moreover, our ability to show that the TNF produced in response to HKLM was IL-18–dependent provided further evidence for the TNF-inducing properties of IL-18 in response to Listeria. Hence we conclude that the role of IL-18 in responses to Listeria encompasses the induction of TNF production by macrophages and IFN-γ production by antigen-specific T cells and NK cells.
Our findings showing that macrophages were triggered by IL-18 to secrete TNF in response to Listeria led us to speculate that other properties of the macrophage may also be IL-18 dependent. We confirm here that IL-18 is required for the macrophage's subsequent release of NO in response to a Listeria infection in vivo. Since NO production is a key mediator of the macrophage's listericidal effects 65 66 67, this may be a major mechanism to explain the profound influence of IL-18 on bacterial clearance.
We also show that upon neutralization of IL-18, Listeria elicited a massive neutrophil influx to the liver with formation of multiple microfoci. This pathology was comparable to that seen in the absence of TNF with fewer and smaller microfoci observed in the absence of either IL-12 or IFN-γ. Hence, the situation in the liver correlated with the relative capacity for bacterial clearance from the spleen in these situations. It has been shown that a “spillover” of Listeria from the granuloma to neutrophilic microabscesses occurs in SCID mice chronically infected with Listeria and depleted of IFN-γ or TNF 72. In our hands, a lack of IL-18 receptor signaling resulted in a similar outcome but in this case in immunocompetent mice, reflecting an inability to clear the organism and a disruption in the equilibrium between the macrophage-rich granulomas and the neutrophilic microabscesses.
Here we show for the first time that in addition to its protective role in primary immunity to Listeria, IL-18 is as important in memory effector responses to reinfection with Listeria. Since IL-18 was required for optimal levels of IFN-γ it may represent the missing element which has been postulated to act in combination with IL-12 for maximal IFN-γ recall responses to Listeria 28. Hence, one role for IL-18 may be to induce optimal Th1 memory effector cell responses to repeat infections with Listeria. However, in our hands IL-18 appeared to have a more extensive role in bacterial clearance than via its effects on IFN-γ. Indeed IL-18 was as potent as TNF in bacterial clearance. Effective recall responses to Listeria are known to require TNF 27 29 but its mechanism of induction and subsequent effects remain to be clearly defined in a recall response to Listeria. The extent to which IL-18 operates via TNF and/or other additional mechanisms in a secondary infection also remains to be determined. As these findings were reminiscent of those observed during a primary immune response to Listeria and indeed the histology was also similar, it seems possible that similar mechanisms may operate to eradicate intracellular pathogens during primary and memory effector responses, with IL-18 playing a dominant role at both stages.
It is also possible that IL-18 influences other responses and/or cell types invoked by a Listeria infection. IL-18 can augment the cytotoxic activity of CD8+ T cells 73 and this cell subset is an essential component of the host defense to Listeria 40. The rapid mortality of Listeria-infected mice given anti–IL-18R showed that IL-18 was important for controlling bacterial expansion at an early stage in the infection. Indeed we found that mice treated with anti–IL-18R did not survive beyond days 3–4 (Fig. 1) even after infecting with only 100 CFU Listeria (data not shown). Since a primary CTL response to Listeria is not detectable until after this time 74 75 we were unable to determine how its induction was affected by anti–IL-18R mAb treatment. Memory effector CTLs on the other hand are invoked more quickly to Listeria 74 75, and we show here that IL-18 is also required for an effective recall response to secondary challenge with Listeria. However, we could find no impairment of the antigen-specific CTL responses during a secondary challenge in Listeria-immune mice treated with anti–IL-18R before reinfection (data not shown). Therefore, we can conclude that IL-18 is not required for the cytotoxic effector function of memory CTLs induced by a secondary infection with Listeria. However, the bacterial load is very high (Fig. 7 A) when Listeria-immune mice are given anti–IL-18R mAbs before reinfection suggesting that the CTL activity may be insufficient for host resistance in the absence of effective innate immunity. Moreover, the possibility still exists that IL-18 is required for the development of memory CTLs. This will be difficult to assess since mice will not survive treatment with anti–IL-18R mAbs during a primary infection to allow the development of memory CTL responses to Listeria.
IL-18 has been shown to be important for organism clearance in a number of acute pathogenic infections. IL-18−/− mice were impaired in their capacity to eliminate Leishmania major 46 and Shigella 47. Moreover, anti–IL-18 treatment exacerbated the bacterial burden after infection with Salmonella 48 or Yersinia 44, while IL-18 protein treatment reduced titers of herpes simplex virus type 1 45 and lowered pock formation after a Vaccinia virus infection 49. Only this latter study suggested an IFN-γ–independent protective role for IL-18 by showing that IL-18 treatment improved symptoms even in mice infected with Vaccinia virus and depleted of IFN-γ. The extent of IFN-γ–independent IL-18–mediated protection had not, to our knowledge, been investigated in bacterial infections. Moreover, only the role of IL-18 in the acute response to an initial infection had been focused on in the existing literature, in contrast to our study demonstrating a role for IL-18 in both primary and recall responses to Listeria infections.
In conclusion, our study shows that IL-18 plays a profound role in protective cell-mediated immune responses to an intracellular pathogen, which extend beyond its known effects on the induction of IFN-γ by NK and T cells, to the induction of TNF and NO production by macrophages. For these reasons we believe that our data have important therapeutic implications for the use of IL-18 to induce appropriate immune responses during vaccination or to eradicate chronic infections arising as a result of immunosuppression. Conversely, it is known that such responses can also be pathology-inducing in autoimmune and inflammatory disorders and indeed IL-18 has been implicated in the development of several autoimmune diseases 76 77 78 79 80. Therefore, our data would predict that antagonizing the global effects of IL-18 might have more impact on the treatment of such diseases than the current strategies of TNF, IL-12, or IFN-γ antagonism. Our novel finding that IL-18 is necessary for a recall cell-mediated immune response further supports the potential for using IL-18 antagonists in preexisting inflammatory pathologies.
Acknowledgments
The authors thank Drs. Emil R. Unanue, Gregory J. Bancroft, and Joan K. Brieland for their comments on the manuscript and Maribel Andonian for her assistance with graphics.
Footnotes
Abbreviation used in this paper: HKLM, heat-killed Listeria monocytogenes.
References
- Barza M. Listeriosis in milk. N. Engl. J. Med. 1985;312:438–440. doi: 10.1056/NEJM198502143120710. [DOI] [PubMed] [Google Scholar]
- North R.J. Cellular mediators of anti-Listeria immunity as an enlarged population of short-lived replicating T cells. J. Exp. Med. 1973;138:342–355. doi: 10.1084/jem.138.2.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray M.L., Killinger A.H. Listeria monocytogenes and listeric infections. Bacteriol. Rev. 1966;30:309–382. doi: 10.1128/br.30.2.309-382.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeliger, H.P.R. 1961. Listeriosis. Hafner, editor. New York. 308 pp.
- Jurado R.L., Farley M.M., Pereira E., Harvey R.C., Schuchat A., Wenger J.D., Stephens D.S. Increased risk of Listeria monocytogenes meningitis and bacteraemia in patients with human immunodeficiency virus infection. Clin. Infect. Dis. 1993;17:224–227. doi: 10.1093/clinids/17.2.224. [DOI] [PubMed] [Google Scholar]
- Mackaness G.B. Cellular resistance to infection. J. Exp. Med. 1962;116:381–390. doi: 10.1084/jem.116.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann, S.H.E. 1994. In Fundamental Immunology, 3rd edition. W.E. Paul, editor. Raven Press, New York. pp. 1287–1308.
- Bancroft G.J., Schrieber R.D., Unanue E.R. Natural immunitya T cell independent pathway of macrophage activation, defined in the SCID mouse. Immunol. Rev. 1991;124:5–24. doi: 10.1111/j.1600-065x.1991.tb00613.x. [DOI] [PubMed] [Google Scholar]
- Rogers H.W., Unanue E.R. Neutrophils are involved in acute, nonspecific resistance to Listeria monocytogenes in mice. Infect. Immun. 1993;61:5090–5096. doi: 10.1128/iai.61.12.5090-5096.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann S.H.E., Blum C., Yamamoto S. Crosstalk between alpha/beta T cells and gamma/delta T cells in vivoactivation of alpha/beta T-cell responses after gamma/delta T-cell modulation with the monoclonal antibody GL3. Proc. Natl. Acad. Sci. USA. 1993;90:9620–9624. doi: 10.1073/pnas.90.20.9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackaness G.B., Hill W.C. The effect of anti-lymphocyte globulin on cell-mediated resistance to infection. J. Exp. Med. 1969;129:993–1012. doi: 10.1084/jem.129.5.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn H., Kaufmann S.H. The role of cell-mediated immunity in bacterial infections. Rev. Infect. Dis. 1981;3:1221–1250. doi: 10.1093/clinids/3.6.1221. [DOI] [PubMed] [Google Scholar]
- Buchmeier N.A., Schreiber R.D. Requirement of endogenous interferon-γ production for resolution of Listeria monocytogenes infection. Proc. Natl. Acad. Sci. USA. 1985;82:7404–7408. doi: 10.1073/pnas.82.21.7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancroft G.J., Schreiber R.D., Bosma G.C., Bosma M.J., Unanue E.R. A T cell-independent mechanism of macrophage activation by interferon-γ. J. Immunol. 1987;139:1104–1107. [PubMed] [Google Scholar]
- Beller D.I., Kiely J.M., Unanue E.R. Regulation of macrophage populations. I. Preferential induction of Ia-rich peritoneal exudates by immunologic stimuli. J. Immunol. 1980;124:1426–1432. [PubMed] [Google Scholar]
- Tripp C.S., Gately M.K., Hakimi J., Ling P., Unanue E.R. Neutralization of IL-12 decreases resistance to Listeria in SCID and C.B-17 mice. J. Immunol. 1994;152:1883–1887. [PubMed] [Google Scholar]
- Bancroft G.J., Sheehan K.C., Schreiber R.D., Unanue E.R. Tumor necrosis factor is involved in the T-cell independent pathway of macrophage activation in SCID mice. J. Immunol. 1989;143:127–130. [PubMed] [Google Scholar]
- Wherry J.C., Schreiber R.D., Unanue E.R. Regulation of γ interferon prodcution by natural killer cells in scid miceroles of tumor necrosis factor and bacterial stimuli. Infect. Immun. 1991;59:1709–1715. doi: 10.1128/iai.59.5.1709-1715.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh C.-S., Macatonia S.E., Tripp C.S., Wolf S.F., O'Garra A., Murphy K.M. Development of Th1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- Shibuya K., Robinson D., Zonin F., Hartley S.B., Macatonia S.E., Somoza C., Hunter C.A., Murphy K.M., O'Garra A. IL-1α and TNF-α are required for IL-12-induced development of Th1 cells producing high levels of IFN-γ in BALB/c but not C57BL/6 mice. J. Immunol. 1998;160:1708–1716. [PubMed] [Google Scholar]
- Harty J.T., Bevan M.J. Specific immunity to Listeria monocytogenes in the absence of IFNγ. Immunity. 1995;3:109–117. doi: 10.1016/1074-7613(95)90163-9. [DOI] [PubMed] [Google Scholar]
- Huang S., Hendriks W., Althage A., Hemmi S., Bluethmann H., Kanifo R., Vulcek J., Zienkernagel R.M., Aguet M. Immune response in mice that lack the interferon-γ receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- Oxenius A., Karrer U., Zinkernagel R.M., Hengartner H. IL-12 is not required for induction of type 1 cytokine responses in viral infections. J. Immunol. 1999;162:965–973. [PubMed] [Google Scholar]
- Pfeffer K., Matsuyama T., Kundig T.M., Wakeham A., Kishihara K., Shahinian A., Wiegmann K., Ohashi P.S., Kronke M., Mak T.M. Mice deficient for the 55kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- Rothe J., Lesslauer W., Lotscher H., Lang Y., Koebel P., Kontgen F., Althage A., Zinkernagel R., Steinmetz M., Bluethmann H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes . Nature. 1993;364:798–801. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- Ehlers S., Mielke M.E.A., Blankenstein T., Hahn H. Kinetic analysis of cytokine gene expression in the livers of naive and immune mice infected with Listeria monocytogenes . J. Immunol. 1992;149:3016–3022. [PubMed] [Google Scholar]
- Nakane A., Minagawa T., Kohanawa M., Chen Y., Sato H., Moriyama M., Tsuruoka N. Interactions between endogenous γ interferon and tumor necrosis factor in host resistance against primary and secondary Listeria monocytogenes infections. Infect. Immun. 1989;57:3331–3337. doi: 10.1128/iai.57.11.3331-3337.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripp C.S., Kanagawa O., Unanue E.R. Secondary response to Listeria infection requires IFN-γ but is partially independent of IL-12. J. Immunol. 1995;155:3427–3432. [PubMed] [Google Scholar]
- Samson J.N., Langermans J.A.M., Savelkoul H.F.J., Furth S.V. Tumor necrosis factor, but not interferon-γ, is essential for acquired resistance to Listeria monocytogenes during a secondary infection in mice. Immunology. 1995;86:256–262. [PMC free article] [PubMed] [Google Scholar]
- Czuprynski C.J., Brown J.F., Wagner R.D., Steinberg H. Administration of antigranulocyte monoclonal antibody RB6-8C5 prevents expression of acquired resistance to Listeria monocytogenes infection in previously immunized mice. Infect. Immun. 1994;62:5161–5163. doi: 10.1128/iai.62.11.5161-5163.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhmilevich A.L. Neutrophils are essential for resolution of primary and secondary infection with Listeria monocytogenes . J. Leukoc. Biol. 1995;57:827–831. doi: 10.1002/jlb.57.6.827. [DOI] [PubMed] [Google Scholar]
- Okamura H., Tsutsi H., Komatsu T., Yutsudo M., Hakura A., Tanimoto T., Torigoe K., Okura T., Nukada Y., Hattori K. Cloning of a new cytokine that induces IFN-γ production by T cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- Okamura H., Nagata K., Komatsu T., Tanimoto T., Nukata Y., Tanabe F., Akita K., Torigoe K., Okura T., Fukuda S., Kurimoto M. A novel costimulatory factor for γ interferon induction found in the livers of mice causes endotoxic shock. Infect. Immun. 1995;63:3966–3972. doi: 10.1128/iai.63.10.3966-3972.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D., Shibuya K., Mui A., Zonin F., Murphy E., Sana T., Hartley S.B., Menon S., Kastelein R., Bazan F., O'Garra A. IGIF does not drive Th1 development but synergizes with IL-12 for interferon-γ production and activates IRAK and NFκB. Immunity. 1997;7:571–581. doi: 10.1016/s1074-7613(00)80378-7. [DOI] [PubMed] [Google Scholar]
- Hunter C.A., Timans J., Pisacane P., Menon S., Cai G., Chizzonitte R., Bazan J.F., Kastelein R.A. Comparison of the effects of interleukin-1α (IL-1α), IL-1β and IGIF (IL-1γ) on the production of interferon-γ by NK cells. Eur. J. Immunol. 1997;27:2787–2792. doi: 10.1002/eji.1830271107. [DOI] [PubMed] [Google Scholar]
- Takeda K., Tsutsui H., Yoshimoto T., Adachi O., Yoshida N., Kishimoto T., Okamura H., Nakanishi K., Akira S. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity. 1998;8:383–390. doi: 10.1016/s1074-7613(00)80543-9. [DOI] [PubMed] [Google Scholar]
- Tsutsui H., Nakanishi K., Matsui K., Higashino K., Okamura H., Miyazawa Y., Kaneda K. IFN-γ-inducing factor up-regulates Fas ligand-mediated cytotoxic activity of murine natural killer cell clones. J. Immunol. 1996;157:3967–3973. [PubMed] [Google Scholar]
- Hyodo Y., Matsui K., Hayashi N., Tsutsui H., Kashiwamura S., Yamauchi H., Hiroishi K., Takeda K., Tagawa Y., Iwakura Y. IL-18 up-regulates perforin-mediated NK activity without increasing perforin messenger RNA expression by binding to constitutively expressed IL-18 receptor. J. Immunol. 1999;162:1662–1668. [PubMed] [Google Scholar]
- Dao T., Ohashi K., Kayano T., Kurimoto M., Okamura H. Interferon-γ-inducing factor, a novel cytokine, enhances Fas ligand-mediated cytotoxicity of murine T helper 1 cells. Cell. Immunol. 1996;173:230–235. doi: 10.1006/cimm.1996.0272. [DOI] [PubMed] [Google Scholar]
- Kagi D., Ledermann B., Burki K., Zinkernagel R.M., Hengartner H. Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Annu. Rev. Immunol. 1996;14:207–232. doi: 10.1146/annurev.immunol.14.1.207. [DOI] [PubMed] [Google Scholar]
- Ushio S., Namba M., Okura T., Hattori K., Nukada Y., Akita Y., Tanabe F., Konishi K., Okamura H., Kurimoto M. Cloning of the cDNA for human IFN-γ-inducing factor, expression in Escherichia coli, and studies on the biologic activities from non-CD14+ human blood mononuclear cells. J. Immunol. 1996;156:4274–4279. [PubMed] [Google Scholar]
- Micallef M., Ohtsuki T., Kohno K., Tanabe F., Ushio S., Namba M., Tanimoto T., Torigoe K., Fujii M., Ikeda M. Interferon-γ inducing factor enhances T helper 1 cytokine production by stimulated human T cellssynergism with IL-12 for interferon-γ production. Eur. J. Immunol. 1996;26:1647–1651. doi: 10.1002/eji.1830260736. [DOI] [PubMed] [Google Scholar]
- Kohno K., Kataoka J., Ohtsuki T., Suemoto Y., Okamoto I., Usui M., Ikeda M., Kurimoto M. IFN-γ-inducing factor (IGIF) is a costimulatory factor on the activation of Th1 but not Th2 cells and exerts its effect independently of IL-12. J. Immunol. 1997;158:1541–1550. [PubMed] [Google Scholar]
- Bohn E., Sing A., Zumbihl R., Biefeldt C., Okamura H., Kurimoto M., Heesemann J., Autenrieth I.B. IL-18 (IFN-γ-inducing factor) regulates early cytokine production in, and promotes resolution of, bacterial infection in mice. J. Immunol. 1998;160:299–307. [PubMed] [Google Scholar]
- Fujioka N., Akazawa R., Ohashi K., Fujii M., Ikeda M., Kurimoto M. Interleukin-18 protects mice against acute herpes simplex virus type 1 infection. J. Virol. 1999;73:2401–2409. doi: 10.1128/jvi.73.3.2401-2409.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X.Q., Leung B.P., Niedbala W., Peidrafita D., Feng G.J., Sweet M., Dobbie L., Smith A.J., Liew F.Y. Altered immune responses and susceptiblity to Leishmania major and Staphylococcus aureus infection in IL-18 deficient mice. J. Immunol. 1999;163:2821–2828. [PubMed] [Google Scholar]
- Sansonetti P.J., Phalipon A., Arondel J., Thirumalai K., Banerjee S., Akira S., Takeda K., Zychlinsky A. Caspase-1 activation of IL-1β and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity. 2000;12:581–590. doi: 10.1016/s1074-7613(00)80209-5. [DOI] [PubMed] [Google Scholar]
- Mastroeni P., Clare S., Khan S., Harrison J.A., Hormaeche C.E., Okamura H., Kurimoto M., Dougan G. Interleukin 18 contributes to host resistance and γ interferon production in mice infected with virulent Salmonella typhimurium . Infect. Immunol. 1999;67:478–483. doi: 10.1128/iai.67.2.478-483.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka-Kataoka M., Kunikata T., Takayama S., Iwaki K., Ohashi K., Ikeda M., Kurimoto M. In vivo antiviral effect of interleukin 18 in a mouse model of vaccinia virus infection. Cytokine. 1999;11:593–599. doi: 10.1006/cyto.1998.0453. [DOI] [PubMed] [Google Scholar]
- Torigoe K., Ushio S., Okaru T., Kobayashi S., Taniai M., Kunikata T., Murakami T., Sanou O., Kojima H., Fujii M. Purification and characterization of the human interleukin-18 receptor. J. Biol. Chem. 1997;272:25737–25742. doi: 10.1074/jbc.272.41.25737. [DOI] [PubMed] [Google Scholar]
- Born T.L., Thomassen E., Bird T.A., Sims J.E. Cloning of a novel receptor subunit, AcPL, required for interleukin-18 signaling. J. Biol. Chem. 1998;273:29445–29450. doi: 10.1074/jbc.273.45.29445. [DOI] [PubMed] [Google Scholar]
- Hoshino K., Tsutsui H., Kawai T., Takeda K., Nakanshi K., Takeda Y., Akira S. Cutting edgegeneration of IL-18 receptor-deficient mice, evidence for IL-1 receptor-related protein as an essential IL-18 binding protein. J. Immunol. 1999;162:5041–5044. [PubMed] [Google Scholar]
- Debets R., Timans J.C., Churakowa T., Zurawski S., Waal-Malefyt R.D., Moore K.W., Abrams J.S., O'Garra A., Bazan J.F., Kastelein R.A. IL-18 receptors, their role in ligand binding and function. Anti-IL18RAcPL antibody, a potent antagonist of IL-18. J. Immunol. 2000;165:4950–4956. doi: 10.4049/jimmunol.165.9.4950. [DOI] [PubMed] [Google Scholar]
- Murphy K.M., Heimberger A.B.A., Loh D.Y. Induction by antigen of intrathymic apoptosis of CD4+CD8+ TCRlo thymocytes in vivo. Science. 1990;250:1720–1724. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- Haskins K., Kobo R., White J., Pigeon M., Kappler J., Marrack P. The MHC-restricted antigen on T cells. I. Isolation of a monoclonal antibody. J. Exp. Med. 1983;157:1149–1169. doi: 10.1084/jem.157.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed L.J., Muench H. A simple method of estimating fifty percent endpoints. The American J. Hygiene. 1938;27:493–497. [Google Scholar]
- Bradley L.M., Duncan D.D., Tonkonogy S., Swain S.L. Characterization of antigen-specific CD4+ effector T cells in vivoimmunization results in a transient population of Mel-14−, CD45RB− helper cells that secretes interleukin 2(IL-2), IL-3, IL-4, and interferon γ. J. Exp. Med. 1991;174:547–559. doi: 10.1084/jem.174.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green S.J., Mellouk S., Hoffman S.L., Meltzer M.S., Nacy C.A. Cellular mechanisms of nonspecific immunity to intracellular infectioncytokine-induced synthesis of toxic nitrogen oxides from L-arginine by macrophages and hepatocytes. Immunol. Lett. 1990;25:15–19. doi: 10.1016/0165-2478(90)90083-3. [DOI] [PubMed] [Google Scholar]
- Slade S.J., Langhorne J. Production of interferon-γ during infection of mice with plasmodium chabaudi chabaudi. Immunobiology. 1989;179:353–365. doi: 10.1016/S0171-2985(89)80041-5. [DOI] [PubMed] [Google Scholar]
- Murphy E., Shibuya K.K., Hosken N., Openshaw P., Maino V., Davis K., Murphy K., O'Garra A. Reversibility of T helper 1 and 2 populations is lost after long-term stimulation. J. Exp. Med. 1996;183:901–913. doi: 10.1084/jem.183.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripp C.S., Wolf S.E., Unanue E.R. Interleukin-12 and tumor necrosis factor a are costimulators of interferon-γ production by natural killer cells in severe combined immunodeficiency mice with listeriosis, and interleukin 10 is a physiologic anatagonist. Proc. Natl. Acad. USA. 1993;90:3725–3729. doi: 10.1073/pnas.90.8.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung B.P., McInnes I.B., Esfandiari E., Wei X.-Q., Liew F.Y. Combined effects of IL-12 and IL-18 on the induction of collagen-induced arthritis. J. Immunol. 2000;164:6495–6502. doi: 10.4049/jimmunol.164.12.6495. [DOI] [PubMed] [Google Scholar]
- Puren A.J., Fantuzzi G., Gu Y., Su M.S.-S., Dinarello C.A. Interleukin-18 (IFN-γ-inducing factor) induces IL-1β and IL-8 via TNFα production from non-CD14 human blood mononuclear cells. J. Clin. Invest. 1998;101:711–721. doi: 10.1172/JCI1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui H., Matsui K., Kawada N., Hyodo Y., Hayashi N., Okamura H., Higashino K., Nakanishi K. IL-18 accounts for both TNF-α- and Fas ligand-mediated hepatotoxic pathways in endotoxin-induced liver injury in mice. J. Immunol. 1997;159:3961–3967. [PubMed] [Google Scholar]
- Beckerman K.P., Rogers H.W., Corbett J.A., Schreiber R.D., McDaniel M.L., Unanue E.R. Release of nitric oxide during the T cell-independent pathway of macrophage activation. Its role in resistance to Listeria monocytogenes . J. Immunol. 1993;150. 3:888–895. [PubMed] [Google Scholar]
- Boockvar K.S., Granger D.L., Poston R.M., Maybodi M., Washington M.K., Hibbs J.B., Jr., Kurlander R.L. Nitric oxide produced during murine listeriosis is protective. Infect. Immun. 1994;62:1089–1100. doi: 10.1128/iai.62.3.1089-1100.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking J.D., Nathan C., Hom G., Chartrain D.S., Fletcher M., Trumbauer K., Stevens Q.W., Xie K., Sokol, Hutchinson N. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- Hunter C.A., Chizzonite R., Remington J.S. IL-1β is required for IL-12 to induce production of IFN-γ by NK cells. A role for IL-1β in the T cell-independent mechanism of resistance against intracellular pathogens. J. Immunol. 1995;155:4347–4354. [PubMed] [Google Scholar]
- Hsieh C.-S., Macatonia S.E., O'Garra A., Murphy K.M. Pathogen-induced Th1 phenotype development in CD4+ αβ-TCR transgenic T cells is macrophage dependent. Int. Immunol. 1993;5:371–382. doi: 10.1093/intimm/5.4.371. [DOI] [PubMed] [Google Scholar]
- Xu D., Chan W.L., Leung B.P., Hunter D., Schulz K., Carter R.W., McInnes I.B., Robinson J.H., Liew F.Y. Selective expression and functions of interleukin 18 receptor on T helper (Th) type 1 but not Th2 cells. J. Exp. Med. 1998;188:1485–1492. doi: 10.1084/jem.188.8.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J.T., Segal B.M., Nakanishi K., Okamura H., Shevach E.M. The costimulatory effect of IL-18 on the induction of antigen-specific IFN-γ production by resting T cells is IL-12 dependent and is mediated by up-regulation of the IL-12 receptor β2 subunit. Eur. J. Immunol. 2000;30:1113–1119. doi: 10.1002/(SICI)1521-4141(200004)30:4<1113::AID-IMMU1113>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Bhardwaj V., Kanagawa O., Swanson P.E., Unanue E.R. Chronic Listeria infection in SCID micerequirements for the carrier state and the dual role of T cells in transferring protection or suppression. J. Immunol. 1998;160:376–384. [PubMed] [Google Scholar]
- Okamoto I., Kohno K., Tanimoto T., Ikegami H., Kurimoto M. Development of CD8+ effector T cells is differentially regulated by IL-18 and IL-12. J. Immunol. 1999;162:3202–3211. [PubMed] [Google Scholar]
- Busch D.H., Pamer E.G. T lymphocyte dynamics during Listeria monocytogenes infection. Immunol. Lett. 1999;65:93–98. doi: 10.1016/s0165-2478(98)00130-8. [DOI] [PubMed] [Google Scholar]
- Seaman M.S., Wang C.R., Forman J. MHC class Ib-restricted CTL provide protection against primary and secondary Listeria monocytogenes infection. J. Immunol. 2000;165:5192–5201. doi: 10.4049/jimmunol.165.9.5192. [DOI] [PubMed] [Google Scholar]
- Rothe H., Jenkins N., Copeland N., Kolb H. Active stage of autoimmune diabetes is associated with the expression of a novel cytokine, IGIF, which is located near Idd2. J. Clin. Invest. 1997;99:469–474. doi: 10.1172/JCI119181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildbaum G., Youssef S., Grabie N., Karin N. Neutralizing antibodies to IFN-γ-inducing factor prevent experimental autoimmune encephalomyelitis. J. Immunol. 1998;161:6368–6374. [PubMed] [Google Scholar]
- Shi F., Takeda K., Akira S., Sarvetnick N., Ljunggren H. IL-18 directs autoreactive T cells and promotes autodestruction in the central nervous system via induction of IFN-γ by NK cells. J. Immunol. 2000;165:3099–3104. doi: 10.4049/jimmunol.165.6.3099. [DOI] [PubMed] [Google Scholar]
- Pizarro T.T., Michie M.H., Bentz M., Woraratanadharm J., Smith M.F., Foley E.J., Moskaluk C.A., Bickston S.J., Cominelli F. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn's diseaseexpression and localization in intestinal mucosal cells. J. Immunol. 1999;162:6829–6835. [PubMed] [Google Scholar]
- Gracie J.A., Forsey R.J., Chan W.L., Gilmour A., Leung B.P., Greer M.R., Kennedy K., Carter R., Wei W.Q. A proinflammatory role for IL-18 in rheumatoid arthritis. J. Clin. Invest. 1999;104:1393–1401. doi: 10.1172/JCI7317. [DOI] [PMC free article] [PubMed] [Google Scholar]