

Abstract
Experimental autoimmune encephalomyelitis (EAE), a model for multiple sclerosis, can be induced by immunization with a number of myelin antigens. In particular, myelin oligodendrocyte glycoprotein, a central nervous system (CNS)-specific antigen expressed on the myelin surface, is able to induce a paralytic MS-like disease with extensive CNS inflammation and demyelination in several strains of animals. Although not well understood, the egress of immune cells into the CNS in EAE is governed by a complex interplay between pro and antiinflammatory cytokines and chemokines. The hematopoietic growth factor, granulocyte macrophage colony-stimulating factor (GM-CSF), is considered to play a central role in maintaining chronic inflammation. The present study was designed to investigate the previously unexplored role of GM-CSF in autoimmune-mediated demyelination. GM-CSF−/− mice are resistant to EAE, display decreased antigen-specific proliferation of splenocytes, and fail to sustain immune cell infiltrates in the CNS, thus revealing key activities for GM-CSF in the development of inflammatory demyelinating lesions and control of migration and/or proliferation of leukocytes within the CNS. These results hold implications for the pathogenesis of inflammatory and demyelinating diseases and may provide the basis for more effective therapies for inflammatory diseases, and more specifically for multiple sclerosis.
Keywords: multiple sclerosis, experimental autoimmune encephalomyelitis, GM-CSF, myelin oligodendrocyte glycoprotein, immune therapy
Introduction
Experimental autoimmune encephalomyelitis (EAE), a CD4+ T cell–mediated inflammatory demyelinating disease of the central nervous system (CNS) serves as an experimental model of the human disease, multiple sclerosis (MS; reference 1). EAE can be induced by immunization with a number of myelin antigens or by adoptive transfer of myelin-reactive CD4+ T cells into naive recipients. Historically, myelin basic protein and proteolipid protein, the major proteins of CNS myelin, were identified as the major encephalitogenic antigens in EAE 2 3. However, a number of laboratories have now focused their attention to the autoimmune response to myelin oligodendrocyte glycoprotein (MOG), a quantitatively minor myelin protein. MOG is of particular interest because it is a CNS-specific antigen expressed on the myelin surface that is able to induce a paralytic MS-like disease with extensive CNS inflammation and demyelination in several strains of animals 4 5 6. Of particular interest is the fact that C57BL/6 mice exhibit a chronic nonremitting disease while NOD/Lt mice develop a severe relapsing-remitting disease 4 7, thus making this model a useful tool to study the two most common clinical forms of MS.
Although the aetiological events and the molecular mechanisms leading to autoimmune demyelination are not fully understood, work in our and other laboratories has indicated that the activation and recruitment of lymphocytic and mononuclear cells to the CNS is a necessary step in the development of pathological inflammatory lesions. The egress of immune cells into the CNS is governed by a complex interplay between cytokines, chemokines, and adhesion molecules. Many studies have shown that the differential production of such pro-(Th1) and anti-(Th2) inflammatory molecules mediates and modulates the course of disease. The hematopoietic growth factor, GM-CSF, was first considered to be proinflammatory because of its ability to stimulate macrophage plasminogen activator activity 8. A number of subsequent studies have shown that it can also regulate the many functions of mature myeloid cells 9, for example, activation of dendritic cells (DCs; reference 10), necessary for T cell activation by foreign proteins, induction of monocyte/macrophage MHC class II expression 11, enhancement of the phagocytic activity and antigen presenting function of macrophages and/or microglia 12 13, priming of monocytes for cytokine production 14 15, and enhancement of macrophage and granulocyte adherence 16 17. A cytokine network of general relevance has been proposed in which GM-CSF has a central role in maintaining chronic inflammation 18.
The proinflammatory role of GM-CSF has been demonstrated using various models of inflammation and immunity. We have shown that administration of rGM-CSF accelerates onset and exacerbates the pathology of murine collagen-induced arthritis (CIA; reference 19) while GM-CSF–deficient mice show decreased susceptibility to CIA 20. GM-CSF–deficient mice also show exacerbated susceptibility to infection with Listeria monocytogenes, which correlates with a poor inflammatory response and failure to maintain a supply of phagocytic cells over the long term 21. Transgenic mice expressing GM-CSF, driven by a constitutive promoter, demonstrate an excessive accumulation and activation of macrophages and granulocytes 22 23 and local expression of GM-CSF in the stomach is sufficient to initiate autoimmune gastritis 24. In light of these observations and given that elevated levels of GM-CSF have been shown to correlate with the active phase of MS 25, it is possible that GM-CSF may play a role in initiating or sustaining immune responses in autoimmune inflammatory and demyelinating diseases such as MS and EAE.
Therefore, this study was designed to examine whether GM-CSF was critical for initiation and/or progression of EAE. For this, we have used GM-CSF–deficient (GM-CSF−/−) mice that have been backcrossed onto an EAE-susceptible NOD/Lt background. Mice with a disrupted GM-CSF gene show no major perturbation of hematopoiesis 26 27 and can mount both primary cell-mediated and humoral immune responses 28. Here, we report that GM-CSF−/− mice are resistant to EAE, display decreased antigen-specific proliferation of splenocytes, and fail to sustain immune cell infiltrates in the CNS.
Materials and Methods
Mice.
NOD/Lt mice (10–16 wk old) were bred at the La Trobe University Central Animal House and the Walter and Eliza Hall Institute. NOD/Lt GM-CSF−/− mice (10–16 wk old) and NOD/Lt wild-type (WT) littermates were bred at the Walter and Eliza Hall Institute. All mice were maintained in the La Trobe University Central Animal House. All experiments were conducted in accordance with the Australian code of practice for the care and use of animals for scientific purposes (NHMRC, 1997), after approval by the La Trobe University Animal Ethics committee.
Induction and Clinical Assessment of EAE.
A total of 150 μg of the encephalitogenic peptide MOG35–55 (MEVGWYRSPFSRVVHLYRNGK; Auspep) emulsified in CFA (Difco) supplemented with 4 mg/ml Mycobacterium tuberculosis was injected subcutaneously into the flanks. Mice were then immediately injected intravenously with 350 ng of pertussis vaccine (List Biological Laboratories) and again 48 h later 4. Animals were monitored daily and neurological impairment was quantified on an arbitrary clinical scale: 0, no detectable impairment; 1, flaccid tail; 2, hind limb weakness; 3, hind limb paralysis; 4, hind limb paralysis and ascending paralysis; and 5, moribund or deceased 7. Under recommendation of the animal ethics committee, mice were euthanized after reaching a clinical score of 4.
Abs and Recombinant Proteins.
The mouse anti-MOG mAb (clone 8-18C5) was purified from hybridoma culture supernatants on a Protein G-Sepharose 4B Fast Flow column (Pharmacia LKB) according to the manufacturer's instructions. Antiserum to MOG35–55 peptide 29 was raised in rabbits by procedures similar to those described previously 30. The extracellular domain of human MOG (amino acid residues 1–121 of the mature protein) 31 (rMOG) containing an amino terminal six histidine tag was produced in M15(pREP4) bacteria using the pQE9 expression vector (QIAGEN). rMOG was purified using Ni-NTA Superflow (QIAGEN) under denaturing conditions (8 M urea) as per the manufacturer's instructions on a BioLogic LP Chromatography System (Bio-Rad Laboratories). The eluted protein was refolded by dialysis against successive dilutions of urea (i.e., from 8 M down to 0.5 M urea) and finally against 10% glycerol in phosphate buffered saline. Murine rGM-CSF was a gift from F.R. Seiler, Behringwerke AG, Marburg, Germany. Purified anti-murine GM-CSF mAb and the IgG2a isotype control (anti-murine β-galactosidase) were from the 22E9.11 and GL117.41 hybridomas, respectively; these hybridomas were obtained from DNAX Research Institute.
rGM-CSF Treatment.
GM-CSF− / − and WT mice immunized with MOG35–55 were injected intraperitoneally with 10 ng of rGM-CSF dissolved in 0.5 ml of PBS every day beginning from the day of MOG35–55 immunization until day 40. GM-CSF− / − and WT mice receiving no treatment served as controls for these experiments.
Anti-GM-CSF Treatment.
Anti–GM-CSF mAb (300 ug diluted in 0.5 ml of PBS) was injected intraperitoneally into WT mice on days 0, 2, 4, 6, 9, 12, 15, 18, 21, and 24 after MOG35–55 immunization (prevention) or every 2 d from the onset of clinical disease (clinical score of 2; see Induction and Clinical Assessment of EAE above) until day 33 (suppression). As controls, mice received an isotype control IgG mAb (anti-βgal) or PBS, using the same injection protocols as above.
CNS Histology.
Brain and spinal cord were carefully dissected and immersion fixed in 4% formaldehyde in PBS. In brief, fixed tissues were embedded in paraffin wax and sections were cut from various locations of the brain, cerebellum, and spinal cord. Sections were stained with H&E and Luxol fast blue for evidence of inflammation and demyelination, respectively 32.
MOG-specific Ig Determination.
Ab activity to rMOG and MOG35–55 in mouse sera was measured by ELISA, as described previously 33. In brief, serum was collected 40 d after sensitization and tested by ELISA with rMOG and MOG35–55 peptide-coated plates. Ig isotype response was assessed by ELISA with MOG35–55 peptide-coated plates.
T Cell Proliferation and Cytokine Production.
Spleens were taken from mice killed 18 and 40 d after MOG35–55 immunization. Cells were gently dispersed through nylon mesh into a single cell suspension, washed, and cultured at 106 cells per milliliter in complete RPMI (RPMI 1640 containing 10% heat-inactivated FCS [CSL Biosciences], 2 mM l-glutamine, 100 U/ml of penicillin, and 100 μg/ml of streptomycin). 200 μl of cell suspensions were then added to 96-well microtiter plates either alone, with MOG35–55 (20 μg/ml) or with Concanavalin A (Con A; 2 μg/ml) and incubated for 90 h at 37°C with 5% CO2. 20 μl of [3H]thymidine (1 μCi per well, diluted 1/20 in RPMI; Amersham Pharmacia Biotech) was added to each well for the last 18 h. Plates were harvested onto glass fibre filter discs and added to vials containing scintillation cocktail. Counts were read using a Wallac 1410 Liquid Scintillation Counter. Presented values are the mean of three wells. For cytokine assays, 2 ml of cells (106 cells per milliliter) from spleens isolated 18 d after immunization were added to 24-well plates either alone or with MOG35–55 (10 μg/ml) or ConA (5 μg/ml). Supernatants were collected at 72 h and Quantitative ELISA was performed for cytokines using paired mAbs as recommended by the manufacturer (BD PharMingen).
Results
GM-CSF−/− Mice Are Resistant to Induction of EAE.
To determine if endogenous GM-CSF was required for the initiation and/or progression of EAE, GM-CSF−/− mice and WT littermate controls were immunized with the encephalitogenic peptide MOG35–55 and monitored for the development of clinical disease. As expected, 21/21 WT mice (on a susceptible NOD/Lt background) developed a typical relapsing remitting disease. In contrast, 20/21 GM-CSF−/− mice failed to develop clinical signs of disease (Fig. 1). The single mouse that displayed clinical symptoms did not progress past a clinical score of 1.
Figure 1.

GM-CSF regulates the onset and progression of clinical EAE provoked by MOG35–55 peptide. Mice were immunized with MOG35–55 peptide emulsified in CFA and monitored daily for the development of clinical disease. Mean clinical disease score in GM-CSF−/− and WT mice (n = 8 for each group). Note this is a representative of three independent experiments; in total 20/21 GM-CSF−/− mice failed to develop any clinical symptoms. The single mouse that developed symptoms did not progress past a clinical score of 1. In contrast all WT mice developed clinical symptoms.
rGM-CSF Treatment Reinstates Susceptibility to EAE in GM-CSF−/− Mice and Alters the Course of Disease in WT Mice.
To verify that the absence of GM-CSF was responsible for the observed resistance to the MOG35–55-induced EAE in GM-CSF−/− mice, GM-CSF−/− mice were treated daily with rGM-CSF, starting at day 0 until day 40 after immunization with MOG35–55. Such treatment restores susceptibility to EAE in 5/5 GM-CSF−/− mice (Fig. 2 A). To further establish the influence of GM-CSF on the progression of EAE, WT mice were treated with rGM-CSF after immunization with MOG35–55. The treated WT mice developed a relapsing remitting disease similar to that of untreated animals (Fig. 2 B). However, they had accelerated onset and exacerbation of clinical disease with more frequent and severe relapses.
Figure 2.
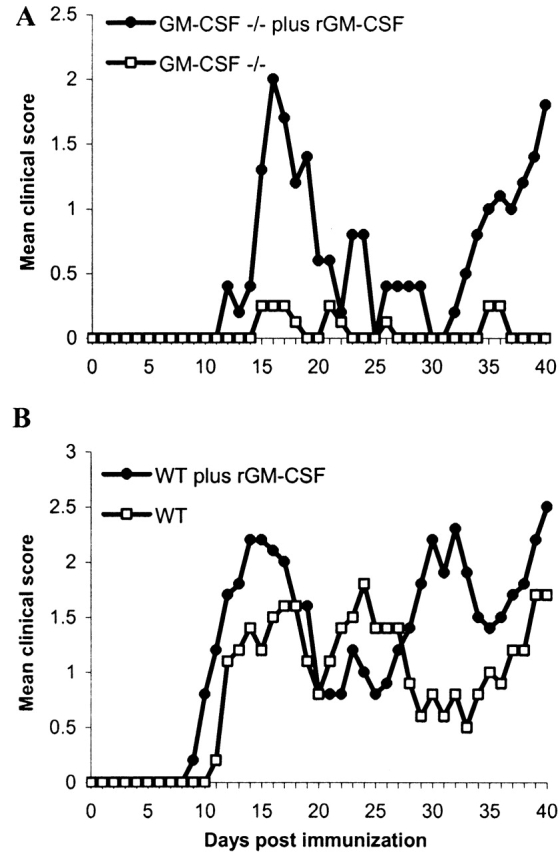
Mean clinical disease score in (A) GM-CSF−/− and (B) WT mice treated with rGM-CSF (n = 5 for each group). rGM-CSF (10 ng/mouse) was injected subcutaneously every day, starting at day 0 until day 40 after immunization with MOG35–55 peptide.
GM-CSF−/− Mice Fail to Sustain Inflammation in the CNS.
The influence of GM-CSF on the formation of CNS inflammatory lesions was examined by histological studies of fixed tissues using haematoxylin eosin staining of cells within the CNS (Fig. 3). At day 18, a time at which the disease in WT mice peaked, there were numerous inflammatory lesions in both WT and GM-CSF−/− mice. However, whilst lesions in WT mice showed typical lymphocytic cuffing around blood vessels with some cellular infiltration into the CNS parenchyma (Fig. 3 A), lesions in GM-CSF−/− mice were, on average, much smaller and exhibited much less diffusion of cells into the CNS parenchyma (Fig. 3 B).
Figure 3.

Brain histopathology of paraffin sections from mice immunized with MOG35–55. H&E staining showing typical inflammatory cuffing around blood vessels with cellular infiltration into the CNS parenchyma of WT mice killed (A) 18 and (C) 40 d after immunization (original magnification: ×10). H&E staining showing small focal inflammatory lesions in GM-CSF−/− mice killed 18 d after immunization (B) and no inflammation in GM-CSF−/− mice killed 40 d after immunization (D) (original magnification: ×10). 26–30 sagital sections per mouse (n = 6 for each group) were examined without knowledge of the injection regime by two independent investigators.
At day 40, there was little alteration in the number of lesions within the CNS of WT mice (Fig. 3 C); however, there was an increase in cellular infiltration into the parenchyma compared with that observed at day 18. Strikingly, the CNS of GM-CSF−/− mice at day 40 was almost free of inflammatory lesions with only a few small lesions found in some of the tissues analyzed (Fig. 3 D). In contrast, after treatment with rGM-CSF for 40 d, lesions within the CNS of GM-CSF−/− mice were abundant (data not shown); a finding correlating with the clinical assessment showing that rGM-CSF treatment induces susceptibility in GM-CSF−/− mice (Fig. 2 A). After treatment with rGM-CSF for 40 d, lesions were more numerous compared with untreated WT mice (data not shown). Lesions exhibited the normal diffuse appearance, with cells moving from the blood vessel into the perivascular space and even further into the CNS parenchyma (data not shown).
The MOG-specific T Cell Response Is Decreased in GM-CSF−/− Mice.
To elucidate the mechanism underlying the failure of GM-CSF−/− mice to develop EAE, the immune response against MOG was investigated. Given that EAE is characterized by the generation of autoreactive T cells, which are thought to infiltrate the CNS parenchyma and initiate the local inflammatory response, the functional activity of autoreactive T cells was assessed. Proliferation assays were performed to quantitate the MOG-specific proliferative response of T cells generated by immunization with MOG35–55. At 18 d after immunization with MOG35–55, splenocytes from WT mice proliferated strongly to MOG35–55 with stimulation indices approximately three times higher than cells from GM-CSF−/− mice (Fig. 4 A). At day 40 the proliferative response in GM-CSF−/− was still markedly less than WT mice (Fig. 4 C). In contrast, in the experiment where mice were treated with rGM-CSF, the proliferative response was restored in the GM-CSF−/− mice and enhanced in WT mice (Fig. 4 C). To address the question as to whether the decreased proliferative response was antigen specific or due to a generalized defect in the activation or function of T cells, splenocytes were stimulated with the polyclonal activator Con A. Splenocytes from GM-CSF−/−, WT, and rGM-CSF-treated mice showed no difference in proliferation to Con A (Fig. 4 B and D).
Figure 4.
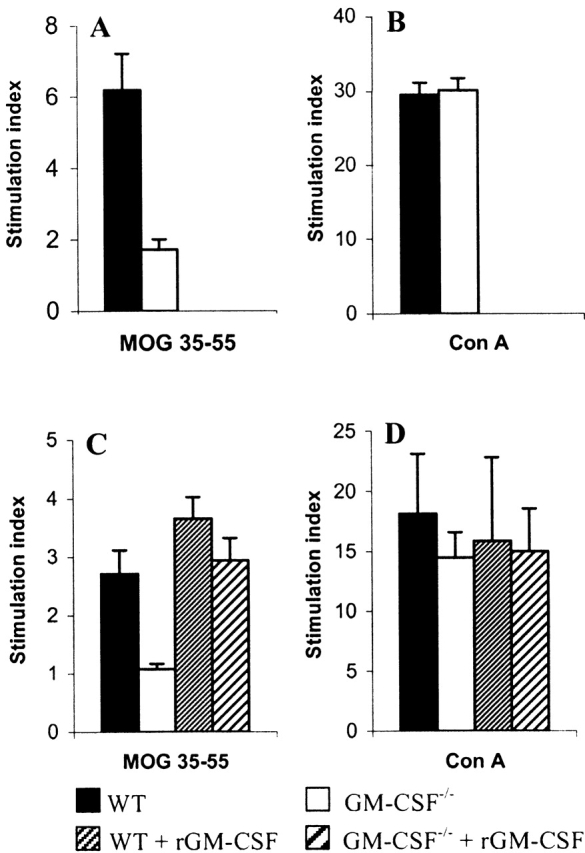
Antigen-specific proliferative responses by spleen cells from mice immunized with MOG35–55 peptide. Cells from GM-CSF−/− and WT mice (n = 6 for both groups) were isolated 18 d after disease induction and stimulated in vitro with (A) MOG35–55 peptide and (B) Con A. At day 40 cells were taken from GM-CSF−/− and WT mice, and GM-CSF−/− and WT mice treated with rGM-CSF (n = 5 for each group) and stimulated in vitro with (C) MOG35–55 peptide and (D) Con A. Each bar represents the mean stimulation indices ± SEM.
MOG35–55-specific T Cells in GM-CSF−/− Mice Exhibit Decreased Th1 Cytokine Expression.
We next ascertained if the phenotype of MOG35–55-specific T cells was altered in GM-CSF−/− mice. Accordingly, cytokine profiles of cultured splenocytes were determined (Fig. 5). As expected, no GM-CSF was detected in supernatants of cultured splenocytes from GM-CSF−/− mice (Fig. 5 A). The level of IFN-γ and IL-6, proinflammatory cytokines known to be involved in the pathogenesis of EAE, were decreased in GM-CSF−/− mice compared with WT mice (Fig. 5 C and D, respectively). IL-2 levels were similar in both groups (Fig. 5 D). TNF-α, IL-4, and IL-10 were undetectable in culture supernatants (data not shown).
Figure 5.

Production of cytokines, GM-CSF (A), IL-2 (B), IFN-γ (C), and IL-6 (D) by spleen cells from GM-CSF−/− and WT mice immunized with MOG35–55 peptide. Cytokine concentrations were measured from supernatants after 72 h of in vitro culture with MOG35–55 peptide. Each bar represents the mean concentration ± SEM (n = 6 for both groups), each tested in triplicate.
The AutoAb Response in EAE Is not Dependent on GM-CSF.
Although considerable attention has been directed to the role of cell-mediated immunity to myelin antigens in MS and EAE, it should be emphasized that primary demyelination could also be mediated by autoAbs 34 35. Therefore, sera were collected from mice at day 40 after immunization with MOG35–55 and analyzed for autoAbs against MOG. GM-CSF−/− mice, WT mice, and mice treated with rGM-CSF all secreted similar levels of total MOG-specific IgG (Fig. 6 A and B) and there was no difference in expression of different Ig isotypes (Fig. 6 C).
Figure 6.

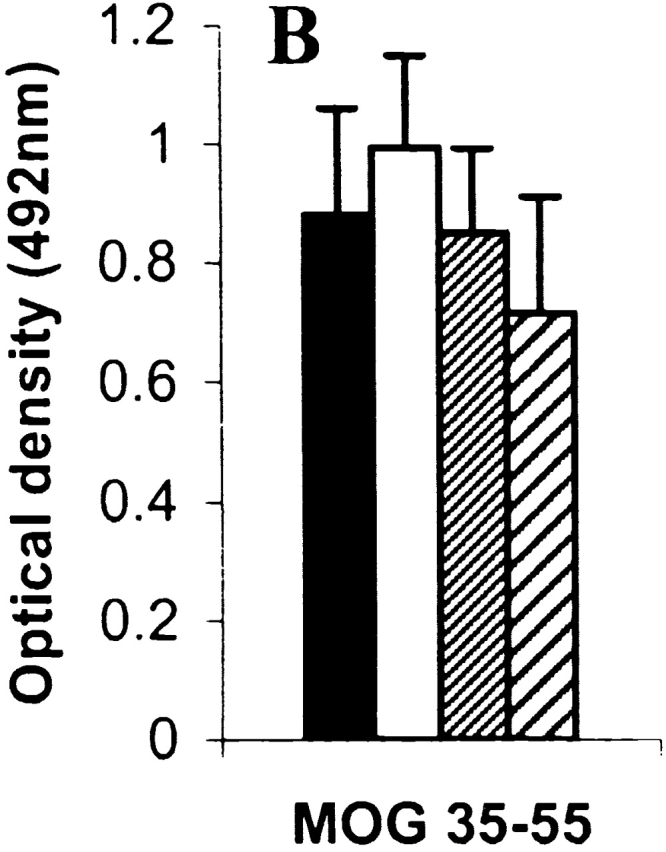

MOG-specific Ab response in mice immunized with MOG35–55 peptide. Serum was collected 40 d after sensitization and tested by ELISA with (A) rMOG and (B) MOG35–55 peptide-coated plates. (C), Ig isotype response was assessed by ELISA 33 with MOG35–55 peptide-coated plates. Each bar represents the mean specific absorbance (corrected against BSA coated plates) ± SEM (n = 5 for each group), each tested in triplicate at 1:100 dilutions. For the GM-CSF−/− mice and WT controls this study was repeated (n = 8) with no difference in results (data not shown).
Anti–GM-CSF Is a Potent Therapeutic Agent for EAE.
As an alternative means of studying the actions of GM-CSF in EAE, GM-CSF activity was blocked in WT mice by administration of anti–GM-CSF. When administered from the time of antigenic challenge, anti–GM-CSF prevented the onset of clinical disease for the period of treatment and for 10 d after treatment was ceased (Fig. 7 A). After this time and with no further treatment, all mice developed severe neurological deficits similar to control mice (data not shown). Mice receiving PBS developed the typical relapsing remitting disease and were killed at the peak of disease to conform with ethical requirements, denoted by a cross in Fig. 7 A. Mice administered with an isotype control Ab exhibited a slightly less severe disease (Fig. 7 A), which is consistent with reports that nonspecific Ig treatment is therapeutic in EAE and MS 36. Histochemical studies showed that mice treated with anti–GM-CSF had a reduction in the total number and severity of lesions (data not shown).
Figure 7.

Influence of anti–GM-CSF mAb on the clinical course of MOG35–55-induced EAE in WT mice. (A) Mean clinical score of MOG35–55-induced EAE in WT mice treated by intraperitoneal injection with PBS (n = 4), anti–GM-CSF mAb (n = 8), or isotype control IgG (n = 8, anti-βgal) from time of sensitization with MOG35–55 peptide until day 24. (B) Mean clinical score of MOG35–55-induced EAE in WT mice treated by intraperitoneal injection with PBS, anti–GM-CSF mAb, or isotype control IgG (n = 6 for each group) from first signs of clinical disease (score = 2) until day 33.
To examine the potential therapeutic action of anti-GM-CSF, mice were treated after the onset of clinical disease (clinical score at least 2). Mice administered with anti-GM-CSF after disease onset completely recovered within 20 d of treatment (Fig. 7 B). Conversely, mice receiving PBS or isotype control Ab exhibited the typical relapsing remitting disease (Fig. 7 B). Again, the isotype control Ab elicited a slight suppression of disease. Histochemical studies showed that lesions within the CNS of mice treated with anti–GM-CSF were small and sparse, compared with PBS treated mice, which mostly exhibited large diffuse lesions. Mice treated with the isotype control Ab also had fewer lesions, compared with the PBS treated controls (data not shown).
Discussion
EAE is a CD4+ T cell–mediated inflammatory demyelinating disease of the CNS that serves as an experimental model of the human disease, MS. It can be induced by a number of myelin antigens, such as myelin basic protein, proteolipid protein, and MOG. While MOG is a quantitatively minor protein of the CNS myelin with an as yet unknown function, this molecule and its immunodominant peptide, MOG35–55, are able to induce a paralytic MS-like disease with extensive CNS inflammation and demyelination in several strains of mice. C57BL/6 mice exhibit a chronic nonremitting disease while NOD/Lt mice develop a severe relapsing-remitting disease 4 7. Here, we examined whether GM-CSF, a pleiotropic cytokine considered to be a critical mediator in the development of chronic inflammation, influenced the development and progression of EAE in NOD/Lt mice.
Our study shows that GM-CSF−/− mice, on a susceptible NOD/Lt background, fail to develop clinical signs of MOG35–55-induced EAE. To address the question of whether resistance was associated with alterations in the MOG-specific immune response, we analyzed the Th1/Th2 phenotype and proliferative response of MOG-specific T cells. T cells from GM-CSF−/− mice proliferated less robustly to MOG and secreted lower levels of IFN-γ and IL-6, compared with WT controls. This suggests that resistance was associated with a decreased MOG-specific Th1 response. It is unclear whether GM-CSF is involved in the activation and expansion of T cells in the periphery, or if it is involved in regulating survival and migration of encephalitogenic T cells into the CNS and/or their reactivation within the CNS. This reduced T cell response is not mediated by a direct effect on T cells because T cells lack receptors for GM-CSF 37. Rather, there is considerable evidence that GM-CSF may play a crucial role in CD4+ T cell–mediated immune responses by activating DCs 28 38.
T cell activation involves the presentation of antigen by DCs to naive antigen-specific T cells in the periphery. Once activated, T cells are able to provide antigen-specific B cells with costimulation, required for clonal expansion, differentiation, Ab production, and isotype switching 39. Hence, in view of the fact that Ab production persists in GM-CSF−/− mice, with no difference between total IgG or Ig isotype response in comparison with WT controls, we can assume that MOG-reactive T cells in GM-CSF−/− mice can be functionally activated 39. Furthermore, T cells from GM-CSF−/− mice proliferated vigorously in response to the polyclonal activator, ConA, indicating that there was no generalized defect in T cell receptor signaling events and the proliferative potential of the T cells was intact. Thus, it is unlikely that inefficient activation of T cells by dendritic APCs in the periphery can account alone for the observed resistance to EAE in GM-CSF−/− mice. Alternatively, GM-CSF may play a role in the effector phase of the disease, particularly within the CNS environment. In the CIA model, a similar intact humoral response to type II collagen was found in GM-CSF−/− mice in spite of a dramatic suppression in arthritis 20.
Histological analysis of CNS tissue from GM-CSF−/− mice revealed development of early lesions at day 18 within the CNS of GM-CSF−/− mice, confirming that there was activation of the inflammatory cascade. However, despite the presence of early lesions within the CNS of GM-CSF−/− mice, where some perivascular congestion of cells around blood vessels was observed, strikingly, inflammation was almost completely absent at day 40. In contrast, numerous lesions in WT mice were present at both time points (days 18 and 40) and appeared more diffuse with cellular infiltration further into the CNS parenchyma. Also, when treated with rGM-CSF, numerous lesions were found within the CNS of GM-CSF−/− mice and even more in rGM-CSF–treated WT mice. On the other hand, GM-CSF inhibition studies using anti–GM-CSF mAb showed that GM-CSF blockade not only prevented the onset of clinical disease when administered from the time of antigenic challenge, but also ameliorated disease when administered after the onset of clinical symptoms. This was associated with a marked reduction in cellular infiltration within the CNS of anti–GM-CSF–treated mice. Taken together, these findings suggest that the normal formation, maintenance, and expansion of inflammatory lesions within the CNS in the effector phase of EAE are dependent on GM-CSF.
Despite initial reports of normal hematopoiesis in GM-CSF–deficient mice, there is increasing evidence for an essential hematopoietic role for GM-CSF after antigenic challenge 21 26. Thus, the inability to maintain inflammation in the CNS could reflect a failure to maintain supply of macrophages and PMNs in the site of inflammation and/or insufficient activation or local proliferation of inflammatory cells within the CNS 40. A number of studies have shown that monocytes, macrophages, and PMNs play a critical role in the effector phase of EAE in mice 41 42 43 44. Lack of antigen presentation by such cells within the CNS could in part explain the diminished MOG-reactive T cell response observed. Besides macrophages and PMNs, resident microglia are also believed to play an important role in antigen presentation and stimulation of T cells within the CNS 45.
There is now a large body of evidence supporting a role for GM-CSF in the activation of macrophages, PMNs, and microglia 46. Indeed, GM-CSF has been shown to induce MHC class II and B7 expression 11 47 as well as enhance the phagocytic and APC function of both macrophages and resident microglia 12 13. In EAE, activated macrophages and/or microglia in the CNS show upregulated expression of MHC class II and B7 molecules 42 48 49. A recent study has also implicated GM-CSF in the differentiation of brain DCs from local CNS progenitors 10. Due to their strong Th1-inducing capacity and their long-lasting presence within the CNS, these brain DCs may play a key role in the exacerbation or maintenance of immune-mediated CNS diseases such as EAE.
Another explanation for the collapse in the inflammatory cascade in GM-CSF−/− mice could be an imbalance in proteinases, causing a failure in the digestion of the extracellular matrix in the CNS, and/or inefficient generation of chemoattractant gradients 50 51 52. In this context, it is noteworthy that recent studies using transformed cells engineered to express GM-CSF have demonstrated a role for GM-CSF in regulating the synthesis of MIP-1α and MCP-1 in murine PMN and macrophages 53. Such chemokines were shown to be largely responsible for accumulation and infiltration of immune cells into the CNS in EAE 54 55. Whether GM-CSF influences the inflammatory response via any one, or a combination of the aforementioned pathways in EAE, is yet to be elucidated.
Narrowing down the precise mechanism(s) involved in the proinflammatory effect of GM-CSF in the pathogenesis of disease is currently under investigation. It is likely that GM-CSF has a central role in regulating the complex network of cytokines and chemokines involved in the activation, recruitment, and local proliferation of DCs, macrophages, PMNs, and microglia both during induction in the periphery and within the CNS during the effector phase of disease. Collectively, the work presented here clearly demonstrates a critical role for GM-CSF in the maintenance of chronic inflammation in the experimental MS-like disease in NOD/Lt mice and opens new avenues for the development of therapeutic strategies for the treatment of MS.
Acknowledgments
We thank Prof. Donald Metcalf for reviewing the manuscript, Melinda Goodyear for assistance in data analysis, and Anne Chow for technical assistance.
This work was supported by a donation from the J.B. Were and Son Trust and grants from MS Australia and the Bethlehem Griffiths Research Foundation.
Footnotes
Abbreviations used in this paper: CIA, collagen-induced arthritis; CNS, central nervous system; DC, dendritic cell; EAE, experimental autoimmune encephalomyelitis; MOG, myelin oligodendrocyte glycoprotein; MS, multiple sclerosis; WT, wild-type.
References
- Steinman L. Assessment of animal models for MS and demyelinating disease in the design of rational therapy. Neuron. 1999;24:511–514. doi: 10.1016/s0896-6273(00)81107-1. [DOI] [PubMed] [Google Scholar]
- Sobel R.A., Greer J.M., Kuchroo V.K. Minireviewautoimmune responses to myelin proteolipid protein. Neurochem. Res. 1994;19:915–921. doi: 10.1007/BF00968701. [DOI] [PubMed] [Google Scholar]
- Bernard C.C.A., Ichikawa M., Menon K., Slavin A., Ewing C., Johns T., Liu J., Bettadapura J. Autoantigens in experimental autoimmune encephalomyelitis and multiple sclerosis Abramsky O., Ovadia H. Frontiers in Multiple SclerosisClinical Research and Therapy 1997. 61 70 Martin Dunitz Limited; London: pp [Google Scholar]
- Bernard C.C., Johns T.G., Slavin A., Ichikawa M., Ewing C., Liu J., Bettadapura J. Myelin oligodendrocyte glycoproteina novel candidate autoantigen in multiple sclerosis. J. Mol. Med. 1997;75:77–88. doi: 10.1007/s001090050092. [DOI] [PubMed] [Google Scholar]
- Kerlero de Rosbo N., Brok H.P., Bauer J., Kaye J.F., Hart B.A., Ben-Nun A. Rhesus monkeys are highly susceptible to experimental autoimmune encephalomyelitis induced by myelin oligodendrocyte glycoproteincharacterization of immunodominant T- and B-cell epitopes. J. Neuroimmunol. 2000;110:83–96. doi: 10.1016/s0165-5728(00)00306-4. [DOI] [PubMed] [Google Scholar]
- Stefferl A., Brehm U., Storch M., Lambracht-Washington D., Bourquin C., Wonigeit K., Lassmann H., Linington C. Myelin oligodendrocyte glycoprotein induces experimental autoimmune encephalomyelitis in the “resistant” Brown Norway ratdisease susceptibility is determined by MHC and MHC-linked effects on the B cell response. J. Immunol. 1999;163:40–49. [PubMed] [Google Scholar]
- Slavin A., Ewing C., Liu J., Ichikawa M., Slavin J., Bernard C.C. Induction of a multiple sclerosis-like disease in mice with an immunodominant epitope of myelin oligodendrocyte glycoprotein. Autoimmunity. 1998;28:109–120. doi: 10.3109/08916939809003872. [DOI] [PubMed] [Google Scholar]
- Hamilton J.A., Stanley E.R., Burgess A.W., Shadduck R.K. Stimulation of macrophage plasminogen activator activity by colony-stimulating factors. J. Cell Physiol. 1980;103:435–445. doi: 10.1002/jcp.1041030309. [DOI] [PubMed] [Google Scholar]
- Metcalf D., Nicola N.A. The hemopoietic colony-stimulating factorsfrom biology to clinical applications 1995. Cambridge University Press; Cambridge: pp. 327 pp [Google Scholar]
- Fischer H.G., Reichmann G. Brain dendritic cells and macrophages/microglia in central nervous system inflammation. J. Immunol. 2001;166:2717–2726. doi: 10.4049/jimmunol.166.4.2717. [DOI] [PubMed] [Google Scholar]
- Alvaro-Gracia J.M., Zvaifler N.J., Firestein G.S. Cytokines in chronic inflammatory arthritis. IV. Granulocyte/macrophage colony-stimulating factor-mediated induction of class II MHC antigen on human monocytesa possible role in rheumatoid arthritis. J. Exp. Med. 1989;170:865–875. doi: 10.1084/jem.170.3.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey P.J., Bressler L., Park L.S., Alpert A., Gillis S. Granulocyte-macrophage colony-stimulating factor augments the primary antibody response by enhancing the function of antigen-presenting cells. J. Immunol. 1987;139:1113–1119. [PubMed] [Google Scholar]
- von Zahn J., Moller T., Kettenmann H., Nolte C. Microglial phagocytosis is modulated by pro- and anti-inflammatory cytokines. Neuroreport. 1997;8:3851–3856. doi: 10.1097/00001756-199712220-00003. [DOI] [PubMed] [Google Scholar]
- Hart P.H., Whitty G.A., Piccoli D.S., Hamilton J.A. Synergistic activation of human monocytes by granulocyte-macrophage colony-stimulating factor and IFN-γ. Increased TNF-α but not IL-1 activity. J. Immunol. 1988;141:1516–1521. [PubMed] [Google Scholar]
- Hamilton J.A. Colony stimulating factors, cytokines and monocyte-macrophages-some controversies. Immunol. Today. 1993;14:18–24. doi: 10.1016/0167-5699(93)90319-G. [DOI] [PubMed] [Google Scholar]
- Gamble J.R., Elliott M.J., Jaipargas E., Lopez A.F., Vadas M.A. Regulation of human monocyte adherence by granulocyte-macrophage colony-stimulating factor. Proc. Natl. Acad. Sci. USA. 1989;86:7169–7173. doi: 10.1073/pnas.86.18.7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaout M.A., Wang E.A., Clark S.C., Sieff C.A. Human recombinant granulocyte-macrophage colony-stimulating factor increases cell-to-cell adhesion and surface expression of adhesion-promoting surface glycoproteins on mature granulocytes. J. Clin. Invest. 1986;78:597–601. doi: 10.1172/JCI112615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton J.A. Rheumatoid arthritisopposing actions of haemopoietic growth factors and slow-acting anti-rheumatic drugs. Lancet. 1993;342:536–539. doi: 10.1016/0140-6736(93)91653-4. [DOI] [PubMed] [Google Scholar]
- Campbell I.K., Bendele A., Smith D.A., Hamilton J.A. Granulocyte-macrophage colony stimulating factor exacerbates collagen induced arthritis in mice. Ann. Rheum. Dis. 1997;56:364–368. doi: 10.1136/ard.56.6.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell I.K., Rich M.J., Bischof R.J., Dunn A.R., Grail D., Hamilton J.A. Protection from collagen-induced arthritis in granulocyte-macrophage colony-stimulating factor-deficient mice. J. Immunol. 1998;161:3639–3644. [PubMed] [Google Scholar]
- Zhan Y., Lieschke G.J., Grail D., Dunn A.R., Cheers C. Essential roles for granulocyte-macrophage colony-stimulating factor (GM-CSF) and G-CSF in the sustained hematopoietic response of Listeria monocytogenes-infected mice. Blood. 1998;91:863–869. [PubMed] [Google Scholar]
- Johnson G.R., Gonda T.J., Metcalf D., Hariharan I.K., Cory S. A lethal myeloproliferative syndrome in mice transplanted with bone marrow cells infected with a retrovirus expressing granulocyte-macrophage colony stimulating factor. EMBO J. 1989;8:441–448. doi: 10.1002/j.1460-2075.1989.tb03396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang R.A., Metcalf D., Cuthbertson R.A., Lyons I., Stanley E., Kelso A., Kannourakis G., Williamson D.J., Klintworth G.K., Gonda T.J. Transgenic mice expressing a hemopoietic growth factor gene (GM-CSF) develop accumulations of macrophages, blindness, and a fatal syndrome of tissue damage. Cell. 1987;51:675–686. doi: 10.1016/0092-8674(87)90136-x. [DOI] [PubMed] [Google Scholar]
- Biondo M., Nasa Z., Marshall A., Hock Toh B., Alderuccio F. Local transgenic expression of granulocyte macrophage-colony stimulating factor initiates autoimmunity. J. Immunol. 2001;166:2090–2099. doi: 10.4049/jimmunol.166.3.2090. [DOI] [PubMed] [Google Scholar]
- Carrieri P.B., Provitera V., De Rosa T., Tartaglia G., Gorga F., Perrella O. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosisa correlation with clinical activity. Immunopharmacol. Immunotoxicol. 1998;20:373–382. doi: 10.3109/08923979809034820. [DOI] [PubMed] [Google Scholar]
- Stanley E., Lieschke G.J., Grail D., Metcalf D., Hodgson G., Gall J.A., Maher D.W., Cebon J., Sinickas V., Dunn A.R. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc. Natl. Acad. Sci. USA. 1994;91:5592–5596. doi: 10.1073/pnas.91.12.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vremec D., Lieschke G.J., Dunn A.R., Robb L., Metcalf D., Shortman K. The influence of granulocyte/macrophage colony-stimulating factor on dendritic cell levels in mouse lymphoid organs. Eur. J. Immunol. 1997;27:40–44. doi: 10.1002/eji.1830270107. [DOI] [PubMed] [Google Scholar]
- Wada H., Noguchi Y., Marino M.W., Dunn A.R., Old L.J. T cell functions in granulocyte/macrophage colony-stimulating factor deficient mice. Proc. Natl. Acad. Sci. USA. 1997;94:12557–12561. doi: 10.1073/pnas.94.23.12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa M., Johns T.G., Liu J., Bernard C.C. Analysis of the fine B cell specificity during the chronic/relapsing course of a multiple sclerosis-like disease in Lewis rats injected with the encephalitogenic myelin oligodendrocyte glycoprotein peptide 35-55. J. Immunol. 1996;157:919–926. [PubMed] [Google Scholar]
- Bernard C.C., Townsend E., Randell V.B., Williamson H.G. Do antibodies to myelin basic protein isolated from multiple sclerosis cross-react with measles and other common virus antigens? Clin. Exp. Immunol. 1983;52:98–106. [PMC free article] [PubMed] [Google Scholar]
- Hilton A.A., Slavin A.J., Hilton D.J., Bernard C.C. Characterization of cDNA and genomic clones encoding human myelin oligodendrocyte glycoprotein. J. Neurochem. 1995;65:309–318. doi: 10.1046/j.1471-4159.1995.65010309.x. [DOI] [PubMed] [Google Scholar]
- Johns T.G., Kerlero de Rosbo N., Menon K.K., Abo S., Gonzales M.F., Bernard C.C. Myelin oligodendrocyte glycoprotein induces a demyelinating encephalomyelitis resembling multiple sclerosis. J. Immunol. 1995;154:5536–5541. [PubMed] [Google Scholar]
- Ichikawa M., Koh C.S., Inaba Y., Seki C., Inoue A., Itoh M., Ishihara Y., Bernard C.C., Komiyama A. IgG subclass switching is associated with the severity of experimental autoimmune encephalomyelitis induced with myelin oligodendrocyte glycoprotein peptide in NOD mice. Cell. Immunol. 1999;191:97–104. doi: 10.1006/cimm.1998.1414. [DOI] [PubMed] [Google Scholar]
- Bernard C.C., Kerlero de Rosbo N. Multiple sclerosisan autoimmune disease of multifactorial etiology. Curr. Opin. Immunol. 1992;4:760–765. doi: 10.1016/0952-7915(92)90058-m. [DOI] [PubMed] [Google Scholar]
- Lassmann H., Bruck W., Lucchinetti C. Heterogeneity of multiple sclerosis pathogenesisimplications for diagnosis and therapy. Trends Mol. Med. 2001;7:115–121. doi: 10.1016/s1471-4914(00)01909-2. [DOI] [PubMed] [Google Scholar]
- Achiron A., Mor F., Margalit R., Cohen I.R., Lider O., Miron S. Suppression of experimental autoimmune encephalomyelitis by intravenously administered polyclonal immunoglobulins. J. Autoimmun. 2000;15:323–330. doi: 10.1006/jaut.2000.0433. [DOI] [PubMed] [Google Scholar]
- Park L.S., Friend D., Gillis S., Urdal D.L. Characterization of the cell surface receptor for granulocyte-macrophage colony-stimulating factor. J. Biol. Chem. 1986;261:4177–4183. [PubMed] [Google Scholar]
- Kelly K.A., Lucas K., Hochrein H., Metcalf D., Wu L., Shortman K. Development of dendritic cells in culture from human and murine thymic precursor cells. Cell. Mol. Biol. 2001;47:43–54. [PubMed] [Google Scholar]
- Smith K.M., Pottage L., Thomas E.R., Leishman A.J., Doig T.N., Xu D., Liew F.Y., Garside P. Th1 and Th2 CD4+ T cells provide help for B cell clonal expansion and antibody synthesis in a similar manner in vivo. J. Immunol. 2000;165:3136–3144. doi: 10.4049/jimmunol.165.6.3136. [DOI] [PubMed] [Google Scholar]
- Juedes A.E., Ruddle N.H. Resident and infiltrating central nervous system APCs regulate the emergence and resolution of experimental autoimmune encephalomyelitis. J. Immunol. 2001;166:5168–5175. doi: 10.4049/jimmunol.166.8.5168. [DOI] [PubMed] [Google Scholar]
- Martiney J.A., Rajan A.J., Charles P.C., Cerami A., Ulrich P.C., Macphail S., Tracey K.J., Brosnan C.F. Prevention and treatment of experimental autoimmune encephalomyelitis by CNI-1493, a macrophage-deactivating agent. J. Immunol. 1998;160:5588–5595. [PubMed] [Google Scholar]
- Tran E.H., Hoekstra K., van Rooijen N., Dijkstra C.D., Owens T. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J. Immunol. 1998;161:3767–3775. [PubMed] [Google Scholar]
- Huitinga I., Ruuls S.R., Jung S., Van Rooijen N., Hartung H.P., Dijkstra C.D. Macrophages in T cell line-mediated, demyelinating, and chronic relapsing experimental autoimmune encephalomyelitis in Lewis rats. Clin. Exp. Immunol. 1995;100:344–351. doi: 10.1111/j.1365-2249.1995.tb03675.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl S.R., Staykova M.A., Wozniak A., Fordham S., Bruce J., Willenborg D.O. Treatment with anti-granulocyte antibodies inhibits the effector phase of experimental autoimmune encephalomyelitis. J. Immunol. 1998;161:6421–6426. [PubMed] [Google Scholar]
- Aloisi F., Ria F., Adorini L. Regulation of T-cell responses by CNS antigen-presenting cellsdifferent roles for microglia and astrocytes. Immunol. Today. 2000;21:141–147. doi: 10.1016/s0167-5699(99)01512-1. [DOI] [PubMed] [Google Scholar]
- Aloisi F., De Simone R., Columba-Cabezas S., Penna G., Adorini L. Functional maturation of adult mouse resting microglia into an APC is promoted by granulocyte-macrophage colony-stimulating factor and interaction with Th1 cells. J. Immunol. 2000;164:1705–1712. doi: 10.4049/jimmunol.164.4.1705. [DOI] [PubMed] [Google Scholar]
- Wei R., Jonakait G.M. Neurotrophins and the anti-inflammatory agents interleukin-4 (IL-4), IL-10, IL-11 and transforming growth factor-β1 (TGF-β1) down-regulate T cell costimulatory molecules B7 and CD40 on cultured rat microglia. J. Neuroimmunol. 1999;95:8–18. doi: 10.1016/s0165-5728(98)00248-3. [DOI] [PubMed] [Google Scholar]
- Owens T., Tran E., Hassan-Zahraee M., Krakowski M. Immune cell entry to the CNS-a focus for immunoregulation of EAE. Res. Immunol. 1998;149:781–789. doi: 10.1016/s0923-2494(99)80005-4. [DOI] [PubMed] [Google Scholar]
- Miller S.D., Vanderlugt C.L., Lenschow D.J., Pope J.G., Karandikar N.J., Dal Canto M.C., Bluestone J.A. Blockade of CD28/B7-1 interaction prevents epitope spreading and clinical relapses of murine EAE. Immunity. 1995;3:739–745. doi: 10.1016/1074-7613(95)90063-2. [DOI] [PubMed] [Google Scholar]
- Godiska R., Chantry D., Dietsch G.N., Gray P.W. Chemokine expression in murine experimental allergic encephalomyelitis. J. Neuroimmunol. 1995;58:167–176. doi: 10.1016/0165-5728(95)00008-p. [DOI] [PubMed] [Google Scholar]
- Gijbels K., Galardy R.E., Steinman L. Reversal of experimental autoimmune encephalomyelitis with a hydroxamate inhibitor of matrix metalloproteases. J. Clin. Invest. 1994;94:2177–2182. doi: 10.1172/JCI117578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements J.M., Cossins J.A., Wells G.M., Corkill D.J., Helfrich K., Wood L.M., Pigott R., Stabler G., Ward G.A., Gearing A.J., Miller K.M. Matrix metalloproteinase expression during experimental autoimmune encephalomyelitis and effects of a combined matrix metalloproteinase and tumor necrosis factor-α inhibitor. J. Neuroimmunol. 1997;74:85–94. doi: 10.1016/s0165-5728(96)00210-x. [DOI] [PubMed] [Google Scholar]
- Shinohara H., Yano S., Bucana C.D., Fidler I.J. Induction of chemokine secretion and enhancement of contact-dependent macrophage cytotoxicity by engineered expression of granulocyte-macrophage colony-stimulating factor in human colon cancer cells. J. Immunol. 2000;164:2728–2737. doi: 10.4049/jimmunol.164.5.2728. [DOI] [PubMed] [Google Scholar]
- Gerard C., Rollins B. Chemokines and disease. Nat. Immunol. 2001;2:108–115. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- Kennedy K.J., Strieter R.M., Kunkel S.L., Lukacs N.W., Karpus W.J. Acute and relapsing experimental autoimmune encephalomyelitis are regulated by differential expression of the CC chemokines macrophage inflammatory protein-1α and monocyte chemotactic protein-1. J. Neuroimmunol. 1998;92:98–108. doi: 10.1016/s0165-5728(98)00187-8. [DOI] [PubMed] [Google Scholar]