

Abstract
The terminal components of the complement system contribute to host defense by forming the multiprotein membrane attack complex (MAC) which is responsible for cell lysis and several noncytotoxic effects. Most of the complement proteins are synthesized in the liver, but the mechanisms controlling their tissue-specific expression have not been elucidated. In this study we show that mice lacking the hepatic transcription factor hepatocyte nuclear factor 1α (HNF1α) fail to transcribe C5 and C8A complement genes. In addition, mRNAs encoding for several other terminal complement components or subunits are expressed at lower levels, including C8β, C8γ, and C9. We next used a reconstitution assay involving human sera with selective complement deficiencies to assess mouse complement activity. Sera from HNF1α-deficient mice showed negligible hemolytic activity of both C5 and C8α-γ subunits. The activity of C8β was severely affected despite only a 50% reduction in C8β mRNA levels in the liver. This is reminiscent of C8α-γ–deficient patients who accumulate extremely low levels of the C8β subunit.
Our results demonstrate that HNF1α plays a key role in the expression of C5 and C8A genes, two terminal complement component genes that are essential for the assembly of MAC as a result of complement activation.
Keywords: HNF1α, complement, C5, C8 α, transcription
Introduction
The complement system is an important component of the innate immunity that protects the host from infectious and other noxious agents collaborating with other components of both innate and acquired immunity. Complement first appeared in the invertebrate deuterostomes, in which it is mainly involved in opsonization of foreign particles, and developed in vertebrates into a complex system that comprises over 30 glycoproteins including complement components, regulators, and receptors (1, 2). Upon activation, the late complement components form a multiprotein complex, that inserts into the phospholipid bilayer of the target cell as membrane attack complex (MAC) causing both cytolysis and noncytotoxic effects (3–5). The liver is the main site of synthesis of most of the complement components circulating in blood, as indicated by allotypic changes in recipients of liver transplants (6).
Tissue-specific regulation of gene activity results from the concerted action of tissue-specific and ubiquitously expressed transcription factors. Transcription factors regulating the constitutive expression and cytokine responsiveness of C4, C2, C3, and factor B in hepatocytes have in part been identified using mostly cell lines (7), but the precise mechanism responsible for the hepatic expression of complement genes, particularly of the late components, remains largely unknown.
Hepatocyte nuclear factor 1 α (HNF1α) is a dimeric transcription factor characterized by the presence of a distantly related homeobox DNA-binding domain which is involved in the transcriptional regulation of hepatic genes (8–12). HNF1α protein is also expressed in highly polarized epithelia in kidney, pancreas, and intestine. HNF1α-deficient mice manifest dysfunctions affecting liver, kidney, and pancreas and develop hyperphenylalaninemia (13, 14), dysfunction in bile acid and cholesterol metabolism (12), insulin secretion defect (15, 16), and renal defective reabsorption of glucose, phosphate, and amino acids (17). Insulin secretion defects and a lower threshold for renal glucose reabsorption have been also observed in maturity onset diabetes of the young type 3 (MODY3) patients (17, 18), who carry mutations in the HNF1α gene. In this study we demonstrate that mice carrying a null mutation in the HNF1α gene fail to express C5 and C8A genes and to synthesize the related proteins. In addition, HNF1α-deficient mice express reduced levels of mRNA encoding for C8β, C8γ, and C9.
Materials and Methods
Animals.
HNF1α-deficient mice were generated by replacing the first exon with a β-Galactosidase cassette as described previously (13). The animals employed in this study were 2–3-wk-old and HNF1α mutant homozygous, heterozygotes, and wild-type mice were sex and age-matched. The genetic background of the animals was either C57/Bl6/129sv or 129sv/DBA2/C57bl/6.
Sera.
Murine sera were obtained by tail bleeding and stored at −80°C until analyzed. Human C5 deficient serum (C5D) was obtained from a patient with a history of meningococcal disease and was identified in our laboratory because of undetectable immunochemical level of C5 and lack of hemolytic activity, that was reconstituted by the addition of purified C5. The C6-deficient (19) and C8α-γ–deficient (20) sera were a gift of Dr. Orren (National University of Ireland, Galway, Ireland) and Dr. Fukumori (Osaka Red Cross Blood Center, Osaka, Japan), respectively. The C8β deficient serum was obtained from a patient previously reported (21) and C9 depleted human serum was purchased from Quidel Corp.
Erythrocyte Intermediates and Hemolytic Assays.
Sheep erythrocytes were sensitized with subagglutinating amount of rabbit IgM antibodies (EA). The intermediate EAC1–3b was prepared by incubating EA with 1/10 dilution of C5D serum in glucose veronal-buffered saline (GVBS) for 70 s at 37°C followed by the addition of Suramin (Bayer) to block the reaction, as described by Harrison and Lachmann (22). Hemolytic titration of the various complement components was performed by mixing dilutions of the sera under test in GVBS with 50 μl of 1% EAC1–3b suspended in 1/20 dilution of C-deficient sera to a final volume of 250 μl. The mixtures were incubated at 37°C for 30 min. Lysis was read at 415 nm and the CH50 units were calculated as described (22)
RNA Analysis.
Total RNA was isolated as described elsewhere (23). Representational difference analysis (RDA) was conducted as described previously (24) except that only 1 cycle of subtraction was performed. The R3 clone that we isolated has been submitted to GenBank/EMBL/DDBJ (AJ312972). For Northern blots 15 μg of total RNA were used. DNA probes were labeled by random priming. Detailed description of the oligonucleotides used for the reverse transcription (RT)-PCR experiments are available upon request. Quantification of signals of Northern blot hybridization was performed by means of PhosphorImager screens.
Screening of a Mouse cDNA Library and Isolation of C8A Gene.
The probe used in the screening of the genomic library was obtained as follows: a 116 nucleotide fragment, corresponding to the first exon of the C8A murine gene, deduced from the comparison with the genomic structure of the human ortholog gene, was derived from the EST sequence AI097851 via PCR amplification from mouse genomic DNA. The fragment was labeled by random priming and used to screen a λDASH mouse genomic library. Two clones were isolated, DNA inserts were restriction mapped and sequenced (AJ409152).
Preparation of Nuclei, Micrococcal Nuclease Sensitivity, and Gel Shift Assay.
Nuclei were prepared from freshly excised livers of mice by homogenization in a Teflon pestle tissue homogenizer or in a glass Dounce homogenizer as described previously (14). Nuclear suspensions corresponding to 300 μg of genomic DNA were digested at room temperature for 0 to 9 min with 0.4 U of micrococcal nuclease as previously described (14) Gel shift assays were performed as described previously (14).
Results
Screening for Hepatic Genes Regulated by HNF1α.
To identify hepatic HNF1α target genes, we employed the representational difference analysis (RDA) (24), a PCR-based technique, to isolate cDNA fragments of genes expressed in the liver of wild animals but not in HNF1α-deficient mice. Although RDA is not as exhaustive as microarray hybridization, it has certain advantages. In fact, this technique does not depend on the previous gene characterization and may detect cDNA that are expressed at low levels. Our screening analysis identified a number of cDNA fragments including R3, a 264 nucleotide clone, with a significant homology (86% identity) with the human complement component C8α (M16974). We then monitored the level of the corresponding mRNA in livers from HNF1α-deficient mice or heterozygous and wild-type animals by Northern blot hybridization using a probe prepared from this R3 fragment. A C8α-specific signal, of an apparent molecular weight of 4 kb, was expressed to roughly similar levels in wild-type and heterozygous animals (Fig. 1). On the contrary, HNF1α-deficient mice failed to express this mRNA, suggesting that HNF1α is absolutely required for the expression of C8A.
Figure 1.

Northern blot analysis of hepatic RNA. Northern blot analysis of total hepatic RNA from wild-type, heterozygous, and mutant homozygous animals. Four animals for each genotype were analyzed. Parallel filters were hybridized with probes specific for the different cDNAs as indicated on the left. C6 mRNA expression along with an internal control for β-actin were monitored with RT-PCR with 12 cycles and subsequently revealed with Southern blot analysis. Histograms on the right indicate the average expression levels for each genotype expressed in arbitrary units and normalized on the expression of the β actin (the value of 100 was attributed to the expression level in the wild-type animals). Error bars represent standard error of the mean and the significance of the differences between wild-type and heterozygotes vs. homozygotes is indicated with (*) for P < 0.05 and (**) for P < 0.01 in a Student's t test (−/− vs. control animals; Student's t test).
Sera from HNF1α-deficient Mice Lack C8α-γ Functional Activity.
To establish whether clone R3 was indeed the murine ortholog of the human C8A gene, we evaluated the function of the corresponding protein in reconstitution experiments of the hemolytic activity of a C8α-γ–deficient human serum. As shown in Fig. 2, sera from wild-type or heterozygous mice restored the lytic activity of the deficient human serum even at high dilution, whereas sera from homozygous HNF1α2/− mice were ineffective even at high concentrations, revealing a severe defect in C8α-γ activity. Similarly, complete reconstitution of lytic activity of a C8β-deficient human serum was obtained with the sera from wild-type and HNF1α heterozygous mice, but 100-fold higher concentration of sera from HNF1α2/− mice was required to produce a similar effect, indicating that the C8β level in the homozygous mice was substantially reduced, though not absent (Fig. 3, and data not shown).
Figure 2.

Complementation assay of C8α-γ specific activities in sera from HNF1α-deficient mice. Increasing dilutions of sera from wild-type, heterozygous, and mutant homozygous animals to a volume of 200 μl were incubated with 50 μl of 1% EAC1–3b suspended in 1/20 of C8α-γ–deficient human sera. The mixtures were incubated for 30 min at 37°C and lysis was monitored by reading optical density at 415 nm. The results are expressed as percentage of lysis. Close circles, open circles, and triangles represent wild-type, heterozygous, and mutant homozygous animals respectively.
Figure 3.
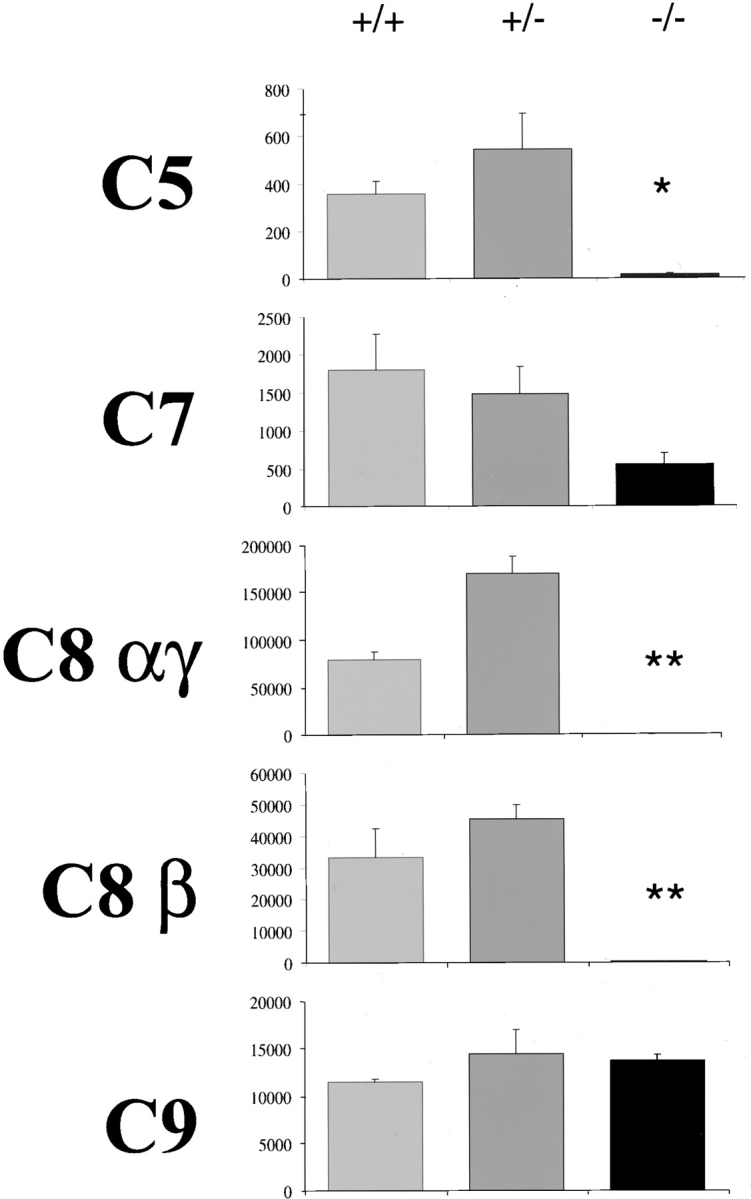
Hemolytic activity of the late complement components in the sera from HNF1α-deficient mice. The sera from wild-type, heterozygous, and mutant homozygous animals were examined for the lytic activity of the late components using human sera with selective deficiencies of the individual components (as in Fig. 2). The results are expressed as CH50 (complement hemolytic units where one unit corresponds to the amount required to lyse 50% red cells in the assay). Error bars represent standard error of the mean and the significance of the differences between wild-type and heterozygotes and homozygotes is indicated with (*) for P < 0.05 and (**) for P < 0.01.
HNF1α2/− Mice Manifest Reduced Hepatic Expression of Several Terminal Complement Components.
To check whether the transcription of other complement components was also abolished or reduced in HNF1α homozygous deficient mice, total hepatic RNA was analyzed by Northern blot for the expression of C3 and the late components from C5 to C9. For C6 we performed RT-PCR, since the signal obtained by Northern blot was too weak. As shown in Fig. 1, the expression of C8β, C8γ, and C9 was significantly reduced in the homozygous-deficient mice. The expression level of C7 mRNA was not affected at all whereas that of C5 mRNA, like that C8A, was undetectable indicating that HNF1α controls the expression of several late complement components, two of which are completely silenced in the absence of HNF1α. The equal loading of each lane was confirmed with the hybridization of Northern blot with β actin and Apolipoprotein E (ApoE) probes (Fig. 1).
We next measured the hemolytic activity of the late components in the sera from homozygous and heterozygous HNF1α-deficient mice and from wild-type littermate controls. The sera of wild-type and heterozygous animals restored the lytic activity of all the deficient sera except that of C6 deficient serum, indicating that mouse C6 cannot be incorporated into human MAC. Conversely, the hemolytic activity of C5 and of the two C8 subunits in the sera of homozygous deficient animals was undetectable, whereas that of C7 was marginally reduced and the level of C9 was within the normal range (Fig. 3).
HNF1α Deficiency Impairs Chromatin Remodeling at the C8α Locus.
HNF1α has been shown to be essential for the chromatin remodeling of the phenylalanine hydroxylase gene during development (14, 25). The complete lack of C8α expression raised the possibility that C8A locus was maintained in an inactive chromatin configuration in HNF1α-deficient livers. As the murine C8A locus had not been cloned previously, we isolated the murine C8A transcriptional control region from a lambda murine genomic library with a probe corresponding to the first exon. Two independent and overlapping clones were isolated and a genomic fragment of 7 kb was restriction mapped and sequenced. The sequenced region included the first exon of 155 nucleotides, 6 kb of promoter, and 2 kb of the first intron (GenBank/EMBL/DDBJ AJ409152). The chromatin structure of the locus was monitored by limited digestion of liver nuclei with endonucleases. In this way we could detect a hypersensitive site at the 5′ end of the transcribed region with both micrococcal nuclease (Fig. 4) and DNaseI digestion (data not shown). This site was absent in HNF1α-deficient mice (Fig. 4). Inspection of the murine genomic sequence encompassing the C8A gene using a computer program (11), revealed a highly scoring HNF1α binding site in the region which was hypersensitive to nucleases. Electrophoresis mobility shift analysis (EMSA) confirmed that this site had a high binding affinity for HNF1α, comparable to, if not better than, a well characterized HNF1 binding site located in the liver-type pyruvate kinase (L-PK) promoter (Fig. 5). In addition, the comparison between the murine and human genomic sequences revealed the presence of a region with a relative high degree of homology that included the HNF1α binding site (data not shown).
Figure 4.
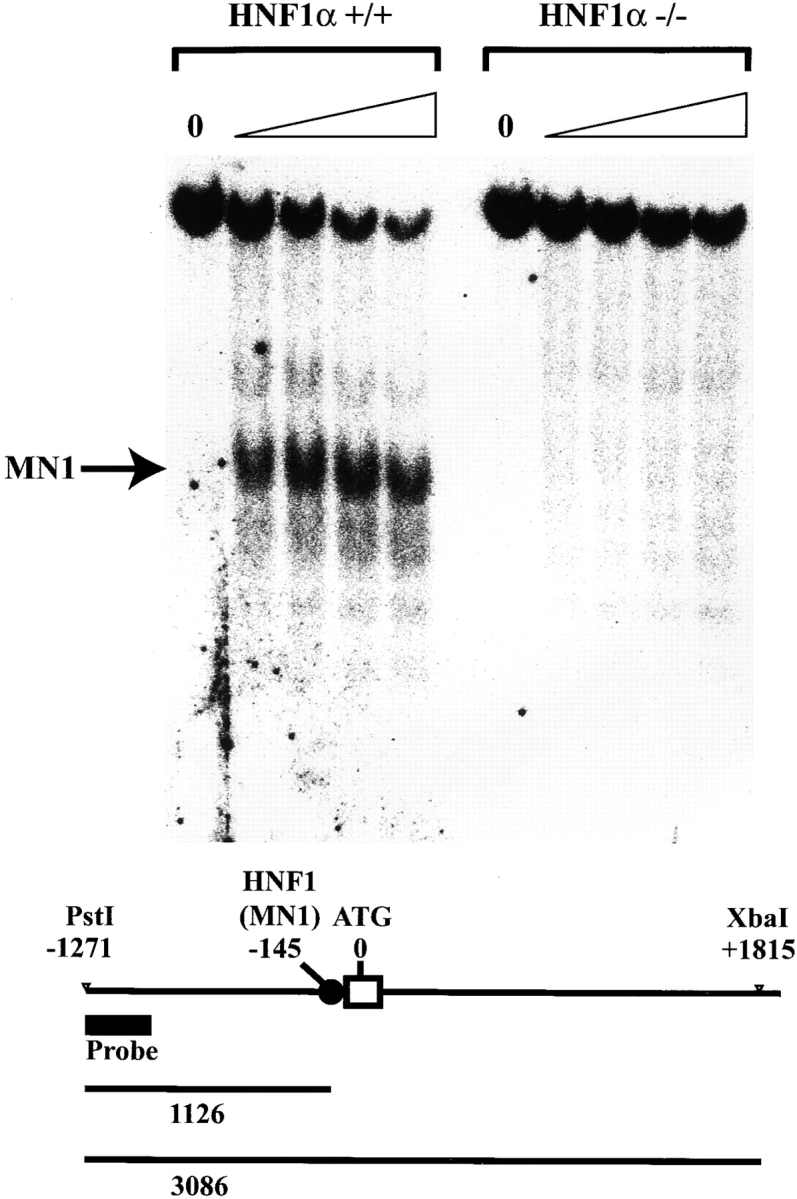
Micrococcal nuclease sensitivity of the C8A locus. Micrococcal nuclease sensitivity over the C8A promoter region. (Top) Nuclei from wild-type or HNF1α2/− mouse liver were treated with micrococcal nuclease for increasing length of time (1 min step) up to 4 min. The DNA was extracted and digested with PstI and XbaI and then subjected to Southern blot analysis. The position of the probe is indicated by the thick bar in the scheme. The length of the PstI-XbaI fragment and the micrococcal nuclease digestion products hybridizing with the probe are indicated below.
Figure 5.

HNF1 binding site to the C8A promoter. 32P-labeled double stranded oligonucleotide containing the putative C8A−HNF1 binding site and a known typical HNF1 binding site located in the human liver-type pyruvate kinase promoter (L-PK), were tested for the ability to bind a COOH-terminal truncated HNF1α (ΔC-HNF1α) that retains its DNA binding specificity or with HNF1α present in liver nuclear extracts. The retarded bands and free probes are indicated with arrows on the right. Excess (100×) cold competitor corresponding to the L-PK probe was added as indicated to verify the specificity of the binding.
Discussion
The data presented in this study show that HNF1α controls the expression of the genes encoding for the complement components C5 and C8α. We demonstrate a dramatic defect in the expression of both genes in the liver and in their functional activity in the sera of homozygous deficient mice. C5 and C8 are late complement components that play a critical role in the activation and in the biological function of the terminal complement complex. C5 initiates the assembly of the MAC upon its activation by the C5 convertases, whereas C8 plays a key role in later steps by binding to the C5b67 complex and recruiting C9 to the nascent complex. C8 and C9 enable the MAC to penetrate more deeply into the cell membrane (26).
C8 is an oligomeric protein composed of three chains (α, β, and γ) encoded by separate genes. C8 α and γ subunits are disulfide-linked and noncovalently associated with the β subunit (27). The use of sera from patients with selective deficiencies of either C8α-γ or C8β in reconstitution experiments led us to conclude that the α-γ subunit of C8 was missing in the sera of the homozygous HNF1α-deficient mice. An interesting finding was the extremely low activity of C8β in the sera of HNF1α2/− mice despite the modest 50% reduction in the expression of C8β mRNA. A similar low level of C8β is observed in C8α-γ–deficient patients ranging between 1 and 3% of that found in normal individuals (28), suggesting that the level of C8β protein both in HNF1α2/− mice and in C8α-γ–deficient individuals depends on the α-γ for secretion or stability. Although the expression of C9 encoding mRNA was reduced to about to 50% in HNF1α-deficient mice, the functional activity of the circulating component was not significantly affected in mutant animals. The reason for this is unclear, but we cannot exclude that an impaired activation of the terminal complement pathway, resulting from lack of C5 and C8, leads to an increased half-life of C9. The mRNA expression level of C7 was not affected by HNF1α deficiency. This results is consistent with the observations made in liver transplanted patients showing that C7 is not predominantly synthesized by hepatocytes but secreted by mononuclear phagocyte lineage, Kupffer, and endothelial cells that do not express HNF1α (29, 30).
We have previously identified a high affinity HNF1 binding site in the promoter region of the human C5 gene (11). Our present data clearly demonstrate that mouse C5 transcription is HNF1α dependent. We also show that the C8A promoter contains an HNF1 binding site and that the lack of HNF1α is associated with a defective chromatin remodeling at the C8A locus. This is somewhat reminiscent of the silencing of phenylalanine hydroxylase gene that results from a defective chromatin remodeling (14). This observation further suggests the possibility that HNF1α could activate the transcription of C5 and C8A via at least two mechanisms. The first is the simple transcriptional activation of promoters by the recruitment of the transcription basal machinery. The second mechanism could rely on remodeling of the chromatin structure allowing additional transcription factors to access target promoters.
Based on the molecular organization of the complement components forming the terminal complex and on phylogenetic studies, it is now widely accepted that the genes for these proteins evolved through rounds of duplications and trimmings from two distinct types of ancestral genes (1, 2, 31, 32). C5, along with C3 and C4, derived from a serum-protease inhibitor that also gave rise to α-2 macroglobulin, whereas the remaining late components C6, C7, C8α, C8β, and C9 derived from a common ancestral gene that also gave rise to perforin-like genes (32, 33). In this respect, it is interesting to note that C8A and C5, the two genes that show the most drastic reduction in expression in HNF1α2/− mice, belong to two evolutionary different gene families. Notably, evolution of the complement system, including a membrane attack complex, and the appearance of HNF1α took place with emergence of the first vertebrates. Hence, it is tempting to speculate that a DNA binding protein along with its recognition sites might have been conserved during evolution to ensure liver expression of C5 and C8A genes. In conclusion, our results show that HNF1α plays a crucial role in the transcriptional activation of two genes, C5 and C8A, which are essential components of the membrane attack complex.
Acknowledgments
We are grateful to Agnès Begue from the Pasteur Institute of Lille for kindly providing a murine lambda genomic library. We also thank Jonathan Weitzman and Mario Tosi for helpful discussions and for reading our manuscript.
This work was supported by grants provided by Human Frontier Science Program (HFSP) (RGP0024/2001-M102) to M. Pontoglio and by the Italian MIUR (60% and cofin) and CNR (Target Project on Biotechnology) to F. Tedesco.
B. Viollet's present address is Institut Cochin de Génétique Moléculaire, Institut National de la Santé et de la Recherche Medicale U129, Port-Royal, 24 rue du Faubourg Saint-Jacques, 75014 Paris, France.
References
- 1.Sunyer, J.O., and J.D. Lambris. 1998. Evolution and diversity of the complement system of poikilothermic vertebrates. Immunol. Rev 166:39–57. [DOI] [PubMed] [Google Scholar]
- 2.Nonaka, M. 1998. The Human Complement System in Health and Disease. J.E. Volanakis and M.M. Frank, editors. Marcel Dekker, New York. 203–215.
- 3.Morgan, B.P. 1989. Complement membrane attack on nucleated cells: resistance, recovery and non-lethal effects. Biochem. J. 264:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicholson-Weller, A., and J.A. Halperin. 1993. Membrane signaling by complement C5b-9, the membrane attack complex. Immunol. Res. 12:244–257. [DOI] [PubMed] [Google Scholar]
- 5.Tedesco, F., M. Pausa, E. Nardon, M. Introna, A. Mantovani, and A. Dobrina. 1997. The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J. Exp. Med. 185:1619–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alper, C.A., D. Raum, Z.L. Awdeh, B.H. Petersen, P.D. Taylor, and T.E. Starzl. 1980. Studies of hepatic synthesis in vivo of plasma proteins, including orosomucoid, transferrin, alpha 1-antitrypsin, C8, and factor B. Clin. Immunol. Immunopathol. 16:84–89. [DOI] [PubMed] [Google Scholar]
- 7.Colten, H.R., and G. Garnier. 1998. Regulation of the complement protein gene expression. In The Human Complement System in Health and Disease. J.E. Volanakis and M.M. Frank, editors. Marcel Dekker, New York. 217–240.
- 8.Frain, M., G. Swart, P. Monaci, A. Nicosia, S. Stampfli, R. Frank, and R. Cortese. 1989. The liver-specific transcription factor LF-B1 contains a highly diverged homeobox DNA binding domain. Cell. 59:145–157. [DOI] [PubMed] [Google Scholar]
- 9.Chouard, T., M. Blumenfeld, I. Bach, J. Vandekerckhove, S. Cereghini, and M. Yaniv. 1990. A distal dimerization domain is essential for DNA-binding by the atypical HNF1 homeodomain. Nucleic Acids Res. 18:5853–5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baumhueter, S., D.B. Mendel, P.B. Conley, C.J. Kuo, C. Turk, M.K. Graves, C.A. Edwards, G. Courtois, and G.R. Crabtree. 1990. HNF-1 shares three sequence motifs with the POU domain proteins and is identical to LF-B1 and APF. Genes Dev. 4:372–379. [DOI] [PubMed] [Google Scholar]
- 11.Tronche, F., F. Ringeisen, M. Blumenfeld, M. Yaniv, and M. Pontoglio. 1997. Analysis of the distribution of binding sites for a tissue-specific transcription factor in the vertebrate genome. J. Mol. Biol. 266:231–245. [DOI] [PubMed] [Google Scholar]
- 12.Shih, D.Q., M. Bussen, E. Sehayek, M. Ananthanarayanan, B.L. Shneider, F.J. Suchy, S. Shefer, J.S. Bollileni, F.J. Gonzalez, J.L. Breslow, and M. Stoffel. 2001. Hepatocyte nuclear factor-1alpha is an essential regulator of bile acid and plasma cholesterol metabolism. Nat. Genet. 27:375–382. [DOI] [PubMed] [Google Scholar]
- 13.Pontoglio, M., J. Barra, M. Hadchouel, A. Doyen, C. Kress, J.P. Bach, C. Babinet, and M. Yaniv. 1996. Hepatocyte nuclear factor 1 inactivation results in hepatic dysfunction, phenylketonuria, and renal Fanconi syndrome. Cell. 84:575–585. [DOI] [PubMed] [Google Scholar]
- 14.Pontoglio, M., D.M. Faust, A. Doyen, M. Yaniv, and M.C. Weiss. 1997. Hepatocyte nuclear factor 1alpha gene inactivation impairs chromatin remodeling and demethylation of the phenylalanine hydroxylase gene. Mol. Cell. Biol. 17:4948–4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pontoglio, M., S. Sreenan, M. Roe, W. Pugh, D. Ostrega, A. Doyen, A.J. Pick, A. Baldwin, G. Velho, P. Froguel, et al. 1998. Defective insulin secretion in hepatocyte nuclear factor 1alpha-deficient mice. J. Clin. Invest. 101:2215–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee, Y.H., B. Sauer, and F.J. Gonzalez. 1998. Laron dwarfism and non-insulin-dependent diabetes mellitus in the Hnf-1alpha knockout mouse. Mol. Cell. Biol. 18:3059–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pontoglio, M., D. Prié, C. Cheret, A. Doyen, C. Leroy, P. Froguel, G. Vehlo, M. Yaniv, and G. Friedlander. 2000. HNF1α controls renal glucose reabsorption in mouse and man. EMBO Rep. 1:359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamagata, K., N. Oda, P.J. Kaisaki, S. Menzel, H. Furuta, M. Vaxillaire, L. Southam, R.D. Cox, G.M. Lathrop, V.V. Boriraj, et al. 1996. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature. 384:455–458. [DOI] [PubMed] [Google Scholar]
- 19.Orren, A., P.C. Potter, R.C. Cooper, and E. du Toit. 1987. Deficiency of the sixth component of complement and susceptibility to Neisseria meningitidis infections: studies in 10 families and five isolated cases. Immunology. 62:249–253. [PMC free article] [PubMed] [Google Scholar]
- 20.Inai, S., Y. Akagaki, T. Moriyama, Y. Fukumori, K. Yoshimura, S. Ohnoki, and H. Yamaguchi. 1989. Inherited deficiencies of the late-acting complement components other than C9 found among healthy blood donors. Int. Arch. Allergy Appl. Immunol. 90:274–279. [DOI] [PubMed] [Google Scholar]
- 21.Tedesco, F., M. Bardare, A.M. Giovanetti, and G. Sirchia. 1980. A familial dysfunction of the eight component of complement (C8). Clin. Immunol. Immunopathol. 16:180–191. [DOI] [PubMed] [Google Scholar]
- 22.Harrison, R.A., and P.J. Lachmann. 1986. Handbook of Experimental Immunology. D.M. Weir, editor. Blackwell, Oxford. 1–49.
- 23.Chomczynski, P., and N. Sacchi. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. l Biochem. 162:156–159. [DOI] [PubMed] [Google Scholar]
- 24.Hubank, M., and D.G. Schatz. 1994. Identifying differences in mRNA expression by representational difference analysis of cDNA. Nucleic Acids Res. 22:5640–5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Viollet, B., M. Yaniv, and M. Pontoglio. 2001. Embryonic but not postnatal re-expression of HNF1α can reactivate the silent PAH gene in HNF1α-deficient hepatocytes. Mol. Cell. Biol. 21:3662–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steckel, E.W., B.E. Welbaum, and J.M. Sodetz. 1983. Evidence of direct insertion of terminal complement proteins into cell membrane bilayers during cytolysis. Labeling by a photosensitive membrane probe reveals a major role for the eighth and ninth components. J. Biol. Chem. 258:4318–4324. [PubMed] [Google Scholar]
- 27.Steckel, E.W., R.G. York, J.B. Monahan, and J.M. Sodetz. 1980. The eighth component of human complement. Purification and physicochemical characterization of its unusual subunit structure. J. Biol. Chem. 255:11997–12005. [PubMed] [Google Scholar]
- 28.Tedesco, F., L. Roncelli, B.H. Petersen, V. Agnello, and J.M. Sodetz. 1990. Two distinct abnormalities in patients with C8 alpha-gamma deficiency. Low level of C8 beta chain and presence of dysfunctional C8 alpha-gamma subunit. J. Clin. Invest. 86:884–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wurzner, R., V.C. Joysey, and P.J. Lachmann. 1994. Complement component C7. Assessment of in vivo synthesis after liver transplantation reveals that hepatocytes do not synthesize the majority of human C7. J. Immunol. 152:4624–4629. [PubMed] [Google Scholar]
- 30.Langeggen, H., M. Pausa, E. Johnson, C. Casarsa, and F. Tedesco. 2000. The endothelium is an extrahepatic site of synthesis of the seventh component of the complement system. Clin. Exp. Immunol. 121:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ng, S.C., A.G. Rao, O.M. Howard, and J.M. Sodetz. 1987. The eighth component of human complement: evidence that it is an oligomeric serum protein assembled from products of three different genes. Biochemistry. 26:5229–5233. [DOI] [PubMed] [Google Scholar]
- 32.Mondragon-Palomino, M., D. Pinero, A. Nicholson-Weller, and J.P. Laclette. 1999. Phylogenetic analysis of the homologous proteins of the terminal complement complex supports the emergence of C6 and C7 followed by C8 and C9. J. Mol. Evol. 49:282–289. [DOI] [PubMed] [Google Scholar]
- 33.Hobart, M.J., B. Fernie, and R.G. DiScipio. 1993. Structure of the human C6 gene. Biochemistry. 32:6198–6205. [DOI] [PubMed] [Google Scholar]