

Abstract
The chemokine receptor CCR5 plays an important role in leukocyte chemotaxis and activation, and also acts as a coreceptor for human and simian immunodeficiency viruses (HIV-1, HIV-2, and SIV). We provide evidence that CCR5 is O-glycosylated on serine 6 in the NH2 terminus. The O-linked glycans, particularly sialic acid moieties, significantly contribute to binding of the chemokine ligands. By contrast, removal of O-linked oligosaccharide exerted little effect on HIV-1 infection. Sulfation of specific tyrosine residues in the CCR5 NH2 terminus was important for efficient β-chemokine binding. Thus, as has been observed for the binding of selectins and their ligands, O-linked carbohydrates and tyrosine sulfates play major roles in promoting the interaction of chemokines with CCR5. The resulting flexible arrays of negative charges on the CCR5 surface may allow specific, high-affinity interactions with diverse chemokine ligands. Although this is the first example of O-linked oligosaccharides and tyrosine sulfates playing a role in chemokine binding, the high density of serines, threonines and tyrosines in the N-termini of many CC chemokine receptors suggests that these posttranslational modifications may commonly contribute to chemokine binding.
Keywords: CCR5, O-Glycosylation, sialic acid, tyrosine sulfation, HIV
Introduction
Chemokines are a large family of cytokines mainly involved in cell traffic coordination and cell activation during immunological processes (1, 2). All chemokines act via a subclass of G protein–coupled, seven-transmembrane-segment receptors (GPCRs)* (3). CC chemokine receptor (CCR)5, a functional receptor with high affinity for macrophage inflammatory protein (MIP)-1α, MIP-1β, regulated on activation normal T cell expressed (RANTES), and some other β-chemokines has been found on several cell types including memory T cells, B cells, macrophages, microglia, mast cell precursors, and dendritic cells (1–4). Together with CD4, CCR5 serves as a coreceptor facilitating the entry of HIV-1, HIV-2, and simian immunodeficiency virus (SIV) into target cells (5, 6). Primary infection with these viruses is predominantly mediated by so-called R5 viral strains that utilize CCR5. Later in the course of HIV-1 infection, X4 viral strains often emerge that utilize another chemokine receptor, CXC chemokine receptor (CXCR)4. The emergence of X4 or dual tropic R5X4 viral subtypes is typically associated with rapid deterioration of the immune system and development of AIDS (7).
Approximately 1% of Caucasians are homozygous for a 32-base pair deletion in the CCR5 gene. These individuals produce a nonfunctional protein and thus are highly resistant to HIV-1 infection, demonstrating the pivotal role of CCR5 for viral transmission (8). Individuals lacking CCR5 are immunologically competent, a phenotype that may reflect the high degree of redundancy in the chemokine system. The ability of healthy individuals to withstand the absence of CCR5 has stimulated efforts to develop specific CCR5 inhibitors for antiretroviral therapy (9). Natural chemokine ligands, monoclonal antibodies, and small nonpeptide compounds like TAK-779 can inhibit HIV-1 entry in vitro (10, 11) and are being considered for in vivo applications.
Precise knowledge of the structural basis for chemokine and HIV-1 envelope glycoprotein binding to CCR5 would advance our understanding of the role of CCR5 in physiology and disease, and inform efforts to develop potent drugs for therapeutic intervention. Like other GPCRs, CCR5 has an amino terminus and three extracellular loops. The NH2 terminus and the second extracellular loop of CCR5 contribute to high affinity binding of chemokines and the HIV-1 gp120 exterior envelope glycoprotein, but other CCR5 domains can also influence these associations (12–14). A current model for chemokine binding suggests a two-step mechanism involving an initial association with the probably more exposed CCR5 NH2 terminus and a subsequent, more specific, interaction with the CCR5 extracellular loops leading to receptor activation (12, 15, 16). Deletion of the NH2-terminal sequences of CCR5 strongly impairs chemokine binding, but substitution of this domain with corresponding regions from related receptors, including CCR1, CCR2b, CXCR2, and CXCR4, can restore chemokine binding and receptor activation (12, 13, 17). Therefore, similar determinants in the NH2 terminus shared among related receptors likely contribute to high affinity binding of chemokines (13). Negatively charged and aromatic residues in the NH2-terminal region of CCR5 are crucial for the association with chemokines and the HIV-1 gp120 envelope glycoprotein (13, 14, 17–20). Sulfate moieties on tyrosines contribute extra negative charges to this region and facilitate HIV-1 entry (21). Inhibition of tyrosine sulfation substantially decreases the affinity of MIP-1α, MIP-1β, and HIV-1 gp120/CD4 complexes for CCR5 (21).
In addition to tyrosine sulfation, CCR5 is also modified by O-linked oligosaccharides (O-glycans), which are typically added to serine or threonine residues in the Golgi compartment of the cell (21). In the cell types examined to date, the O-glycans on CCR5 are not sulfated themselves but contain negatively charged terminal sialic acid residues (21). The functions of O-glycans are as diverse as their structure. These include the maintenance of protein conformation, protection of proteins from proteases, control of active epitopes and antigenicity, blood clotting, embryogenesis, development, participation in cell-egg binding, and microbe attachment to cells (22, 23). Of special interest is the contribution of sialylated O-glycans to the adhesion of activated immune cells and to the metastatic potential of cancer cells (24).
In this paper, we report the location of the major O-glycosylation site of CCR5 expressed in several cell lines as well as in primary macrophages. We analyze the contribution of CCR5 O-glycosylation to chemokine binding and HIV-1 entry. Finally, we extend our studies on tyrosine sulfation in the CCR5 NH2 terminus and investigate its role in chemokine binding.
Materials and Methods
Cells.
Cf2Th cells (canine thymocytes), HeLa, and HEK 293T cells were obtained from the American Type Culture Collection. Cells were maintained in Dulbecco's modified Eagle's medium containing 10% fetal calf serum, 100 U of penicillin/ml, and 100 U of streptomycin/ml. LdlD and Chinese hamster ovary (CHO) cells were kindly provided by Dr. M. Krieger (Massachusetts Institute of Technology, Cambridge, MA) and maintained in F12 medium supplemented with 10% FCS and antibiotics. 3 d before an experiment, ldlD cells were split into two flasks and cultured in F12 medium containing antibiotics, 1× ITS-supplement, and 2% dialyzed FCS. The following day the medium in one flask was supplemented with 500 μM N-acetylgalactosamine (GalNAc) and 50 μM galactose (Gal).
Monocyte-derived rhesus macrophages were obtained and cultured as described previously (25). Approximately 5 × 106 mononuclear cells, isolated on a Ficoll-Paque gradient, were seeded in each well of a 6-well plate and cultured in macrophage-driving medium (25) containing 50 U GM-CSF and 5 U M-CSF. Nonadherent cells were removed every second day by extensive washes with PBS.
Plasmids.
A previously described pcDNA3.1-based expression plasmid (26) for human codon-optimized CCR5 with a COOH-terminal C9 tag (TETSQVAPA) was used as a template to generate CCR5 mutants. The mutations specifying changes in the CCR5 NH2 terminus were introduced by PCR amplification using Pfu-polymerase and 5′-primers with changes of serine/threonine codons into alanine codons, along with appropriate restriction sites for cloning. The 5′ primers were as follows, with the mutated nucleotides underlined: AATS mutant: GAATTCAAGCTTGCCGCCGCCATGGACTACCAGGTGGCGGCGCCCATCTACGACATCAACTACTAC; SSAA mutant: GAATTCAAGCTTGCCGCCGCCATGGACTACCAGGT-GTCCTCCCCATCTACGACATCAACTACTACGCGGCG- GAGCCCTGCCAGAAGATC; AAAA mutant: GAATTCAAGCTTGCCGCCGCCATGGACTACCAGGTGGCGGCG-CCCATCTACGACATCAACTACTACGCGGCGGAGCCCTGCCAGAAGATC; ASTS mutant: GAATTCAAGCTTGCCGCCGCCATGGACTACCAGGTGGCGTCCCCCATCTACGACATCAACTACTAC; SATS mutant: GAATTC-AAGCTTGCCGCCGCCATGGACTACCAGGTGTCCGCGCCCATCTACGACATCAACTACTAC.
The 3′ primer for all of the above mutants anneals to the region encoding the C9 tag of the CCR5 gene and has the following sequence: CCTCTAGACTCGAGCGGCCGCTTAGGCGGGGGC. The mutated CCR5 genes were amplified by PCR and ligated into the pcDNA 3.1 vector using the HindIII and NotI restriction sites. Analogous amplified fragments were also cloned into the pSIvec1Δenv plasmid (25) using AgeI and NotI restriction sites and sequenced to confirm the alterations. The plasmids pSIvec1ΔenvhuCCR5, pHCM-vesicular stomatitis virus G protein (VSV-G), and psrev used to generate VSV G-pseudotyped viruses encoding huCCR5 are described in (25) and references therein.
The QuikChange Mutagenesis kit (Stratagene) was used to generate the CCR5 mutants with serine/threonine residues in the second and third extracellular loops changed to alanine. The primers were as follows (only one primer for each mutant is shown, as the second primer was simply the reverse complement): second extracellular loop mutant: CAGAAGGAGGGCCTGCACTACGCGTGCGCCGCCCACTTCCCCTACTC-CCAG; third extracellular loop mutant: CAGGAGTTCTTCGGCCTGAACAATTGCGCCGCCGCCACCGCCTGG-ACCAGGC. MluI and MfeI restriction sites were engineered into the respective primers to allow for identification of positive clones, which were subsequently sequenced. The mutant CCR5Δ2–17 was generated in a similar way, using a 5′-primer encoding a deletion of the sequence coding for amino acids 2–17.
Cf2Th cells stably expressing human CCR5 variants were established by transfection using the calcium phosphate method and G418 selection. Cells expressing high and comparable surface levels of wild-type and CCR5 mutants, based on staining with dialyzed phycoerythrin-conjugated 2D7 antibody (BD PharMingen), were selected by sorting on a MoFlo FACS® (Cytomatics) machine and cultured in the presence of G418. To maintain consistent expression levels, it was necessary to resort the stable cell lines intermittently. LdlD cells stably expressing human CCR5 were established using the same approach.
Transient expression of CCR5 variants was achieved by transfecting Cf2Th, Cf2Th-CD4, HEK 293T, HeLa, and CHO cells with 25 μg of each expression vector using the calcium phosphate method. Two days after transfection, the surface expression level of CCR5 was determined using phycoerythrin-conjugated 2D7 or 3A9 antibodies (BD PharMingen). Transfectants showing less than 25% variation in expression of the different CCR5 variants were used for immunoprecipitation, chemokine binding, or virus entry experiments.
To express human CCR5 and glycosylation mutants in primary rhesus macrophages, recombinant VSV G-pseudotyped viruses were produced in HEK 293T cells. Recombinant viruses were generated using the SHIV-derived vector pSIvec1ΔenvhuCCR5 or analogous vectors (25) with changes of serines/threonines in the CCR5 sequence, as described above. Macrophages were transduced overnight with 30,000 reverse transcriptase (RT) units of virus. 48 h later, the macrophages were lysed and the CCR5 protein was immunoprecipitated.
Labeling, Immunoprecipitation, and Glycosidase Treatment of CCR5 Variants.
To label and immunoprecipitate the CCR5 proteins, all cells but macrophages were grown to confluency in 10-cm dishes. Primary macrophages were cultured in 6-well plates and 2–3 wells were used for each CCR5 variant. Cells were washed with PBS and labeled overnight with [35S]cysteine/methionine in cysteine- and methionine-free medium or with [3H]galactose in glucose-free medium. Cells were then washed with PBS and lysed using 1 ml lysis buffer consisting of 100 mM (NH4)2SO4, 20 mM Tris-HCl (pH 7.5), 10% glycerol, 1% Cymal-5 (Anatrace), 0.1% SDS, and one tablet of Complete™ Protease Inhibitor Mixture (Roche) per 50 ml. Labeled receptor was immunoprecipitated using the C9 tag-specific 1D4 antibody (National Cell Culture Center) covalently linked to Sepharose beads (Amersham Pharmacia Biotech). Precipitates were washed five times in lysis buffer, incubated at 56°C for 20 min in reducing sample buffer, analyzed by 10% SDS-PAGE, and visualized by autoradiography.
For removal of O-glycan chains, Sepharose beads containing the immunoprecipitated CCR5 proteins were washed twice with PBS to remove detergent, divided, and one half was treated overnight at 37°C with an O-glycosidase mix (α2–3,6,8,9-neuraminidase, β1,4-galactosidase, endo-α-N-acetylgalactosaminidase, and β-N-acetylglucosaminidase present in the Glycoprotein Deglycosylation Kit from Calbiochem.
To remove sialidase moieties from CCR5 present on the cell surface, 2 × 107 cells were detached with 5 mM EDTA in PBS, washed once with medium, and 50% of the cells were treated for 1.5 h at 37°C with 0.3 U of sialidase from Arthrobacter ureafaciens (Roche) in 200 μl medium without FCS and 0.1% sodium azide. The other 50% were incubated in the same medium under the same conditions, but without enzyme. Cells were then used for immunoprecipitation or for chemokine binding assays.
Chemokine Binding Assay.
Cells were detached from the flasks with 5 mM EDTA in PBS, washed with medium without FCS, counted, and resuspended in binding buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM CaCl2, 5 mM MgCl2, and 5% dialyzed FCS). Competition binding assays were performed for 1 h at 37°C with 5 × 105 cells, 0.1 nM 125I-labeled chemokine (Dupont NEN) and varying concentrations of cold chemokine (100 pM-1 μM) in 100 μl volumes. Cells with bound chemokines were washed twice with binding buffer and counted in a gamma counter. In parallel with the binding experiment, the expression level of each CCR5 variant was analyzed with phycoerythrin-conjugated 2D7 and/or 3A9 antibody on a FACScan™ cytometer (Becton Dickinson) with CELLQuest™ software. Binding data were analyzed using GraphPad Prism software running an algorithm for nonlinear regression of homologous one-site competition binding with ligand depletion (27) to obtain relative Kd values.
Calcium Mobilization by CCR5 Variants.
Chemokine-induced Ca2+ influx in Cf2Th cells stably expressing different CCR5 variants was measured after transfecting these cells with a plasmid encoding the G protein component Gα16. Preliminary experiments showed that Gα16 expression strongly enhanced ligand-triggered Ca2+ influx into CCR5-expressing Cf2Th cells. Cells were loaded with the indicator dye Fura 2/AM (Molecular Probes) for 1 h at 37°C in 20 mM Hepes, pH 7.4, 125 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM KCl, 0.5 mM glucose, and 0.2% BSA. Cells were then washed twice in the same buffer without dye and resuspended in new buffer at a density of 2 × 106/ml. Changes in intracellular Ca2+ concentration in response to chemokine binding were measured by monitoring the emission at 510 nm of the excitations at 340 and 380 nm as a function of time.
Virus Entry Assay.
The efficiency of HIV-1 entry into cells was determined using pseudotyped single-round viruses encoding a chloramphenicol acetyltransferase (CAT) reporter gene in a previously published env complementation assay (6). The recombinant HIV-1 were generated in HeLa cells by using the pHXBH10ΔenvCAT vector and plasmids encoding HIV-1 or SIVmac envelope glycoproteins as previously described (6, 25). Cf2Th cells stably expressing human CD4 were transiently transfected with plasmids encoding the CCR5 variants to be tested. Cells expressing similar CCR5 levels, as determined by staining a cell sample with the 2D7 antibody at 48 h after transfection, were seeded in 6-well plates and infected with 5,000 RT units of a single-round virus for 1 h. 3 d after infection, CAT activity was measured to assess the efficiency of virus entry.
Results
Identification of Major O-linked Glycosylation Sites on CCR5.
A common posttranslational modification of GPCRs is the addition of carbohydrate chains (28). It has been recently shown that human CCR5 is not N-glycosylated, but is modified by O-glycosylation (21). To localize major O-glycosylation sites, we established Cf2Th cells stably expressing wild-type CCR5 and CCR5 mutants, all of which contain a COOH-terminal 9-amino acid tag derived from bovine rhodopsin. Cells were radiolabeled with [35S]cysteine/methionine or [3H]galactose, lysed, and CCR5 was immunoprecipitated with the tag-specific 1D4 antibody and analyzed by SDS-PAGE. While sequence or structural motifs for O-glycosylation have yet to be fully characterized, it has been observed that this modification often occurs within surface-exposed clusters of serine or threonine residues. Four serine/threonine clusters, two within the NH2 terminus, one within the second extracellular loop (residues 177, 179, 180), and one within the third extracellular loop (residues 270–272) were identified (Fig. 1 A). The CCR5 NH2 terminus contains serines at positions 6 and 7 (cluster 1) as well as threonine 16 and serine 17 (cluster 2) (Fig. 1 A). Deletion of amino acids 2–17 resulted in a protein (CCR5Δ2–17) that was efficiently expressed on the cell surface, as documented by staining with the 2D7 anti-CCR5 antibody (data not shown). The CCR5Δ2–17 protein migrated with an apparent molecular mass that is 4–5 kD less than the 43 kD mass of the wild-type CCR5 (Fig. 1 B). The CCR5Δ2–17 mutant did not incorporate galactose, a sugar found in most O-glycosylated mammalian membrane proteins. This implies that in Cf2Th cells the extracellular loops of CCR5 are not modified by galactose-containing carbohydrate chains. This interpretation was supported by our analysis of CCR5 mutants with alterations of clusters of serines/threonines in the second and third extracellular loops (residues 177/179/180 and 270–272, respectively; Fig. 1 A). When these residues were altered to alanines, no change in the migration of the CCR5 protein on SDS-polyacrylamide gels was observed (data not shown). We conclude that efficient O-linked glycosylation of CCR5 is dependent upon NH2-terminal sequences.
Figure 1.

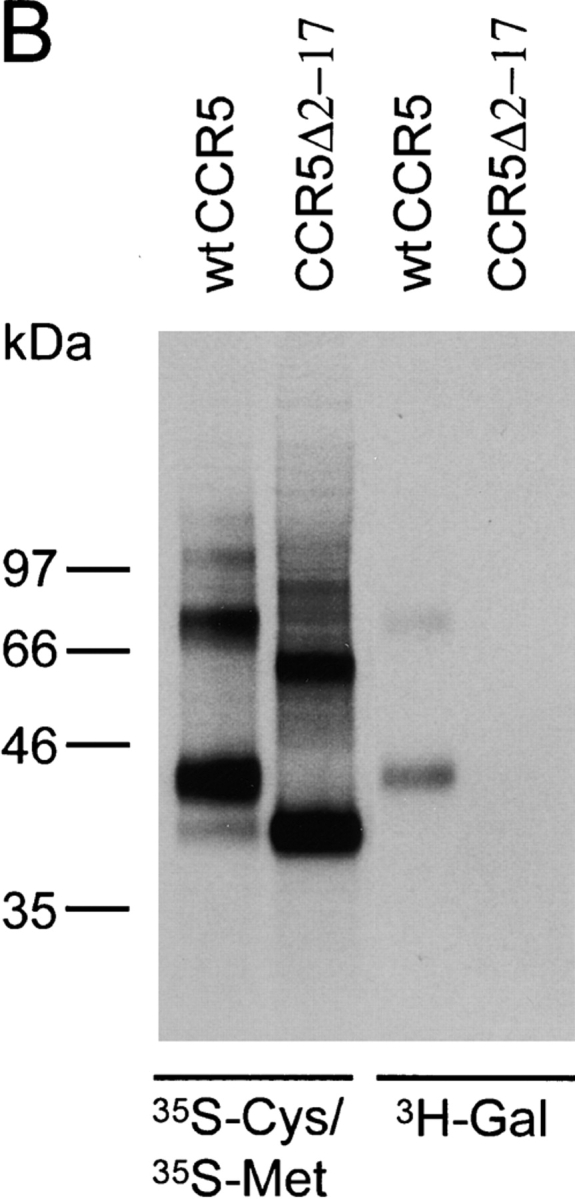

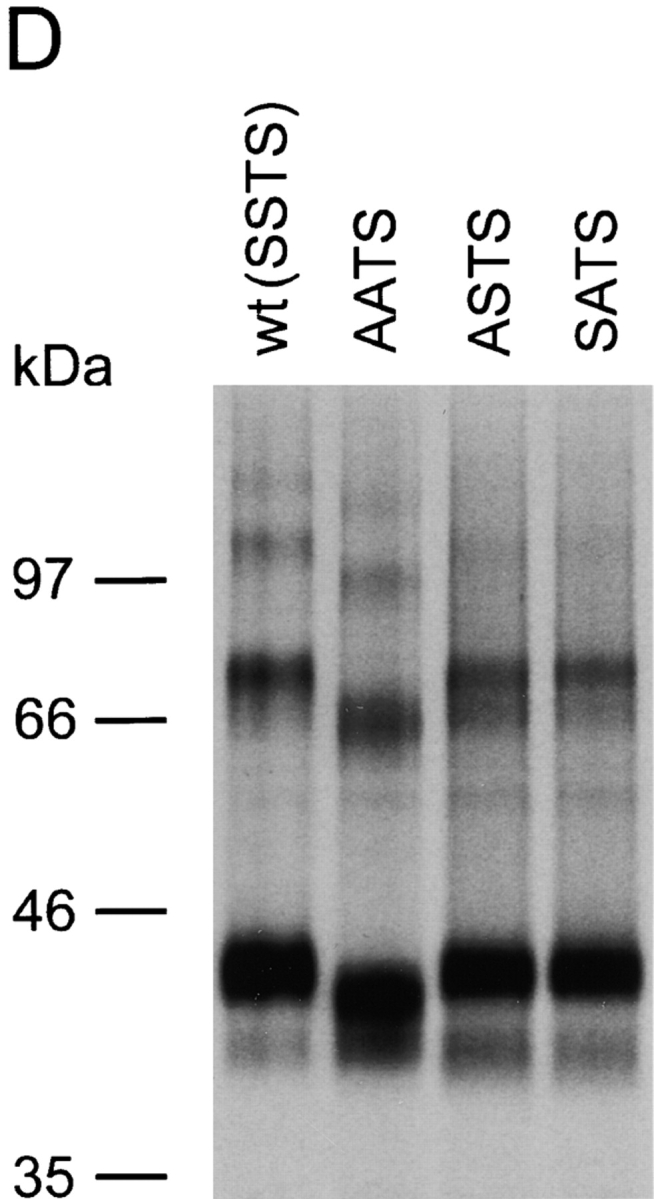

Identification of the major site of CCR5 O-glycosylation in Cf2Th cells. (A) Primary sequence of the CCR5 extracellular domains that contain potential O-linked glycosylation sites (indicated by boxes). (B) Cf2Th cells expressing wild-type CCR5 or the CCR5Δ2–17 mutant, which lacks residues 2–17, were labeled with [35S]cysteine/methionine or [3H]galactose, as indicated. Cell lysates were precipitated with the 1D4 antibody that recognizes a COOH-terminal epitope tag. Precipitates were analyzed by SDS-PAGE. (C) Cf2Th cells stably expressing CCR5 variants were labeled overnight with [35S]cysteine/methionine, lysed and incubated with the 1D4 antibody. Precipitates were divided into equal parts, which were treated overnight with a glycosidase mixture to remove O-linked carbohydrate chains or with enzyme buffer alone. The CCR5 variants are named according to the residues at positions 6, 7, 16, and 17, with SSTS being the wild-type CCR5. (D) Cf2Th cells stably expressing CCR5 variants were labeled with [35S]cysteine/methionine, lysed and incubated with the 1D4 antibody. Precipitates were analyzed by SDS-PAGE. (E) [3H]galactose-labeled Cf2Th cells expressing similar surface levels of wild-type CCR5 (SSTS) and the indicated mutants were used for immunoprecipitation with the 1D4 antibody. Precipitates were analyzed by SDS-PAGE. The experiments were performed at least three times with similar results.
To determine which of the putative O-glycosylation sites in the NH2-terminal domain of CCR5 is used, we generated mutants in which the serine/threonine residues at positions 6, 7, 16, and 17 were changed to alanines. In one mutant, designated AATS to denote the identity of the amino acids at positions 6, 7, 16, and 17, serines 6 and 7 were converted to alanines. A second mutant (SSAA), with threonine 16 and serine 17 altered to alanines, was created. A third mutant (AAAA) had all of the serines and threonines in the CCR5 NH2 terminus changed to alanines. Cf2Th cells expressing similar surface levels of these mutants, as judged by staining with the 2D7 antibody directed against the second extracellular loop of CCR5, were established. The cells were labeled with [35S]cysteine/methionine and lysed. The wild-type and mutant CCR5 proteins were precipitated with the 1D4 antibody, which recognizes the C9 peptide tag at the COOH terminus of the CCR5 variants. Half of the precipitated proteins were treated with a mixture of sialidase and O-glycosidases before analysis by SDS-PAGE. Fig. 1 C shows that the 43-kD mature form of the wild-type CCR5 was evident, as well as smaller amounts of the 40-kD CCR5 precursor and multimeric forms. Glycosidase treatment resulted in an increased mobility corresponding to an ∼2-kD decrease in molecular mass. The identical result was observed for the SSAA mutant, indicating that threonine 16 and serine 17 are not required for O-glycosylation of CCR5 in Cf2Th cells. The AATS and AAAA mutants migrated with an apparent molecular mass that was ∼2 kD less than that of wild-type CCR5. Compared with the wild-type protein, an increase in the amount of the CCR5 precursor was observed for these mutants. The migration of the AATS and AAAA mutants was not affected by glycosidase treatment. Similar results were obtained by labeling the cells with [35S]sulfate (data not shown), indicating that the introduced amino acid changes and consequent effects on O-glycosylation do not prevent tyrosine sulfation of CCR5. The results suggest that a major O-glycosylation site on human CCR5 expressed in Cf2Th cells involves serines 6 and 7.
To characterize the site of CCR5 O-glycosylation further, we created the ASTS and SATS mutants, which have single serine residues at positions 6 and 7, respectively, converted to alanines. Cf2Th cells expressing comparable surface levels of CCR5 variants were labeled either with [35S]cysteine/methionine or [3H]galactose, lysed and used for precipitation by the 1D4 antibody directed against the C9 peptide tag. Comparison of Figs. 1 D ([35S]cysteine/methionine labeling) and 1 E ([3H]galactose labeling) indicates that the ASTS and SATS mutants resembled the wild-type CCR5 both in mobility characteristics and in the intensity of labeling with [3H]galactose. These results suggest that either serine 6 or 7 can be modified by O-linked glycans, and that only one of these two potential sites is extensively modified on the wild-type CCR5 protein.
As expected, the AAAA mutant did not incorporate [3H]galactose (Fig. 1 E), consistent with the results obtained with the CCR5Δ2–17 mutant suggesting that all of the galactose-containing glycans on CCR5 are located in the NH2 terminus of the protein. Of interest, the AATS mutant did incorporate [3H]galactose, but much less efficiently than wild-type CCR5. The lack of [3H]galactose labeling of the AAAA mutant indicates that galactose addition to the AATS mutant is dependent upon threonine 16 and/or serine 17. The lower level of galactose incorporation and the absence of detectable migration differences after glycosidase treatment of the AATS mutant suggest that glycosylation at threonine 16 and/or serine 17 is much less efficient or involves a smaller O-glycan with fewer galactose moieties, compared with O-glycosylation of serines 6 and 7.
O-glycosylation of CCR5 in Different Cells.
The O-glycosylation pattern of a protein may vary, depending on the expressing cell type (24). To examine the O-glycosylation pattern in other cell types and compare it with the results obtained in Cf2Th cells, we transiently expressed wild-type CCR5 as well as the AATS and SSAA mutants in the human embryonic kidney line 293T (HEK 293T), in HeLa cells (a human cervical carcinoma line), and in primary rhesus monkey macrophages. The expressed proteins were precipitated from lysates of [35S]cysteine/methionine-labeled cells and analyzed by SDS-PAGE. Similar to the results seen in Cf2Th cells, the AATS variant migrated, in all cells tested, with an apparent molecular mass ∼2 kD less than that of the wild-type protein (Fig. 2). We conclude that in all the cell types examined, the CCR5 serines at positions 6 and 7 are major sites of O-glycosylation.
Figure 2.



Characterization of O-glycosylation of CCR5 expressed in HEK 293T, HeLa, and primary macrophages. Wild-type (wt) and mutant CCR5 proteins were expressed transiently in HEK 293T cells (A) and HeLa cells (B). To express CCR5 variants in primary rhesus monkey macrophages (C), cells were cultured for 7 d and then transduced with 30,000 RT units of VSV G-pseudotyped SHIV vectors containing the CCR5 genes in place of the nef gene. All cells were labeled with [35S]cysteine/methionine overnight, lysed, and used for precipitation by the 1D4 antibody. Precipitated proteins were analyzed by SDS-PAGE.
Contribution of O-glycosylation of CCR5 to HIV-1 and SIV Entry.
The NH2 terminus of CCR5 plays an important role in HIV-1 and SIV entry. We therefore examined the ability of HIV-1 and SIVmac strains to use CCR5 mutants deficient in O-glycosylation for entry into Cf2Th cells stably expressing human CD4. The experiments were performed with recombinant HIV-1 expressing CAT and pseudotyped with HIV-1 or SIV envelope glycoproteins. As shown in Fig. 3, the entry of all HIV and SIV pseudotypes into cells expressing similar levels of AASS, SSAA, and AAAA variants was consistently ∼15–25% reduced compared with that of wild-type CCR5. Because virus entry associated with even the glycosylated SSAA mutant was affected, this marginal reduction is not necessarily caused by a lack of O-glycosylation and might represent a result of the amino acid alterations per se.
Figure 3.

Contribution of O-glycosylation to HIV-1 and SIV entry. Cf2Th cells stably expressing CD4 were transfected with plasmids encoding wild-type CCR5 or the indicated mutants. 2 d after transfection, the CCR5 expression level was assessed by FACS® analysis of a cell sample stained with phycoerythrin-conjugated 2D7 antibody. Cells expressing similar levels of the CCR5 variants were infected with recombinant CAT-expressing viruses pseudotyped with different HIV-1 and SIV envelope glycoproteins. 3 d later, the CAT activity in cell lysates was measured. The results represent means and standard deviations of triplicate samples. One representative experiment of four performed is shown.
Effects of Changes in CCR5 O-glycosylation Site(s) on Chemokine Binding.
Previous studies indicated that negatively charged amino acids and sulfated tyrosines in the CCR5 NH2 terminus are important for chemokine binding (13, 21). To investigate the impact of preventing O-glycosylation on chemokine binding, we performed homologous competitive binding studies using the CCR5 ligands MIP-1α and MIP-1β. Cf2Th cells stably expressing comparable levels of wild-type, AATS, SSAA, and AAAA variants of CCR5 were selected by fluorescence-activated cell sorting, using a phycoerythrin-conjugated 2D7 antibody that recognizes a conformation-dependent epitope in the second extracellular loop of CCR5. The expression level of CCR5 on these cells was tested in parallel with each binding experiment, using the 2D7 antibody and the 3A9 antibody, which recognizes a linear epitope in the CCR5 NH2 terminus (data not shown). As shown in Fig. 4 A, [125I]MIP-1α bound to cells expressing the SSAA mutant with a slightly higher affinity than to cells expressing wild-type CCR5 (measured dissociation constants of 0.91 ± 0.8 nM and 1.6 ± 0.9 nM for SSAA and wild-type CCR5, respectively). More interestingly, MIP-1α exhibited dramatically impaired binding to cells expressing the AATS and AAAA mutants, which are not efficiently O-glycosylated (Fig. 4 A). Almost identical binding results were obtained using MIP-1β (Fig. 4 B). MIP-1β also bound slightly more efficiently to the SSAA mutant (Kd value of 2.06 ± 0.5 nM) than to the wild-type receptor (Kd of 7.2 ± 0.6 nM). Binding of MIP-1β to the AATS and AAAA mutants was significantly impaired but was higher than the negligible binding of MIP-1α to these variants.
Figure 4.





Effects of changes in CCR5 O-glycosylation sites on binding and signaling of MIP-1α and MIP-1β. Cf2Th cells stably expressing CCR5 variants were incubated with 0.1 nM 125I-MIP-1α (A) or 125I-MIP-1β (B) and increasing amounts of unlabeled competitor. Cells were washed and bound labeled chemokine was quantitated in a β-counter. The binding data were analyzed with Prism software (GraphPad) (reference 27). FACS® measurements were done in parallel on the same cells using phycoerythrin-conjugated 2D7 antibody. Mean fluorescence for cells expressing wild-type (wt) CCR5 was 469 (100%); for SSAA, 434 (92.5%), for AATS, 375 (80%), and for AAAA, 399 (85.1%). The experiments shown are representative of four binding assays performed with similar results. The same cells expressing wild-type CCR5 or the SSAA or AATS mutants, as well as CCR5-negative parental Cf2Th cells, were transfected with a plasmid encoding the G protein subunit Gα16, loaded with the indicator dye Fura-2, and used to measure changes in intracellular Ca2+ concentrations after stimulation (arrowhead) with 10 nM MIP-1α (C) or 10 nM MIP-1β (D). Ca2+ influx is shown as the ratio of the fluorescence signals observed at 510 nm after excitation at 340 and 380 nm. The curves were superimposed for comparison and are representative of the results obtained in the two experiments performed. (E) Homologous competition experiment with MIP-1β similar to B except that Cf2Th cells transiently expressing CCR5 variants were used. The mean fluorescence intensities of the cells stained with the 2D7 antibody were: wild-type, 174 (100%); AATS, 174 (100%); SATS, 139 (79.9%); ASTS, 152 (87.4%). A representative experiment of three, all with similar results, is shown.
The dramatically reduced interaction of MIP-1α and MIP-1β with the glycosylation-deficient AATS mutant resulted in reduced efficiency of Ca2+ mobilization. As shown in Fig. 4, C and D, the peak Ca2+ influx after a stimulation of Cf2Th cells expressing the AATS variant with 10 nM chemokine is ∼3–5 times lower than that seen in cells expressing similar levels of wild-type receptor or the SSAA mutant.
We also examined the binding affinities of MIP-1β to the SATS and ASTS mutants, which can be O-glycosylated at serine 6 and 7, respectively. Cf2Th transfectants transiently expressing similar surface levels of the CCR5 variants were used for binding assays. The SATS variant bound MIP-1β with an affinity comparable to that of wild-type CCR5, but MIP-1β association with the ASTS mutant was significantly less efficient (relative Kd value of 19.9 ± 1.3 nM) (Fig. 4 E). Thus, O-glycosylation of serine 6 on CCR5 is optimal for MIP-1β binding.
Chemokine Binding to O-glycosylation–defective CHO Cells Expressing Human CCR5.
The CHO-derived ldlD cells lack the epimerase needed to synthesize UDP-galactose and UDP-N-acetylgalactosamine from UDP-glucose or UDP-N-acetylglucosamine, respectively (29). This defect completely prevents O-glycosylation of proteins and impairs maturation of N-linked carbohydrate chains (29). This phenotype can be reversed by addition of Gal and GalNAc to the cell culture media. To confirm that O-glycosylation of CCR5 is necessary for high affinity binding of chemokines, we generated ldlD cells stably expressing human CCR5 with a C9 tag to perform binding experiments. The 1D4 antibody directed against the C9 peptide tag was used to precipitate CCR5 from the lysates of labeled ldlD-CCR5 cells cultured in Gal/GalNAc-deficient medium. CCR5 in these cells migrated with an apparent molecular mass 2–3 kD smaller than that of CCR5 from cells grown in the same medium supplemented with 50 μM Gal/500 μM GalNAc (Fig. 5 A). The CCR5 from the ldlD-CCR5 cells cultured in Gal/GalNAc-supplemented medium migrated comparably to CCR5 expressed in the parental CHO cells. These results are consistent with the expected O-glycosylation of CCR5 in the ldlD-CCR5 cells that is conditionally dependent upon the availability of Gal/GalNAc.
Figure 5.



Effect of O-glycosylation of CCR5 expressed in ldlD cells on chemokine association. The addition of O-glycans in ldlD cells depends on the presence of GalNAc and Gal in the culture media. Parental CHO cells expressing CCR5 were grown in normal F12 medium, as described in Materials and Methods. ldlD cells stably expressing CCR5 were grown for 48 h in F12 medium supplemented with 2% dialyzed FCS and ITS, either with or without 500 μM GalNAc and 50 μM Gal. (A) Cells were labeled for 8 h with [35S]cysteine/methionine. CCR5 was precipitated with the 1D4 antibody from cell lysates and subjected to SDS-PAGE. Homologous competitive binding assays with MIP-1α (B) and MIP-1β (C) were performed on CCR5-expressing ldlD-CCR5 cells and, to assess nonspecific binding, on CCR5-negative ldlD cells. The mean fluorescence values determined with phycoerythrin-conjugated 2D7 antibody in parallel to the binding assays were 3.6 for ldlD cells, 176.1 (100%) and 134 (76.1%) for ldlD-CCR5 cells grown with or without GalNAc and Gal, respectively. Experiments are representatives of two (B) or three (C) assays performed with similar results.
Competitive binding assays with MIP-1α and MIP-1β were performed with the ldlD-CCR5 cells. Addition of Gal/GalNAc to the medium did not significantly change the staining of the cells with phycoerythrin-conjugated 2D7 antibody (Fig. 5 legend), suggesting that the glycosylation state of CCR5 does not greatly affect surface expression of the protein in ldlD cells. Binding of both MIP-1α and MIP-1β was significantly lower to ldlD-CCR5 cells grown in Gal/GalNAc-deficient medium than to these cells cultured in Gal/GalNAc-containing medium (Fig. 5, B and C). No specific binding of either chemokine was detected to ldlD cells lacking CCR5. These results confirm the importance of O-glycosylation for the ability of wild-type CCR5 to bind its chemokine ligands.
Contribution of Sialic Acid Moieties on the CCR5 O-glycan to Chemokine Binding.
A common modification of O-linked carbohydrate chains is the addition of sialic acid residues by sialyltransferases. The important role that negative charges in the NH2 terminus of CCR5 play with respect to ligand binding prompted us to examine the effect on chemokine binding of removing only sialic acids from the CCR5 O-glycan. A 1.5-h treatment of 35S-labeled Cf2Th cells expressing wild-type CCR5 with 0.3 units of sialidase from Arthrobacter ureafaciens, which hydrolyzes terminal α2–3, α2–6, and α2–8-linked acylneuraminic acids, led to a visible shift in migration of the immunoprecipitated CCR5 band on SDS-PAGE (Fig. 6 A). In the presence of 0.1% sodium azide to prevent receptor internalization, sialidase treatment had no effect on the expression level of CCR5, as determined by staining cells with phycoerythrin-conjugated 2D7 antibody (data not shown). Binding of MIP-1α and MIP-1β to sialidase-treated Cf2Th cells expressing wild-type CCR5 was almost completely abolished (Fig. 6, B and C). As expected, no specific binding of chemokines to the CCR5 NH2-terminal deletion mutant, CCR5Δ2–17, could be detected (Fig. 6 B, and data not shown).
Figure 6.
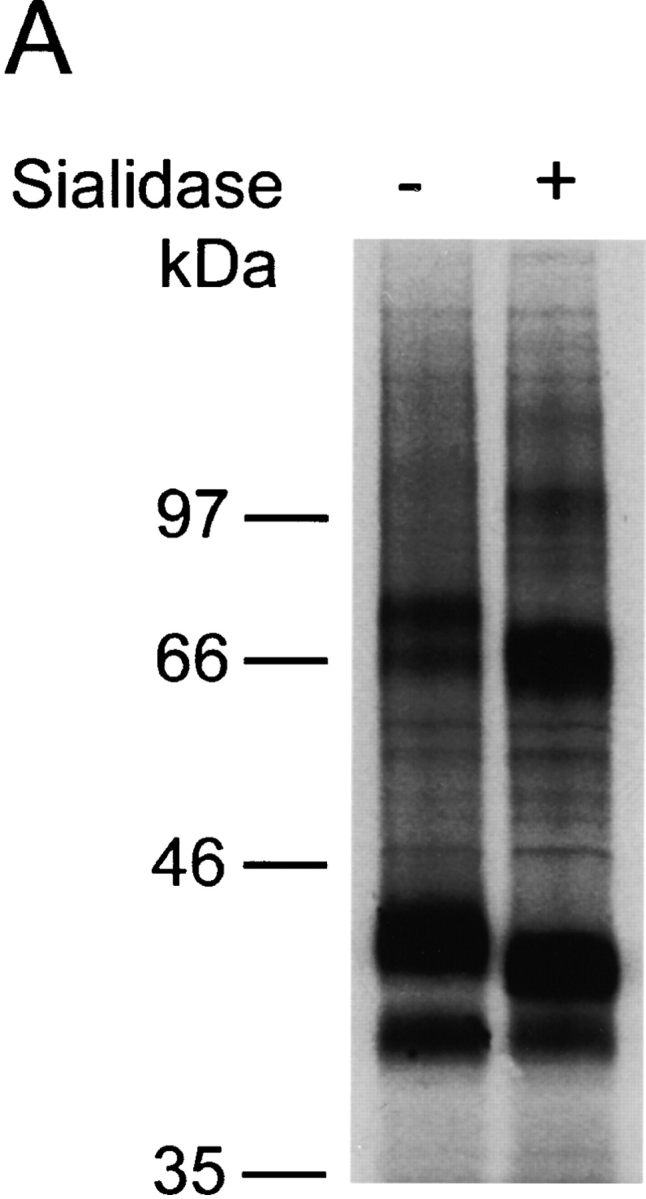


Effect of sialidase treatment of Cf2Th cells expressing CCR5 on chemokine binding. CCR5-expressing Cf2Th cells were divided into two groups and incubated with either sialidase or buffer without enzyme. (A) Sialidase treatment increased CCR5 mobility on a reducing SDS-polyacrylamide gel. Cells were labeled with 35S-cysteine/methionine overnight before incubation with buffer alone (−) or sialidase (+). Subsequently, cells were lysed and the lysates precipitated with the 1D4 antibody. Homologous competitive binding assays with MIP-1α (B) and MIP-1β (C) were performed on Cf2Th cells expressing wild-type CCR5 after sialidase treatment. Cells expressing the deletion mutant CCR5Δ2–17 were included in (B) to determine nonspecific chemokine binding. Sialidase treatment did not change the mean fluorescence of cells stained with the phycoerythrin-conjugated 2D7 antibody (data not shown). wt, wild-type.
Contribution of Sulfation of NH2-terminal CCR5 Tyrosines to Chemokine Binding.
Sulfation of tyrosines 10, 14, and 15 in the CCR5 NH2 terminus has been shown to be important for HIV-1 binding and entry (21). Treatment of CCR5-expressing cells with sodium chlorate, an inhibitor of tyrosine sulfation, lowered the affinity of CCR5 for MIP-1α and MIP-1β (21). To examine the contribution of specific CCR5 NH2-terminal tyrosine sulfates to chemokine binding, CCR5 mutants with alterations in one or more of the tyrosines at positions 3, 10, 14, and 15 were used. Cf2Th cell lines expressing comparable levels of the mutants were established, and equivalent surface expression of the CCR5 variants was verified by 2D7 antibody staining in parallel with the binding assays. The CCR5 mutants are named according to the amino acid residues located at positions 3, 10, 14, and 15. Previous studies (21) demonstrated that alteration of all four NH2-terminal CCR5 tyrosines to phenylalanine (FFFF) or to aspartic acid (DDDD) resulted in a loss of sulfate incorporation. Restoration of individual tyrosines in the context of the DDDD mutant resulted in mutant proteins (YDDD, DYDD, DDYD, and DDDY) that incorporated sulfate (21). Thus, all of the CCR5 NH2-terminal tyrosines can potentially be sulfated, and the study of mutants (FDDD, DFDD, DDFD, and DDDF) in which the restored tyrosines are converted to phenylalanine allows an assessment of the contribution of sulfation of a specific tyrosine to a phenotype. Changes in all four NH2-terminal CCR5 tyrosines resulted in ∼70–200-fold decreases in the affinity of MIP-1α and RANTES (Table I). Restoration of tyrosine 3 in the YDDD mutant did not restore the efficient binding of MIP-1α. Restoration of tyrosines 10, 14, and 15 all resulted in improved chemokine binding in the context of the DDDD mutant. In the case of tyrosine 14, this improvement was significantly better for the restoration of tyrosine than for a phenylalanine substitution. This suggests that, for both MIP-1α and RANTES, the presence of sulfation on tyrosine 14 contributes to the interaction with CCR5. By similar reasoning, the data in Table I suggest that the sulfation of tyrosine 15 contributes to the binding of RANTES but not that of MIP-1α. Thus, chemokine binding is facilitated by sulfation of NH2-terminal CCR5 tyrosines, particularly tyrosine 14 and, for RANTES, tyrosine 15.
Table I.
Chemokine Binding to CCR5 Mutants with Changes in NH2-terminal Tyrosines
| Ligand CCR5 variantsa | MIP-1α | RANTES |
|---|---|---|
| Wild-type (YYYY) | 0.33 ± 0.2 | 0.38 ± 0.12 |
| FFFF | 32 ± 2.5 | 74 ± 18 |
| DDDD | 22 ± 5.4 | 83 ± 22 |
| YDDD | 25 ± 4.4 | ND |
| FDDD | 98 ± 40 | ND |
| DYDD | 4.5 ± 1.2 | ND |
| DFDD | ND | ND |
| DDYD | 3.0 ± 1.2 | 3.6 ± 1.8 |
| DDFD | 14 ± 4.0 | 20 ± 5.0 |
| DDDY | 2.4 ± 0.7 | 8.9 ± 1.0 |
| DDDF | 3.0 ± 1.8 | 25 ± 6.8 |
The Kd values (nM) and standard errors were calculated from the results of experiments in which the CCR5 variants exhibited comparable levels of surface expression. The data were analyzed with GraphPad Prism software, using an algorithm for homologous one-site competition binding with ligand depletion (reference 27). The average Kd from two or three experiments is shown, with the highest standard error of each experiment reported. ND, not determined.
The names of the CCR5 mutants reflect the identity of the amino acids at positions 3, 10, 14, and 15.
Discussion
The interaction of chemokines and their receptors is characterized by promiscuity and redundancy, properties that allow these proteins to be involved in a wide variety of immunological processes (1–3, 30). How do chemokine receptors, whose extracellular domains are characterized by limited protein surface, achieve specific, high-affinity interactions while maintaining the flexibility to bind different ligands? Here we show that posttranslational modification of the CCR5 chemokine receptor by the addition of O-linked glycans and tyrosine sulfates contributes to chemokine binding. These modifications provide not only a larger and potentially more flexible binding surface, but also supply an array of negative charges that allow electrostatic interactions with the generally positive receptor-binding face of the chemokines.
As an initial step in evaluating the functional significance of CCR5 glycosylation, we characterized the location and extent of this modification. Glycan chains are commonly added to chemokine receptors, either by N-linkage to an asparagine residue in the sequon asparagine-X-serine/threonine or by O-linkage to a serine or threonine. CCR5 contains one potential N-linked glycosylation site (asparagine 268 - cysteine 269 - serine 270) in the third extracellular loop, but this site is not used (21, 31). Cysteine 269 in the consensus sequence is thought to be involved in the formation of a disulfide bond with cysteine 20 in the NH2 terminus, perhaps limiting the accessibility of this region to glycosyltransferases. CCR5 has been shown to be O-glycosylated (21) and has four serine/threonine-rich regions in its extracellular domains. Our mutagenic analysis of these regions indicates that the major CCR5 O-glycosylation site in all cells tested, including primary macrophages, involves a pair of serines at positions 6 and 7. A mutant CCR5 protein with serine to alanine changes at these positions demonstrated an ∼2-kD shift on SDS-PAGE and migrated with the same mobility as enzymatically deglycosylated wild-type protein. The ASTS and SATS mutants, in which serine 6 and 7, respectively, are individually converted to alanine, migrated similarly to the wild-type CCR5 protein. This result indicates that either serine 6 or serine 7 can be glycosylated but only one of them is modified in wild-type CCR5.
Our studies suggest that a second potential O-glycosylation site in the CCR5 NH2 terminus, at threonine 16/serine 17, can be modified by galactose-containing glycans under some circumstances. The AATS mutant, in which serines 6 and 7 are converted to alanines, incorporated galactose at a reduced level compared with the wild-type CCR5. This galactose incorporation was abolished by the additional conversion of threonine 16 and serine 17 to alanines in the AAAA mutant, indicating the dependence of this modification on the second potential O-glycosylation site in the CCR5 NH2 terminus. The AATS and AAAA mutants labeled with [35S]cysteine/methionine migrated indistinguishably on SDS-polyacrylamide gels. Furthermore, enzymatic deglycosylation of the AATS mutant does not result in a detectable shift in its migration. These observations suggest that the sugar moiety added to either threonine 16 or serine 17 in the AATS mutant is relatively small and likely consists of a T antigen structure (Gal β1–3 GalNAcα - Ser/Thr).
It is unclear whether O-glycosylation of either threonine 16 or serine 17 occurs in wild-type CCR5. The comparable migration and efficiency of galactose incorporation observed for wild-type CCR5 and the SSAA mutant in which threonine 16 or serine 17 are both converted to alanines, indicate that any O-glycosylation at this site involves short glycan chains. It is also quite possible that preventing glycosylation at the preferred site, serines 6 and 7, in the AATS mutant promotes aberrant glycosylation at threonine 16/serine 17. A similar effect has been reported for vasopressin receptor 2, where the removal of one glycosylation site resulted in other sites becoming available to glycosyltransferases (32). Our most extensive studies were performed in Cf2Th cells and it should be kept in mind that utilization of the potential O-glycosylation site at threonine 16 and serine 17 could vary with cell type or the differentiation or activation state of the cell (24).
Efficient O-glycosylation of CCR5 appears to be limited to the NH2 terminus, based on the phenotypes of the CCR5Δ2–17 and AAAA mutants. The serine/threonine-rich regions in the CCR5 second and third extracellular loops occur adjacent to cysteine residues involved in disulfide bonds. Like the potential N-glycosylation site, these potential sites of O-glycosylation may not be accessible due to close interactions of these regions with other elements of the CCR5 ectodomain.
To date, only a few instances of O-glycosylation of GPCRs have been documented (32) and little is known about the presence of this modification on chemokine receptors and its potential contribution to the physiologic function of these proteins. The results of our homologous competition binding experiments and Ca2+ mobilization assays performed with MIP-1α and MIP-1β demonstrate that O-glycosylation of CCR5 at serine 6 or 7 is necessary for high affinity binding of both chemokines. Results analogous to those obtained with CCR5 mutants were observed for the wild-type CCR5 expressed in ldlD cells, which are conditionally defective for O-linked sugar addition. Most of the contribution of the O-linked glycans on CCR5 to chemokine binding appears to depend upon the negatively charged sialic acids. The sialic acids likely make specific contacts with basic residues on the chemokine surface thought to interact with CCR5 (33, 34). The binding of MIP-1α was disrupted more than that of MIP-1β by removal of the CCR5 O-glycans or sialic acid, suggesting different degrees of contact between the negatively charged glycans and the binding surfaces of different chemokine ligands.
The chemokine binding studies provide clues that serine 6 is the preferred site of O-glycosylation on wild-type CCR5. The SATS mutant, in which serine 7 is changed to alanine, bound chemokine with an affinity comparable to that of wild-type CCR5. This is consistent with other mutagenic studies of CCR5 in which serine 7 was altered (13). By contrast, replacement of serine 6 with alanine (ASTS mutant) resulted in a decreased chemokine binding affinity of ∼3–4-fold, despite the O-glycosylation of this mutant at serine 7. A similar decrease in chemokine binding has been observed for a CCR5 mutant in which serine 6 is converted to proline (33). Thus, although O-glycosylation at serine 7 contributes somewhat to chemokine binding, the presence of O-linked glycans on serine 6 is optimal for the interaction of CCR5 with chemokine ligands. Because MIP-1β binds comparably to wild-type CCR5 and the SATS mutant, but binds less well to the ASTS mutant, the wild-type CCR5 probably is O-glycosylated predominantly on serine 6.
Further studies will be required to define the structure of the O-glycans added to CCR5. CHO cells do not express core 2 β1,6-N-acetylglucosaminyltransferase and α1,3-fucosyltransferase (35) and, as a result, O-linked carbohydrates added to proteins produced in CHO cells are typically relatively short, core-1 O-glycans (36–43). Our data demonstrating MIP-1α and MIP-1β binding to CCR5 expressed in CHO or IdID cells indicate that the addition of such core-1 structures is sufficient to allow the interaction with chemokines.
It has been shown previously that glycosylation is required to maintain high-affinity SDF-1α binding (44) and influences the ability of CXCR4 to serve as a coreceptor for T-tropic (X4) and dual-tropic (R5X4) HIV-1 strains (45). Removal of a site of N-linked glycosylation allows CXCR4 to serve as a more universal coreceptor, allowing efficient entry of several HIV-1 isolates that normally use only CCR5 (45). We tested the impact of O-glycosylation on the ability of CCR5 to serve as a coreceptor for HIV-1 and SIV. The absence of O-glycans on CCR5 did not significantly influence HIV-1 and SIV entry efficiency. It is possible that such an influence might be revealed in target cells with lower receptor density. O-glycosylation–deficient CCR5 mutants did not support the entry of viruses pseudotyped with the T-tropic (X4) HXBc2 envelope glycoproteins (data not shown).
The binding of HIV-1 envelope glycoproteins to CCR5 and the entry of R5 viruses into CCR5-expressing cells is greatly influenced by the sulfation of tyrosines 10, 14, and 15 in the CCR5 NH2 terminus (21). Treatment of CCR5-expressing cells with sodium chlorate, an inhibitor of tyrosine sulfation, decreased the binding affinity of the HIV-1 gp120 envelope glycoprotein and β-chemokines (21). Here, using a mutagenic approach, we provide evidence for the contribution of sulfation of tyrosine 14 in the CCR5 NH2 terminus to the binding of both MIP-1α and RANTES. Although the presence of a phenyl group in the side chain of CCR5 residue 15 contributes to both MIP-1α and RANTES binding, the sulfate moiety on tyrosine 15 appears to be more important for the binding of RANTES than that of MIP-1α. Our data are also consistent with a contribution of the tyrosine sulfate at position 10 to MIP-1α binding, although the specific contribution of sulfate was not addressed. Finally, as was previously seen for HIV-1 gp120 binding (21), the tyrosine sulfate at position 3 of CCR5 makes little contribution to chemokine binding.
Together, our results demonstrate a significant role of posttranslational modification of the CCR5 NH2 terminus in binding the natural chemokine ligands. Sialylated O-linked oligosaccharides and sulfated tyrosines provide an array of negative charges on a potentially flexible substructure, allowing electrostatic interactions with the structurally diverse, basic surfaces of different chemokines. High-affinity binding must, in addition, require more specific contacts between the chemokine and the CCR5 sulfotyrosines and sialic acids; this conclusion follows from the observation that acidic amino acid residues cannot effectively substitute for these moieties. A potential β-turn involving proline 8 on CCR5 may bring the O-glycans attached to serine 6 into proximity with the tyrosine sulfate–rich stretch at residues 10–15, facilitating such specific contacts. The NH2 termini of several chemokine receptors are rich not only in tyrosines that can be potentially sulfated (21), but also in serines and threonines. This suggests that post-translational modifications by sulfate and O-linked oligosaccharides may be common among the chemokine receptors and play roles in ligand binding similar to those demonstrated for CCR5.
Striking parallels exist between the structures implicated in chemokine binding by CCR5 and those contributing to selectin-mediated intercellular adhesion. Sialyl lewis X structures on core 2 branched O-linked oligosaccharides, together with the sulfation of closely located amino-terminal tyrosines, provide the basis for specific interaction of some type of selectin ligands with their counter-receptors. For example, PSGL-1 (P-selectin glycoprotein ligand-1) contains three tyrosines close to the NH2-terminal mucin-like glycosylation site. Sulfation of one of these tyrosines in addition to the sialyl lewis X glycan(s) is essential for P-selectin binding (46–49). In this case, the position, structure, and chirality of the O-linked carbohydrate can influence the affinity of the interaction (49–52).
All α- and β-chemokines, including MIP-1α, MIP-1β, and RANTES, bind sulfated glycosaminoglycans (53, 54). These less specific electrostatic associations may serve to concentrate the chemokines near the target cell surface, setting the stage for more specific interactions with the chemokine receptor. It is possible that the properties of individual chemokines may similarly impact binding to both glycosaminoglycans and chemokine receptors. In this context, it is interesting that Ali and colleagues reported that MIP-1α, but not MIP-1β, bound to CCR5-expressing cells much more efficiently when glycosaminoglycans were present on the cell surface (55). This might reflect some of the differences that we observed in the impact of O-glycosylation on MIP-1α and MIP-1β binding. Additional work should further elucidate the molecular interactions that allow chemokines to exert their effects on diverse targets.
Acknowledgments
We acknowledge Monty Krieger for CHO and ldlD cells. We thank Maris Handley at the Dana-Farber Cancer Institute flow cytometry core facility for excellent technical support and Yvette McLaughlin and Sheri Farnum for manuscript preparation.
This work was supported by grants from the National Institutes of Health AI41851 and by a Center for AIDS Research grant AI28691. Additional support was provided by the G. Harold and Leila Y. Mathers Foundation, the late William F. McCarty-Cooper, the Friends 10, and Douglas and Judith Krupp. N. Bannert was supported by a fellowship from the Deutsche Forschungsgemeinschaft (DFG).
Footnotes
Abbreviations used in this paper: CAT, chloramphenicol acetyltransferase; CCR, CC chemokine receptor; CHO, Chinese hamster ovary; GPCR, G protein–coupled receptor; MIP, macrophage inflammatory protein; RANTES, regulated on activation normal T cell expressed; RT, reverse transcriptase; SIV, simian immunodeficiency virus; VSV-G, vesicular stomatitis virus G protein.
References
- 1.Baggiolini, M. 1998. Chemokines and leukocyte traffic. Nature. 392:565–568. [DOI] [PubMed] [Google Scholar]
- 2.Strieter, R.M., T.J. Standiford, G.B. Huffnagle, L.M. Colletti, N.W. Lukacs, and S.L. Kunkel. 1996. “The good, the bad, and the ugly.” The role of chemokines in models of human disease. J. Immunol. 156:3583–3586. [PubMed] [Google Scholar]
- 3.Rollins, B.J. 1997. Chemokines. Blood. 90:909–928. [PubMed] [Google Scholar]
- 4.Ochi, H., W.M. Hirani, Q. Yuan, D.S. Friend, K.F. Austen, and J.A. Boyce. 1999. T helper cell type 2 cytokine-mediated comitogenic responses and CCR3 expression during differentiation of human mast cells in vitro. J. Exp. Med. 190:267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alkhatib, G., C. Combadiere, C.C. Broder, Y. Feng, P.E. Kennedy, P.M. Murphy, and E.A. Berger. 1996. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 272:1955–1958. [DOI] [PubMed] [Google Scholar]
- 6.Choe, H., M. Farzan, Y. Sun, N. Sullivan, B. Rollins, P.D. Ponath, L. Wu, C.R. Mackay, G. LaRosa, W. Newman, et al. 1996. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 85:1135–1148. [DOI] [PubMed] [Google Scholar]
- 7.Connor, R.I., K.E. Sheridan, D. Ceradini, S. Choe, and N.R. Landau. 1997. Change in coreceptor use correlates with disease progression in HIV-1-infected individuals. J. Exp. Med. 185:621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu, R., W.A. Paxton, S. Choe, D. Ceradini, S.R. Martin, R. Horuk, M.E. MacDonald, H. Stuhlmann, R.A. Koup, and N.R. Landau. 1996. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 86:367–377. [DOI] [PubMed] [Google Scholar]
- 9.Dean, M., M. Carrington, C. Winkler, G.A. Huttley, M.W. Smith, R. Allikmets, J.J. Goedert, S.P. Buchbinder, E. Vittinghoff, E. Gomperts, et al. 1996. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE study. Science. 273:1856–1862. [DOI] [PubMed] [Google Scholar]
- 10.Cocchi, F., A.L. DeVico, A. Garzino-Demo, S.K. Arya, R.C. Gallo, and P. Lusso. 1995. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science. 270:1811–1815. [DOI] [PubMed] [Google Scholar]
- 11.Baba, M., O. Nishimura, N. Kanzaki, M. Okamoto, H. Sawada, Y. Iizawa, M. Shiraishi, Y. Aramaki, K. Okonogi, Y. Ogawa, et al. 1999. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc. Natl. Acad. Sci. USA. 96:5698–5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samson, M., G. LaRosa, F. Libert, P. Paindavoine, M. Detheux, G. Vassart, and M. Parmentier. 1997. The second extracellular loop of CCR5 is the major determinant of ligand specificity. J. Biol. Chem. 272:24934–24941. [DOI] [PubMed] [Google Scholar]
- 13.Blanpain, C., B.J. Doranz, J. Vakili, J. Rucker, C. Govaerts, S.S. Baik, O. Lorthioir, I. Migeotte, F. Libert, F. Baleux, et al. 1999. Multiple charged and aromatic residues in CCR5 amino-terminal domain are involved in high affinity binding of both chemokines and HIV-1 Env protein. J. Biol. Chem. 274:34719–34727. [DOI] [PubMed] [Google Scholar]
- 14.Farzan, M., H. Choe, L. Vaca, K. Martin, Y. Sun, E. Desjardins, N. Ruffing, L. Wu, R. Wyatt, N. Gerard, et al. 1998. A tyrosine-rich region in the N terminus of CCR5 is important for human immunodeficiency virus type 1 entry and mediates an association between gp120 and CCR5. J. Virol. 72:1160–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerard, C., and N.P. Gerard. 1994. The pro-inflammatory seven-transmembrane segment receptors of the leukocyte. Curr. Opin. Immunol. 6:140–145. [DOI] [PubMed] [Google Scholar]
- 16.Monteclaro, F.S., and I.F. Charo. 1996. The amino-terminal extracellular domain of the MCP-1 receptor, but not the RANTES/MIP-1alpha receptor, confers chemokine selectivity. Evidence for a two-step mechanism for MCP-1 receptor activation. J. Biol. Chem. 271:19084–19092. [DOI] [PubMed] [Google Scholar]
- 17.Baik, S.S., R.W. Doms, and B.J. Doranz. 1999. HIV and SIV gp120 binding does not predict coreceptor function. Virology. 259:267–273. [DOI] [PubMed] [Google Scholar]
- 18.Dragic, T., A. Trkola, S.W. Lin, K.A. Nagashima, F. Kajumo, L. Zhao, W.C. Olson, L. Wu, C.R. Mackay, G.P. Allaway, et al. 1998. Amino-terminal substitutions in the CCR5 coreceptor impair gp120 binding and human immunodeficiency virus type 1 entry. J. Virol. 72:279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rabut, G.E., J.A. Konner, F. Kajumo, J.P. Moore, and T. Dragic. 1998. Alanine substitutions of polar and nonpolar residues in the amino-terminal domain of CCR5 differently impair entry of macrophage- and dualtropic isolates of human immunodeficiency virus type 1. J. Virol. 72:3464–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ross, T.M., P.D. Bieniasz, and B.R. Cullen. 1998. Multiple residues contribute to the inability of murine CCR-5 to function as a coreceptor for macrophage-tropic human immunodeficiency virus type 1 isolates. J. Virol. 72:1918–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farzan, M., T. Mirzabekov, P. Kolchinsky, R. Wyatt, M. Cayabyab, N.P. Gerard, C. Gerard, J. Sodroski, and H. Choe. 1999. Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell. 96:667–676. [DOI] [PubMed] [Google Scholar]
- 22.Varki, A. 1993. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 3:97–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brockhausen, I. 1999. Pathways of O-glycan biosynthesis in cancer cells. Biochim. Biophys. Acta. 1473:67–95. [DOI] [PubMed] [Google Scholar]
- 24.Fukuda, M., S.R. Carlsson, J.C. Klock, and A. Dell. 1986. Structures of O-linked oligosaccharides isolated from normal granulocytes, chronic myelogenous leukemia cells, and acute myelogenous leukemia cells. J. Biol. Chem. 261:12796–12806. [PubMed] [Google Scholar]
- 25.Bannert, N., D. Schenten, S. Craig, and J. Sodroski. 2000. The level of CD4 expression limits infection of primary rhesus monkey macrophages by a T-tropic simian immunodeficiency virus and macrophagetropic human immunodeficiency viruses. J. Virol. 74:10984–10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mirzabekov, T., N. Bannert, M. Farzan, W. Hofmann, P. Kolchinsky, L. Wu, R. Wyatt, and J. Sodroski. 1999. Enhanced expression, native purification, and characterization of CCR5, a principal HIV-1 coreceptor. J. Biol. Chem. 274:28745–28750. [DOI] [PubMed] [Google Scholar]
- 27.Swillens, S. 1995. Interpretation of binding curves obtained with high receptor concentrations: practical aid for computer analysis. Mol. Pharmacol. 47:1197–1203. [PubMed] [Google Scholar]
- 28.Ludwig, A., J.E. Ehlert, H.D. Flad, and E. Brandt. 2000. Identification of distinct surface-expressed and intracellular CXC-chemokine receptor 2 glycoforms in neutrophils: N-glycosylation is essential for maintenance of receptor surface expression. J. Immunol. 165:1044–1052. [DOI] [PubMed] [Google Scholar]
- 29.Kingsley, D.M., K.F. Kozarsky, L. Hobbie, and M. Krieger. 1986. Reversible defects in O-linked glycosylation and LDL receptor expression in a UDP-Gal/UDP-GalNAc 4-epimerase deficient mutant. Cell. 44:749–759. [DOI] [PubMed] [Google Scholar]
- 30.Blanpain, C., I. Migeotte, B. Lee, J. Vakili, B.J. Doranz, C. Govaerts, G. Vassart, R.W. Doms, and M. Parmentier. 1999. CCR5 binds multiple CC-chemokines: MCP-3 acts as a natural antagonist. Blood. 94:1899–1905. [PubMed] [Google Scholar]
- 31.Rucker, J., M. Samson, B.J. Doranz, F. Libert, J.F. Berson, Y. Yi, R.J. Smyth, R.G. Collman, C.C. Broder, G. Vassart, et al. 1996. Regions in beta-chemokine receptors CCR5 and CCR2b that determine HIV-1 cofactor specificity. Cell. 87:437–446. [DOI] [PubMed] [Google Scholar]
- 32.Sadeghi, H., and M. Birnbaumer. 1999. O-Glycosylation of the V2 vasopressin receptor. Glycobiology. 9:731–737. [DOI] [PubMed] [Google Scholar]
- 33.Zhou, N., Z. Luo, J.W. Hall, J. Luo, X. Han, and Z. Huang. 2000. Molecular modeling and site-directed mutagenesis of CCR5 reveal residues critical for chemokine binding and signal transduction. Eur. J. Immunol. 30:164–173. [DOI] [PubMed] [Google Scholar]
- 34.Laurence, J.S., C. Balanpain, A. De Leener, M. Parmentier, and P.J. Wang. 2001. Importance of basic residues and quaternary structure in the function of MIP-1 beta: CCR5 binding and cell surface sugar interactions. Biochemistry. 40:4990–4999. [DOI] [PubMed] [Google Scholar]
- 35.Datti, A., and J.W. Dennis. 1993. Regulation of UDP-GlcNAc:Gal beta 1-3GalNAc-R beta 1-6-N-acetylglucosaminyltransferase (GlcNAc to GalNAc) in Chinese hamster ovary cells. J. Biol. Chem. 268:5409–5416. [PubMed] [Google Scholar]
- 36.Sasaki, H., B. Bothner, A. Dell, and M. Fukuda. 1987. Carbohydrate structure of erythropoietin expressed in Chinese hamster ovary cells by a human erythropoietin cDNA. J. Biol. Chem. 262:12059–12076. [PubMed] [Google Scholar]
- 37.Seguchi, T., R.K. Merkle, M. Ono, M. Kuwano, and R.D. Cummings. 1991. The dysfunctional LDL receptor in a monensin-resistant mutant of Chinese hamster ovary cells lacks selected O-linked oligosaccharides. Arch. Biochem. Biophys. 284:245–256. [DOI] [PubMed] [Google Scholar]
- 38.Pahlsson, P., D.P. Blackall, M. Ugorski, M. Czerwinski, and S.L. Spitalnik. 1994. Biochemical characterization of the O-glycans on recombinant glycophorin A expressed in Chinese hamster ovary cells. Glycoconj. J. 11:43–50. [DOI] [PubMed] [Google Scholar]
- 39.Orita, T., M. Oh-eda, M. Hasegawa, H. Kuboniwa, K. Esaki, and N. Ochi. 1994. Polypeptide and carbohydrate structure of recombinant human interleukin-6 produced in Chinese hamster ovary cells. J. Biochem. 115:345–350. [DOI] [PubMed] [Google Scholar]
- 40.Hokke, C.H., A.A. Bergwerff, G.W. van Dedem, J.P. Kamerling, and J.F. Vliegenthart. 1995. Structural analysis of the sialylated N- and O-linked carbohydrate chains of recombinant human erythropoietin expressed in Chinese hamster ovary cells. Sialylation patterns and branch location of dimeric N-acetyllactosamine units. Eur. J. Biochem. 228:981–1008. [DOI] [PubMed] [Google Scholar]
- 41.Chapman, B.S., M.R. Eckart, S.E. Kaufman, and G.R. Lapointe. 1996. O-linked oligosaccharide on the 75-kDa neurotrophin receptor. J. Neurochem. 66:1707–1716. [DOI] [PubMed] [Google Scholar]
- 42.Ushida, Y., S. Natsuka, Y. Shimokawa, Z. Takatsu, S. Shimamura, and S. Hase. 1997. Structures of the sugar chains of recombinant macrophage colony-stimulating factor produced in Chinese hamster ovary cells. J. Biochem. 122:148–156. [DOI] [PubMed] [Google Scholar]
- 43.Nakahara, Y., T. Miyata, T. Hamuro, A. Funatsu, M. Miyagi, S. Tsunasawa, and H. Kato. 1996. Amino acid sequence and carbohydrate structure of a recombinant human tissue factor pathway inhibitor expressed in Chinese hamster ovary cells: one N- and two O-linked carbohydrate chains are located between Kunitz domains 2 and 3 and one N-linked carbohydrate chain is in Kunitz domain 2. Biochemistry. 35:6450–6459. [DOI] [PubMed] [Google Scholar]
- 44.Zhou, H., and H.H. Tai. 1999. Characterization of recombinant human CXCR4 in insect cells: Role of extracellular domains and N-glycosylation in ligand binding. Arch. Biochem. Biophys. 369:267–276. [DOI] [PubMed] [Google Scholar]
- 45.Chabot, D.J., H. Chen, D.S. Dimitrov, and C.C. Broder. 2000. N-linked glycosylation of CXCR4 masks coreceptor function for CCR5-dependent human immunodeficiency virus type 1 isolates. J. Virol. 74:4404–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sako, D., K.M. Comess, K.M. Barone, R.T. Camphausen, D.A. Cumming, and D. Shaw. 1995. A sulfated peptide segment at the amino terminus of PSGL-1 is critical for P-selectin binding. Cell. 83:323–331. [DOI] [PubMed] [Google Scholar]
- 47.Pouyani, T., and B. Seed. 1995. PSGL-1 recognition of P-selectin is controlled by a tyrosine sulfation consensus at the PSGL-1 amino terminus. Cell. 83:333–343. [DOI] [PubMed] [Google Scholar]
- 48.Wilkins, P.P., K.L. Moore, R.P. McEver, and R.D. Cummings. 1995. Tyrosine sulfation of P-selectin glycoprotein ligand-1 is required for high affinity binding to P-selectin. J. Biol. Chem. 270:22677–22680. [DOI] [PubMed] [Google Scholar]
- 49.Somers, W.S., J. Tang, G.D. Shaw, and R.T. Camphausen. 2000. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe(X) and PSGL-1. Cell. 103:467–479. [DOI] [PubMed] [Google Scholar]
- 50.Leppanen, A., S.P. White, J. Helin, R.P. McEver, and R.D. Cummings. 2000. Binding of glycosulfopeptides to P-selectin requires stereospecific contributions of individual tyrosine sulfate and sugar residues. J. Biol. Chem. 275:39569–39578. [DOI] [PubMed] [Google Scholar]
- 51.Leppanen, A., P. Mehta, Y.B. Ouyang, T. Ju, J. Helin, K.L. Moore, I. van Die, W.M. Canfield, R.P. McEver, and R.D. Cummings. 1999. A novel glycosulfopeptide binds to P-selectin and inhibits leukocyte adhesion to P-selectin. J. Biol. Chem. 274:24838–24848. [DOI] [PubMed] [Google Scholar]
- 52.Liu, W., V. Ramachandran, J. Kang, T.K. Kishimoto, R.D. Cummings, and R.P. McEver. 1998. Identification of N-terminal residues on P-selectin glycoprotein ligand-1 required for binding to P-selectin. J. Biol. Chem. 273:7078–7087. [DOI] [PubMed] [Google Scholar]
- 53.Tanaka, Y., D.H. Adams, S. Hubscher, H. Hirano, U. Siebenlist, and S. Shaw. 1993. T-cell adhesion induced by proteoglycan-immobilized cytokine MIP-1 beta. Nature. 361:79–82. [DOI] [PubMed] [Google Scholar]
- 54.Koopmann, W., C. Ediriwickrema, and M.S. Krangel. 1999. Structure and function of the glycosaminoglycan binding site of chemokine macrophage-inflammatory protein-1 beta. J. Immunol. 163:2120–2127. [PubMed] [Google Scholar]
- 55.Ali, S., A.C. Palmer, B. Banerjee, S.J. Fritchley, and J.A. Kirby. 2000. Examination of the function of RANTES, MIP-1alpha, and MIP-1beta following interaction with heparin-like glycosaminoglycans. J. Biol. Chem. 275:11721–11727. [DOI] [PubMed] [Google Scholar]