

Abstract
CD4+ T cells play an important role in orchestrating host immune responses against cancer, particularly by providing critical help for priming and extending the survival of CD8+ T cells. However, relatively little is known about major histocompatibility complex class II–restricted human tumor antigens capable of activating CD4+ T cells. Here, we describe the identification of a mutated fibronectin (FN) as a tumor antigen recognized by human histocompatibility leukocyte antigen-DR2–restricted CD4+ T cells. Deoxyribonucleic acid (DNA) sequencing analysis indicated that this gene contains a mutation that results in the substitution of lysine for glutamic acid and gives rise to a new T cell epitope recognized by CD4+ T cells. Tumor cells harboring the mutant FN resulted in the loss of FN matrix formation and the gain of metastatic potential based on the migration pattern compared with that of tumor cells that express wild-type FN. Additional experiments using cell lines stably expressing the mutated FN cDNA demonstrated that the point mutation in FN was responsible for the loss of FN staining in extracellular matrices and the enhancement of tumor cell migration. These findings represent the first demonstration that a mutated gene product recognized by CD4+ T cells is directly involved in tumor metastasis, which indicates the importance of CD4+ T cells in controlling the spread of tumor cells to distant anatomic sites.
Keywords: cancer vaccines, cancer biology, CD4+ T cells, antitumor immunity, immunotherapy
Introduction
Cancer cells are derived from a cell with accumulated genetic mutations or alterations that make them more immunogenic than normal cells. Although a number of tumor antigens recognized by CD8+ T cells have been identified in melanomas as well as other types of cancers, the majority of these class I–restricted antigens are nonmutated self-proteins (1–3). Few mutated antigens, including CDK4, β-catenin, and caspase 8, have been identified and implicated in the involvement of cell cycle regulation, tumorigenesis, or apoptosis (4–6). To facilitate the identification of MHC class II–restricted antigens, we recently developed a novel genetic approach to cloning the genes encoding MHC class II–restricted tumor antigens (7). Three class II–restricted tumor antigens were successfully identified by this method: fusion protein LDFP (LDLR and FUT fusion protein) resulting from chromosomal rearrangement, and the mutated antigens CDC27 and triosephosphate isomerase, of which the latter is being independently identified by a biochemical approach (7–9). Of particular interest, the mutated human CDC27 protein, an important component of an anaphase-promoting complex involved in cell cycle regulation, could give rise to a melanoma target antigen, although the point mutation itself does not constitute a T cell epitope. Instead, the missense mutation in a putative phosphorylation site allows a nonmutated peptide within CDC27 to be presented to T cells by MHC class II molecules (7). Indeed, the majority of MHC class II–restricted tumor antigens currently identified with the use of tumor-reactive T cells are mutated or fusion proteins and therefore may represent immunogenic targets recognized by CD4+ T cells.
The biological functions of the mutated antigens identified by tumor-reactive CD4+ and CD8+ T cells suggest that these mutated gene products not only contribute to tumor development, but also to tumor metastasis and progression. Although melanoma is a highly invasive skin cancer, few genetic mutations have been linked to its biological behavior. It is widely accepted that after the onset of oncogenesis, tumor cells with metastatic potential migrate away from the primary tumor, and invade and implant in distant sites where they reestablish tumor growth (10). Gene products involved in the formation of extracellular matrix (ECM),*including fibronectin (FN), have been implicated in tumorigenesis and metastasis (11–13), but so far no direct evidence for this association has been reported (10).
Here we describe the identification and characterization of mutant FN as a class II–restricted tumor antigen recognized by tumor-reactive CD4+ T cells derived from a melanoma patient. A point mutation resulted in the substitution of a Lys (positively charged) for Glu (negatively charged) residue in FN, giving rise to an epitope for T cell recognition. Importantly, we found that the mutation in FN was directly responsible for the loss of FN matrix formation, leading to the enhanced migration of tumor cells. Thus, the identification of such MHC class II–restricted tumor antigens not only provides potentially important immune targets for effective cancer immunotherapy, but also improves understanding of the mechanisms by which these antigens participate in tumor development and metastasis.
Materials and Methods
Chemicals and Reagents.
The following chemicals and reagents were used: RPMI 1640, AIM-V medium, Lipofectamine, and G418 (GIBCO BRL); the eukaryotic expression vector pcDNA3.1 (Invitrogen); anti–HLA-DR2 mAb (One Lambda); and anti-immunoglobulin mAb conjugated with fluorescent isothiocyanate (Vector Laboratories).
Cell Lines and Cultures.
CD4+ F27 tumor-infiltrating lymphocytes (TIL) were cultured from a subcutaneous metastasis obtained from a melanoma patient by fine needle aspiration. T cell clones or lines were grown in AIM-V medium containing 10% human AB serum and recombinant IL-2 (6,000 IU/ml; Chiron Corp.). Melanoma cell lines and EBV-transformed B cell lines were maintained in RPMI 1640 with 10% FCS. 293IMDR2 cells were grown in DMEM containing 10% FCS. The T cell clones were generated by limiting dilution methods (one cell/well) from the CD4+ F27TIL cell line, and feeder allogeneic PBMCs in RPMI 1640 containing 10% human AB serum and 300 IU IL-2. To obtain optimal expansion, we used the OKT3 expansion method as previously described (14, 15). Melanoma cell lines and EBV-transformed B cell lines were cultured in RPMI 1640 medium containing 10% FCS. 293IMDR2 cells were established by transfecting plasmid DNA encoding DRB1*1501 cDNA into 293IMA cells expressing the Ii, DMA, DMB, and DRA genes, and selected with RPMI 1640/10% FCS containing blasticidin S (25 μg/ml). HLA-DR2–positive cells were sorted by FACS® using DR2-specific Abs.
cDNA Library Construction.
Total RNA was extracted from F27mel cells using Trizol reagent (GIBCO BRL). PolyA RNA was purified from total RNA by the polyATract system (Promega) and converted to cDNA using a cDNA construction kit (GIBCO BRL) with an oligo-dT primer. The cDNA inserts were then ligated to a pTSX vector containing an Ii fragment (amino acid 1–80) (7), and cDNA libraries were electroporated into DH10B cells (GIBCO BRL). Plasmid DNA for cDNA library pools was prepared from bacteria, each consisting of ∼100 cDNA clones.
cDNA Library Screening and GM-CSF Secretion Assay.
DNA transfection and GM-CSF assays were performed as previously described (7, 8). In brief, 200 ng cDNA pools were mixed with 2 μl lipofectamine in 100 μl serum-free DMEM for 15–45 min. The DNA–lipofectamine mixture was then added to the 293IMDR2 cells (5 × 104) and incubated overnight. The next day, cells were washed twice with AIM-V medium. CD4+ T cells were then added at a concentration of 5 × 104 cells/well in AIM-V medium containing 120 IU/ml IL-2. After 18–24 h of incubation, 100 μl supernatant was collected and GM-CSF concentrations were measured in a standard ELISA assay (R&D Systems). To test peptide recognition, 888EBV were incubated with peptides at 37°C for 90 min and then washed three times with AIM-V medium containing 120 IU/ml IL-2. T cells were added and incubated for an additional 18–24 h.
Northern Blot Analysis.
Total RNA from human normal tissue was purchased from CLONTECH Laboratories, Inc. 20 μg total RNA was subjected to electrophoresis in a 1.2% formaldehyde agarose gel and transferred to a nylon membrane. DNA fragments of the FN gene were labeled with [α-32P]dCTP by the random priming method. Prehybridization and hybridization were performed according to the QuickHyb protocol (Stratagene). Membranes were washed twice with 2× SSC/0.1% SDS at room temperature for 15 min and twice with 0.1× SSC/0.1% SDS at 60°C for 30 min. Autoradiography was performed at −70°C. An actin probe was used to serve as the internal control for the amount of total RNA loading onto the gel.
Peptide Synthesis and T Cell Epitopes.
The peptides were synthesized by a solid phase method using a peptide synthesizer (model AMS 422; Gilson Co.). Some peptides were purified by HPLC and had >98% purity. The mass of some peptides was confirmed by mass spectrometry analysis. Peptides reactive with CD4+ F27TIL-T1 cells were identified and characterized as previously described (7, 8).
Cloning of Full-length FN cDNA.
A SuperScript II reverse transcription kit (Invitrogen) was used. 20 μl reverse transcription mixture contained 2 μg total RNA and was incubated at 42°C for 1 h. After reverse transcription, 2.5 μl reverse transcription mixture was used in PCR. 50 μl PCR reaction mixture contained 5 μl 10× ThermalAce buffer, 1 μl 50× dNTP mixture, 200 μM primer FN5P2 (5′ CTCAACATGGTTAGGGGTCCGGGGCCCGGGCTG), 200 μM primer FN3P1 (5′ AGAGACATGCTTGTTCCTCTGGA), and 2 μl ThermalAce DNA polymerase (Invitrogen). Primer FN5P2 is a sense primer that corresponds to the 5′-end sequence of the FN gene. Primer FN3P1 is an antisense primer that corresponds to the 3′-end sequence of the FN gene. The PCR amplification program included an initial step of 2 min at 95°C, 30 cycles of 30 s at 95°C, 30 s at 65°C, 10 min at 72°C, and a final step of 15 min at 72°C. 50 μl PCR product was then supplemented with 1 μl Taq DNA polymerase (Invitrogen) and incubated at 72°C for 30 min. 50 μl PCR product was separated on a 0.8% (wt/vol) agarose gel. The desired bands of PCR product was gel purified and then cloned into pCR-XL-TOPO vector (Invitrogen). Individual plasmid DNAs were prepared and sequenced to confirm the correctness of full-length wild-type and point-mutated FN cDNAs.
Transfection of FN Genes into Tumor Cells.
Both wild-type and mutated FN gene fragments were released with Hind III and NotI from the pCR-XL-TOPO vector and subcloned into a pcDNA3.1/zeocin vector (Invitrogen). Plasmid DNAs encoding either wild-type or mutated FN cDNA, as well as a pcDNA3.1/zeocin empty vector, were transfected into 1143mel tumor cells in six-well plates with lipofectAMINE (GIBCO BRL). Transfected tumor cells were selected in RPMI 1640/10% FCS containing 250 μg/ml zeocin (Invitrogen).
Immunostaining of ECM with Anti-FN Ab.
1143mel, 1195mel, F27mel, and FN-transfected tumor cells were cultured overnight in six-well plates containing a coverslip. Cells attached to the coverslips were fixed in PBS/4% formaldehyde at room temperature for 20 min and washed three times with PBS. The coverslips were kept in acetone at −20°C for 3 min, air-dried, and washed three times with PBS. They were then blocked in PBS/5% FCS in a moist chamber at 37°C for 20 min, washed with PBS for 5 min, and incubated with 1:500 anti-FN mAb (Ab-9; NeoMarkers) in a moist chamber at 37°C for 1 h. Similar experiments with other anti-FN Abs (Ab-1, Ab-7, Ab-8, and Ab-9; NeoMarkers) were also performed. The slides were washed three times with PBS, blocked with PBS/5% FCS at room temperature for 20 min, washed two times with PBS, incubated with 1:20 Texas red–conjugated anti–mouse IgG in the dark in a moist chamber at 37°C for 30 min, and then washed five times with PBS. They were kept in PBS overnight at 4°C, mounted in PBS/50% glycerol, viewed with a fluorescence microscope, and photographed at ×40.
Migration Assays.
2.5 × 104 1143mel and transfected tumor cells were seeded into the upper chamber with 0.75 ml RPMI 1640 serum-free medium in Biocoat Matrigel cells (Becton Dickinson). The bottom chamber contained 0.75 ml RPMI 1640 serum-free medium (control medium) or 0.75 ml RPMI 1640/10% FCS (complete medium). After incubation at 37°C with 5% CO2 for 48 h, cells in the lower chambers were fixed in 4% formaldehyde in PBS at room temperature for 20 min, stained with crystal violet, and examined and counted from four different fields under a compound microscope.
Immunoprecipitation and Western Blotting.
1143mel, 1195mel, and F27 were lysed in lysis buffer on ice. After spinning, the cell lysates were immunoprecipitated with an anti-β1 integrin Ab and pulled down with protein A beads. The immunoprecipitated complexes were separated on SDS-PAGE. After membrane transfer, proteins were detected with anti-FN or anti-β1 integrin Abs. For the detection of FN in whole cell lysates, 1143mel, 1195mel, and F27mel tumor cells from T75 flasks with 80–90% confluence were trypsinized. After spinning, the cell pellets were collected and redissolved in SDS loading buffer (50 mM Tris-HCl, pH 6.8, 100 mM DTT, 2% SDS, and 10% glycerol). The protein concentration was determined with Bio-Rad protein assay dye reagent (Bio-Rad Laboratories). 15 μg total protein for each sample was loaded onto an 8% SDS polyacrylamide gel, separated at 100 V constant for 1 h, and transblotted onto a nitrocellulose membrane (BA83; Schleicher & Schuell) at a constant 100 V for 1 h. The membrane was blocked with 3% milk for 1 h, incubated with 1:200 anti-FN mAb for 1 h, and washed three times with TBST (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 0.05% Tween-20) for 5 min. The membrane was incubated with 1:3,000 goat anti–mouse horseradish peroxidase–conjugated IgG for 1 h, washed three times with TBST for 5 min, rinsed with water, detected with LumiGLO chemiluminescent substrate (Kirkegaard & Perry Laboratories), exposed to X-OMAT AR film (Eastman Kodak Co.), developed automatically, and photographed.
Results
Recognition of Autologous Tumor Cells by HLA-DR2–restricted CD4+ T Cells.
In a search for tumor-reactive CD4+ T cells, we recently established four CD4+ T cell clones from F27TIL cells. They recognized the autologous tumor cell line F27mel, but did not respond to autologous PBMC, MHC class II–matched or –mismatched tumor cell lines, or 293 cells expressing DR2 molecules. Representative data from one such clone (F27TIL-T1) are shown in Fig. 1 A. To determine the restriction elements used by T cells, we tested T cell recognition in the presence or absence of specific Abs. As shown in Fig. 1 B, T cell reactivity against F27mel cells was specifically blocked by mAb against HLA-DR, but not by mAb against HLA-DQ, HLA-DP, or MHC class I molecules. HLA typing analysis of F27mel cells indicated that only HLA-DR2 was expressed in tumor cells (unpublished data), which suggests that CD4+ F27TIL-T1 and other clones recognized a mutated or unique tumor antigen presented by HLA-DR2. Therefore, HLA-DR2 was selected as the restriction element for the initial cDNA library screening.
Figure 1.

Specific recognition of autologous melanoma cells by CD4+ F27TIL-T1. (A) Specific antitumor recognition of CD4+ F27TIL-T1. F27TIL-T1 recognized the autologous F27mel cells, but not EBV-B cell lines and allogeneic melanoma cell lines or 293-derived cell lines. 888mel, 1011mel, 1195mel, 1280mel, 1297mel, and 1390mel shared the DR2 molecule with F27mel. T cell recognition was evaluated by GM-CSF release from CD4+ F27TIL-T1. (B) HLA restriction of T cell recognition. CD4+ F27TIL-T1 cells were cocultured with autologous F27mel cells in the presence or absence of various anti-MHC Abs. GM-CSF release was determined after an 18-h incubation. T cell recognition of F27mel was specifically blocked by an anti-DR Ab, but not by anti-MHC class I, anti-DQ, or anti-DP Abs.
Identification of Mutated FN as a Tumor Antigen Recognized by CD4+ T Cells.
To isolate the gene encoding a tumor antigen recognized by CD4+ F27TIL-T1, we used a genetic targeting expression system that has been successfully used to identify several MHC class II–restricted tumor antigens (7). This system uses a 293-based cell line as professional APC and an Ii-fused cDNA library. To generate 293IMDR2 cells expressing Ii, DMA, DMB, DRA, and DRB1*1501, we first cloned HLA-DRB1*1501 cDNA from the autologous F27mel cells into a pEF6/V5-His/TOPO expression vector containing a blasticidin S resistance gene. The cDNA sequence was confirmed to be that of DRB1*1501. 293 cells stably expressing the other four genes (Ii, DMA, DMB, and DRA, but not DRB1*1501), were then transfected with the DR2-expressing plasmid DNA. After selection with blasticidin S for 2 wk, the 293IMDR2 cell line was screened by FACS® analysis using a DR2-specific Ab, and further expanded for use as APC for library screening.
We next constructed a cDNA library by fusing a targeting sequence of invariant chain to the 5′ end of cDNAs derived from F27mel tumor cells. As previously demonstrated, this strategy will target Ii fusion proteins translated from the Ii fusion library to the endosomal/lysosomal compartment for efficient antigen processing and presentation to T cells (7). cDNA subpools with ∼100 cDNA clones per pool were prepared. The F27mel cDNA library was then introduced into 293IMDR2-expressing DMA, DMB, Ii, DRA, and DRB1*1501 molecules. After screening a total of 2 × 105 cDNA clones, we identified five positive cDNA pools that conferred T cell recognition by CD4+ F27TIL-T1 cells when transfected into 293IMDR2 cells. The individual positive clones were then isolated from the positive cDNA pools and tested for recognition by CD4+ F27TIL-T1 cells (Fig. 2 A). Evaluation of three other CD4+ T cell clones showed that they were capable of recognizing the same positive cDNA clone isolated from the F27 cDNA library with F27TIL-T1 cells.
Figure 2.

Screening of an Ii-cDNA library from RNA of F27mel using CD4+ F27TIL-T1 cells. (A) Identification of positive cDNA clones encoding a tumor antigen recognized by CD4+ T cells. After the screening of 2 × 105 Ii-cDNA fusion library clones generated from F27mel RNA, positive cDNA pools were identified on the basis of GM-CSF release from CD4+ F27TIL-T1. CD4+ F27TIL-T1 recognized 293IMDR2 transfected with the cDNA clone 3, but not with a control cDNA or green fluorescent protein. (B) Schematic presentation of FN protein and the position of the mutated amino acid.
DNA sequencing analysis revealed that cDNA clone 3 contained a DNA fragment encoding the FN protein involved in ECM formation, tumor transformation, and metastasis (Fig. 2 B). All DNA sequences from five different cDNA pools were identical to the published sequence with the exception of one nucleotide A substitution for G at position 6435, based on the numbering of the published FN sequence. These sequence data are available from GenBank/EMBL/DDBJ under accession no. NM-002026. (16). This point mutation (G to A) at amino acid position 2053 was found in all positive cDNA clones, resulting in a nonconservative substitution of Lys (positively charged residue) for Glu (negatively charged residue) (Fig. 2 B). This mutation was located in the region of type III repeats (IIICS) of cellular FN that is involved in the formation of ECM (17). In addition, DNA sequences from the amplified DNA fragments from autologous T cells or PBMC were identical to the published wild-type sequence, suggesting that this is a somatic mutation occurring in cancer cells.
Mutated Peptides Recognized by CD4+ T Cells.
To confirm that T cell recognition was restricted by HLA-DR2, we transfected cDNA clone 3 into both 293IMDR2 and 293IMDR4 cells and then tested for recognition by CD4+ F27TIL-T1 cells. CD4+ T cells recognized 293DR2 cells transfected with the mutant FN cDNA, but not 293DR4 cells transfected with the wild-type FN cDNA nor 293DR4 cells transfected with the mutant FN cDNA (unpublished data), which suggests that T cell epitopes are located in the region with a point mutation. To identify the antigenic epitopes recognized by CD4+ T cells, we made a wild-type peptide and three overlapping 20-mer peptides containing the mutated residue and tested their ability to activate T cells. Two out of three mutant peptides were recognized by CD4+ F27TIL-T1 (Fig. 3 A). These two mutated peptides shared 13 amino acids (MIFEKHGFRRTTPP), suggesting that this peptide sequence contained a T cell epitope for T cell recognition. By contrast, T cells failed to recognize the wild-type peptide, which lacked the mutant residue but was otherwise identical to the mutated peptides. Peptide titration experiments showed that T cell recognition could readily be detected at a peptide concentration of ∼500 nM (Fig. 3 B).
Figure 3.

Identification and characterization of peptides capable of stimulating CD4+ T cells. (A) Identification of peptides recognized by F27TIL-T1 cells. Two out of three overlapping peptides that contain the mutated amino acid residue were recognized by T cells, whereas the corresponding peptide with a wild-type residue failed to stimulate CD4+ T cells. (B) Determination of peptide concentrations required for T cell recognition. 293IMDR2 cells were incubated with different concentrations of FN-P2 peptide for 90 min and then washed three times with T cell assay medium. T cells were added to peptide-pulsed 293IMDR2 cells overnight. GM-CSF release from T cells was determined with a GM-CSF ELISA kit.
Mutant FN Disrupts FN Matrix Formation and Enhances the Metastatic Potential of Tumor Cells.
Because tumor cells are a heterogeneous population, we conjectured that some cells within the primary tumor cells might not contain the FN mutation, whereas this G to A substitution should be found in all metastatic lesions provided that the mutation affects the metastatic potential of tumor cells. To test this prediction, we generated 53 tumor cell clones by the limiting dilution method from fresh tumor cells derived from different metastatic lesions. After PCR amplification and sequencing of genomic DNA, we found the presence of the mutant FN gene in all tumor cell clones, which strongly suggests that the mutation in FN plays a role in subsequent tumor metastasis. By contrast, two control tumor cell lines, 1143mel and 1195mel, contained the wild-type FN allele exclusively. Representative data are shown in Fig. 4 B.
Figure 4.


Loss of FN matrix formation in F27mel cells harboring a mutated FN. (A) Immunostaining of FN matrix of three melanoma cell lines (1143mel, 1195mel, and F27mel) with Ab-9 anti-FN Ab. FN was detected by indirect immunofluorescence. Phase contrast and fluorescence images were taken at ×40. (B) Genomic DNA sequence analysis of FN in three melanoma cell lines. Genomic DNA fragments were amplified by FN-specific primers. The PCR products were sequenced to identify mutated FN. (C) Comparison of immunostaining for FN matrix in 1143mel and F27mel cells with different anti-FN (Ab-1, Ab-7, AB-8, and Ab-9) Abs that recognize epitopes in the different regions of FN. DAPI staining was used as controls for cell density. Staining of FN matrix in 1143mel and F27mel cells with all four Abs showed these similar patterns: intensive staining of FN in 1143mel cells, but little or weak staining in F27mel cells. 1143 and F27 stand for 1143mel and F27mel, respectively.
Because FN plays a significant role in FN matrix formation (13, 18), we examined FN matrix formation in F27mel as well as in 1143mel and 1195mel by staining with an anti-FN Ab-9 Ab. As shown in Fig. 4 A, intense FN staining was evident in 1143mel and 1195mel, but was significantly reduced in F27mel cells. In contrast to 1143mel and 1195mel cell lines, which only contained wild-type FN, F27mel cells contained both wild-type and mutant FN (Fig. 4 B). Thus, the point mutation in FN results in the loss of ability to form FN matrices. We also stained for extracellular FN in tumor cell lines with three anti-FN Abs (Ab-1, Ab-7, and Ab-8) that recognize epitopes located in different regions of the FN molecule. The resulting staining patterns were either identical or similar to those obtained with Ab-9 (Fig. 4 C), indicating that the point mutation in FN did not alter its recognition by anti-FN Abs.
The loss of FN matrix formation has been implicated in tumor transformation (12, 13). Tumor cells with metastatic potential must migrate away from the primary tumor and invade and implant in a distant site for the reestablishment of a new tumor. Thus, the modulation of FN matrix formation is a critical step in tumor metastasis (10). We postulated that the loss or reduction of FN matrix formation in F27mel cells may enhance tumor metastasis. To test this possibility, we performed migration assays to measure the ability of F27mel, 1143mel, and 1195mel tumor cells to migrate through matrigel from one chamber containing control medium to another chamber containing complete growth medium. Interestingly, we found that relatively few 1143mel and 1195mel tumor cells with the wild-type FN gene and normal FN matrix formation were capable of migrating to the well containing the complete medium. In contrast, the F27mel tumor cells readily migrated to the well with complete medium in >20-fold higher numbers than 1143mel and 1195mel (Fig. 5) . These results demonstrate that the FN mutation contributes to the subsequent loss of FN matrix formation, leading to the enhanced metastatic potential of tumor cells.
Figure 5.

The migratory capacity of F27mel compared with 1143mel and 1195mel cells. The poorly metastatic tumor cell lines 1143mel and 1195mel have little or no capacity to migrate from the seeded chamber to the chamber with complete medium, whereas F27mel readily migrated to the chamber with complete medium. Cells were stained with crystal violet, examined under a compound microscope, and photographed.
Dominant-negative Effect on FN Matrix Formation.
Because the interaction of FN with its receptor, an integrin heterodimer (α5β1), is important to ECM formation (12), we next tested whether mutant FN may alter protein interaction between FN and integrin. We first immunoprecipitated FN–integrin complexes with anti-β1 integrin Ab. As shown in Fig. 6 A, similar amounts of integrin were pulled down by anti-β1 integrin in the lysates of F27mel and 1143mel cells, but the amount of FN that formed complexes with integrins in F27mel was much less than that in 1143mel tumor cells (Fig. 6 A). Because the Ab we used could not distinguish wild-type from mutant FN proteins, we also checked the total amount of FN in F27mel and 1143mel lysates. We found that the total amount of both wild-type and mutant FN in F27mel was much lower than that in 1143mel (Fig. 6 B). Thus, the overall difference in the total amount of FN in the cell lysates of F27mel and 1143mel may account for the difference in the amount of FN in the FN–integrin complexes immunoprecipitated from F27mel and 1143mel.
Figure 6.

Western and Northern blot analyses of FN in different tumor cell lines. (A) Tumor cell lysates of 1143mel and F27mel were first immunoprecipitated with an anti-β1 integrin. The immunoprecipitated proteins were separated on an SDS-PAGE. After the transfer to membrane, proteins were detected with anti-FN or anti-β integrin Abs. (B) Whole tumor cell lysates of 1143mel and F27mel cells were separated by SDS-PAGE and analyzed with the anti-FN Ab to determine the total FN protein in the tumor cell lysates. (C) Northern blot analysis of total RNA isolated from 1143mel and F27mel cells. Hybridization of blots with the probe of FN detected an 8-kb band in 1143mel and F27mel tumor cells. An actin probe was used to verify that equal amounts of total RNA were loaded for each well. FN-specific RNA in F27mel cells was at least three- to fourfold higher than that in 1143mel cells.
To exclude the possibility that the low level of FN in F27mel cells is due to low expression at the mRNA level, we performed Northern blot analysis. Interestingly, the mRNA level of FN in F27mel was much higher than in 1143mel cells, whereas actin probe hybridization showed an equal amount of RNA loading (Fig. 6 C), which suggests that the mutation in FN, rather than RNA expression level, reduces the amount of FN and its complexes in F27mel cells. In other words, mutant FN appears to have a dominant-negative effect on FN matrix formation.
To determine whether the point mutation in FN is responsible for the loss of FN Ab staining in FN matrices and the enhanced migration of tumor cells, we cloned wild-type and mutant FN cDNAs from F27mel in a pcDNA3.1Z expression vector (Fig. 7 A). The sequences of both wild-type and mutant cDNA clones were confirmed by DNA sequencing analysis. These clones were full-length cDNAs containing ED-A, ED-B, and IIICS segments (Fig. 2 B), and the mutation (G to A) at amino acid position 2053 was found in the mutated cDNA, but otherwise was identical to the wild-type cDNA. With these constructs, we were able to test our hypothesis that the point mutation in FN was responsible for the observed phenotypic changes. We transfected 1143mel with pcDNA3-FN (wt) or pcDNA3-FN (mt) and selected them in culture medium containing zeocin (250 μg/ml). 12 clones expressing the wild-type FN construct were established for 1143mel, 22 clones expressing mutant FN for 1143mel, and 9 clones for empty vector (control cell line). The presence and expression of the mutant FN gene were confirmed by reverse transcription–PCR and sequencing analysis (unpublished data). FN staining was performed to assess the effect of the transfected FN gene on matrix formation and cell migration. Representative data from three independent studies with different 1143mel cell clones stably expressing wild-type FN, mutant FN, or empty vector are shown in Fig. 7 B. The similar staining patterns of FN matrices were found for all three independent clones examined for each construct. The parental cell line 1143mel and 1143mel/pcDNA (empty vector) exhibited a comparable staining intensity of FN matrices, whereas 1143mel/pcDNA3-FN (wt) cells resulted in more intense staining. By contrast, 1143mel cells stably expressing pcDNA3-FN (mt) showed little or no staining of FN matrices compared with other cell lines/clones containing wild-type FN, demonstrating that expression of mutant FN in 1143mel cells abolishes immunostaining for endogenous wild-type FN/ectopic mutant FN in the matrix assembly of tumor cells.
Figure 7.

Determination of the FN mutation responsible for the dominant-negative phenotype of FN matrix formation. (A) PCR products (7,114 bp) of full-length FN were obtained after amplification using a pair of primers and the first strand cDNA generated from F27mel tumor RNA. Cloning of both wild-type and mutant FN in a pcDNA3 expression vector. Wild-type and mutant FN were verified by DNA sequencing. (B) Immunostaining of FN matrices in 1143mel and derivative cell lines expressing wild-type FN, mutant FN, or empty vector. DAPI staining was used as a control for cell density.
We next evaluated the migratory ability of these cell lines expressing wild-type or mutant FN. As shown in Fig. 8 A, 1143mel, 1143mel/pcDNA3-FN (wt), and 1143mel/pcDNA3 cells displayed little or no migration in a matrigel assay. However, 1143mel/pcDNA3-FN (mt) cells greatly gained the ability to migrate from one chamber containing control medium to another chamber containing growth medium, indicating that the mutant FN enhances the metastatic potential of tumor cells. To rule out clonal variation, we repeated similar experiments with four sets of independent tumor cell clones expressing wild-type FN, mutant FN, or empty vector. In Fig. 8 B, a summary of results obtained from these experiments shows that all tumor cell clones expressing mutant FN had an increased ability to migrate compared with cell clones expressing wild-type FN or an empty vector. The number of migrating cells in 1143mel/pcDNA-FN (mt) clones was at least fourfold higher than that of other groups that express either wild-type FN or empty vector. The P value for significant difference is 0.0007 among the two comparison groups of cell clones expressing mutant FN and other cell clones expressing either wild-type FN or empty vector. There were no statistically significant differences among groups of parental 1143mel and its derivative clones expressing wild-type or empty vector. These results indicate that expression of mutant FN greatly enhances the migratory capacity of tumor cells.
Figure 8.
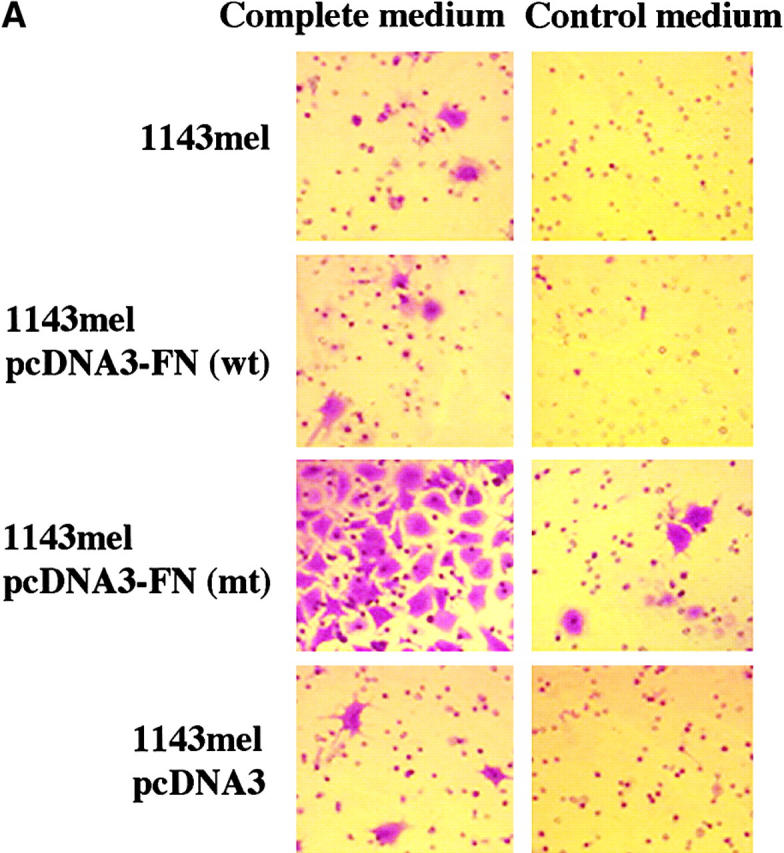

Mutant FN is responsible for the enhanced metastatic potential of tumor cells. (A) Stable expression of mutant FN in 1143mel cells resulted in enhanced migration ability in matrigel assays. Untreated 1143mel and 1143mel cells expressing wild-type FN or empty vector showed little or no migration from one chamber to another containing complete medium. (B) Migration assays were repeated in three independent experiments, each comprising four sets of independent 1143mel-derived tumor cell clones expressing wild-type FN, mutant FN, or empty vector. The numbers of cells that migrated to the bottomed chamber containing complete medium were counted in four representative fields (20×) per chamber. The data are presented as means of the number of cells migrated and standard deviations. A P value of 0.0007 was obtained from Student's t test for the group expressing mutant FN and any other groups.
Discussion
Because of the importance of CD4+ T cells in antitumor immunity, much effort has been directed toward identifying MHC class II–restricted tumor antigens. So far, only DR1- or DR4-restricted tumor antigens have been identified with tumor-reactive CD4+ T cells established from TIL (3, 19). Due to the polymorphic properties of MHC class II molecules, DR1 or DR4 expression accounts for 15–20% of the population (20). Identification of tumor antigens presented by other relatively dominant alleles such as DR2, DR3, or R7 (each accounting for 20–25% of the population) remains critically important for the development of effective vaccines by recruiting both CD4+ and CD8+ T cells. Indeed, T cell recognition of potentially shared tumor antigens presented by DR2 (DRβ1*1501) molecules has been reported (21), but the identity of such antigens remains unknown.
In this study, we identified a novel mutated form of FN as a tumor antigen recognized by CD4+ F27TIL using a genetic targeting expression approach. The findings presented in this study strongly suggest that tumor-reactive CD4+ T cells may play a significant role in eliminating metastatic cancer cells. FN is a gene product critical for ECM formation and indispensable for vertebrate embryogenesis (22). FN forms a complex with its receptor integrins (α5β1) for the initiation of ECM formation. The loss of capacity to form an FN containing ECM has been suggested to be a feature of the transformed phenotype of cancer cells, and restoration of ECM formation correlates with the reduced malignancy of cancer cells (11–13). Recently, several groups, using microarray technology, demonstrated a link between FN expression at the RNA level and tumor metastasis (10, 23, 24). Interestingly, our Northern blot data are consistent with these published results, which show that FN mRNA is approximately fourfold higher in metastatic versus poorly metastatic tumor cells. However, the protein level of FN in highly metastatic tumor cells (F27mel) was much lower than in poorly metastatic tumor cells (1143mel). The reduced FN protein in metastatic tumor cells was associated with the significant reduction of FN matrix formation. Because introduction of the mutant FN into tumor cells expressing wild-type FN resulted in a significant loss of FN matrix formation whereas the empty vector did not, we believed that the point mutation, which resulted in the substitution of Lys for Glu, was responsible for the observed phenotype. More importantly, we demonstrated that this point mutation in FN converted poorly metastatic tumor cells to highly metastatic ones. To our knowledge, this is the first report showing that mutated FN directly affects extracellular FN matrix formation and therefore enhances the metastatic potential of tumor cells.
Our results indicate that CD4+ F27TIL-T1 cells recognized a mutated FN presented by DR2 molecules. Although several mutated forms of proteins such as CDC27 (nuclear protein) involved in cell cycle regulation, triosephosphate isomerase (cytosolic protein) required for energy production, and the secretory fusion protein LDFP (LDLR and FUT fusion protein) resulting from chromosomal rearrangement have been identified as class II–restricted tumor antigens, they are presented by either DR1 or DR4 molecules. Of particular interest is that the majority of MHC class II–restricted tumor antigens identified by tumor-reactive CD4+ T cells derived from cancer patients are mutated or fusion proteins. By contrast, the majority of the MHC class I–restricted tumor antigens recognized by CD8+ CTLs are nonmutated self-antigens (2). All of the known MHC class I–restricted human melanoma antigens, such as tyrosinase, gp100, MAGE-3, and NY-ESO-1, also contained CD4+ T cell epitopes, but they were identified either by the stimulation of human PBMC with dendritic cells pulsed with peptides or proteins (25–29), or by the use of HLA-DR4 transgenic mice (30, 31). None, with the exception of tyrosinase, were identified by tumor-reactive CD4+ T cells derived from patients, although the mechanism responsible for this is not understood. Because both animal and human studies indicate that CD4+ T cells play a central role in initiating and maintaining host immune responses against cancer, the presence of such self-reactive CD4+ T cells may cause self-tissue destruction. Self-antigens such as gp100- and tyrosinase-specific CD4+ T cells could be induced after the stimulation of PBMCs with peptides in vitro. However, such T cells exhibited a relatively low affinity for MHC–peptide complexes compared with T cells specific for mutated tumor antigens and required a high concentration of peptide for T cell recognition (31, 32).
Although vaccines containing nonmutated, shared class II–restricted tumor antigens may be useful in a broad coverage of cancer patients, mutated tumor antigens may have a limited clinical application. Nonetheless, several MHC class II–restricted mutated murine tumor antigens have been identified by a biochemical approach (33, 34). Immunization with T helper peptides from viral or the mutated proteins can effectively reject MHC class II–negative tumor cells, which suggests the importance of CD4+ T cell response in antitumor immunity (34–37). Moreover, in addition to providing help for CD8+ T cells (38), CD4+ T cells may play a far broader role in orchestrating the host response to tumor (39) and autoimmune diseases (40). Additional definition of MHC class II–restricted antigens may provide new opportunities for developing effective immunotherapy against cancer.
Acknowledgments
We would like to thank Dr. Malcolm Brenner for critical reading of the manuscript, and Dr. Steven A. Rosenberg for his support and many cell lines established in Surgery Branch, National Cancer Institute (NCI), National Institutes of Health (NIH), and Dr. Peter Emtage for the initial involvement in this project.
This work is supported in part by the funds from the Baylor College of Medicine and the NCI, NIH (R01-CA90327).
H. Wang and J. Zhou contributed equally to this work.
Footnotes
Abbreviations used in this paper: ECM, extracellular matrix; FN, fibronectin; TIL, tumor-infiltrating lymphocytes.
References
- 1.Boon, T., J.-C. Cerottini, B. Van Den Eynde, P. Van der Bruggen, and A. Van Pel. 1994. Tumor antigens recognized by T lymphocytes. Annu. Rev. Immunol. 12:337–365. [DOI] [PubMed] [Google Scholar]
- 2.Wang, R.-F., and S.A. Rosenberg. 1999. Human tumor antigens for cancer vaccine development. Immunol. Rev. 170:85–100. [DOI] [PubMed] [Google Scholar]
- 3.Houghton, A.N., J.S. Gold, and N.E. Blachere. 2001. Immunity against cancer: lessons learned from melanoma. Curr. Opin. Immunol. 13:134–140. [DOI] [PubMed] [Google Scholar]
- 4.Wolfel, T., M. Hauer, J. Schneider, M. Serrano, C. Wolfel, E. Klehmann-Hieb, E. De Plaen, T. Hankeln, K.-H. Meyer Zum Buschenfelde, and D. Beach. 1995. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 269:1281–1284. [DOI] [PubMed] [Google Scholar]
- 5.Robbins, P.F., M. El-Gamil, Y.F. Li, Y. Kawakami, D. Loftus, E. Appella, and S.A. Rosenberg. 1996. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J. Exp. Med. 183:1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mandruzzato, S., F. Brasseur, G. Andry, T. Boon, and P. van der Bruggen. 1997. A CASP-8 mutation recognized by cytolytic T lymphocytes on a human head and neck carcinoma. J. Exp. Med. 186:785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang, R.-F., X. Wang, A.C. Atwood, S.L. Topalian, and S.A. Rosenberg. 1999. Cloning genes encoding MHC class II-restricted antigens: mutated CDC27 as a tumor antigen. Science. 284:1351–1354. [DOI] [PubMed] [Google Scholar]
- 8.Wang, R.-F., X. Wang, and S.A. Rosenberg. 1999. Identification of a novel MHC class II–restricted tumor antigen resulting from a chromosomal rearrangement recognized by CD4+ T cells. J. Exp. Med. 189:1659–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pieper, R., R.E. Christian, M.I. Gonzales, M.I. Nishimura, G. Gupta, R.E. Settlage, J. Shabanowitz, S.A. Rosenberg, D.F. Hunt, and S.L. Topalian. 1999. Biochemical identification of a mutated human melanoma antigen recognized by CD4+ T cells. J. Exp. Med. 189:757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark, E.A., T.R. Golub, E.S. Lander, and R.O. Hynes. 2000. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 406:532–535. [DOI] [PubMed] [Google Scholar]
- 11.Hynes, R.O., I.U. Ali, A.T. Destree, V. Mautner, M.E. Perkins, D.R. Senger, D.D. Wagner, and K.K. Smith. 1978. A large glycoprotein lost from the surfaces of transformed cells. Ann. NY Acad. Sci. 312:317–342. [DOI] [PubMed] [Google Scholar]
- 12.Giancotti, F.G., and E. Ruoslahti. 1990. Elevated levels of the alpha 5 beta 1 fibronectin receptor suppress the transformed phenotype of Chinese hamster ovary cells. Cell. 60:849–859. [DOI] [PubMed] [Google Scholar]
- 13.Akamatsu, H., K. Ichihara-Tanaka, K. Ozono, W. Kamiike, H. Matsuda, and K. Sekiguchi. 1996. Suppression of transformed phenotypes of human fibrosarcoma cells by overexpression of recombinant fibronectin. Cancer Res. 56:4541–4546. [PubMed] [Google Scholar]
- 14.Wang, R.-F., E. Appella, Y. Kawakami, X. Kang, and S.A. Rosenberg. 1996. Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J. Exp. Med. 184:2207–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang, R.-F., S.L. Johnston, G. Zeng, D.J. Schwartzentruber, and S.A. Rosenberg. 1998. A breast and melanoma-shared tumor antigen: T cell responses to antigenic peptides translated from different open reading frames. J. Immunol. 161:3596–3606. [PubMed] [Google Scholar]
- 16.Kornblihtt, A.R., K. Vibe-Pedersen, and F.E. Baralle. 1983. Isolation and characterization of cDNA clones for human and bovine fibronectins. Proc. Natl. Acad. Sci. USA. 80:3218–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gutman, A., and A.R. Kornblihtt. 1987. Identification of a third region of cell-specific alternative splicing in human fibronectin mRNA. Proc. Natl. Acad. Sci. USA. 84:7179–7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roman, J., R.M. LaChance, T.J. Broekelmann, C.J. Kennedy, E.A. Wayner, W.G. Carter, and J.A. McDonald. 1989. The fibronectin receptor is organized by extracellular matrix fibronectin: implications for oncogenic transformation and for cell recognition of fibronectin matrices. J. Cell Biol. 108:2529–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang, R.-F. 2001. The role of MHC class II-restricted tumor antigens and CD4+ T cells in antitumor immunity. Trends Immunol. 22:269–276. [DOI] [PubMed] [Google Scholar]
- 20.Zeng, G., X. Wang, P.F. Robbins, S.A. Rosenberg, and R.-F. Wang. 2001. CD4+ T cell recognition of MHC class II-restricted epitopes from NY-ESO-1 presented by a prevalent HLA-DP4 allele: association with NY-ESO-1 antibody production. Proc. Natl. Acad. Sci. USA. 98:3964–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi, T., P.B. Chapman, S.Y. Yang, I. Hara, S. Vijayasaradhi, and A.N. Houghton. 1995. Reactivity of autologous CD4+ T lymphocytes against human melanoma. Evidence for a shared melanoma antigen presented by HLA-DR15. J. Immunol. 154:772–779. [PubMed] [Google Scholar]
- 22.George, E.L., E.N. Georges-Labouesse, R.S. Patel-King, H. Rayburn, and R.O. Hynes. 1993. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 119:1079–1091. [DOI] [PubMed] [Google Scholar]
- 23.Maniotis, A.J., R. Folberg, A. Hess, E.A. Seftor, L.M. Gardner, J. Pe'er, J.M. Trent, P.S. Meltzer, and M.J. Hendrix. 1999. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am. J. Pathol. 155:739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacDonald, T.J., K.M. Brown, B. LaFleur, K. Peterson, C. Lawlor, Y. Chen, R.J. Packer, P. Cogen, and D.A. Stephan. 2001. Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat. Genet. 29:143–152. [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi, H., T. Kokubo, K. Sato, S. Kimura, K. Asano, H. Takahashi, H. Iizuka, N. Miyokawa, and M. Katagiri. 1998. CD4+ T cells from peripheral blood of a melanoma patient recognize peptides derived from nonmutated tyrosinase. Cancer Res. 58:296–301. [PubMed] [Google Scholar]
- 26.Chaux, P., V. Vantomme, V. Stroobant, K. Thielemans, J. Corthals, R. Luiten, A.M. Eggermont, T. Boon, and P. van der Bruggen. 1999. Identification of MAGE-3 epitopes presented by HLA-DR molecules to CD4+ T lymphocytes. J. Exp. Med. 189:767–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manici, S., T. Sturniolo, M.A. Imro, J. Hammer, F. Sinigaglia, C. Noppen, G. Spagnoli, B. Mazzi, M. Bellone, P. Dellabona, et al. 1999. Melanoma cells present a MAGE-3 epitope to CD4+ cytotoxic T cells in association with histocompatibility leukocyte antigen DR11. J. Exp. Med. 189:871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zarour, H.M., J.M. Kirkwood, L.S. Kierstead, W. Herr, V. Brusic, C.L. Slingluff, Jr., J. Sidney, A. Sette, and W.J. Storkus. 2000. Melan-A/MART-1(51-73) represents an immunogenic HLA-DR4-restricted epitope recognized by melanoma-reactive CD4(+) T cells. Proc. Natl. Acad. Sci. USA. 97:400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jager, E., D. Jager, J. Karbach, Y.T. Chen, G. Ritter, Y. Nagata, S. Gnjatic, E. Stockert, M. Arand, L.J. Old, et al. 2000. Identification of NY-ESO-1 epitopes presented by human histocompatibility antigen (HLA)-DRB4*0101-0103 and recognized by CD4+ T lymphocytes of patients with NY-ESO-1–expressing melanoma. J. Exp. Med. 191:625–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeng, G., C.E. Touloukian, X. Wang, N.P. Restifo, S.A. Rosenberg, and R.-F. Wang. 2000. Identification of CD4+ T cell epitopes from NY-ESO-1 presented by HLA-DR molecules. J. Immunol. 165:1153–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Touloukian, C.E., W.W. Leitner, S.L. Topalian, Y.F. Li, P.F. Robbins, S.A. Rosenberg, and N.P. Restifo. 2000. Identification of a MHC class II-restricted human gp100 epitope using DR4-IE transgenic mice. J. Immunol. 164:3535–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Topalian, S.L., M.I. Gonzales, M. Parkhurst, Y.F. Li, S. Southwood, A. Sette, S.A. Rosenberg, and P.F. Robbins. 1996. Melanoma-specific CD4+ T cells recognize nonmutated HLA-DR–restricted tyrosinase epitopes. J. Exp. Med. 183:1965–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monach, P.A., S.C. Meredith, C.T. Siegel, and H. Schreiber. 1995. A unique tumor antigen produced by a single amino acid substitution. Immunity. 2:45–59. [DOI] [PubMed] [Google Scholar]
- 34.Matsutake, T., and P.K. Srivastava. 2001. The immunoprotective MHC II epitope of a chemically induced tumor harbors a unique mutation in a ribosomal protein. Proc. Natl. Acad. Sci. USA. 98:3992–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ossendorp, F., E. Mengede, M. Camps, R. Filius, and C.J. Melief. 1998. Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors. J. Exp. Med. 187:693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toes, R.E., F. Ossendorp, R. Offringa, and C.J. Melief. 1999. CD4 T Cells and their role in antitumor immune responses. J. Exp. Med. 189:753–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mumberg, D., P.A. Monach, S. Wanderling, M. Philip, A.Y. Toledano, R.D. Schreiber, and H. Schreiber. 1999. CD4(+) T cells eliminate MHC class II-negative cancer cells in vivo by indirect effects of IFN-gamma. Proc. Natl. Acad. Sci. USA. 96:8633–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalams, S.A., and B.D. Walker. 1998. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J. Exp. Med. 188:2199–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hung, K., R. Hayashi, A. Lafond-Walker, C. Lowenstein, D. Pardoll, and H. Levitsky. 1998. The central role of CD4+ T cells in the antitumor immune response. J. Exp. Med. 188:2357–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van de Keere, F., and S. Tonegawa. 1998. CD4+ T cells prevent spontaneous experimental autoimmune encephalomyelitis in anti-myelin basic protein T cell receptor transgenic mice. J. Exp. Med. 188:1875–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]