

Abstract
Rac2 is a hematopoietic-specific GTPase acting as a molecular switch to mediate both transcriptional activation and cell morphological changes. We have examined the effect of Rac2 deficiency during T cell activation. In Rac2−/− T cells, proliferation was reduced upon stimulation with either plate-bound anti-CD3 or T cell receptor–specific antigen. This defect is accompanied with decreased activation of mitogen activated protein kinase extracellular signal–regulated kinase (ERK)1/2 and p38, and reduced Ca2+ mobilization. TCR stimulation–induced actin polymerization is also reduced. In addition, anti-CD3 cross-linking–induced T cell capping is reduced compared with wild-type T cells. These results indicate that Rac2 is important in mediating both transcriptional and cytoskeletal changes during T cell activation. The phenotypic similarity of Rac2−/− to Vav−/− cells implicates Rac2 as a downstream mediator of Vav signaling.
Keywords: T cell activation, Rac2, VAV, MAPK, cytoskeleton
Introduction
After the engagement of TCR and its specific ligand, peptide/MHC complexes, a cascade of biochemical changes occur leading to transcriptional activation and cell cytoskeletal reorganization. Engagement of TCRs cause the activation of Src and Syk/ZAP-70 family proteins and the tyrosine phosphorylation of various cellular substrates, calcium (Ca2+) mobilization, and activation of mitogen-activated protein kinases (MAPKs) including extracellular signal–regulated kinase (ERK), c-Jun NH2-terminal kinase (JNK), and p38, thus translating the receptor signal to transcriptional activation of effector genes. Simultaneously, TCR engagement also causes cytoskeleton reorganization, results in T cell polarization toward APCS, enhanced T cell APC interaction, and supramolecular activation clusters (SMACs) formation 1. These biochemical and cytoskeletal changes are well orchestrated and ensure sustained TCR signaling, leading to T cell proliferation, differentiation, and cytokine production. Compromising either biochemical or cytoskeletal changes results in dampened T cell response to stimulation 2. Thus, molecules orchestrating these two aspects play a key role in T cell activation.
Rac proteins are members of the Rho GTPase family and cycle between the active GTP-bound form and the inactive GDP bound form. Activated Rac binds to a variety of down stream effector molecules thereby mediating multiple cellular functions in different cell types 3. Specifically, Rac is well known for its involvement in cytoskeleton reorganization. Activation of Rac is associated with changes in actin polymerization, causing lamellipodia formation and membrane ruffles in fibroblasts 4 5, morphological changes in platelets 6, and facilitating cell adhesion and chemotaxis response in macrophages and neutrophils 7 8 9. Rac also participates in signal transduction via mitogen-activated kinase pathways. In vitro studies indicate that by activating PAK1, a well known Rac target, Rac can activate ERK in T cells upon TCR stimulation 10, and in NK cells upon interaction with the target cell 11. Rac also activates c-Jun NH2-terminal kinase and p38 kinase pathways through PAK1 12 13. Moreover, Rac GTPases play an important role in cell cycle progression and survival in response to mitogenic stimulation 14 15, and are essential for cellular transformation by Ras 16 17. Furthermore, Rac is essential for the activation of a multiprotein complex that produces superoxide in phagocytic cells, the NADPH oxidase 9 18 19 20.
While the function and biochemical properties have been well studied in many cell types, the role of Rac GTPases during T cell activation is less clear. Studies from Vav-deficient mice suggest that Rac could play an important role in T cell activation 21 22 23. Vav is a guanine nucleotide exchange factor (GEF), enhancing the exchange of GDP to GTP, thus promoting the activation of Rho GTPase. In vitro, Rac GTPase has been shown to be a major substrate of Vav GEF activity 24. Upon T cell stimulation, Vav is rapidly activated through phosphorylation. In the absence of Vav, T cells exhibit a profound activation defect, with decreased proliferation in response to anti-CD3 in the presence and absence of anti-CD28, and decreased IL-2 production 21 22 23. Biochemical analysis of Vav−/− T cells showed that although early tyrosine phosphorylation of Zap70, Cbl-c, SH2 domain–containing leukocyte protein of 76 kD (SLP-76), and linker for activation of T cells (LAT) are normal, ERK and nuclear factor (NF)-κB activation are defective upon TCR stimulation. TCR induced Ca2+ flux is also defective. Moreover, Vav−/− T cells showed defects in actin polymerization and cap formation in response to TCR stimulation 21 22 23. As the Rac GTPase is a major substrate for Vav GEF activity and has demonstrated its function in both transcriptional activation and cytoskeleton reorganization, it is conceivable that at least part of the phenotype of Vav knockouts may derive from the defective activation of Rac. Most recently, using an in vitro system, Arrieumerlou et al. have demonstrated that dominant negative Rac affects Ca2+ mobilization and actin polymerization in response to TCR cross-linking 25. These results indicate that Rac could play an important role in T cell activation. However, as dominant negative small GTPases are pleiotropic in action, it is possible that this effect is mediated through other proteins such as Ras or Rho. Moreover, there are three members within the Rac subfamily, Rac1, Rac2, and Rac3. While Rac1 is universally expressed, the expression of Rac2 is restricted to the hematopoietic lineage 26. Rac3 is a relative newly described member of Rac family and is highly expressed in brain 27. As Rac proteins are highly homologous to each other, sharing 89–92% sequence homology, it is plausible that there are functional overlaps between Rac proteins.
In this report, we studied the role of the hematopoietic-specific GTPase Rac2 in T cell activation using Rac2-deficient mice 9. T cell activation was compromised in response to either anti-CD3 or TCR-specific antigen. Costimulation with anti-CD28 or adding IL-2 partially compensates for the proliferation defect. Biochemically, Rac2−/− T cells showed decreased phosphorylation of ERK1/2 and p38. Antigen-induced Ca2+ flux is also reduced. Moreover, actin polymerization upon TCR cross-linking or antigen stimulation is decreased. This defective cytoskeletal change is accompanied by decreased cap formation. These results indicated that deficiency of Rac2 affects both transcriptional activation and cytoskeleton reorganization during T cell stimulation, and suggests that defective Rac activation is partially responsible for the observed defects in Vav−/− mice.
Materials and Methods
Mice.
B10. BR mice were obtained from The Jackson Laboratory. AND TCR transgenic mice with CD4+ T cells expressing a TCR specific for the COOH terminus of pigeon cytochrome c have been described 28 and were maintained on the C57BL/6 background in our facility. Rac2 knockout mice (Rac2KO) on the C57BL/6 background have been described 9. Rac2KO mice were further bred with AND TCR transgenic mice. All mice were maintained in pathogen-free condition and were used at 6–12 wk of age.
Preparation of APC, CD4+, CD8+ Mature T Cells and Thymocytes Subpopulations.
T cell–depleted APC were prepared by incubating B10BR splenocytes with anti–Thy-1, anti-CD8, anti-CD4, and anti-NK1.1 antibody, followed by complement mediated lysis. CD4+ T cells from spleens and lymph nodes were isolated by immunomagnetic negative selection using antibodies against CD8, NK1.1, and MHC class II, followed by incubation with anti–mouse and anti–rat Ig-coated magnetic beads (PerSeptive Biosystems). Mature CD8+ T cells were isolated in the similar manner as CD4+ T cells. CD4+CD8+ and CD4+ thymocytes were isolated by flow cytometry sorting. CD4−CD8− and CD8+ thymocytes were isolated by depleting CD4+ cells in total thymocytes first, followed by separation of CD8+ and CD4−CD8− cells using MACs column according to the manufacturer's suggestion (Miltenyi Biotec).
T Cell Proliferation and IL-2 Production.
105 purified CD4+ T cells were cultured in triplicate in round-bottom plate precoated with anti-CD3 antibody (clone 145–2C11) with or without anti-CD28. Alternatively, 105 AND TCR transgenic CD4+ cells were cocultured with 105 mitomycin C treated I-Ek+ CH27 cells or B10BR APCs in the presence of different doses of MCC peptide (VFAGLKKANERADLIAYLKQATK). T cells were pulsed with 1 μCi/well [3H]thymidine 48 h after stimulation and were harvested 12 h later. The level of [3H]thymidine incorporation was determined by scintillation counting. IL-2 production was measured by ELISA (BD PharMingen) 24 h after stimulation.
Immunoblotting.
AND TCR transgenic T cells were stimulated with either fresh or paraformaldehyde fixed B10BR APCs prepulsed with MCC peptide. At the indicated time point, cells were harvested and lysed in ice-cold lysis buffer (20 mM Tris, pH 7.2, 1% NP-40, 150 mM NaCl, 1 mM MaCl2, 1 mM EDTA) containing protease and phosphatase inhibitors. Nuclear material was removed by centrifuging at 13,000 rpm for 15 min. Supernatant of the cell lysate was electrophoresed on a 12.5% SDS-PAGE gel, transferred to immunoblot membrane, and blotted with the indicated antibody. Antibody against phospho-tyrosine 4G10 was obtained from Upstate Biotechnology. Antibodies against phospho-ERK, ERK, phospho-p38, p38, Phospho-JNK, and JNK were obtained from New England Biolabs, Inc. and were used as directed. To check for rac protein expression in different population of mature T cell and thymocytes, 50 μg of cell lysates from each cell population were used for Western blotting. Rac1 antibody was from Santa Cruz Biotechnology, Inc., and Rac2 antibody was provided by Gary M. Bokoch from the Scripps Research Institute, La Jolla, CA.
ERK kinase assay AND TCR transgenic T cells were stimulated with fresh B10BR APCs prepulsed with MCC peptide for the indicated time. Cells were then lysed in the aforementioned lysis buffer. ERK kinase activity were assayed using MAPK Immunoprecipitation Kinase Assay Kit (Upstate Biotechnology) following the manufacturer's protocol.
Ca2+ Mobilization.
Calcium signaling after Ag-specific stimulation was monitored as described previously 29. Briefly, CD4+ T cells loaded with 5 μM fluo3/AM ester (Molecular Probes) were plated by centrifugation in flat 96-well plates at a concentration of 5 × 105 cell/50 μl. The cells were then scanned using the ACAS 570 video laser cytometer (Meridian Instruments). After initiation of scanning, 4 × 106 T cell–depleted splenocytes pulsed with peptide was added to the CD4+ T cells. To examine antibody cross-linking–induced Ca2+ flux, fluo3/AM loaded T cells were incubated with the indicated amount of anti-CD3 antibody for 5 min, then recovered by centrifugation in a microcentrifuge (2,000 rpm for 3 min), resuspended, and plated in a flat 96-well plate. The cells were then scanned for 1 min followed by addition of cross-linking antibody anti–hamster IgG (100 μg/ml). The initial average fluorescence of each cell was digitized and normalized to 1, and the results are expressed as changes in normalized fluorescence intensity of individual cells over time. The percentage of responding cells was determined by dividing the number of cells demonstrating an increase in intracellular calcium of >50% by the total number of scanned cells. The time from the addition of the antibody to the start of Ca2+ flux was recorded among 100 Rac2−/− and Rac2+/+ cells studied.
Actin Polymerization.
To measure changes in actin polymerization upon TCR cross-linking, CD4+ T cells were incubated with anti-CD3 antibody (1 μg/ml) on ice for 30 min, followed by washing and cross-linking with anti–hamster IgG (3 μg/ml) for the indicated time. Alternatively, AND TCR transgenic T cells were mixed with equal number of CH27 APCs preloaded with various amounts of MCC peptide. T cell APC interaction was initiated by a quick spin of the cell mixture followed by incubation at 37°C for the indicated time. Activation was terminated by addition of 4% paraformaldehyde. After fixation, cells were incubated with FITC-conjugated phalloidin (Sigma-Aldrich) for 30 min, washed three times in PBS, and analyzed using a FACSCalibur™ flow cytometer.
Capping and Immunofluorescence Study.
106 CD4+ T cells were incubated with anti-CD3 at the indicated concentrations on ice for 30 min. After washing, cells were further incubated with biotinylated anti–hamster Ig (3 μg/ml) at 37°C for 30 min, followed by addition of 4% paraformaldehyde to fix cells. Fixed cells were stained with Avidin-Texas Red (BD PharMingen) and were cytospun onto slides and mounted with Vectorshield (Vector Laboratories). The cells were visualized with a ZEISS fluorescence microscope. The percentage of capped cells was obtained by counting cells from 10–15 random fields and a total of more than 100 cells were counted per experimental condition in each experiment.
Results
Rac2 Deficiency Affect T Cell Activation.
Rac2 is a GTPase specifically expressed in hematopoietic lineage cells. To study the role of Rac2 during T cell activation using Rac2KO mice, we first examined the expression pattern of Rac proteins among subpopulation of thymocytes and mature peripheral T cells in wild-type and Rac2KO mice. As shown in Fig. 1 A, while Rac1 showed lower level of expression in CD4+ cells, Rac2 showed higher expression in CD4+ thymocytes and mature CD4 and CD8 single-positive cells. Upon disruption of Rac2, there is compensatory overexpression of Rac1 in CD4+ thymocytes and mature T cells (Fig. 1 A). Analysis of peripheral T cell subsets demonstrated no significant alterations in cell number or distribution. Thymocyte cellularity and subsets were also similar to wild-type mice 9. Thus, T cell development was not grossly altered in Rac2−/− mice (Fig. 1 B). This normal T cell development may partly due to the compensatory effect of Rac1 in the Rac2KO mice. To study the functional consequences of the Rac2 deficiency in T cell activation, we first examined the proliferation of total splenocytes stimulated with a dose range of plate bound anti-CD3. As shown in Fig. 2 A, decreased proliferation was observed in Rac2−/− total splenocytes. Purified CD4+ T cells were further studied by stimulating with a dose range of plate-bound anti-CD3. Rac2−/− CD4 T cell proliferation was reduced about twofold upon stimulation with plate-bound anti-CD3 (Fig. 2 B). The addition of IL-2 partially compensated for the defect in T cell activation, suggesting that IL-2 production is compromised in the Rac2−/− T cells and is partially responsible for the proliferation defect (Fig. 2 C). Costimulation with anti-CD28 also compensated for the T cell proliferation defect (Fig. 2 C). As anti-CD28 stimulation alone can cause Vav phosphorylation 30 31, it is plausible that costimulation of anti-CD28 further activates other Rho GTPases such as Rac1 and Rac3, thus compensating for the Rac2 deficiency. Interestingly, Rac2-deficient B cells also showed reduced proliferation upon stimulation with anti-IgM plus IL-4 (P < 0.01; Fig. 2 D).
Figure 1.


Rac protein expression and T cell development in Rac2−/− and Rac2+/+ mice. (A) Rac1 and Rac2 protein expression in subpopulation of thymocytes and mature peripheral T cells. 50 μg of cleared cell lysate from each cell population was used for Western blotting. The same blot probed for Rac1 was stripped and reprobed for Rac2. Lane 1 and 7, mature CD4+ cells; lane 2 and 8, mature CD8 cells; lane 3 and 9, CD4+CD8+ thymocytes; lane 4 and 10, CD4−CD8− thymocytes; lane 5 and 11, CD4+ thymocytes; lane 6 and 12, CD8+ thymocytes. The rac2 antibody weakly cross reacts with another protein in lysates from Rac2−/− cells and the control cells (reference 9). Data represents one of two experiments. (B) Lymph node cells or thymocytes were isolated from Rac2−/− mice and its littermate Rac2+/+ mice. The expression of CD4 and CD8 was examined by two-color flow cytometry.
Figure 2.

Defective T cell proliferation upon stimulation of plate-bound anti-CD3. (A) Splenocytes from Rac2−/− and control mice were stimulated with varying amounts of plate-bound anti-CD3. WT, wild-type. (B) Purified CD4 T cells stimulated with a dose range of plate-bound anti-CD3. (C) Purified T cells were stimulated with plate-bound anti-CD3 (5 μg/ml) alone, in the presence of IL-2 (50 μ/ml), or plate-bound anti-CD28 (2.5 μg/ml). (D) Purified B cells were stimulated with anti-IgM (10μg/ml), anti-IgM (10 μg/ml) + IL-4 (1,000 U/ml), or LPS (1 μg/ml). [3H]Thymidine was pulsed 48 h after stimulation and harvested 12 h later. Data represent one of three separate experiments.
To study Rac2 function during T cell activation in a more physiologically relevant context, Rac2−/− mice was crossed with AND TCR transgenic mice on the C57B/6 background. The AND TCR transgenic mouse expresses Vα11/Vβ3 TCR recognizing a peptide of cytochrome c (pigeon cytochrome c fragment) presented on IEk-bearing APCs 28. Although the AND TCR can be positively selected on I-Ab in the thymus, it cannot recognize MCC peptide on H-2b APC 32, therefore effectively eliminating any contribution of APC contaminants in the T cell preparation. The expression of TCR transgene was assessed by flow cytometry using Vα11 and Vβ3 antibody. A similar percentage of Vα11/Vβ3 CD4+ cells in the lymph node and spleen was observed in Rac2−/− TCR transgenic compared with the wild-type mice (data not shown), indicating that Rac2 deficiency does not significantly affect T cell–positive selection in the thymus. Rac2−/− CD4+ TCR transgenic cells were stimulated with I-EK+ splenic APCs with varying amount of MCC peptide. Proliferation was two- to threefold lower in Rac2−/− T cells compared with the control (Fig. 3 A). I-EK+ CH27 cells, which constitutively express both CD80 and CD86 molecules, were also used to stimulate TCR transgenic CD4 cells. Although the proliferation was much more vigorous compared with the stimulation with resting splenic APCs, Rac2−/− T cells still showed a two- to threefold lower level of proliferation (Fig. 3 B), accompanied by a lower level of IL-2 production (Fig. 3 C). Similar defects were also observed when stimulating T cells with fibroblast APCs (DCEK-Hi7; reference 33) expressing I-Ek and B7 molecules (data not shown). This result suggests that the activation defect in Rac2−/− T cells cannot be overcome using activated APCs. It is also consistent with previous findings that plate-bound anti-CD28 stimulation is different from stimulation with nature B7 ligand 30.
Figure 3.



Defective T cell proliferation and IL-2 production upon stimulation with TCR-specific antigen. Purified CD4+ T cells from AND TCR transgenic Rac2−/− or control mice were stimulated with various doses of MCC peptide in the presence of B10BR splenic APCs (A) or I-EK + CH27 cells (B and C). [3H]Thymidine was pulsed 48 h after stimulation and harvested 12 h later (A and B). To measure IL-2 production, supernatant were harvested 24 h after stimulation and the amount of IL-2 were determined by ELISA (C). Data represent one of four (A) or one of two (B and C) separate experiments.
Decreased ERK, p38 Activation upon T Cell Stimulation.
To understand the mechanism that accounts for the T cell functional defect in Rac2−/− mice, we first studied the early biochemical changes upon T cell stimulation. Total tyrosine phosphorylation was examined in CD4+ T cells after stimulation with antigen pulsed APCs for the indicated time. There was no significant change in the total tyrosine phosphorylation pattern in Rac2−/− T cells compared with the wild-type controls (Fig. 4 A). Further examination of the phosphorylation pattern of Zap70 and PLC-γ1 by immunoprecipitation showed no significant difference between Rac2−/− and control T cells (data not shown). TCR stimulation has been reported to cause activation of the MAPK pathway 34 35 36 37. As a signaling molecule, Rac has been shown to contribute to the activation of the Ras pathway through PAK 10 11. In addition, overexpression of Rac has been demonstrated to activate the JNK and p38 pathways 12. Thus, we further examined the activation of ERK, JNK, and p38 in Rac2−/− T cells during antigen-specific stimulation. As shown in Fig. 4 B, although Rac2−/− T cells showed increased ERK1/2 phosphorylation upon stimulation, the level of ERK1/2 activation is reduced compared with the control. We further examined ERK kinase activity upon stimulation. Consistent with what was observed in ERK1/2 phosphorylation, decreased ERK activity was observed 1 min after stimulation (Fig. 4 C). A slight decrease in p38 phosphorylation was also observed in Rac2−/− T cells (Fig. 4 D). Little phosphorylated JNK was detected upon stimulation in both wild-type and Rac2−/− T cells (data not shown). This is consistent with previous studies from our laboratory which showed that JNK protein is only expressed at low levels in naive T cells, and is upregulated during T cell activation and therefore functions during the differentiation rather than activation of T cells 38 39.
Figure 4.




Reduced MAPK activation upon TCR stimulation. Purified CD4+ T cells (2 × 106) from AND TCR transgenic Rac2−/− or control mice were stimulated with B10BR splenic APCs (2 × 106) pulsed with 50 μg/ml MCC peptide for 1, 5, and 15 min at 37°C. Cell lysates were separated by 12.5% SDS-PAGE and subject to immunoblot analysis. (A) Analysis of total phosphotyrosine proteins after stimulation using anti-phosphotyrosine antibody. (B) Analysis of phosphorylation of ERK1/2 using phospho-ERK antibody. The same blot was stripped and reprobed with ERK kinase antibody to confirm equal protein loading. (C) Analysis of ERK kinase activity. (D) Analysis of phospho-p38. Protein loading was confirmed by reprobing with antibody against total p38 (data represent one of four separate experiments except ERK kinase analysis represent one of two separate assays).
Reduced Ca2+ Mobilization upon TCR Stimulation.
Ca2+ mobilization is an early event during T cell activation and is responsible for the activation of Ca2+ sensitive phosphatase Calcineurin and nuclear factor of activated T cells (NFAT) mobilization 40. We further examined Ca2+ mobilization during antigen-specific stimulation. As shown in A, upon addition of antigen-pulsed APCs, the number of Rac2−/− CD4+ T cells to undergo Ca2+ flux was decreased compared with the CD4 T cells from wild-type mice at both high and low antigen doses tested (Fig. 5 A). To rule out that the decreased number of Ca2+ responsive cells was due to decreased T cell–APC interaction rather than defective Ca2+ mobilization, we also examined Ca2+ flux in response to TCR cross-linking. At a low dose of anti-CD3, there is lower Ca2+ flux among the responding cells in Rac2−/− T cells compared with the wild-type control (Fig. 5 B). In addition, Rac2−/− cells exhibited a longer lag time to Ca2+ increase compared with Rac2+/+ cells, which were more synchronized in their response. The average responding time was 217 ± 72 s for Rac2−/− T cells versus 155 ± 60 s for wild-type stimulated with 0.1 μg/ml anti-CD3 (P < 0.01). At higher doses of anti-CD3, Rac2−/− T cells showed similar pattern of Ca2+ mobilization as wild-type T cells (Fig. 5 B, and data not shown).
Figure 5.

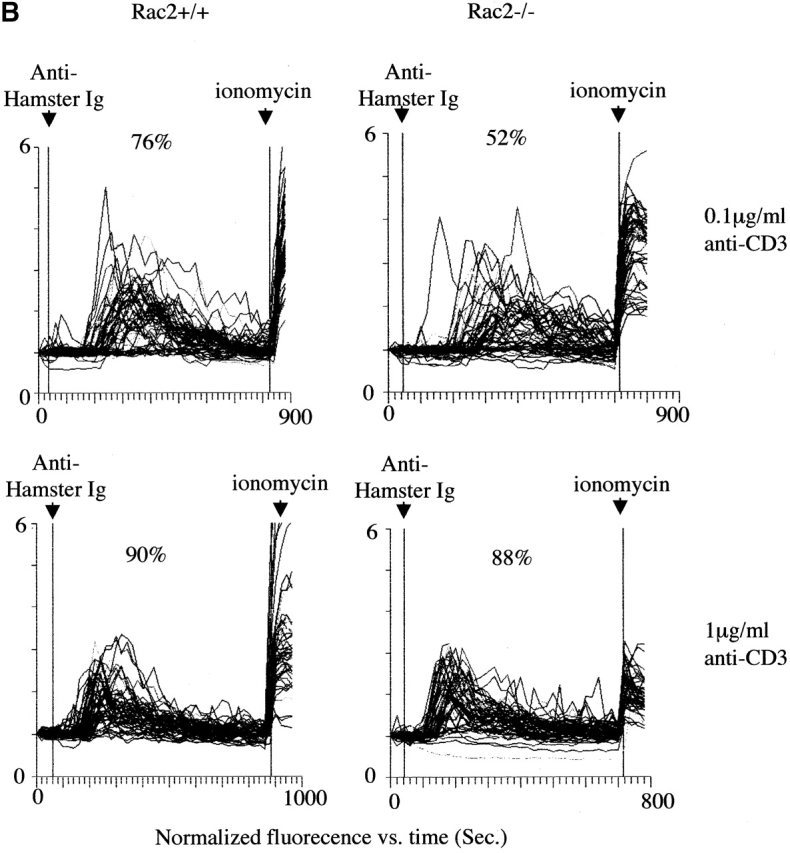
Reduced calcium mobilization in Rac2−/− T cells. (A) AND TCR transgenic CD4 cells were loaded with the Ca2+ sensitive fluorochrome, fluo-3, and stimulated with splenic APCs prepulsed with 50 μg/ml or 0.5 μg/ml MCC peptide. (B) CD4 T cells loaded with fluo-3 were stimulated with 0.1 or 1 μg /ml anti-CD3. The percentage of responding cells and the pattern of response in individual T cells was examined using a video laser cytometer. Each graph indicates the pattern of Ca2+ mobilization in a field of 40 to 50 cells, in which each line represents the average fluorescent intensity of an individual T cell over time. The first arrow indicates the addition of APC pulsed with peptide or cross-linking antibody, and the second arrow indicates the addition of ionomycin (666 ng/ml). S stands for sustained response, i.e., increased calcium concentration lasting >5 min. T stands for transient response, i.e., calcium increase lasting less than 5 min.
Defective Cytoskeleton Changes.
TCR activation leads to cytoskeletal reorganization in T cells. As Rac proteins have been shown to mediate actin polymerization and cell shape changes 3 4 5, we further investigated whether deficiency of Rac2 affects TCR-induced actin polymerization. Purified CD4+ cells were cross-linked with anti-CD3 antibody and actin polymerization was analyzed quantitatively using phalloidin labeled with FITC. Phalloidin preferentially binds to polymerized filamentous actin (F-actin). Although a significant increase in actin polymerization was observed in Rac2−/− T cells, the level of polymerization was at a reduced level in comparison to the wild-type control (Fig. 6 A). Similar results were obtained when TCR transgenic T cells were stimulated with APC pulsed with various amount of antigen (Fig. 6 B, and data not shown). These results indicate that Rac2 plays a role in mediating TCR-induced cytoskeleton changes.
Figure 6.


Impaired actin polymerization upon T cell stimulation. CD4+ T cells were stimulated by anti-CD3 cross-linking (A) or with CH27 cells (B) prepulsed with 0.5 μg /ml of MCC peptide. Cells were fixed after stimulation for the indicated time and polymerized actin was measured by phalloidin staining. Data showing one of three representative experiments.
Defective Cap Formation and T Cell APC Interaction.
We further investigated an actin dependent event upon TCR stimulation: TCR capping. Upon antibody cross-linking, TCR molecules cluster into a cap structure 1. Capping has been demonstrated to require actin polymerization and is blocked by Cytochalasin D 22 41. To investigate whether antibody cross-linking–induced TCR capping is affected in Rac2−/− cells, purified T cells were incubated with a range of concentrations of anti-CD3 at 4°C, and was subsequently cross-linked with anti–hamster IgG and incubated at 37°C for 30 min. At all doses of anti-CD3 antibody, a reduced number of cells with capped TCR was observed in Rac2−/− T cells (Fig. 7).
Figure 7.


Reduced cap formation in Rac2−/− T cells. Purified CD4 T cells coated with a dose range of anti-CD3 were cross-linked with biotinylated anti–hamster antibody and further incubated at 37°C for 30 min followed by fixation. TCR cap formation was visualized by staining with Texas-red-Avidin and analyzed by immunofluorescence microscopy. (A) Representative T cell capping in Rac2−/− and control cells. (B) Percentage of capping at different dose of anti-CD3. Data represents at least three separate experiments.
Discussion
Rac GTPase plays important roles in both transcriptional activation and cytoskeletal reorganization in a variety of cell types. In this study, we examined the function of hematopoietic-specific Rac2 GTPase during T cell activation. In the absence of Rac2, T cell activation stimulated by either antibody or TCR-specific antigen was defective. This defective T cell activation was accompanied by aberrant biochemical changes manifested as decreased ERK, p38 activation, Ca2+ mobilization, and defective actin polymerization which affects TCR capping. Consistent with these in vitro observations, reduced delayed-type hypersensitivity response was observed (data not shown).
Upon T cell stimulation, the small GTPase Ras is rapidly activated and leads to Raf activation, which in turn activates MEK1, and ERK. ERK activation is involved in mediating cellular proliferation, transformation, and differentiation. Although Rac GTPase is well known for its ability to activate the JNK and p38 pathways, activated Rac alone has little effect in activating the ERK pathway 12 42. However, accumulating evidence suggests that Rac can also influence ERK activation through its target PAK 10 11 43. In 293 cells, overexpression of either a dominant negative Rac2 or PAK inhibits Ras-mediated activation of ERK2, whereas constitutively active Rac synergizes with active Raf to increase ERK2 activity 43. In T cells, dominant negative PAK also inhibits TCR-mediated ERK2 activation 10. Activated PAK may influence ERK activation through phosphorylation of MEK1 and/or Raf1 44. Similar to the findings in Vav−/− T cells and Rac2−/− neutrophils 9 23, we have observed decreased ERK1/2 phosphorylation upon TCR stimulation. As in many other studies on early T cell activation 45 46 47, biochemical changes were observed in T cells stimulated with live antigen-pulsed APCs. To rule out the possibility that decreased ERK phosphorylation reflects changes in APCs rather than in T cells, we used fixed APCs prepulsed with antigen and obtained the same result (data not shown). Furthermore, reduced ERK activation was also observed in Rac2−/− T cells stimulated in the absence of APCs, by anti-CD3 cross-linking (data not shown). This result is consistent with the aforementioned in vitro findings that Rac influences the activation of the ERK pathway. In reference to results from Vav−/− T cells showing decreased ERK activation 23, our result is also consistent with the hypothesis that TCR-stimulated tyrosine phosphorylation of Vav promotes its GEF activity toward Rac, which in turn activates PAK and ERK 10. As the activation of the ERK pathway plays an important role in TCR-mediated AP-1 induction and cell cycle progression, defective ERK activation may be partially accountable for the observed proliferation defect in Rac2−/− T cells.
MAPK p38 is activated by cellular stresses, inflammatory cytokines, LPS, and G protein–coupled receptors 48 49 50 51. During T cell activation, p38 is activated with either anti-CD3 or anti-CD28 in primary lymphocytes and its activation correlates closely with T cell proliferation 37. However, how p38 influence primary T cell activation is less clear. As T cell proliferation is normal in response to anti-CD3 or ConA in dominant-negative p38 transgenic mice, activated p38 may mainly affect T helper 1 cell differentiation rather than directly affect T cell activation 52. Consistent with these studies, we observed decreased p38 activation in Rac2−/− T cells, accompanied with decreased T cell proliferation and T helper 1 cell differentiation 53.
Calcium mobilization is a crucial event during T cell activation. This process is achieved through the activation of phospholipase C-γ (PLC-γ) to catalyze the hydrolysis of phosphatidylinositol 4,5 bisphosphate (PIP2) to diacylglycerol and inositol 1,4,5-triphosphate (IP3), which activate protein kinase C and elevates intracellular calcium, respectively. In this study, we have observed decreased Ca2+ flux upon TCR stimulation by antigen or antibody cross-linking. Several potential mechanisms may explain this Ca2+ mobilization defect. First, Rac may affect Ca2+ mobilization by modulating the level of PIP2, the substrate for PLC-γ. Previously, Rho and Rac have been implicated in the activation of phosphatidylinositol 4-phosphate 5-kinase (PIP5K), which in turn phosphorylates phosphatidylinositol 4 phosphate (PIP), resulting in the production of PIP2 6 54. Decreased activation of PIP5K may leads to reduced level of PIP2, decreased production of IP3, hence decreased Ca2+ mobilization. Alternatively, Rac may affect the activation of PLC-γ through phosphatidylinositol 3-kinase (PI3K) activation. Recent studies suggests that phosphatidylinositol 3,4,5-trisphosphate (PIP3), a catalytic product of PI3K, can activate and synergize with phosphorylated PLC-γ to achieve full-scale Ca2+ mobilization 55 56. As PI3K may act downstream of Rac 57, Rac may affect Ca2+ flux through PI3K activation. Lastly, inhibition of actin polymerization in thymocytes and human T cell lines also reduces Ca2+ flux during activation mediated by specific peptide/MHC complex on APCs 58 59. We have observed decreased actin-polymerization in Rac2−/− T cells upon stimulation, which may account for the decreased Ca2+ mobilization.
T cell activation is accompanied by rapid cytoskeletal changes and formation of a cap upon antibody cross-linking, or SMACs at the interfaces of physical contact between T cells and APCs 1 60. It has been proposed that receptor clustering could favor sustained T cell signaling in three ways: first, by increasing the likelihood of contact between the TCR and MHC-bound ligand; second, by increasing the concentration of cytosolic signaling molecules and second messengers at regionally organized focal points in the proximity of TCRs; and last, by excluding negative regulatory molecules such as phosphatases from the zone of antigen receptor signaling 1. The formation of cap or SMAC requires actin polymerization in T cells 61. Rac has long been recognized to play a role in receptor-mediated cytoskeletal changes 62. We found that in the absence of Rac2, T cells showed decreased level of actin polymerization upon stimulation, accompanied with reduced antibody cross-linking–induced capping. These defects are similar to the findings in Vav−/− T cells, and are further evidence of the importance of actin in TCR signaling.
In summary, our results indicate that Rac2 affects ERK and p38 activation, Ca2+ mobilization, and the cytoskeleton reorganization related event, capping, during T cell activation. These functional defects are similar in nature but lesser in magnitude compared with the Vav−/− T cell phenotype 21 22 23. In light of the high homology among Rac proteins (sharing 89–92% sequence homology), functional overlap among Rho family GTPases 3, and the compensatory overexpression of Rac1 in Rac2−/− CD4+ T cells, it is conceivable that the moderate phenotypic defect could be due to the compensatory effect of Rac1, Rac3, and other Rho family GTPases such as CDC42. In any event, our results corroborate the concept that Rac is a major substrate of Vav GEF activity, and may relay Vav signaling during T cell activation. The correlation between defective T cell activation, defective actin polymerization, and defective capping in Rac2−/− T cells confirms the importance of Rac as a signaling molecule connecting the TCR signal to cytoskeleton reorganization.
Acknowledgments
We thank David A. Williams for providing Rac2−/− mice, Gary M. Bokoch for providing Rac2 antibody; Elizabeth Eynon and Charles A. Janeway for helpful discussions, Juli Unternaehrer for help in fluorescent staining, Judy Stein for technical help and Fran Manzo for help with manuscript preparation; and Li Wen and Victor Tybulewicz for critical reading of the manuscript.
H. Yu was an associate, and R.A. Flavell is an investigator of the Howard Hughes Medical Institute. H. Yu is supported by National Research Service Award (NRSA) fellowship GM20885-01.
Footnotes
D. Leitenberg's present address is George Washington University, Department of Immunology, Ross Hall Rm. 411, 2300 Eye St. NW, Washington, DC 20037.
B. Li's present address is Pfizer, Inc., East Point Rd., Groton, CT 06340.
Abbreviations used in this paper: ERK, extracellular signal–regulated kinase; JNK, c-Jun NH2-terminal kinase; KO, knockout; MAPK, mitogen-activated protein kinase; SMAC, supramolecular activation cluster; GEF, guanine nucleotide exchange factor.
References
- Penninger J.M., Crabtree G.R. The actin cytoskeleton and lymphocyte activation. Cell. 1999;96:9–12. doi: 10.1016/s0092-8674(00)80954-x. [DOI] [PubMed] [Google Scholar]
- Acuto O., Cantrell D. T cell activation and the cytoskeleton. Annu. Rev. Immunol. 2000;18:165–184. doi: 10.1146/annurev.immunol.18.1.165. [DOI] [PubMed] [Google Scholar]
- Van Aelst L., D'Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- Ridley A.J., Paterson H.F., Johnston C.L., Diekmann D., Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Nobes C.D., Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Hartwig J.H., Bokoch G.M., Carpenter C.L., Janmey P.A., Taylor L.A., Toker A., Stossel T.P. Thrombin receptor ligation and activated Rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell. 1995;82:643–653. doi: 10.1016/0092-8674(95)90036-5. [DOI] [PubMed] [Google Scholar]
- Allen W.E., Jones G.E., Pollard J.W., Ridley A.J. Rho, Rac and Cdc42 regulate actin organization and cell adhesion in macrophages. J. Cell Sci. 1997;110:707–720. doi: 10.1242/jcs.110.6.707. [DOI] [PubMed] [Google Scholar]
- Cox D., Chang P., Zhang Q., Reddy P.G., Bokoch G.M., Greenberg S. Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J. Exp. Med. 1997;186:1487–1494. doi: 10.1084/jem.186.9.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A.W., Kim C., Zhen L., Lowe J.B., Kapur R., Petryniak B., Spaetti A., Pollock J.D., Borneo J.B., Bradford G.B. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity. 1999;10:183–196. doi: 10.1016/s1074-7613(00)80019-9. [DOI] [PubMed] [Google Scholar]
- Yablonski D., Kane L.P., Qian D., Weiss A. A Nck-Pak1 signaling module is required for T-cell receptor-mediated activation of NFAT, but not of JNK. EMBO J. 1998;17:5647–5657. doi: 10.1093/emboj/17.19.5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kun Jiang B.Z., Gilvary D.L., Corliss B.C., Hong-Geller E., Wei S., Dieu J.Y. Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in nature killer cells. Nat. Immunol. 2000;1:419–425. doi: 10.1038/80859. [DOI] [PubMed] [Google Scholar]
- Coso O.A., Chiariello M., Yu J.C., Teramoto H., Crespo P., Xu N., Miki T., Gutkind J.S. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- Zhang S., Han J., Sells M.A., Chernoff J., Knaus U.G., Ulevitch R.J., Bokoch G.M. Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pak1. J. Biol. Chem. 1995;270:23934–23936. doi: 10.1074/jbc.270.41.23934. [DOI] [PubMed] [Google Scholar]
- Olson M.F., Ashworth A., Hall A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science. 1995;269:1270–1272. doi: 10.1126/science.7652575. [DOI] [PubMed] [Google Scholar]
- Yang F.-C., Kapur R., King A.J., Tao W., Kim C., Borneo J., Breese R., Marshall M., Dinauer M.C., Williams D.A. Rac2 stimulates Akt activation affecting BAD/Bcl-XL expression while mediating survival and actin function in primary mast cells. Immunity. 2000;12:557–568. doi: 10.1016/s1074-7613(00)80207-1. [DOI] [PubMed] [Google Scholar]
- Qiu R.G., Chen J., Kirn D., McCormick F., Symons M. An essential role for Rac in Ras transformation. Nature. 1995;374:457–459. doi: 10.1038/374457a0. [DOI] [PubMed] [Google Scholar]
- Khosravi-Far R., Solski P.A., Clark G.J., Kinch M.S., Der C.J. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol. Cell. Biol. 1995;15:6443–6453. doi: 10.1128/mcb.15.11.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus U.G., Heyworth P.G., Evans T., Curnutte J.T., Bokoch G.M. Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science. 1991;254:1512–1515. doi: 10.1126/science.1660188. [DOI] [PubMed] [Google Scholar]
- Diekmann D., Abo A., Johnston C., Segal A.W., Hall A. Interaction of Rac with p67phox and regulation of phagocytic NADPH oxidase activity. Science. 1994;265:531–533. doi: 10.1126/science.8036496. [DOI] [PubMed] [Google Scholar]
- Abo A., Pick E., Hall A., Totty N., Teahan C.G., Segal A.W. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature. 1991;353:668–670. doi: 10.1038/353668a0. [DOI] [PubMed] [Google Scholar]
- Holsinger L.J., Graef I.A., Swat W., Chi T., Bautista D.M., Davidson L., Lewis R.S., Alt F.W., Crabtree G.R. Defects in actin-cap formation in Vav-deficient mice implicate an actin requirement for lymphocyte signal transduction. Curr. Biol. 1998;8:563–572. doi: 10.1016/s0960-9822(98)70225-8. [DOI] [PubMed] [Google Scholar]
- Fischer K.D., Kong Y.Y., Nishina H., Tedford K., Marengere L.E., Kozieradzki I., Sasaki T., Starr M., Chan G., Gardener S. Vav is a regulator of cytoskeletal reorganization mediated by the T-cell receptor. Curr. Biol. 1998;8:554–562. doi: 10.1016/s0960-9822(98)70224-6. [DOI] [PubMed] [Google Scholar]
- Costello P.S., Walters A.E., Mee P.J., Turner M., Reynolds L.F., Prisco A., Sarner N., Zamoyska R., Tybulewicz V.L. The Rho-family GTP exchange factor Vav is a critical transducer of T cell receptor signals to the calcium, ERK, and NF-kappaB pathways. Proc. Natl. Acad. Sci. USA. 1999;96:3035–3040. doi: 10.1073/pnas.96.6.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo P., Schuebel K.E., Ostrom A.A., Gutkind J.S., Bustelo X.R. Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature. 1997;385:169–172. doi: 10.1038/385169a0. [DOI] [PubMed] [Google Scholar]
- Arrieumerlou C., Randriamampita C., Bismuth G., Trautmann A. Rac is involved in early TCR signaling. J. Immunol. 2000;165:3182–3189. doi: 10.4049/jimmunol.165.6.3182. [DOI] [PubMed] [Google Scholar]
- Reibel L., Dorseuil O., Stancou R., Bertoglio J., Gacon G. A hemopoietic specific gene encoding a small GTP binding protein is overexpressed during T cell activation. Biochem. Biophys. Res. Commun. 1991;175:451–458. doi: 10.1016/0006-291x(91)91585-z. [DOI] [PubMed] [Google Scholar]
- Haataja L., Groffen J., Heisterkamp N. Characterization of RAC3, a novel member of the Rho family. J. Biol. Chem. 1997;272:20384–20388. doi: 10.1074/jbc.272.33.20384. [DOI] [PubMed] [Google Scholar]
- Kaye J., Hsu M.L., Sauron M.E., Jameson S.C., Gascoigne N.R., Hedrick S.M. Selective development of CD4+ T cells in transgenic mice expressing a class II MHC-restricted antigen receptor. Nature. 1989;341:746–749. doi: 10.1038/341746a0. [DOI] [PubMed] [Google Scholar]
- Leitenberg D., Boutin Y., Constant S., Bottomly K. CD4 regulation of TCR signaling and T cell differentiation following stimulation with peptides of different affinities for the TCR. J. Immunol. 1998;161:1194–1203. [PubMed] [Google Scholar]
- Nunes J.A., Collette Y., Truneh A., Olive D., Cantrell D.A. The role of p21ras in CD28 signal transductiontriggering of CD28 with antibodies, but not the ligand B7-1, activates p21ras. J. Exp. Med. 1994;180:1067–1076. doi: 10.1084/jem.180.3.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehner S.P., Hofmann T.G., Dienz O., Droge W., Schmitz M.L. Tyrosine-phosphorylated Vav1 as a point of integration for T-cell receptor- and CD28-mediated activation of JNK, p38, and interleukin-2 transcription. J. Biol. Chem. 2000;275:18160–18171. doi: 10.1074/jbc.275.24.18160. [DOI] [PubMed] [Google Scholar]
- Kaye J., Vasquez N.J., Hedrick S.M. Involvement of the same region of the T cell antigen receptor in thymic selection and foreign peptide recognition. J. Immunol. 1992;148:3342–3353. [PubMed] [Google Scholar]
- Dubey C., Croft M., Swain S.L. Costimulatory requirements of naive CD4+ T cells. ICAM-1 or B7-1 can costimulate naive CD4 T cell activation but both are required for optimum response. J. Immunol. 1995;155:45–57. [PubMed] [Google Scholar]
- Izquierdo Pastor M., Reif K., Cantrell D. The regulation and function of p21ras during T-cell activation and growth. Immunol. Today. 1995;16:159–164. doi: 10.1016/0167-5699(95)80134-0. [DOI] [PubMed] [Google Scholar]
- Robinson M.J., Cobb M.H. Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- Salmon R.A., Foltz I.N., Young P.R., Schrader J.W. The p38 mitogen-activated protein kinase is activated by ligation of the T or B lymphocyte antigen receptors, Fas or CD40, but suppression of kinase activity does not inhibit apoptosis induced by antigen receptors. J. Immunol. 1997;159:5309–5317. [PubMed] [Google Scholar]
- Zhang J., Salojin K.V., Gao J.X., Cameron M.J., Bergerot I., Delovitch T.L. p38 mitogen-activated protein kinase mediates signal integration of TCR/CD28 costimulation in primary murine T cells. J. Immunol. 1999;162:3819–3829. [PubMed] [Google Scholar]
- Dong C., Yang D.D., Tournier C., Whitmarsh A.J., Xu J., Davis R.J., Flavell R.A. JNK is required for effector T-cell function but not for T-cell activation. Nature. 2000;405:91–94. doi: 10.1038/35011091. [DOI] [PubMed] [Google Scholar]
- Weiss L., Whitmarsh A.J., Yang D.D., Rincon M., Davis R.J., Flavell R.A. Regulation of c-Jun NH(2)-terminal kinase (Jnk) gene expression during T cell activation. J. Exp. Med. 2000;191:139–146. doi: 10.1084/jem.191.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A., Luo C., Hogan P.G. Transcription factors of the NFAT familyregulation and function. Annu. Rev. Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Kammer G.M., Smith J.A., Mitchell R. Capping of human T cell specific determinantskinetics of capping and receptor re-expression and regulation by the cytoskeleton. J. Immunol. 1983;130:38–44. [PubMed] [Google Scholar]
- Minden A., Lin A., Claret F.X., Abo A., Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- Frost J.A., Xu S., Hutchison M.R., Marcus S., Cobb M.H. Actions of Rho family small G proteins and p21-activated protein kinases on mitogen-activated protein kinase family members. Mol. Cell. Biol. 1996;16:3707–3713. doi: 10.1128/mcb.16.7.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagrodia S., Cerione R.A. Pak to the future. Trends Cell Biol. 1999;9:350–355. doi: 10.1016/s0962-8924(99)01618-9. [DOI] [PubMed] [Google Scholar]
- Combadiere B., e Sousa C.R., Germain R.N., Lenardo M.J. Selective induction of apoptosis in mature T lymphocytes by variant T cell receptor ligands. J. Exp. Med. 1998;187:349–355. doi: 10.1084/jem.187.3.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas B., Stefanova I., Yasutomo K., Dautigny N., Germain R.N. Divergent changes in the sensitivity of maturing T cells to structurally related ligands underlies formation of a useful T cell repertoire. Immunity. 1999;10:367–376. doi: 10.1016/s1074-7613(00)80036-9. [DOI] [PubMed] [Google Scholar]
- Kersh G.J., Kersh E.N., Fremont D.H., Allen P.M. High- and low-potency ligands with similar affinities for the TCRthe importance of kinetics in TCR signaling. Immunity. 1998;9:817–826. doi: 10.1016/s1074-7613(00)80647-0. [DOI] [PubMed] [Google Scholar]
- Read M.A., Whitley M.Z., Gupta S., Pierce J.W., Best J., Davis R.J., Collins T. Tumor necrosis factor alpha-induced E-selectin expression is activated by the nuclear factor-kappaB and c-JUN N-terminal kinase/p38 mitogen- activated protein kinase pathways. J. Biol. Chem. 1997;272:2753–2761. doi: 10.1074/jbc.272.5.2753. [DOI] [PubMed] [Google Scholar]
- Raingeaud J., Gupta S., Rogers J.S., Dickens M., Han J., Ulevitch R.J., Davis R.J. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- Rouse J., Cohen P., Trigon S., Morange M., Alonso-Llamazares A., Zamanillo D., Hunt T., Nebreda A.R. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- Yamauchi J., Nagao M., Kaziro Y., Itoh H. Activation of p38 mitogen-activated protein kinase by signaling through G protein-coupled receptors. Involvement of Gbetagamma and Galphaq/11 subunits. J. Biol. Chem. 1997;272:27771–27777. doi: 10.1074/jbc.272.44.27771. [DOI] [PubMed] [Google Scholar]
- Rincon M., Enslen H., Raingeaud J., Recht M., Zapton T., Su M.S., Penix L.A., Davis R.J., Flavell R.A. Interferon-gamma expression by Th1 effector T cells mediated by the p38 MAP kinase signaling pathway. EMBO J. 1998;17:2817–2829. doi: 10.1093/emboj/17.10.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Yu H., Zheng W., Voll R., Na S., Roberts A.W., Williams D.A., Davis R.J., Ghosh S., Flavell R.A. Role of the guanosine triphosphatase Rac2 in T helper 1 cell differentiation. Science. 2000;288:2219–2222. doi: 10.1126/science.288.5474.2219. [DOI] [PubMed] [Google Scholar]
- Chong L.D., Traynor-Kaplan A., Bokoch G.M., Schwartz M.A. The small GTP-binding protein Rho regulates a phosphatidylinositol 4- phosphate 5-kinase in mammalian cells. Cell. 1994;79:507–513. doi: 10.1016/0092-8674(94)90259-3. [DOI] [PubMed] [Google Scholar]
- Bae Y.S., Cantley L.G., Chen C.S., Kim S.R., Kwon K.S., Rhee S.G. Activation of phospholipase C-gamma by phosphatidylinositol 3,4,5- trisphosphate. J. Biol. Chem. 1998;273:4465–4469. doi: 10.1074/jbc.273.8.4465. [DOI] [PubMed] [Google Scholar]
- Rameh L.E., Rhee S.G., Spokes K., Kazlauskas A., Cantley L.C., Cantley L.G. Phosphoinositide 3-kinase regulates phospholipase Cgamma-mediated calcium signaling. J. Biol. Chem. 1998;273:23750–23757. doi: 10.1074/jbc.273.37.23750. [DOI] [PubMed] [Google Scholar]
- Genot E.M., Arrieumerlou C., Ku G., Burgering B.M., Weiss A., KrAm I.M. The T-cell receptor regulates Akt (protein kinase B) via a pathway involving Rac1 and phosphatidylinositide 3-kinase. Mol. Cell. Biol. 2000;20:5469–5478. doi: 10.1128/mcb.20.15.5469-5478.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valitutti S., Dessing M., Aktories K., Gallati H., Lanzavecchia A. Sustained signaling leading to T cell activation results from prolonged T cell receptor occupancy. Role of T cell actin cytoskeleton. J. Exp. Med. 1995;181:577–584. doi: 10.1084/jem.181.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y.Y., Fischer K.D., Bachmann M.F., Mariathasan S., Kozieradzki I., Nghiem M.P., Bouchard D., Bernstein A., Ohashi P.S., Penninger J.M. Vav regulates peptide-specific apoptosis in thymocytes. J. Exp. Med. 1998;188:2099–2111. doi: 10.1084/jem.188.11.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks C.R., Kupfer H., Tamir I., Barlow A., Kupfer A. Selective modulation of protein kinase C-theta during T-cell activation. Nature. 1997;385:83–86. doi: 10.1038/385083a0. [DOI] [PubMed] [Google Scholar]
- Wulfing C., Sjaastad M.D., Davis M.M. Visualizing the dynamics of T cell activationintracellular adhesion molecule 1 migrates rapidly to the T cell/B cell interface and acts to sustain calcium levels. Proc. Natl. Acad. Sci. USA. 1998;95:6302–6307. doi: 10.1073/pnas.95.11.6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarche N., Tapon N., Stowers L., Burbelo P.D., Aspenstrom P., Bridges T., Chant J., Hall A. Rac and Cdc42 induce actin polymerization and G1 cell cycle progression independently of p65PAK and the JNK/SAPK MAP kinase cascade. Cell. 1996;87:519–529. doi: 10.1016/s0092-8674(00)81371-9. [DOI] [PubMed] [Google Scholar]