

Abstract
We have previously shown that small B16 melanomas can be successfully treated using a combination of anti–cytotoxic T lymphocyte antigen (CTLA)-4 monoclonal antibody with a granulocyte/macrophage colony-stimulating factor (GM-CSF) producing irradiated tumor cell vaccine. Regression of tumors results in long-lasting immunity and is frequently accompanied by autoimmune depigmentation. Here we examine the cellular and molecular mechanisms of this combined treatment. Histological examination of depigmented lesions revealed infiltration of polymorphonuclear cells and deposition of antibody. The combination therapy also induced tumor rejection and skin depigmentation in B cell–deficient and in CD4+ T cell–depleted mice. Both effects of the treatment absolutely required CD8+ T cells. Analysis of the response in successfully treated mice revealed elevated levels of CD8+ T cells specific for a nonameric peptide consisting of residues 180–188 of the melanocyte differentiation antigen tyrosinase-related protein (TRP)2. There was no evidence of reactivity to the melanocyte antigens gp100, tyrosinase, Mart1/MelanA, or TRP1. Fas–FasL interactions and perforin played a role in mounting the effector response, whereas the tumor necrosis factor pathway was not required. The cellular requirements for tumor rejection in this therapeutic setting were strikingly different from those in a prophylactic setting. In particular, if mice received a prophylactic vaccine consisting of anti–CTLA-4 and B16–GM-CSF before tumor challenge, full protection was obtained even in the absence of CD8+ T cells. Our data demonstrate that therapeutic autoreactive CD8+ T cell responses can effectively be generated in tumor-bearing mice and stresses the value of studying tumor immunity in a therapeutic rather than a prophylactic setting.
Keywords: immunotherapy, prophylaxis, T lymphocyte, TRP-2, depigmentation
Introduction
In the past decade, a wealth of tumor-associated antigens have been identified that provide targets for CTL and CD4+ T cells in cancer patients (for reviews, see references 1 and 2). Human melanoma has provided a paradigm in this respect, demonstrating that such tumor antigens can be derived from tissue-specific self-antigens, from tumor-specific mutated proteins, or from aberrantly expressed proteins that normally function only during embryonic development. Depigmentation or vitiligo (spontaneous or treatment related) appears to correlate with favorable prognosis and successful rejection of metastatic melanoma, indicating that self-antigens might play a role in antitumor responses 3 4. Melanocyte proteins involved in pigment synthesis have frequently been shown to be targets for CTLs, CD4+ T cells, and antibodies in melanoma patients or in healthy donors. In vitiligo and melanoma patients, tumor-associated and potentially autoimmune T cell–mediated reactivity can be linked directly to destruction of pigmented cells in the skin 5 6. As these pigmentation antigens are expressed by most melanomas even in advanced stages, they are considered to be good candidate targets for specific immunotherapy, provided that damage to healthy tissue is limited. Importantly, immunological tolerance to these melanocyte antigens is clearly incomplete or absent providing the rationale for clinical testing of vaccines consisting of pigmentation proteins or their T cell epitopes. A satisfactory animal model for melanoma treatment is essential for assessment of the risks and requirements for such immunotherapy to be successful.
T cell responses are regulated not only by antigen receptor signals, but also by positive and inhibitory costimulatory signals mediated by the interactions of CD28 and CTLA-4, respectively, with their cognate B7 ligands on antigen-presenting cells 7. We and others have shown that administration of antibodies to block the inhibitory effects of CTLA-4 can enhance antitumor responses in several murine tumor models 8 9 10 11 12 13 14. Of particular relevance to melanoma is our demonstration that mice carrying a small load of B16 melanoma cells can be successfully treated with a combination vaccine consisting of GM-CSF–producing, irradiated B16 cells and CTLA-4 blockade 13. Approximately 60% of the surviving mice developed lesions of depigmentation reminiscent of vitiligo. B16 therapy in this model absolutely requires the presence of CD8+ cells. Our finding that the combination therapy was CD8 dependent and resulted in depigmentation was especially interesting in light of previous studies using the GM-CSF–producing B16 vaccine as a single agent 15. In these studies it was found that the vaccine was capable of inducing prophylactic immunity mediated mainly by CD4+ T cells with no requirement for CD8+ T cells 15 16. Also, this cell-based vaccine by itself was not effective in a therapeutic setting, and depigmentation was not reported even when prophylactic immunity had been successfully obtained.
We now report an extensive examination of the mechanisms involved in the generation of immunity to B16 using the combination therapy. We have found that neither CD4+ T cells, nor antibody responses are required for treatment effect, as tumor rejection and depigmentation occur in B cell–deficient mice. Both the Fas and perforin pathways are required, but TNF-α is dispensable. T cells responding to the melanocyte differentiation antigen tyrosinase-related protein 2 (TRP-2) antigen were found in spleen cell cultures and in peripheral blood from depigmented mice. Depletion studies confirm that CD8+ T cells are required for both the antitumor effect and depigmentation in a therapeutic setting. In the setting of prophylaxis, CD8+ T cells are not required for tumor immunity, but are required for depigmentation. These findings demonstrate that the cellular mechanisms involved in protection against subsequent tumor challenge are different from those observed in a therapeutic setting. This underscores the added value of studying tumor immunity in tumor-bearing subjects.
Materials and Methods
Mice.
C57Bl/6 female mice, B cell–deficient (from The Jackson Laboratory), MHC class I− (β2m−/−), and class II–deficient mice (I-Aβ2/−; Taconic) as well as perforin-deficient, TNF-α knockout, gld, and lpr mice (all bred into the C57Bl/6 background) were maintained and treated in accordance with institute guidelines. Mice were used for tumor experiments when 8–16 wk old.
Antibodies and Cell Lines.
Generation and purification of anti–CTLA-4 (9H10) has been described previously 17. Control hamster IgG, control rat IgG, and control mouse IgG were purchased from Jackson ImmunoResearch Laboratories. Anti–H-2Db, anti–H-2Kb, anti-CD4 (GK1.5), anti-CD8 (2.43), anti-NK1.1 (PK136), and anti-Lyt2.1 (116.3) were isolated from hybridoma culture supernatants, or grown as ascites by standard procedures. Antisera specific for TRP-1 (TA99) and TRP-2 (αPEP8) were generously provided by Alan Houghton (Memorial Sloan-Kettering Institute, New York, NY) and Vincent Hearing (National Institutes of Health, Bethesda, MD). The C57Bl/6-derived tumor cell lines B16-BL6, B16-F0, B16-F10 (obtained from I. Fidler, M.D. Anderson Cancer Center, Houston, TX), EL-4, MC38, RMA-S, as well as the immortalized dendritic cell line DC2.4 18 were cultured in DMEM or IMDM supplemented with 1 U/ml penicillin, 1 μg/ml streptomycin, 50 μg/ml gentamycin, 2 μM l-glutamine, 20 μM β-mercaptoethanol, and 8% fetal calf serum (hereafter referred to as CM). GM-CSF–producing B16-BL6 clones BL6/GM-E, BL-6/GM-18, and the CD80-expressing variant B16-BL6/B7.1 13 were similarly cultured in CM.
Tumor Challenge and Treatment.
Subcutaneous tumor challenge and treatment experiments were performed as described previously 13. Briefly, mice were challenged subcutaneously with 1–2 × 104 B16-BL6 cells in PBS. At the same day or later as indicated, treatment was initiated by injecting 106 irradiated (16,000 rad) GM-CSF–producing cells (in PBS) subcutaneously into the left flank, and repeated 3 and 6 d later. The vaccine consisted of a 1:1 mixture of clones BL6/GM-E and BL6/GM-18. Treatment with 9H10 or control hamster IgG started 3 d later. Antibodies were delivered intraperitoneally at 100 μg in PBS, followed by two injections of 50 μg 3 and 6 d later. Tumor growth was scored by measuring perpendicular diameters. Mice were killed when the tumors displayed severe ulceration or reached a size of 250 mm2. Depletion of lymphoid subsets was done as described earlier, starting a week before tumor challenge. Depletions were maintained for at least 3 wk by weekly injecting the appropriate antibodies. Prophylactic experiments were done as follows. Mice were immunized by injecting 106 irradiated GM-CSF–producing B16-BL6 cells subcutaneously on days −12, −9, and −6, in combination with anti–CTLA-4 given on days −9, −6, and −3 (100, 50, and 50 μg per mouse). On day 0, mice were challenged with B16-BL6. Depletions of lymphoid subsets in the prophylactic model were started at day −3 by three daily injections of 500 μg of depleting antibody, and maintained for 3 wk by biweekly administration of antibody.
Generation of T Cell Cultures from Spleen and IFN-γ Release Assays.
Spleens were harvested from mice rejecting B16-BL6 and restimulated in vitro with B16-BL6/B7.1 or a mixture of B16-F0 and the dendritic cell line DC2.4 after overnight coculture. 5 × 106 spleen cells were mixed with 105 irradiated (16,000 rad) stimulator cells and recombinant human IL-2 was added to a final concentration of 30 IU/ml. After 7 d, cells were collected and purified by Histopaque gradient centrifugation. Live cells (2.5 × 105 per well) were stimulated with target cells (5 × 104 per well) in 96-well round-bottom plates for 24 h, after which supernatant was collected and tested for the presence of IFN-γ by sandwich ELISA (BD PharMingen). As target cells, several variants derived from parental line B16-F0, as well as the nonpigmented colon tumor line MC38 were used. MC38 cells were transduced as described earlier 19 with recombinant vaccinia virus (rVV) expressing murine Pmel-17/gp100, Mart-1/Melan-A, tyrosinase, gp75/TRP-1, TRP-2, or β-galactosidase to serve as targets in a cytokine release assays. As a control, wild-type vaccinia (VVwt) was used in each experiment. Peptide pulsed targets were prepared by washing MC38 cells threefold in serum-free media, and incubating up to 2 × 106 cells per ml with 10 μg/ml peptide, for 90 min at 37°C. Unbound peptide was washed away by three washes with serum-free media, after which cells were diluted in CM and dispensed into 96-well plates for the cytokine release assays.
Peptide Selection, Synthesis, and Testing.
Peptides were selected from the murine TRP-2 protein sequence based on the published motifs for binding to H-2Kb or H-2Db. For H-2Kb, both 8- and 9-mer sequences were included, whereas for H-2Db this included 9- and 10-mer sequences. Peptides were synthesized by standard Fmoc chemistry, and purity was checked by HPLC. Fractions routinely contained >95% of the expected sequence. Peptides were dissolved in DMSO at 50 mg/ml, and diluted into PBS for binding assays and peptide pulsing onto MC38 or RMA-S cells. Peptide binding to H-2Kb and H-2Db was determined as follows. RMA-S cells, precultured at 26°C for 48 h, were washed three times in serum-free media and incubated with peptide at 26°C for 2 h, followed by 2 h at 37°C. Cells were washed with ice-cold PBS/0.5% BSA, stained for expression of H-2Kb and H-2Db, and analyzed on a FACScan™ (Becton Dickinson).
Tetramer Staining.
TRP-2180–188/Kb tetramers labeled with allophycocyanin (APC) were a gift from Ton Schumacher and John Haanen (Netherlands Cancer Institute, Amsterdam, Netherlands). Blood leukocytes were collected in heparin coated tubes and erythrocytes were lysed using standard procedures. Remaining leukocytes were washed and incubated with FITC-labeled CD8β-specific antibodies and APC-labeled tetramers in PBS/1% BSA, and after two washes in PBS/1% BSA analyzed in PBS/1% BSA/1 μg/ml propidium iodide (PI). PI-negative cells were gated for the lymphocyte population and further analyzed.
Histology.
Formalin-fixed, paraffin-embedded sections were prepared from the depigmenting skin and control skin and stained with hematoxylin and eosin. Alternatively, sections were deparaffinized and reacted with biotinylated anti–mouse IgG/IgM antibody followed by streptavidin-peroxidase, and the reaction visualized with diaminobenzidine (brown precipitate). Sections were counterstained with hematoxylin.
Results
Lack of Functional B Cell Responses Does Not Impair B16 Treatment or Depigmentation.
Both tumor rejection and depigmentation occurred in mice bearing B16 tumors after combination therapy with anti–CTLA-4 and GM-CSF B16 vaccine, even in the absence of CD4+ T cells 13. This suggested that neither CD4 T cell help, nor by extension antibody responses, were required for either effect. We examined the depigmented skin of successfully treated mice for hallmarks of autoimmunity. Polymorphonuclear cells were clearly detected infiltrating the dermis concentrating around the hair follicles (Fig. 1). Also, pigmented granules were found deposited around the hair follicles and Ig deposits were detected (Fig. 1). These hallmarks of autoimmune response were absent from nonaffected skin in these depigmented mice and in control animals. Antibody responses directed against melanocyte antigens such as TRP-1/gp75 were shown previously to be involved in depigmentation in humans and in mice 20 21. However, we were unable to detect antibodies against TRP1/gp75 or other B16 specific proteins by staining intact or permeabilized cells using sera collected from depigmented mice (data not shown). As a definitive determination of the role of B cells in tumor rejection and depigmentation using our combination therapy of B16, we attempted to treat B16 tumors in B cell knockout (μMT) mice bred onto the C57Bl/6 background. At the usual tumor dose of 104 cells per mouse, B cell–deficient mice displayed ∼75% tumor take (Fig. 2 A). The reduced tumor take compared with wild-type mice is consistent with earlier studies showing that B cell–deficient mice displayed enhanced immunity to several transplantable tumor cell lines 22. All B6.μMT mice receiving combination therapy rejected their tumors and most of these (6 of 9) displayed signs of depigmentation within 4 wk after challenge (Fig. 2 B). These results demonstrate that B cells are not required to initiate or maintain immunity against B16 or normal melanocytes in this model and further suggest that the antibody deposition detected in the skin of depigmented wild-type mice may be a secondary effect of the autoimmune response.
Figure 1.

Histological signs of autoimmunity in depigmented skin of mice surviving B16 challenge. Transverse sections from depigmented (B and D) or uninvolved skin (A and C) from the same mouse were stained with hematoxylin and eosin to detect cellular infiltrate (A and B), or stained for the presence of Ig deposition (C and D) following procedures described in Materials and Methods.
Figure 2.


Rejection of B16-BL6 and subsequent depigmentation in B−/− mice, after combination treatment with anti–CTLA-4 and B16/GM-CSF. B cell–deficient mice were challenged with B16-BL6 and treated following the general scheme as outlined before. Tumor-free survival (A) as well as signs of autoimmune depigmentation were scored. Of nine B−/− mice receiving combination treatment, six developed depigmentation of which an example is shown (B). wt, wild-type.
Role of Fas, Perforin, and TNF-dependent Pathways of Killing in Mounting a Productive Immune Response to B16.
The perforin, Fas/Fas-L, or TNF-α–dependent pathways of killing have all been implicated in controlling tumor outgrowth 23 24 25. To delineate the relative roles of these pathways in the combination therapy of B16, we tested our combination B16 vaccine protocol in mice deficient in perforin, TNF-α, Fas (lpr), or Fas-L (gld) functions bred into the B6 background. The results are presented in Table . In the absence of treatment all mice developed B16 tumors appearing macroscopically between days 10 and 12 and growing at comparable rates. The incidence and rate of tumor growth in lpr or gld receiving the combination therapy was similar to untreated controls, demonstrating that functional Fas/Fas-L interactions are necessary for the therapeutic effects of the treatment. In the absence of perforin, B16 tumor outgrowth was delayed by the combination therapy, but all of the mice eventually succumbed to tumor. In contrast, all TNF-α–deficient mice rejected their tumors upon combination treatment, and 4 of the 7 survivors developed autoimmune depigmentation, demonstrating that this pathway of killing is not required for therapeutic effect. These results underscore the importance of both Fas/FasL interactions and perforin for obtaining tumor rejection in our B16 model of combination therapy.
Table 1.
B16 Treatment Fails in Mice Lacking Perforin, Fas, or Fas-L Expression
| Tumor rejection (rejecting/challenged) | |||||
|---|---|---|---|---|---|
| Wild-type | Perforin−/− | lpr | gld | TNF-α−/− | |
| No treatment | 0/8 | 0/5 | 0/5 | 0/6 | 0/6 |
| Anti–CTLA-4 + B16/GM-CSF | 6/9 | 0/6 | 0/5 | 0/7 | 7/7 |
Mice were immunized and challenged with tumor as described in Materials and Methods. Tumors in wild-type mice became palpable by 10–14 d after implant. Euthanasia or death usually occurred by 50 d. Surviving mice were followed for signs of tumor growth for about 50 d.
Definition of TRP-2180–188 as a T Cell Target in Mice Receiving Combination Treatment.
Depletion studies had indicated that successful treatment of B16 by the combination therapy required CD8+ T cells (13; see Fig. 5). As most of the surviving mice developed depigmentation, we reasoned that melanocyte antigens involved in pigment synthesis could be serving as targets for cytotoxic T cells. We generated short-term T cell cultures by stimulating spleen cells with a mixture of a syngeneic dendritic cell line DC2.4 and B16 (cocultivated for 24 h). These cultures were tested for reactivity against syngeneic MC38 cells that had been transduced with rVV expressing each of the aforementioned melanocyte antigens. T cell cultures from mice rejecting B16-BL6 subcutaneous tumor challenge (mouse 12K54), or B16-F10 lung metastases (mouse 44C1; see, for example, Fig. 4 in reference 13), displayed a restricted reactivity toward the TRP-2 antigen (Fig. 3).
Figure 5.
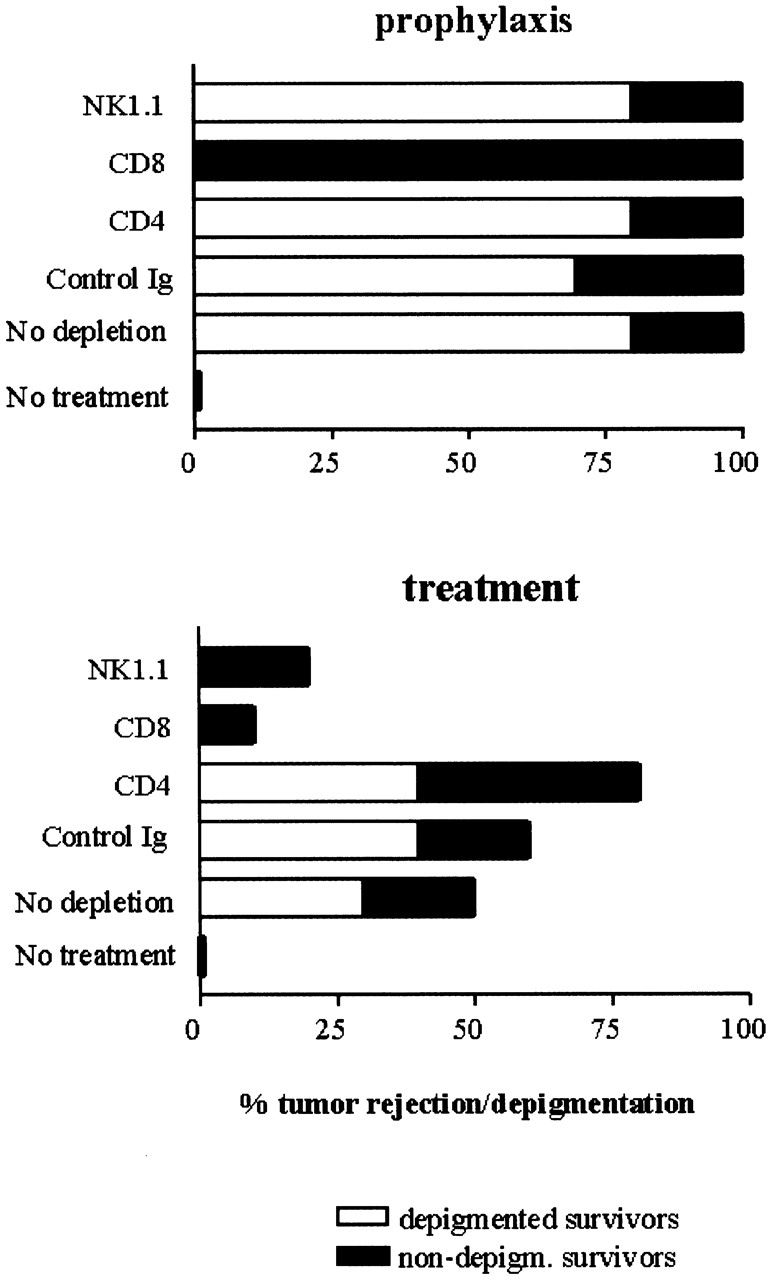
Differential requirements for lymphoid subpopulations in therapeutic vs. prophylactic treatment of B16. C57Bl/6 mice received treatment with anti–CTLA-4 plus B16/GM-CSF vaccine started either before challenge with B16-BL6 (“prophylaxis,” top graph), or simultaneously with challenge (“treatment,” bottom graph). Before tumor challenge mice were depleted from CD4+, CD8+, or NK1.1+ cells or treated with control IgG (as described in Materials and Methods). The percentage of mice rejecting the tumor was scored (total of each bar) as well as the fraction of surviving mice developing depigmentation, as indicated by the unfilled (“depigmented”) part of each bar. This is a representative of two experiments.
Figure 4.

Detection of TRP-2180–188/Kb reactive CD8+ T cells in PBL of mice receiving combination treatment. PBL taken at day 17 from mice challenged with B16-BL6 receiving no further treatment (left) or treated with anti–CTLA-4 plus B16/GM-CSF (right) were gated for live cells in the presence of PI, and CD8β1 TRP-2180–188/Kb tetramer-positive T cells were analyzed. Indicated in the top right quadrant is the percentage of CD8b+ T cells that are detected with the tetramer reagent.
Figure 3.
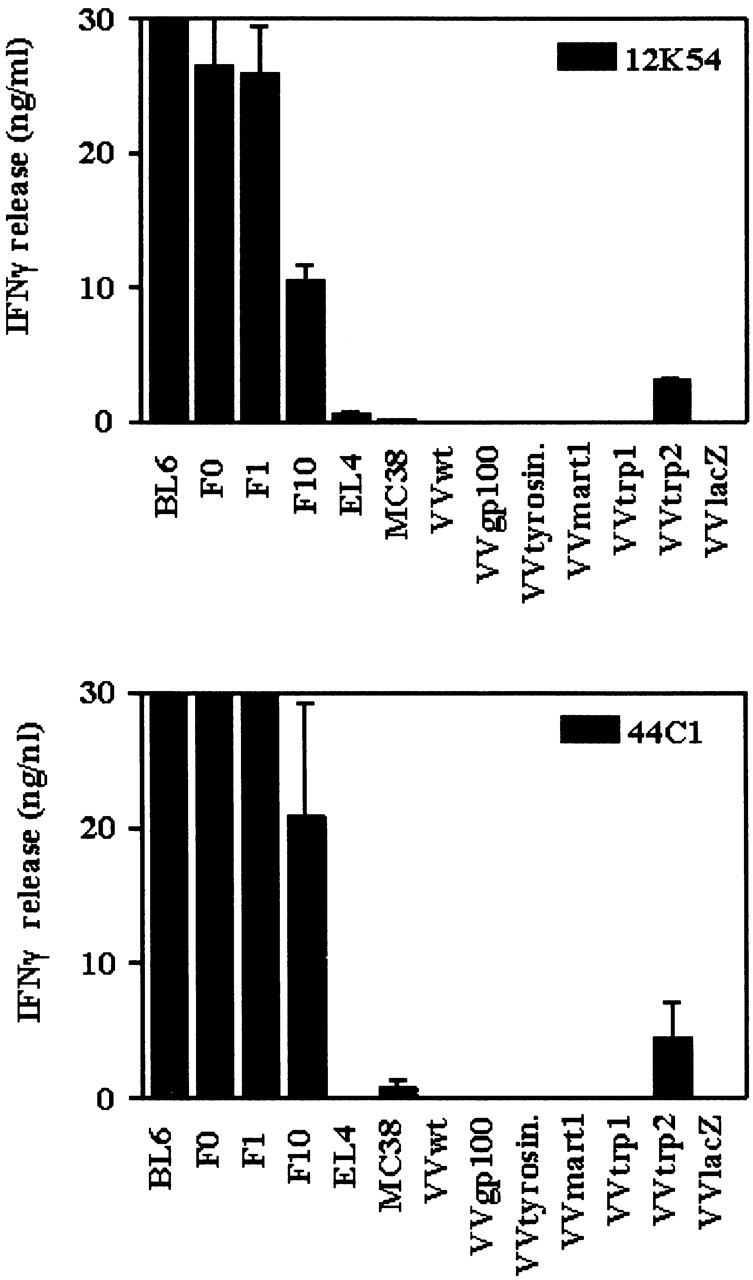
T cell lines derived from mice surviving B16-BL6 challenge react to VV-TRP-2 transduced targets. T cell lines established from mice surviving B16-BL6 challenge after combination therapy were tested for reactivity toward B16 variants BL6, F0, and F10, as well as MC38 cells expressing murine pigmentation antigens gp100, tyrosinase, Mart-1, TRP-1, and TRP-2 after transduction with recombinant vaccinia virus. Reactivity was quantified by measuring IFN-γ release after 24 h coculture of T cells and targets. T cell culture 12K54 (top) was derived from a mouse surviving B16-BL6 subcutaneous tumor upon combination treatment, whereas culture 44C1 (bottom) was established after treatment of B16-F10 lung metastases (see text).
The murine TRP-2 protein sequence was scanned for potential Kb or Db binding peptides. Over 60 peptides (8 to 10 amino acids [aa]) were synthesized and tested for binding to Kb or Db on RMA-S cells (Table ). Peptides were pulsed onto RMA-S cells and tested for the ability to stimulate splenic T cells from depigmented mice that had rejected B16 tumors. Strong stimulation was found only with a single peptide comprised of aa 180 to 188 of TRP-2, SVYDFFVWL (Table ), a nonamer variant of a previously identified TRP-2–derived B16 epitope 26. Interestingly, this variant had been shown to bind to H-2Kb with much higher affinity than the 8-mer minimal epitope, which may explain why we only detected reactivity against the nonamer 27. Mice rejecting established B16 tumors upon combination treatment were evaluated for the presence of TRP-2–specific CD8+ T cells in peripheral blood samples using TRP-2180–188/Kb tetramers. Indeed, a clearly detectable fraction of CD8β1 cells (1.7%) was stained using the TRP-2/Kb–specific reagent (Fig. 4). As these cells were not detected in significant numbers (<0.2%) in untreated mice, this implies that the combination treatment of CTLA-4 blocking antibody plus GM-CSF producing whole cell vaccine was responsible for inducing this reactivity.
Table 2.
TRP-2180–188 Encodes the Optimal Epitope Recognized by T Cells Derived from Treated Mice
| Binding to | |||||
|---|---|---|---|---|---|
| aa (TRP-2) | Sequence | Motif | K | D | Recognition(IFN-γ ng/ml) |
| 180 | SVYDFFVWL | K | + | − | 73.4 |
| 181 | VYDFFVWL | K | + | − | 11.3 |
| 185 | FVWLHYYSV | K | + | − | 10.3 |
| 186 | VWLHYYSV | K | + | − | 1.3 |
| OVA8 | K | +++ | + | Not tested | |
The data shown are only for the Kb binding peptides relevant to the actual epitope that stimulate T cells from the immunized mice. The data concerning the other Kb and Db peptides can be found on the lab website: http://mcb.Berkeley.edu/labs/allison.
Differential Requirements for Lymphocyte Subsets in Prophylactic versus Therapeutic B16 Model.
In our combination therapy model, CD8+ and NK1.1+ cells were required for successful treatment of B16 melanoma 13. Previous studies using the GM-CSF vaccine alone had clearly demonstrated a requirement for CD4+ T cells and Th2 cytokines for protection against subsequent challenge with B16 cells 16. CD8+ cells were shown to contribute to the response, but were not essential. As these two studies differed not only in the use of anti–CTLA-4 but also in the setting of the treatments, we compared the requirements for CD4+CD8+ cells and NK1.1+ cells in prophylactic and therapeutic application of the combination treatment protocol.
Confirming our previous findings in the therapeutic setting, depletion of CD8+ and NK1.1+ cells hampered rejection of small established tumors (Fig. 5 B). CD4+ T cells were not required or either tumor rejection or depigmentation. In fact, the data suggest that the combination therapy might be slightly more effective in inducing tumor rejection and depigmentation when CD4+ T cells are not present. This suggests that CD4+ T cells might play a role in inhibiting the antitumor response. This possibility has been examined in another study 27a.
In contrast to therapy, in the prophylactic setting full protection to subsequent challenge was conferred by the combination treatment in the absence of either CD4, CD8, or NK1.1+ cells at the time of B16 challenge (Fig. 5 A). Thus, CTLA-4 blockade in combination with the prophylactic application of the GM-CSF vaccine allows the generation of an effective antitumor response that is not dependent on any single lymphocyte subset, including CD4+ T cells. Interestingly, despite the fact that the CD8+ T cell subset was not required for tumor rejection in the prophylactic setting, CD8+ T cells were essential for depigmentation. This showed that in prophylaxis, in contrast to therapy, effective tumor immunity could be generated without subsequent autoimmunity.
Discussion
CD8+ T Cells, but Not B Cells or CD4+ T Cells, Are Required for Tumor Rejection and Associated Autoimmunity after Combination Therapy.
The results presented here confirm our earlier report that CD4+ T cells are not required for successful therapy of B16 using the B16–GM-CSF and anti–CTLA-4 combination therapy and, in addition, demonstrate that B cells are also dispensable for both tumor rejection and depigmentation. These findings are of interest for several reasons. One concerns the mechanism of depigmentation. The fact that tumor rejection was followed by autoimmune depigmentation suggests that a major component of the immune response was directed against antigen(s) shared between the tumor, vaccine, and normal melanocytes. Several previous studies have implicated antibody responses to pigmentation antigens as playing a central causal role in vitiligo or melanoma-associated hypopigmentation 20 21 28 29 30 31. Although we detected the presence of Ig depositions in the depigmented skins of surviving wild-type B6 mice, we did not find evidence of specific B cell responses toward pigmentation antigens expressed by B16. Our findings do not rule out the possibility that the antibody deposited on depigmentation lesions in wild-type mice is secondary to the antitumor and autoimmune response and is the consequence of a process similar to intermolecular epitope spreading. However, the fact that B cell knockout mice (Fig. 2) and MHC class II−/− mice (unpublished data) develop depigmentation after receiving combination therapy and rejecting tumor clearly demonstrates that antibody is not required for either effect. This, together with the demonstration that CD8+ T cells are absolutely required for depigmentation in either the therapeutic or prophylactic settings in our system, indicates that this effect involves recognition of MHC class I–restricted peptides present in or derived from normal melanocytes.
A second important point made by the data is that there is no requirement for CD4+ T cell help in the induction of either tumor rejection or depigmentation in the combination therapy. The basis for this is not clear, but may reflect an effect of CTLA-4 on the threshold of costimulation needed for activation of naive CD8+ T cells. It is tempting to speculate that in this system, blockade of CTLA-4/B7 interactions allows effective activation and expansion of CD8+ T cells even by dendritic cells that have not been licensed by activated CD4+ T cells 32 33 34. This possibility is supported by a previous report that CTLA-4 blockade resulted in the enhancement of CD8+ T cell responses to peptide independently of CD4+ T cell help 35. Whatever the basis for the effect, it is clear that the addition of anti–CTLA-4 antibody frees the GM-CSF–transduced tumor cell vaccine from an absolute dependence on CD4+ T cells 16.
A third point that emerges from the data is that tumor rejection requires both the perforin and the Fas pathways of killing (Table ). The importance of Fas in immune control of tumors has only been recognized recently 23 36 37 and was confirmed in our therapy model. As both lpr and gld mice displayed impaired therapeutic responses in our treatment model, Fas/FasL interactions may primarily be required during the generation of an effective immune response. From these data we cannot rule out the possibility that Fas/FasL interactions are also involved in direct effector mechanisms controlling tumor outgrowth. In perforin-deficient mice, B16 tumors initially displayed delayed outgrowth after combination treatment. Ultimately, however, all the mice succumbed to progressive B16 tumors. This suggests that the first wave of immune effector cells might not depend on perforin for their immune control of tumor growth, whereas ultimately both Fas/Fas-L and perforin dependent lytic pathways are required for tumor rejection (possibly involving both T cells and NK cells).
CD8+ T Cell Response toward TRP-2180–188 Generated by Combination Therapy.
As both antitumor and autoimmunity were dependent on the presence of CD8+ cells (e.g. Fig. 5), we sought to identify the antigenic target(s) of this response. In studies on CTL reactivity against human melanoma, several candidate antigens were identified, including tyrosinase, Pmel-17/gp100, Melan-A/Mart-1, TRP-1, and TRP-2. In response to combination therapy, a T cell population directed to TRP-2 is specifically activated and expanded in our model (Fig. 3 and Fig. 4). Mice surviving for up to 18 mo after the initial subcutaneous or intravenous tumor challenge only reacted against TRP-2 of the melanoma differentiation antigens tested. It is unlikely that this would reflect CD4 T cell reactivity against the TRP-2–derived nonamer, as the vaccinia-infected or peptide-pulsed target cells (MC38 or RMA-S) are MHC class II negative. The murine TRP-2181–188 epitope, VYDFFVWL, presented by H-2Kb was previously characterized as a B16 tumor rejection antigen 26. In our system, only minimal reactivity against the octamer epitope was found. Instead, we found potent reactivity toward a nonamer variant of this epitope SVYDFFVWL (TRP-2180–188). This result is consistent with the idea that the nonamer epitope binds much better to H-2Kb than the 8-mer epitope 27.
Combination Therapy Evokes Different Responses in Prophylactic or Therapeutic Setting.
Applying combination treatment in mice depleted of different lymphoid populations, we compared the requirements for successful B16 rejection in a prophylactic setting and a therapeutic setting. In the therapeutic setting, immunity to B16 tumors was found to be critically dependent on CD8+ T cells and to a lesser extent on NK1.1+ cells. In contrast, prophylactic combination treatment induced sufficient protection to a subsequent B16 challenge in the absence of CD4, CD8, or NK1.1+ cells. However, autoimmune depigmentation still required CD8+ T cells. One interpretation could be that compared with prophylaxis, in the therapeutic setting a more vigorous CTL response is required to fight progressive disease. This suggests that successful treatment of B16 is accompanied by an enhanced risk of concomitant autoimmunity, and that the window between antitumor immunity and autoimmunity will be different in tumor-free or tumor-bearing subjects 38. These observed differences in requirements for lymphoid subpopulations may also explain discrepancies with respect to the mechanism required for immunity to B16 as described by others. A clear involvement of CD4+ T cells in control of B16 tumors was demonstrated earlier 16. Similarly, generation of anti–TRP-2 CTL by genetic immunization required the presence of functional CD4 T cell help, and did not depend on NK cells, Fas, or perforin 29. These results were obtained in prophylactic studies, and our results suggest that they might have led to different conclusions in a setting of treatment. As the requirements for lymphoid subpopulations when compared between the prophylactic and the therapeutic setting were different, studying the therapeutic model for T cell–dependent immunotherapy of B16 is highly valuable for further development of strategies to immunotherapy of cancer in man.
Acknowledgments
We thank members of the Melief and Allison labs for valuable discussions and Michael Curran for a critical review of the manuscript. Alan Houghton and Vincent Hearing are gratefully acknowledged for providing valuable reagents. We thank Leonie van Duivenvoorde for technical help. Ton Schumacher, John Haanen, and Mireille Toebes are gratefully acknowledged for generous provision of tetramer reagents and advice on their use.
A. van Elsas was a recipient of a postdoctoral fellowship of the Dutch Cancer Society (Nederlandse Kankerbestrijding), A.A. Hurwitz is CaPCURE Young Investigator, and J.P. Allison is an investigator of the Howard Hughes Medical Institute. This work was supported in part by National Cancer Institute grant CA57986.
Footnotes
Abbreviations used in this paper: aa, amino acid(s); PI, propidium iodide; TRP, tyrosinase-related protein.
A. van Elsas' present address is Department of Pharmacology, NV Organon, 5340 BH Oss, Netherlands.
A.A. Hurwitz's present address is Department of Microbiology and Immunology, SUNY Upstate Medical University, Syracuse, NY 13210.
W.W. Overwijk's present address is Department of Immunology, Netherlands Cancer Institute, 1066 CX Amsterdam, Netherlands.
References
- Offringa R., van der Burg S.H., Ossendorp F., Toes R.E., Melief C.J. Design and evaluation of antigen-specific vaccination strategies against cancer. Curr. Opin. Immunol. 2000;12:576–582. doi: 10.1016/s0952-7915(00)00145-x. [DOI] [PubMed] [Google Scholar]
- Pardoll D.M., Topalian S.L. The role of CD4+ T cell responses in antitumor immunity. Curr. Opin. Immunol. 1998;10:588–594. doi: 10.1016/s0952-7915(98)80228-8. [DOI] [PubMed] [Google Scholar]
- Richards J.M., Mehta N., Ramming K., Skosey P. Sequential chemoimmunotherapy in the treatment of metastatic melanoma. J. Clin. Oncol. 1992;10:1338–1343. doi: 10.1200/JCO.1992.10.8.1338. [DOI] [PubMed] [Google Scholar]
- Rosenberg S.A., White D.E. Vitiligo in patients with melanomanormal tissue antigens can be targets for cancer immunotherapy. J. Immunother. Emphas. Tumor Immunol. 1996;19:81–84. [PubMed] [Google Scholar]
- Ogg G.S., Rod D.P., Romero P., Chen J.L., Cerundolo V. High frequency of skin-homing melanocyte-specific cytotoxic T lymphocytes in autoimmune vitiligo. J. Exp. Med. 1998;188:1203–1208. doi: 10.1084/jem.188.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C., Thompson J.A., Roche P., Byrd D.R., Lee P.P., Piepkorn M., Kenyon K., Davis M.M., Riddell S.R., Greenberg P.D. Melanocyte destruction after antigen-specific immunotherapy of melanoma. Direct evidence of T cell–mediated vitiligo. J. Exp. Med. 2000;192:1637–1644. doi: 10.1084/jem.192.11.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers C.A., Kuhns M.S., Egen J.G., Allison J.P. CTLA-4-mediated inhibition in regulation of T cell responsesmechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001;19:565–594. doi: 10.1146/annurev.immunol.19.1.565. [DOI] [PubMed] [Google Scholar]
- Leach D., Krummel M., Allison J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- Kwon E.D., Hurwitz A.A., Foster B.A., Madias C., Feldhaus A.L., Greenberg N.M., Burg M.B., Allison J.P. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc. Natl. Acad. Sci. USA. 1997;94:8099–8103. doi: 10.1073/pnas.94.15.8099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz A.A., Yu T.F., Leach D.R., Allison J.P. CTLA-4 blockade synergizes with tumor-derived GM-CSF for treatment of an experimental mammary carcinoma. Proc. Natl. Acad. Sci. USA. 1998;95:10067–10071. doi: 10.1073/pnas.95.17.10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokyr M.B., Kalinichenko T., Gorelik L., Bluestone J.A. Realization of the therapeutic potential of CTLA-4 blockade in low-dose chemotherapy-treated tumor-bearing mice. Cancer Res. 1998;58:5301–5304. [PubMed] [Google Scholar]
- Kwon E.D., Foster B.A., Hurwitz A.A., Madias C., Allison J.P., Greenberg N.M., Burg M.B. Elimination of residual metastatic prostate cancer following surgery and adjunctive CTLA-4 blockade immunotherapy. Proc. Natl. Acad. Sci. USA. 1999;96:15074–15079. doi: 10.1073/pnas.96.26.15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Elsas A., Hurwitz A.A., Allison J.P. Combination immunotherapy of B16 melanoma using anti-CTLA-4 and GM-CSF producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J. Exp. Med. 1999;190:355–366. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz A.A., Foster B.A., Kwon E.D., Trong T., Choi E.M., Greenberg N.M., Burg M.B., Allison J.P. Combination immunotherapy of primary prostate cancer in a transgenic model using CTLA-4 blockade. Cancer Res. 2000;60:2444–2448. [PubMed] [Google Scholar]
- Dranoff G., Jaffee E., Lazenby A., Golumbek P., Levitsky H., Brose K., Jackson V., Hamada H., Pardoll D., Mulligan R.C. Vaccination with irradiated tumor cells engineered to secrete GM-CSF stimulates potent, specific, and long lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung K., Hayashi R., Lafond-Walker A., Lowenstein C., Pardoll D., Levitsky H. The central role of CD4+ T cells in the antitumor immune response. J. Exp. Med. 1998;188:2357–2368. doi: 10.1084/jem.188.12.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel M.F., Allison J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z., Reznikoff G., Dranoff G., Rock K.L. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J. Immunol. 1997;158:2723–2730. [PubMed] [Google Scholar]
- Overwijk W.W., Tsung A., Irvine K.R., Parkhurst M.R., Goletz T.J., Tsung K., Carroll M.W., Liu C., Moss B., Rosenberg S.A., Restifo N.P. gp100/pmel 17 is a murine tumor rejection antigeninduction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J. Exp. Med. 1998;188:277–286. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J., Bystryn J.C. Melanoma and vitiligo are associated with antibody responses to similar antigens on pigment cells. Arch. Dermatol. 1995;131:314–318. [PubMed] [Google Scholar]
- Hara I., Takechi Y., Houghton A.N. Implicating a role for immune recognition of self in tumor rejectionpassive immunization against the brown locus protein. J. Exp. Med. 1995;182:1609–1614. doi: 10.1084/jem.182.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z., Richter G., Schuler T., Ibe S., Cao X., Blankenstein T. B cells inhibit induction of T cell-dependent tumor immunity. Nat. Med. 1998;4:627–630. doi: 10.1038/nm0598-627. [DOI] [PubMed] [Google Scholar]
- Medema J.P., de Jong J., van Hall T., Melief C.J., Offringa R. Immune escape of tumors in vivo by expression of cellular FLICE- inhibitory protein. J. Exp. Med. 1999;190:1033–1038. doi: 10.1084/jem.190.7.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Broek M.E., Kagi D., Ossendorp F., Toes R., Vamvakas S., Lutz W.K., Melief C.J., Zinkernagel R.M., Hengartner H. Decreased tumor surveillance in perforin-deficient mice. J. Exp. Med. 1996;184:1781–1790. doi: 10.1084/jem.184.5.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxevanis C.N., Voutsas I.F., Tsitsilonis O.E., Tsiatas M.L., Gritzapis A.D., Papamichail M. Compromised anti-tumor responses in tumor necrosis factor-alpha knockout mice. Eur. J. Immunol. 2000;30:1957–1966. doi: 10.1002/1521-4141(200007)30:7<1957::AID-IMMU1957>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Bloom M.B., Perry-Lalley D., Robbins P.F., Li Y., el Gamil M., Rosenberg S.A., Yang J.C. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J. Exp. Med. 1997;185:453–459. doi: 10.1084/jem.185.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreurs M.W., Eggert A.A., de Boer A.J., Vissers J.L., van Hall T., Offringa R., Figdor C.G., Adema G.J. Dendritic cells break tolerance and induce protective immunity against a melanocyte differentiation antigen in an autologous melanoma model. Cancer Res. 2000;60:6995–7001. [PubMed] [Google Scholar]
- Sutmuller R.P.M., van Duivenvoorde L.M., van Elsas A., Schumacher T.N.M., Wildenberg M.E., Allison J.P., Toes R.E.M., Offringa R., Melief C.J.M. Synergism of CTLA-4 blockade and depletion of CD25+ regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive CTL responses. J. Exp. Med. 2001;In press doi: 10.1084/jem.194.6.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwijk W.W., Lee D.S., Surman D.R., Irvine K.R., Touloukian C.E., Chan C.C., Carroll M.W., Moss B., Rosenberg S.A., Restifo N.P. Vaccination with a recombinant vaccinia virus encoding a “self” antigen induces autoimmune vitiligo and tumor cell destruction in micerequirement for CD4(+) T lymphocytes. Proc. Natl. Acad. Sci. USA. 1999;96:2982–2987. doi: 10.1073/pnas.96.6.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowne W.B., Srinivasan R., Wolchok J.D., Hawkins W.G., Blachere N.E., Dyall R., Lewis J.J., Houghton A.N. Coupling and uncoupling of tumor immunity and autoimmunity. J. Exp. Med. 1999;190:1717–1722. doi: 10.1084/jem.190.11.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp E.H., Gawkrodger D.J., Watson P.F., Weetman A.P. Immunoprecipitation of melanogenic enzyme autoantigens with vitiligo seraevidence for cross-reactive autoantibodies to tyrosinase and tyrosinase-related protein-2 (TRP-2) Clin. Exp. Immunol. 1997;109:495–500. doi: 10.1046/j.1365-2249.1997.4781381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T., Irie R.F., Fujii S., Huang S.K., Nizze A.J., Morton D.L., Hoon D.S. Anti-tyrosinase-related protein-2 immune response in vitiligo patients and melanoma patients receiving active-specific immunotherapy. J. Invest. Dermatol. 1998;111:1034–1039. doi: 10.1046/j.1523-1747.1998.00411.x. [DOI] [PubMed] [Google Scholar]
- Schoenberger S.P., Toes R.E., van der Voort E.I., Offringa R., Melief C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- Bennett S.R.M., Carbone F.R., Karamalis F., Flavell R.A., Miller J.F., Heath W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signaling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- Ridge J.P., Di Rosa F., Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- McCoy K.D., Hermans I.F., Fraser J.H., Le Gros G., Ronchese F. Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) can regulate dendritic cell–induced activation and cytotoxicity of CD8+ T cells independently of CD4+ T cell help. J. Exp. Med. 1999;189:1157–1162. doi: 10.1084/jem.189.7.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen-Schaub L.B., van Golen K.L., Hill L.L., Price J.E. Fas and Fas ligand interactions suppress melanoma lung metastasis. J. Exp. Med. 1998;188:1717–1723. doi: 10.1084/jem.188.9.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djerbi M., Screpanti V., Catrina A.I., Bogen B., Biberfeld P., Grandien A. The inhibitor of death receptor signaling, FLICE-inhibitory protein defines a new class of tumor progression factors. J. Exp. Med. 1999;190:1025–1032. doi: 10.1084/jem.190.7.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronte V., Apolloni E., Ronca R., Zamboni P., Overwijk W.W., Surman D.R., Restifo N.P., Zanovello P. Genetic vaccination with “self” tyrosinase-related protein 2 causes melanoma eradication but not vitiligo. Cancer Res. 2000;60:253–258. [PMC free article] [PubMed] [Google Scholar]
- Parker K.C., Bednarek M.A., Coligan J.E. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J. Immunol. 1994;152:163–175. [PubMed] [Google Scholar]