

Abstract
CpG oligodeoxynucleotides (ODN) have potent effects on innate and adaptive cellular immune responses. In this report, the ability of CpG ODN to confer long-term immunity and protection when used as a vaccine adjuvant with a clinical grade of leishmanial antigen, autoclaved Leishmania major (ALM), or a recombinant leishmanial protein was studied. In two different mouse models of L. major infection, vaccination with ALM plus CpG ODN was able to control infection and markedly reduce lesion development in susceptible BALB/c and resistant C57BL/6 (B6) mice, respectively, up to 12 wk after immunization. Moreover, B6 mice immunized with ALM plus CpG ODNs were still protected against infectious challenge even 6 mo after vaccination. In terms of immune correlates of protection, ALM plus CpG ODN-vaccinated mice displayed L. major–specific T helper cell 1 and CD8+ responses. In addition, complete protection was markedly abrogated in mice depleted of CD8+ T cells at the time of vaccination. Similarly, mice vaccinated with a recombinant leishmanial protein plus CpG ODN also had long-term protection that was dependent on CD8+ T cells in vivo. Together, these data demonstrate that CpG ODN, when used as a vaccine adjuvant with either a recombinant protein or heat-killed leishmanial antigen, can induce long-term protection against an intracellular infection in a CD8-dependent manner.
Keywords: CD4+ T cells, CD8+ T cells, DNA vaccines, parasitic infection, Th cells
Introduction
The induction and maintenance of cellular immune responses is the primary goal of vaccines for a variety of intracellular infections including Mycobacterium tuberculosis, Plasmodium species, and Leishmania major. Effective primary immunity against L. major, the causative agent of cutaneous leishmaniasis, is known to require IL-12–dependent production of IFN-γ from CD4+ T cells (1–3). Resistant C57BL/6 (B6)*mice develop protective Th1 responses that control infection, whereas susceptible BALB/c mice are unable to control infection due to an aberrant Th2 response produced by a restricted population of Vβ4Vα8 CD4+ T cells (4, 5). The treatment of BALB/c mice with IL-12 protein or neutralizing Ab to IL-4 at the time of infection has been shown to shift the immune response to a Th1 profile and prevent susceptibility to infection (1, 6). This ability to alter Th responses in BALB/c mice and to assess the functionality of these responses with a biologic correlate has made this a useful model for vaccine development against diseases requiring Th1 immunity.
Previous vaccine studies in BALB/c mice have demonstrated that long-lasting control (up to 12 wk after vaccination) of L. major infection can result from vaccination with plasmid DNA encoding of a specific leishmanial Ag (7). In contrast, vaccination with either soluble leishmanial Ag (SLA) or a recombinant leishmanial protein plus IL-12 protein conferred short-term (4, 8) but not long-term protection (9). However, if leishmanial protein plus IL-12 protein–vaccinated mice were repeatedly boosted with IL-12 protein, control of infection was better sustained (10). Furthermore, mice vaccinated with leishmanial protein plus IL-12 DNA also had long-term Th1 immunity and protection (9). These data suggested that persistent IL-12 is necessary to sustain immunity sufficient for protection against L. major when using a protein vaccine. Similarly, in a low dose challenge model that more closely mimics the human disease in terms of route and dose of infection, vaccination with DNA again has been shown superior to vaccination with leishmanial protein plus IL-12 in protecting against lesion development and parasite burden in resistant B6 mice challenged 12 wk after immunization (11, 12).
To better understand why protein and DNA vaccines have varying efficacy in the Leishmania mouse model, the immune correlates of protection were analyzed. As previously noted, protein vaccines that primarily elicit CD4+ T cell responses require persistent IL-12 for control of infection in this model (10). In contrast, DNA vaccination induces Ag-specific CD4+ and CD8+ T cells that are required for long-term immunity (7, 13). In addition, plasmid DNA vaccination may offer an advantage over protein vaccines by inducing a qualitatively and/or quantitatively different type of cellular immune response through specific immunostimulatory CpG sequences contained within the vector (14, 15). In this regard, nucleotide sequences containing these CpG motifs have been synthesized and studied as possible immune adjuvants for diseases requiring Th1 immune responses (16, 17). CpG oligodeoxynucleotides (ODN) have been shown to stimulate macrophages and dendritic cells (DC) to synthesize several cytokines including IL-12, IL-18, TNF-α, IFN-α, IFN-β, and IFN-γ and to up-regulate costimulatory molecules such as CD40 and MHC class II (18–20). The range and level of cytokine production vary according to each ODN sequence and its particular modifications (21). Moreover, CpG ODN have been shown to activate DC, leading to the presentation of soluble protein to class I–restricted T cells and the induction of CTL responses (20, 22, 23). This ability of CpG ODN to induce both innate and adaptive cellular immune responses has made it a potential treatment and/or prophylactic vaccine adjuvant, respectively, for diseases requiring cellular immunity.
In studying the role of CpG ODN as a prophylactic vaccine adjuvant, leishmanial proteins plus CpG ODN have been reported to confer some protection after a challenge with L. major (24, 25). In addition, susceptible BALB/c mice treated only with CpG ODN up to 2 wk before or 20 d after infection with L. major were able to control infection (26, 27). It is notable that in the latter study, a Th1 response was induced in the course of an ongoing Th2 response in mice treated with CpG ODN after infection (27). In striking contrast, treatment of susceptible BALB/c mice with IL-12 protein alone was only sufficient to control infection if administered at the time of infection with L. major and continued for up to 1 wk (1). Together, these data suggest that CpG ODN is more potent and durable than IL-12 protein in terms of immune and biologic effects in susceptible BALB/c mice after infection with L. major.
In view of the potent in vivo effects on the cellular immune response elicited by CpG ODN, the data presented here determined whether mice vaccinated with CpG ODN and either a clinical grade of autoclaved (heat-killed) L. major (ALM) Ag or recombinant leishmanial protein had sustained immunity and protection in two different mouse models of L. major infection.
Materials and Methods
Mice.
Female BALB/c mice were purchased from Taconic Farms, Inc., and female B6 mice were purchased from the Division of Cancer Treatment, National Cancer Institute. All mice were maintained in the National Institute of Allergy and Infectious Diseases Animal Care Facility or Vaccine Research Center Animal Care Facility (Bethesda, MD) under pathogen-free conditions.
ODN.
Phosphorothioate-modified ODN sequence 1826 containing two CpG motifs (underlined: TCCATGACGTTCCTGACGTT) was provided by Coley Pharmaceutical Group and used in most experiments. ODN sequence 1982 was used as a control in some experiments (TCCAGGACTTCTCTCAG GTT). The ODN contained endotoxin levels <0.1 EU/mg using the limulus amebocyte lysis assay (Associates of Cape Cod, Inc.). For the 6-mo challenge study in B6 mice, CpG ODN were synthesized at the Center for Biologics Evaluation and Research Core Facility (Bethesda, MD). Sequences were TCAACGTTGA and GCTAGACGTTAGCGT.
Immunization.
BALB/c and B6 mice were injected subcutaneously in their hind footpad with either 50 μg ALM prepared from whole cell, heat-killed L. major promastigotes or 25 μg of a recombinant leishmanial protein containing three Ag: LmSTI1 (28), TSA (29), and LeIF (30). This three-Ag leishmanial protein vaccine is referred to as Trifusion (Corixa Corporation). ALM is a clinical grade reagent and contained <10 EU/dose. Trifusion protein contained levels of endotoxin at the lower limits of detection in the limulus lysate assay. ALM was given alone or with 1 μg recombinant IL-12 (Genetics Institute) or 50 μg CpG ODN. Trifusion protein was given alone or with 25 μg CpG ODN. Each injection was suspended in sterile PBS and given in a volume of 50 μl. In some experiments, as a positive control, naive BALB/c mice were treated with 1 μg neutralizing murine Ab against IL-4 (clone 11B11; provided by W. Paul, NIH, Bethesda, MD) 2 d before, and at the time of, infection. In other experiments, as an additional positive control for long-term protection, mice were vaccinated with a “cocktail” of DNA containing 33 μg each of plasmid DNA encoding LACK Ag, LmSTI1 Ag, or TSA Ag, combined for a total of 99 μg DNA/dose (11). In the 6-mo challenge study, B6 mice were vaccinated intradermally in a volume of 10 μl into the ventral surface of the ear. In all experiments, mice were boosted 2 wk after the initial injection into the same site with their initial regimen. To determine the role of CD8+ T cells in mediating protection with ALM plus CpG ODN or Trifusion protein plus CpG ODN, mice were treated 1 d before, and at the time of, each vaccination with 0.5 μg of a rat Ig control Ab or rat anti–mouse CD8 Ab (2.43) that has been shown to deplete >95% of CD8+ T cells and was sufficient to abrogate protection to DNA vaccination in this model (7, 13). In addition, treatment with anti-CD8 did not diminish the number or function (e.g., IL-12 production) of CD11c+ cells (13).
Infectious Challenge.
L. major clone V1 (MHOM/IL/80/Friedlin) promastigotes were grown as previously described (11). Infective stage promastigotes (metacyclics) of L. major were isolated from stationary cultures (4–5 d old) by negative selection using peanut agglutinin (Vector Laboratories). B6 mice were challenged intradermally at 2 or 12 wk after vaccination in both ears using 500 metacyclic promastigotes. In the 6-mo challenge experiment, infection was done in the ear opposite to vaccination. The lesions were monitored by measuring the diameter of the induration of the ear lesion with a metric caliper. BALB/c mice were infected either 2 or 12 wk after the boost with 105 metacyclic promastigotes. Parasites were injected into the footpad subcutaneously contralateral from the site of vaccination at a volume of 50 μl. Weekly footpad swelling measurements were recorded using a metric caliper.
Parasite Quantitation.
Parasite loads in the ears of B6 mice were determined as previously described (11). The number of viable parasites in each ear was determined from the highest dilution that promastigotes could be grown out after 7 d of incubation at 26°C. The number of parasites was also determined in the local draining LN (popliteal) of BALB/c mice. The LN from each mouse was recovered and mechanically dissociated using a pellet pestle and then serially diluted as previously described. For most experiments, ears or draining LN from at least three individual mice were analyzed for parasite burden. Statistical analysis was done as previously described (11).
Measurement of Leishmania-specific Production of IFN-γ from CD4+ T Cells after Infection.
4 wk after infection, draining LN were harvested from different groups of vaccinated BALB/c mice. The nodes from a minimum of three mice in each group were pooled and then purified into CD4+ T cell subpopulations using magnetic-activated cell sorting columns (Miltenyi Biotec). Flow cytometry confirmed >95% purity of CD4+ T lymphocytes. The cells were then plated in triplicate in a 96-well microtiter plate at 2 × 105 cells/200 μl and cultured with media, macrophages (5 × 104), or macrophages plus L. major. Macrophages were obtained by intraperitoneal washings of naive BALB/c mice and then pulsed with or without L. major before being added to the CD4+ T cell cultures. Supernatants were collected 48 h later and assessed for production of IFN-γ by specific ELISA (BD PharMingen). The lower limit of detection of IFN-γ was 31.3 pg/ml.
Assessment of the Frequency of Leishmania-specific CD4+ and CD8+ Cytokine-producing Cells after Vaccination.
6 mo after vaccination, B6 mice were injected in both ears with a combination of living and killed Ag comprising 106 metacyclic L. major promastigotes and 12.5 μg SLA prepared from 33 freeze-thawed stationary phase L. major promastigotes. 48 h later, three mice per group were killed and cells from the local draining LN were obtained as previously described. For intracellular staining of IFN-γ, cells were stimulated with DC or DC pulsed with L. major for 6 h and then 10 μg/ml brefeldin A was added. The cells were cultured for an additional 18 h and then fixed in 4% paraformaldehyde. Staining of surface and cytoplasmic markers was performed as previously described (11). For each sample, at least 100,000 cells were analyzed.
Results
Vaccination with ALM plus CpG ODN Confers Durable Protection Against L. major in BALB/c Mice.
In previous reports, vaccination with SLA or recombinant leishmanial and IL-12 protein was sufficient for the protection of susceptible BALB/c mice against challenge with L. major at 2 but not 12 wk after vaccination (9). To compare CpG ODN with IL-12 protein as a vaccine adjuvant with a heat-killed preparation of leishmanial Ag for long-term immunity, BALB/c mice were vaccinated and boosted with ALM alone, ALM plus IL-12 protein, ALM plus CpG ODN, or normal saline and infected 2 or 12 wk later. Consistent with previous reports (9, 24, 25), mice vaccinated with ALM plus CpG ODN or ALM plus IL-12 protein were able to control the infection when challenged in the opposite footpad 2 wk after vaccination (Fig. 1 A). In contrast, mice vaccinated with ALM alone or with normal saline developed ulcerations and were killed 3–4 wk after infection. Mice vaccinated with ALM plus CpG ODN but not ALM plus IL-12 protein were able to control the infection when challenged 12 wk after vaccination (Fig. 1 B). Moreover, in the same experiment, mice that received ALM plus CpG ODN were able to sustain control of infection up to 3 mo after challenge. The control of infection after ALM plus CpG ODN vaccination was comparable to or better than that in mice treated with anti–IL-4 (an additional positive control) at the time of infection (Fig. 1 C).
Figure 1.

Footpad swelling of BALB/c mice after infection with L. major 2 or 12 wk after vaccination. Mice (n = 6–8 per group) were immunized in the footpad and boosted 2 wk later with ALM, ALM plus IL-12, ALM plus CpG ODN, or normal saline. Mice were then challenged in the contralateral footpad with 105 L. major (A) 2 or (B) 12 wk after the boost and footpad swelling measurements were recorded weekly. These data are representative of three independent experiments. (C) As a positive control in the same experiment, a group of naive mice (n = 6) was administered anti–IL-4 3 d before, and at the time of, infection. (*) Two mice in the anti–IL-4 treatment group were killed 4 wk due to ulceration. (D) In a separate experiment, mice were immunized with CpG ODN alone, ALM, ALM plus CpG ODN, a cocktail of DNA plasmids encoding different leishmanial Ag, or normal saline (n = 6–8 per group). Challenge was then done 12 wk after the boost. Data represent the mean size of the individual footpads ± SEM.
To compare the protection from CpG ODN to that elicited by a DNA vaccine, an additional group of mice was given a cocktail of DNA plasmids encoding the leishmanial Ag LACK, LmSTI1, and TSA previously shown to elicit long-term protection (Fig. 1 D). Mice vaccinated with ALM plus CpG ODN were equally able to control infection, as measured by footpad swelling, as mice vaccinated with the DNA vaccine. To verify that CpG ODN alone had no protective effect, mice vaccinated and boosted only with CpG ODN were not protected when challenged 12 wk after vaccination (Fig. 1 D). In additional experiments, mice vaccinated with CpG ODN alone or ALM plus control ODN (without CpG) were not able to control infection when challenged 2 wk after boost (unpublished data).
Vaccination with ALM plus CpG ODN Confers Durable Protection Against L. major in B6 Mice.
In contrast to the BALB/c model, it has been demonstrated that vaccination with ALM plus IL-12 confers durable protection against lesion development in the innately resistant B6 mouse (11). As previously noted, the route and dose of infection in the B6 mouse differ from those in the BALB/c mouse, offering another model to assess the utility of CpG ODN as a vaccine adjuvant. B6 mice were vaccinated with ALM alone, ALM plus IL-12, ALM plus CpG ODN, or normal saline and then challenged intradermally in both ears 2 or 12 wk after the boost (Fig. 2) . Mice vaccinated with ALM plus CpG ODN or IL-12 protein developed minimal lesions when challenged 2 or 12 wk after vaccination (Fig. 2, A and B). As a control in a separate experiment, mice given CpG ODN alone at the time of vaccination and boost were not protected and developed lesions comparable to those of nonvaccinated mice when challenged 2 wk after the boost (Fig. 2 C). Finally, in an additional experiment (Fig. 2 D), the durability of ALM plus CpG ODN vaccination was comparable to that of DNA vaccination in eliciting protection when challenged 12 wk after vaccination.
Figure 2.

Ear lesion size of B6 mice after infection with L. major 2 or 12 wk after vaccination. Mice (n = 6–8 per group) were immunized and boosted 2 wk later with ALM, ALM plus IL-12, ALM plus CpG ODN, or normal saline in the footpad. Mice were challenged (A) 2 or (B) 12 wk after the boost by intradermal inoculation of 500 L. major metacyclic promastigotes in both ears, and lesion diameter was monitored. These data are representative of three independent experiments. (C) In a separate experiment, mice were vaccinated with CpG ODN, ALM, ALM plus CpG ODN, or normal saline and then challenged 2 wk after the boost. (D) In another experiment, mice were immunized with ALM plus CpG ODN, a cocktail of DNA plasmids encoding different leishmanial Ag, or normal saline and challenged 12 wk after the boost. Data represent the mean lesion diameter ± SEM of four to eight mice, 8–16 ears per group.
Mice Vaccinated with ALM plus CpG ODN Have a Reduction in Parasite Burden Compared with ALM plus IL-12 Protein after Infectious Challenge with L. major.
To determine whether the reduction in footpad swelling (BALB/c mice) or dermal lesions (B6 mice) previously shown correlated with a decrease in parasite burden, mice in each of the vaccine groups were killed 4 wk after infection and parasite burden was assessed. First, BALB/c mice vaccinated with ALM plus CpG ODN had a 40-fold decrease in the parasite burden from draining the LN of mice infected 12 wk after vaccination compared with other groups (1.6 × 103 in the ALM plus CpG ODN group compared with 6.3 × 104 in the saline group and 1.3 × 105 in the ALM group). ALM plus CpG ODN–vaccinated mice were the only group that was significantly different (P < 0.05) from control saline-vaccinated mice in terms of a decrease in parasite burden (Fig. 3 A). Of note, there was also an ∼20-fold decrease in the number of parasites in ALM plus CpG ODN compared with ALM plus IL-12–vaccinated mice. Second, in the low-dose dermal challenge model in B6 mice, both the ALM plus IL-12 and ALM plus CpG ODN groups demonstrated fewer parasites compared with the ALM and saline groups (2.0 × 105 for ALM plus IL-12 and 3.2 × 104 for ALM plus CpG ODN vs. 1.5 × 106 for ALM and 2.5 × 106 for saline; Fig. 3 B). Most significantly, the mice that received ALM plus CpG ODN had a greater than 2-log decrease in parasite burden compared with mice that only received saline (P < 0.05). Moreover, such mice had approximately a sevenfold decrease in parasite burden compared with ALM plus IL-12 protein–vaccinated mice. Together, these data establish the enhanced biologic potency of ALM plus CpG ODN compared with ALM plus IL-12 vaccination in terms of efficient parasite killing in both mouse models.
Figure 3.
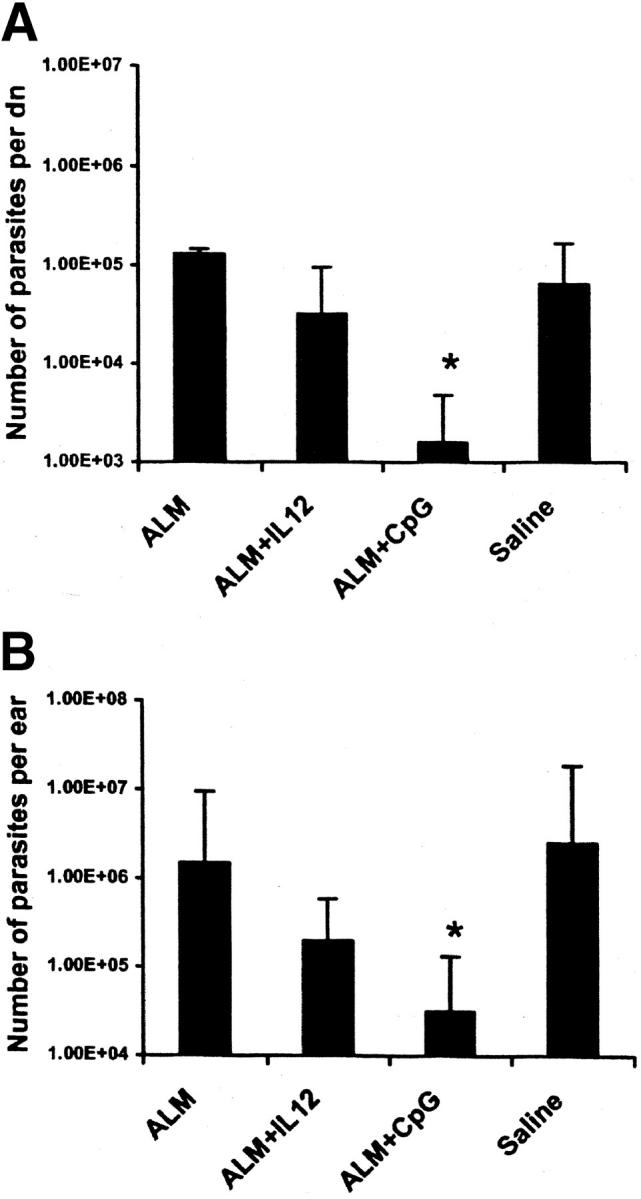
Parasite burden in the draining LN and ears of infected BALB/c and B6 mice after vaccination and infection. (A) BALB/c mice were vaccinated and then challenged 12 wk after boost with L. major in the footpad (groups shown in Fig. 1 B). Parasite quantitation in the draining LN of three individual mice was done 4 wk after challenge. (B) B6 mice were vaccinated and then challenged 12 wk after boost with L. major in both ears (groups shown in Fig. 2 B). Parasite loads in the ears were determined 4 wk after challenge from four individual mice in each group (n = 8 ears). Results are expressed as geometric mean ± SEM. *, P < 0.05 when ALM plus CpG ODN is compared with saline-vaccinated mice.
ALM plus CpG ODN Immunization Induces CD4+ T Cell Production of IFN-γ after Infection with L. major.
Protection against L. major is dependent on Ag-specific production of IFN-γ from CD4+ T cells. To test whether the administration of ALM plus CpG ODN elicits Th1 responses, production of IFN-γ from CD4+ T cells was assessed from draining the LN 4 wk after infection from mice that were challenged 12 wk after vaccination. As shown in Fig. 4 , CD4+ T cells from BALB/c mice vaccinated with ALM plus CpG ODN stimulated in vitro with macrophages pulsed with L. major secreted substantially higher levels of IFN-γ compared with CD4+ T cells from the other groups (Fig. 4). Although not shown in this experiment, in previous studies (9, 10) as well as in the experiment shown in Fig. 1 A, vaccination with ALM or recombinant leishmanial protein plus IL-12 protein does not confer long-term protection or induce sustained Th1 responses in BALB/c mice.
Figure 4.

Leishmania-specific production of IFN-γ from CD4+ T cells after infection with L. major. BALB/c mice were vaccinated and then challenged with L. major 12 wk after boost. CD4+ T cells were isolated from the draining LN 4 wk after challenge and stimulated in culture with media alone, macrophages, or macrophages plus L. major. IFN-γ levels were measured 48 h later by ELISA of culture supernatants. Data are the mean concentrations of triplicate assays ± SD.
ALM plus CpG ODN Sustains Protection Against L. major up to 6 mo after Vaccination in B6 Mice.
To further evaluate the longevity of ALM plus CpG ODN immunization in eliciting protection against L. major, B6 mice were vaccinated intradermally in the ear and then infected 6 mo later in the opposite ear. First, to assess the immune correlates of protection, we evaluated the immune responses of mice 6 mo after the last vaccination by measuring the frequency of IFN-γ–producing T cells after a brief in vivo infection (48 h) followed by in vitro stimulation. This method of in vivo followed by in vitro Ag stimulation provides a sensitive way to measure low level memory immune responses after vaccination with ALM (11). As shown in Fig. 5 A, there was a demonstrable number of both CD4+ and CD8+ T cells producing IFN-γ after stimulation with DC pulsed with L. major. In contrast, nonvaccinated mice demonstrated very low frequencies of IFN-γ–producing CD4+ or CD8+ T cells (0.1–0.2%). It was striking that the ALM plus CpG ODN–vaccinated group had similar or higher frequencies of CD4+ and CD8+ IFN-γ–producing cells than the definitive positive control (previously infected mice that had healed; unpublished data). To correlate these findings with an infectious challenge, mice were injected with L. major intradermally in the ear 6 mo after vaccination. Mice that received ALM plus CpG ODN demonstrated little or no pathology after challenge as assessed by lesion size (Fig. 5 B). Furthermore, the degree of protection induced by ALM plus CpG ODN was comparable to that in previously infected mice that had healed or mice that had been vaccinated with a cocktail of plasmid DNA encoding recombinant leishmanial Ag (unpublished data).
Figure 5.

Assessment of cellular immune responses and protection in B6 mice 6 mo after vaccination. (A) IFN-γ responses by CD4+ and CD8+ T cells in B6 mice were assessed 6 mo after intradermal vaccination with ALM plus CpG ODN or saline. Draining LN cells were obtained 48 h after intradermal injection of metacyclic promastigotes and SLA into the nonvaccinated ear and 24 h after in vitro restimulation with uninfected DC or infected DC (three mice per group, six LN). Analyses are gated on CD3+ cells. Numbers represent the percentage of CD4+ or CD8+ T cells positive for IFN-γ. Nonvaccinated mice were included as negative controls. (B) In the same experiment, mice vaccinated with ALM plus CpG ODN were challenged 6 mo after boost with L. major in the ear opposite from which they were vaccinated and ear lesions were monitored. Data represent the mean lesion diameter ± SEM of six to eight mice.
CD8+ T Cell Depletion at the Time of Vaccination Mitigates Protection Conferred by Vaccination with ALM or Recombinant Leishmanial Protein plus CpG ODN.
We have previously shown that the depletion of CD8+ T cells at the time of vaccination or infection abrogates protective immunity in BALB/c mice vaccinated with plasmid DNA encoding LACK Ag (7, 13) and B6 mice vaccinated with a cocktail of DNA Ag (unpublished data). Because ALM plus CpG ODN immunization was able to induce CD8+ T cell responses 6 mo after vaccination (Fig. 5), the role of CD8+ T cells in mediating protection was determined by depleting CD8+ T cells in vivo at the time mice were vaccinated. In these experiments, the depletion of CD8+ T cells at the time of vaccination rather than at the time of infection was done to limit induction of Ag-specific CD8+ T cells but not limit the role of the endogenous repertoire of CD8+ T cells during the natural course of infection, which we have recently shown to be important in mediating primary protection in the B6 mouse model (31). As shown in Fig. 6 A, depletion of CD8+ T cells at the time of vaccination with ALM plus CpG ODN resulted in an increase in ear lesion swelling compared with that in mice vaccinated with ALM plus CpG ODN alone or treated with a control rat Ig Ab. Because the peak number of parasites in the site is found just before the onset of lesion development in the natural course of the low-dose intradermal challenge model (12), parasite loads were quantitated 4 wk after infection when there was as yet little difference in the size of the dermal lesions between the anti-CD8–treated and control-treated mice. There was, however, a 1–2-log increase in parasite load in the ears and draining LN (Fig. 6 B) in ALM plus CpG ODN–vaccinated mice treated with anti-CD8.
Figure 6.

Effect of CD8+ T cell depletion on lesion development and parasite load in mice vaccinated with ALM plus CpG ODN. (A) Mice (n = 6–8 per group) were immunized and boosted 2 wk later with ALM, ALM plus CpG ODN, or normal saline in the footpad. In some groups, mice were treated 1 d before, and at the time of, vaccination with anti-CD8 (2.43) or rat Ig Ab. Mice were then challenged 2 wk later in the ear and monitored as described in Fig. 2. (B) Parasite loads in the ears and draining LN were determined 4 wk after challenge from four mice in each group (n = 8 ears) or pooled draining LN from four mice. Results for the parasite load in the ears are expressed as geometric mean ± SEM. *, P < 0.05 in comparing parasites per ear harvested from ALM plus CpG ODN–vaccinated mice depleted of CD8+ T cells.
These findings suggest that CpG ODN induces CD8+ T cell responses by enhancing the processing of proteins contained within the ALM. Because ALM is an autoclaved preparation of whole L. major, it was of interest to perform similar studies using a recombinant leishmanial protein to provide additional evidence that CD8+ T cells were important in mediating protection using CpG ODN as a vaccine adjuvant. In this regard, mice were immunized with CpG ODN and a recombinant Trifusion protein comprising three different leishmanial proteins in the presence or absence of CD8 depletion at the time of immunization. These recombinant leishmanial proteins have been previously shown to confer protection in both mouse and primate models of L. major infection (11, 28–30, 32). As shown in Fig. 7 , B6 mice immunized with Trifusion protein plus CpG ODN had minimal lesions when challenged with L. major 2 (Fig. 7 A) or 12 wk (Fig. 7 B) after vaccination. Similar to the results shown above, the treatment of Trifusion plus CpG ODN–immunized mice with anti-CD8 at the time of vaccination resulted in an increase in the lesion size and parasite load (Fig. 7 C) after infection. Finally, mice vaccinated with Trifusion DNA had minimal ear lesions after infection 12 wk after immunization, which was comparably altered by treatment with anti-CD8 at the time of vaccination (unpublished data). Thus, vaccination with either leishmanial DNA (7, 13) or leishmanial protein or killed Ag plus CpG ODN induces long-term protection against L. major in a CD8+ T cell–dependent manner.
Figure 7.
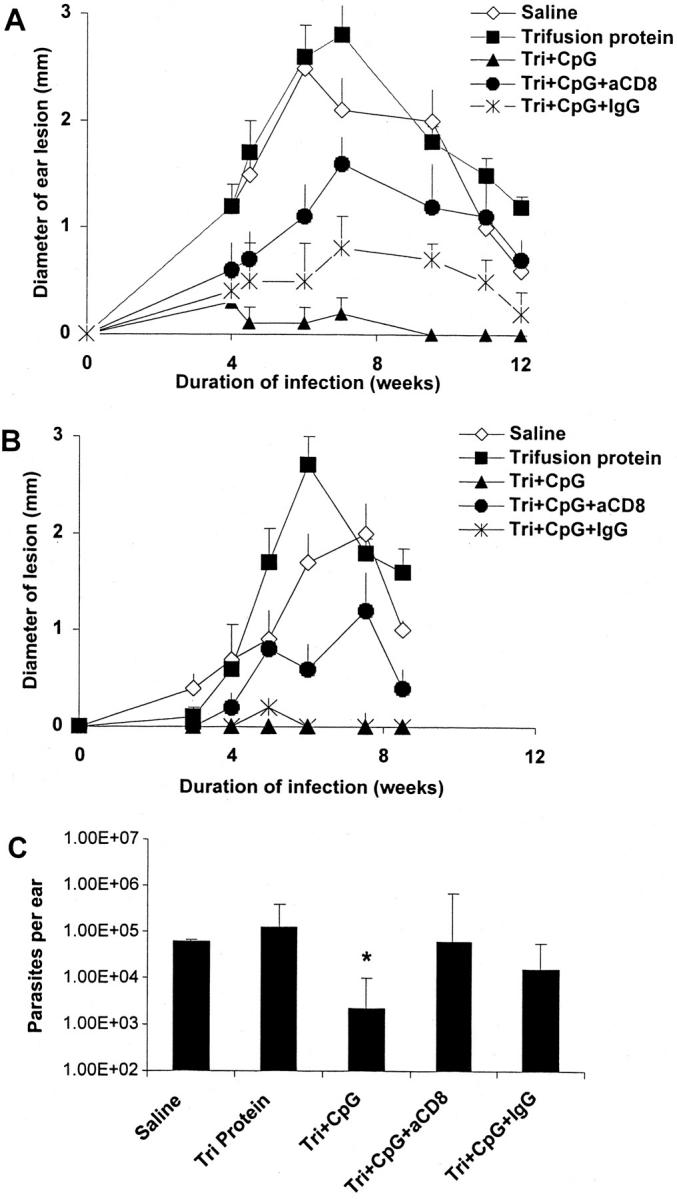
Effect of CD8 depletion in mice vaccinated with recombinant leishmanial protein plus CpG ODN. Mice (n = 6–8 per group) were immunized and boosted 2 wk later with Trifusion protein, Trifusion protein plus CpG ODN, or normal saline (nonvaccinated) in the footpad. In some groups, mice were treated 1 d before, and at the time of, vaccination with anti-CD8 (2.43) or rat Ig control Ab. Mice were challenged (A) 2 or (B) 12 wk after the last immunization in the ear and lesion size was monitored as described in Fig. 2. (C) Parasite loads in the ears from mice challenged 2 wk after vaccination were determined as described in Fig. 6. Tri, Trifusion; *, P < 0.05 in comparing parasites per ear harvested from Trifusion plus CpG ODN–vaccinated mice depleted of CD8+ T cells.
Discussion
This study demonstrates in two different mouse models of L. major infection that CpG ODN provides long-term protection when used as a vaccine adjuvant with either ALM or recombinant leishmanial protein. In addition, similar to our previous studies with DNA vaccines, CpG ODN conferred more potent and durable protection when compared with IL-12 protein as an adjuvant with ALM. These data strongly suggest that a heat-killed Ag and/or a recombinant protein vaccine can be useful for inducing durable immunity for diseases requiring cellular immune responses depending on the adjuvant used.
Our data demonstrating that ALM or Trifusion protein plus CpG ODN provides long-term protection are in contrast to previous studies using CpG ODN as a vaccine adjuvant with other leishmanial Ag preparations. In the study by Stacey and Blackwell (24), partial protection was demonstrated in BALB/c mice infected 6 wk or 6 mo after vaccination, but the quality of protection at any time after vaccination was moderate because all of the mice developed significant footpad swelling. Similarly, Walker et al. (25) demonstrated that the effects of CpG ODN as an adjuvant were partial and limited, as protection was demonstrated in only 40% of BALB/c mice challenged 3 wk after vaccination. The greater efficacy of the leishmanial protein plus CpG ODN in our study may be due to several factors. First, the leishmanial Ag used in the other studies was a preparation of SLA, which might be less immunogenic than ALM, a clinical grade Ag that has been used in human vaccine trials against L. major (33, 34). Second, the parasite dose and strain differed in our studies compared with the other studies. Third, the CpG ODN sequence we used was different than that used in one of the previous studies showing partial long-term immunity. Nevertheless, the results presented here showing that CpG ODN provide long-term protection have been repeated multiple times in both BALB/c and B6 mouse models of leishmania infection with consistent results and little variability.
With regard to the mechanism by which CpG ODN mediate their function in vivo and why they are better than IL-12 protein as a vaccine adjuvant, their role in enhancing APC function needs to be considered. At the APC level, CpG ODN through toll-like receptor 9, augments both the activation and maturation of DC as well as the induction of proinflammatory cytokines (35). Thus, in comparison to the relatively short-lived effect of exogenous IL-12 protein as an adjuvant, the endogenous production of IL-12, IL-18, and other soluble mediators from activated DC induced by CpG ODN are likely to result in a more physiologic cognate interaction between the DC and T cell, resulting in both a qualitatively and quantitatively different type of CD4+ and CD8+ T cell response. These data highlight the potential importance of targeting DC, especially for vaccines against diseases requiring cellular immune responses.
In terms of immune correlates of protection, it is well established that CpG ODN are potent inducers of Th1 responses, which is consistent with our findings that mice vaccinated with ALM plus CpG ODN had striking enhancement in the frequency and production of IFN-γ from CD4+ T cells 6 mo after vaccination in B6 mice (Fig. 5) and after infection in BALB/c mice, respectively (Fig. 4). Moreover, an additional possibility relating to the effectiveness of CpG ODN as a vaccine adjuvant compared with IL-12 protein in the BALB/c and B6 models is the role of CD8+ T cells. As previously mentioned, CD8+ T cell responses are not seen in vaccination with recombinant leishmanial protein plus IL-12 but have an important role in maintaining the protection elicited by DNA vaccination (7, 13). In this regard, CpG ODN have been shown to elicit CD8+ T cell responses in mice when given with a protein Ag such as OVA (20). Thus, our findings that there were significant frequencies of Ag-specific CD8+ IFN-γ+ cells in B6 mice 6 mo after vaccination with ALM plus CpG ODN and that the depletion of CD8+ T cells at the time of vaccination enhances the size of the lesions and the parasite load in B6 mice, provide compelling evidence for the importance of CD8+ T cells in mediating long-term immunity. Furthermore, the data showing that lesion development is altered in B6 mice immunized with a recombinant leishmanial protein plus CpG ODN depleted of CD8+ T cells at the time of vaccination provides additional support for CpG ODN inducing CD8+ T cell responses with a defined parasitic protein Ag. Finally, although lesion size in leishmanial Ag plus CpG ODN–vaccinated mice depleted of CD8+ T cells was increased, these mice had demonstrably smaller lesions than control vaccinated mice. This is consistent with the induction of both CD4+ and CD8+ T cells by such a vaccine. Together, these data suggest that vaccines that elicit both CD4+ and CD8+ T cell responses are sufficient to confer long-term protection against L. major in mice.
Although the vaccine approach described here potentially has immediate clinical use because ALM is already a widely tested vaccine Ag in humans, it remains to be determined whether the effects of CpG ODN on the cellular immune response in humans will be comparable to those observed in mice. One encouraging recent report related to this issue showed that there was a decrease in lesion size after infectious challenge with L. major in primates vaccinated with a killed preparation of leishmanial Ag and a specific type of CpG ODN compared with vaccination with Ag alone (36). To conclude, it is worth noting that the historical standard for effective vaccination against L. major in humans is leishmaniazation (attenuated live infection). Moreover, the fact that previous infection to L. major confers life-long protection in humans and that there is evidence for the persistence of parasitic Ag raises the critical issue of whether the induction of potent CD4+ and CD8+ T cell responses induced after DNA or a killed and/or recombinant leishmanial Ag plus CpG ODN will be sufficient to confer life-long protection in humans. This distinction between the apparent lack of an Ag requirement to maintain cellular immune memory for CD4+ and CD8+ T cell responses for nonliving vaccines versus the potential requirement of Ag for mediating biologic protection will be the critical determinant in whether specific vaccines will be successful over the course of a lifetime against many infections requiring cellular immunity in humans.
Acknowledgments
We thank Brenda Marchal for editorial assistance.
E. Rhee and J. Shah are Howard Hughes Medical Institute-NIH Research Scholars.
E.G. Rhee and S. Mendez contributed equally to this work.
Footnotes
Abbreviations used in this paper: ALM, autoclaved Leishmania major; B6, C57BL/6 mice; DC, dendritic cells; ODN, oligodeoxynucleotides; SLA, soluble leishmanial antigen.
References
- 1.Heinzel, F.P., D.S. Schoenhaut, R.M. Rerko, L.E. Rosser, and M.K. Gately. 1993. Recombinant interleukin 12 cures mice infected with Leishmania major. J. Exp. Med. 177:1505–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sypek, J.P., C.L. Chung, S.E. Mayor, J.M. Subramanyam, S.J. Goldman, D.S. Sieburth, S.F. Wolf, and R.G. Schaub. 1993. Resolution of cutaneous leishmaniasis: interleukin 12 initiates a protective T helper type 1 immune response. J. Exp. Med. 177:1797–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scharton-Kersten, T., L.C. Afonso, M. Wysocka, G. Trinchieri, and P. Scott. 1995. IL-12 is required for natural killer cell activation and subsequent T helper 1 cell development in experimental leishmaniasis. J. Immunol. 154:5320–5330. [PubMed] [Google Scholar]
- 4.Julia, V., M. Rassoulzadegan, and N. Glaichenhaus. 1996. Resistance to Leishmania major induced by tolerance to a single antigen. Science. 274:421–423. [DOI] [PubMed] [Google Scholar]
- 5.Launois, P., I. Maillard, S. Pingel, K.G. Swihart, I. Xenarios, H. Acha-Orbea, H. Diggelmann, R.M. Locksley, H.R. MacDonald, and J.A. Louis. 1997. IL-4 rapidly produced by Vβ4Vα8 CD4+ T cells instructs Th2 development and susceptibility to Leishmania major in BALB/c mice. Immunity. 6:541–549. [DOI] [PubMed] [Google Scholar]
- 6.Sadick, M.D., F.P. Heinzel, B.J. Holaday, R.T. Pu, R.S. Dawkins, and R.M. Locksley. 1990. Cure of murine leishmaniasis with anti-interleukin 4 monoclonal antibody. Evidence for a T cell–dependent, interferon-γ–independent mechanism. J. Exp. Med. 171:115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gurunathan, S., D.L. Sacks, D.R. Brown, S.L. Reiner, H. Charest, N. Glaichenhaus, and R.A. Seder. 1997. Vaccination with DNA encoding the immunodominant LACK parasite antigen confers protective immunity to mice infected with Leishmania major. J. Exp. Med. 186:1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Afonso, L.C., T.M. Scharton, L.Q. Vieira, M. Wysocka, G. Trinchieri, and P. Scott. 1994. The adjuvant effect of interleukin-12 in a vaccine against Leishmania major. Science. 263:235–237. [DOI] [PubMed] [Google Scholar]
- 9.Gurunathan, S., C. Prussin, D.L. Sacks, and R.A. Seder. 1998. Vaccine requirements for sustained cellular immunity to an intracellular parasitic infection. Nat. Med. 4:1409–1415. [DOI] [PubMed] [Google Scholar]
- 10.Stobie, L., S. Gurunathan, C. Prussin, D.L. Sacks, N. Glaichenhaus, C.Y. Wu, and R.A. Seder. 2000. The role of antigen and IL-12 in sustaining Th1 memory cells in vivo: IL-12 is required to maintain memory/effector Th1 cells sufficient to mediate protection to an infectious parasite challenge. Proc. Natl. Acad. Sci. USA. 97:8427–8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mendez, S., S. Gurunathan, S. Kamhawi, Y. Belkaid, M.A. Moga, Y.A. Skeiky, A. Campos-Neto, S. Reed, R.A. Seder, and D. Sacks. 2001. The potency and durability of DNA- and protein-based vaccines against Leishmania major evaluated using low-dose, intradermal challenge. J. Immunol. 166:5122–5128. [DOI] [PubMed] [Google Scholar]
- 12.Belkaid, Y., S. Mendez, R. Lira, N. Kadambi, G. Milon, and D. Sacks. 2000. A natural model of Leishmania major infection reveals a prolonged “silent” phase of parasite amplification in the skin before the onset of lesion formation and immunity. J. Immunol. 165:969–977. [DOI] [PubMed] [Google Scholar]
- 13.Gurunathan, S., L. Stobie, C. Prussin, D.L. Sacks, N. Glaichenhaus, A. Iwasaki, D.J. Fowell, R.M. Locksley, J.T. Chang, C.Y. Wu, et al. 2000. Requirements for the maintenance of Th1 immunity in vivo following DNA vaccination: a potential immunoregulatory role for CD8+ T cells. J. Immunol. 165:915–924. [DOI] [PubMed] [Google Scholar]
- 14.Klinman, D.M., G. Yamshchikov, and Y. Ishigatsubo. 1997. Contribution of CpG motifs to the immunogenicity of DNA vaccines. J. Immunol. 158:3635–3639. [PubMed] [Google Scholar]
- 15.Sato, Y., M. Roman, H. Tighe, D. Lee, M. Corr, M.D. Nguyen, G.J. Silverman, M. Lotz, D.A. Carson, and E. Raz. 1996. Immunostimulatory DNA sequences necessary for effective intradermal gene immunization. Science. 273:352–354. [DOI] [PubMed] [Google Scholar]
- 16.Chu, R.S., O.S. Targoni, A.M. Krieg, P.V. Lehmann, and C.V. Harding. 1997. CpG oligodeoxynucleotides act as adjuvants that switch on T helper 1 (Th1) immunity. J. Exp. Med. 186:1623–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roman, M., E. Martin-Orozco, J.S. Goodman, M.D. Nguyen, Y. Sato, A. Ronaghy, R.S. Kornbluth, D.D. Richman, D.A. Carson, and E. Raz. 1997. Immunostimulatory DNA sequences function as T helper-1-promoting adjuvants. Nat. Med. 3:849–854. [DOI] [PubMed] [Google Scholar]
- 18.Klinman, D.M., A.K. Yi, S.L. Beaucage, J. Conover, and A.M. Krieg. 1996. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon γ. Proc. Natl. Acad. Sci. USA. 93:2879–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartmann, G., G.J. Weiner, and A.M. Krieg. 1999. CpG DNA: a potent signal for growth, activation, and maturation of human dendritic cells. Proc. Natl. Acad. Sci. USA. 96:9305–9310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sparwasser, T., R.M. Vabulas, B. Villmow, G.B. Lipford, and H. Wagner. 2000. Bacterial CpG-DNA activates dendritic cells in vivo: T helper cell-independent cytotoxic T cell responses to soluble proteins. Eur. J. Immunol. 30:3591–3597. [DOI] [PubMed] [Google Scholar]
- 21.Ballas, Z.K., A.M. Krieg, T. Warren, W. Rasmussen, H.L. Davis, M. Waldschmidt, and G.J. Weiner. 2001. Divergent therapeutic and immunologic effects of oligodeoxynucleotides with distinct CpG motifs. J. Immunol. 167:4878–4886. [DOI] [PubMed] [Google Scholar]
- 22.Davila, E., and E. Celis. 2000. Repeated administration of cytosine-phosphorothiolated guanine-containing oligonucleotides together with peptide/protein immunization results in enhanced CTL responses with anti-tumor activity. J. Immunol. 165:539–547. [DOI] [PubMed] [Google Scholar]
- 23.Cho, H.J., K. Takabayashi, P.M. Cheng, M.D. Nguyen, M. Corr, S. Tuck, and E. Raz. 2000. Immunostimulatory DNA-based vaccines induce cytotoxic lymphocyte activity by a T-helper cell-independent mechanism. Nat. Biotechnol. 18:509–514 [DOI] [PubMed] [Google Scholar]
- 24.Stacey, K.J., and J.M. Blackwell. 1999. Immunostimulatory DNA as an adjuvant in vaccination against Leishmania major. Infect. Immun. 67:3719–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker, P.S., T. Scharton-Kersten, A.M. Krieg, L. Love-Homan, E.D. Rowton, M.C. Udey, and J.C. Vogel. 1999. Immunostimulatory oligodeoxynucleotides promote protective immunity and provide systemic therapy for leishmaniasis via IL-12- and IFN-γ-dependent mechanisms. Proc. Natl. Acad. Sci. USA. 96:6970–6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lipford, G.B., T. Sparwasser, S. Zimmermann, K. Heeg, and H. Wagner. 2000. CpG-DNA-mediated transient lymphadenopathy is associated with a state of Th1 predisposition to antigen-driven responses. J. Immunol. 165:1228–1235. [DOI] [PubMed] [Google Scholar]
- 27.Zimmermann, S., O. Egeter, S. Hausmann, G.B. Lipford, M. Rocken, H. Wagner, and K. Heeg. 1998. CpG oligodeoxynucleotides trigger protective and curative Th1 responses in lethal murine leishmaniasis. J. Immunol. 160:3627–3630. [PubMed] [Google Scholar]
- 28.Webb, J.R., D. Kaufmann, A. Campos-Neto, and S.G. Reed. 1996. Molecular cloning of a novel protein antigen of Leishmania major that elicits a potent immune response in experimental murine leishmaniasis. J. Immunol. 157:5034–5041. [PubMed] [Google Scholar]
- 29.Webb, J.R., A. Campos-Neto, P.J. Ovendale, T.I. Martin, E.J. Stromberg, R. Bodaro, and S.G. Reed. 1998. Human and murine immune responses to a novel Leishmania major recombinant protein encoded by members of a multicopy gene family. Infect. Immun. 66:3279–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coler, R.N., Y.A.W. Sheiky, K. Bernards, K. Greeson, D. Carter, C. Cornellison, F. Modabber, A. Campos-Neto, and S.G. Reed. 2002. Immunization with a poly-protein vaccine consisting of the T cell antigens TSA, LmSTI1 and LeIF protects against leishmaniasis. Infect. Immun. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belkaid, Y., E. Von Stebut, S. Mendez, R. Lira, E. Caler, S. Bertholet, M.C. Udey, and D. Sacks. 2002. CD8+ T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major. J. Immunol. 168:3992–4000. [DOI] [PubMed] [Google Scholar]
- 32.Campos-Neto, A., R. Porrozzi, K. Greeson, R.N. Coler, J.R. Webb, Y.A. Skeiky, S.G. Reed, and G. Grimaldi, Jr. 2001. Protection against cutaneous leishmaniasis induced by recombinant antigens in murine and nonhuman primate models of the human disease. Infect. Immun. 69:4103–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Satti, I.N., H.Y. Osman, N.S. Daifalla, S.A. Younis, E.A. Khalil, E.E. Zijlstra, A.M. El Hassan, and H.W. Ghalib. 2001. Immunogenicity and safety of autoclaved Leishmania major plus BCG vaccine in healthy Sudanese volunteers. Vaccine. 19:2100–2106. [DOI] [PubMed] [Google Scholar]
- 34.Khalil, E.A., A.M. El Hassan, E.E. Zijlstra, M.M. Mukhtar, H.W. Ghalib, B. Musa, M.E. Ibrahim, A.A. Kamil, M. Elsheikh, A. Babiker, et al. 2000. Autoclaved Leishmania major vaccine for prevention of visceral leishmaniasis: a randomised, double-blind, BCG-controlled trial in Sudan. Lancet. 356:1565–1569. [DOI] [PubMed] [Google Scholar]
- 35.Hemmi, H., O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsumoto, K. Hoshino, H. Wagner, K. Takeda, et al. 2000. A Toll-like receptor recognizes bacterial DNA. Nature. 408:740–745. [DOI] [PubMed] [Google Scholar]
- 36.Verthelyi, D., R.T. Kenney, R.A. Seder, A.A. Gam, B. Freidag, and D.M. Klinman. 2002. CpG oligodeoxynucleotides as vaccine adjvuants in primates. J. Immunol. 168:1659–1663. [DOI] [PubMed] [Google Scholar]