

Abstract
Using transgenic mice that replicate hepatitis B virus (HBV) at high levels in the liver as recipients of HBV-specific cytotoxic T lymphocytes (CTLs), we showed that the chemokines responsive to γ–2/IFN-γ inducible protein ([Crg2]IP-10) and monokine induced by interferon-γ (Mig) are rapidly and strongly induced in the liver after CTL transfer. The transferred CTLs produce neither chemokine; rather, they activate (via the secretion of IFN-γ) hepatocytes and nonparenchymal cells of the liver to produce (Crg2)IP-10 and Mig. Importantly, blocking these chemokines in vivo reduces the recruitment of host-derived lymphomononuclear cells into the liver and the severity of the liver disease without affecting the IFN-γ–dependent antiviral potential of the CTLs. The finding that neutralization of these chemokines is associated with maintenance of antiviral effects but diminished tissue damage may be significant for the development of immunotherapeutic approaches for the treatment of chronic HBV infection.
Keywords: chemokines, hepatitis B virus, infectious immunity-virus, liver
Introduction
The hepatitis B virus (HBV)* is an enveloped DNA virus which causes acute and chronic liver disease characterized by a necroinflammatory lymphomononuclear cell infiltrate and Kupffer cell hyperplasia (1). Since HBV is not directly cytopathic for the hepatocyte, the immune response to viral antigens is thought to be responsible for both liver disease and viral clearance after HBV infection. Indeed, patients with acute viral hepatitis, who successfully clear the virus, mount a multispecific polyclonal CTL response to several HBV-encoded antigens (1, 2). In contrast, this response is absent or extremely weak in chronically infected patients who do not clear the virus (1, 2) and thus, it is believed that the outcome of HBV infection (viral clearance versus viral persistence) is determined primarily by the vigor and quality of the cellular immune response (1, 2).
The experimental approaches to HBV pathogenesis have been difficult because the host range of HBV is limited to man and chimpanzees, and because in vitro systems for the propagation of HBV do not exist. To overcome these limitations, we developed and characterized transgenic mice that replicate HBV at high levels in the liver (3). These animals are profoundly immunologically tolerant to the virus (4). We examined in this model the antiviral and immunopathological consequences of antigen recognition by administration of H2d-restricted CTL clones that recognize a dominant CTL epitope located between residues 28–39 of HBsAg (5, 6). Surprisingly, the antiviral potential of the CTLs was shown to be primarily mediated by noncytolytic mechanisms that involve the intrahepatic production of IFN-γ by the CTLs after antigen recognition (6, 7). The noncytolytic potential of the CTLs was confirmed by demonstrating that HBsAg-specific perforinless CTLs abolished viral replication in the complete absence of liver disease (6). By crossing HBV transgenic mice with mice genetically deficient for IFN-γ, recent studies have shown that this cytokine mediates most of the antiviral effect of the CTLs (8).
We also showed that the cytopathic potential of the CTLs is quite limited, involving the apoptotic death of a small number of hepatocytes and resulting in widely scattered, acidophilic, Councilman bodies (apoptotic hepatocytes) that are characteristic of acute viral hepatitis in man (5). As time progresses, however, many host-derived inflammatory cells are recruited into the liver, thereby contributing to the formation of necroinflammatory foci in which apoptotic hepatocytes and CTLs are outnumbered by host-derived lymphomononuclear and polymorphonuclear cells (5, 9). These recruited inflammatory cells could be responsible for antigen-nonspecific amplification mechanisms that may cause much of the liver damage initiated by the CTLs. The recruitment of inflammatory cells that are involved in this amplification process is likely to be mediated by the secretion of chemokines by either the antigen-activated CTLs or by other cellular components of the liver.
Chemokines are small proteins that regulate the trafficking of inflammatory cells to tissue sites of inflammation (10, 11). Most chemokines share four cysteine residues that are thought to be crucial for the tertiary structure of the proteins. Chemokines have been divided into four different families (C-X-C, CC, CX3C, and C) depending on the position of the first two cysteine residues (10, 11). Since CTLs are very efficient in their property of specifically recognizing virally infected cells in the periphery, it is tempting to speculate that their capacity of secreting chemokines coupled with their capacity of inducing chemokine production by neighboring cells may help to orchestrate the trafficking, homing, and effector functions of the cellular immune response (12). This may turn out to be particularly relevant in the case of infections with noncytopathic viruses (like HBV) which are not known to elicit the strong, nonspecific inflammatory responses that are common during infections with viruses that directly cause tissue damage (12).
Recent reports have shown that several chemokines are induced in infected tissues during different viral infections. Among them, C-X-C chemokines such as chemokine responsive to γ–2/IFN-γ inducible protein, CXCL10 ([Crg2]IP-10) and monokine induced by IFN-γ (Mig), CXCL9 are thought to play an important role in viral immunity. Both chemokines are known to be produced by macrophages, bind the same chemokine receptor (CXCR3), and chemoattract lymphocytes and monocyte/macrophages (10, 11). A recent report has also shown that these same chemokines can be produced by primary hepatocyte cultures derived from mice treated with IL-2/IL-12 (13). (Crg2)IP-10 and Mig are induced in the mouse liver during vaccinia virus infection (14) and mice infected with recombinant vaccinia virus encoding either chemokine resolve the infection much more successfully than mice infected with control virus (15). Similarly, mouse hepatitis virus–infected mice treated with Abs specific for (Crg2)IP-10 or Mig showed decreased T cell infiltration in the brain, delayed viral clearance, and increased mortality (16, 17). Recent studies have also shown that Mig contributes to protection against MCMV infection (18). Finally, it is worth mentioning that IP-10 and Mig have been detected in the liver of patients chronically infected with hepatitis C virus (HCV; references 19 and 20). To our knowledge, no information is available about the role of (Crg2)IP-10 or Mig in HBV pathogenesis.
Based on the aforementioned studies, it is possible that (Crg2)IP-10 and/or Mig might play a role in the intrahepatic recruitment, antiviral, and pathogenetic effector functions of HBV-specific CTLs and/or other inflammatory cells during HBV infection. To test this hypothesis, we took advantage of our HBV transgenic mouse model to perform a series of experiments aimed at monitoring (i) the ability of passively transferred CTLs to induce (Crg2)IP-10 and Mig in the liver; (ii) the source, kinetics, and regulation of (Crg2)IP-10 and Mig expression in our system; and (iii) the ability of chemokine-specific neutralizing Abs to modulate their function(s).
Materials and Methods
Mice.
HBV transgenic mouse lineages 1.3.32 and 1.3.46 used in this study have been described previously (3). Lineage 1.3.32 (inbred C57BL/6, H-2b) was bred one generation against BALB/c mice (H-2d) to produce H-2bxd F1 hybrids before injection of H-2d restricted HBsAg-specific CTL lines and clones. Lineage 1.3.46 (inbred B10D2, H-2d) was backcrossed against mice genetically deficient for IFN-γ (IFN-γ−/−) as described previously (8). In all experiments, the mice were matched for age (8 wk), sex, and hepatitis Be antigen (HBeAg) levels in their serum before experimental manipulations. All animals were housed in pathogen-free rooms under strict barrier conditions.
Injection of HBsAg-specific CTL Lines and Clones.
HBV transgenic mice were injected with a HBsAg-specific, H-2d restricted, CD8+ CTL clone (designated 6C2) that recognizes an epitope (IPQSLDSWWTSL) located between residues 28–39 of HBsAg (9). Clone 6C2 was maintained as described previously (9). 5 d after the last stimulation, the cells were washed, counted, and injected intravenously into HBV transgenic mice. Mice were killed at different time points after injection and their livers were perfused and harvested for histological, histochemical, and flow cytometry analyses, or they were snap frozen in liquid nitrogen and stored at −80°C for subsequent molecular analyses (see below).
Anticytokine and Antichemokine Abs.
Hamster mAbs H22 specific for murine IFN-γ (21) and control hamster IgG (Jackson ImmunoResearch Laboratories) were used. Mice were injected intraperitoneally (250 μg/mouse) 16 h before the intravenous injection of the CTLs. The rabbit polyclonal Abs specific for mouse (Crg2)IP-10 or Mig and the preimmune normal rabbit serum used in this study have been described previously (16, 17). 1 ml of either a cocktail of anti–(Crg2)IP-10 and anti-Mig rabbit Ig or normal rabbit preimmune serum (NRS) was administered intraperitoneally into HBV transgenic mice twice, first 16 h before and then simultaneously with the intravenous injection of the CTLs.
Tissue DNA and RNA Analyses.
Total DNA and RNA were isolated from frozen livers (left lobe) and analyzed for HBV DNA by Southern blot analysis and chemokine, chemokine receptor, cytokine, T lymphocyte, and macrophage marker mRNAs by RNase protection exactly as described previously (3, 8, 22). The relative abundance of specific DNA and RNA molecules was determined by PhosphorImaging analysis, using the Optiquant™ image analysis software (Packard Instrument Co.).
Biochemical and Histological Analyses.
The extent of hepatocellular injury was monitored by measuring serum alanine aminotransferase (sALT) activity at multiple time points after treatment. sALT activity was measured in a Paramax chemical analyzer (Baxter Diagnostics, Inc.) exactly as described previously (6). For histological analysis, liver was fixed in 10% zinc-buffered formalin (Anatech), embedded in paraffin, sectioned (3 μm), and stained with H&E (6). Quantitative morphometric analysis of the number and size of intrahepatic inflammatory foci (scored as apoptotic hepatocytes and inflammatory cells) was performed by counting ∼100 high power (original magnification: ×400) fields (hpf) representing 4 mm2 of liver tissue.
In Situ Hybridization.
This procedure was performed exactly as described previously (3). For (Crg2)IP-10, a cDNA fragment (726-bp) was synthesized and cloned in pGEM4 (Promega) as described previously (22). For Mig, a cDNA fragment spanning nucleotides-101–402 of the murine Mig gene (GenBank/EMBL/DDBJ M34815) was synthesized by RT-PCR and after sequence verification cloned in pGEM4. The 33[P]-labeled RNA probes used for in situ hybridization were generated by T7-driven transcription of 1 μg of linearized plasmids containing (Crg2)IP-10– or Mig-specific sequences.
Isolation and Analysis of the Intrahepatic Leukocytes.
Mouse livers were weighed at the time of autopsy. intrahepatic leukocytes (IHLs) were isolated from two liver lobes of a known weight and analyzed by flow cytometry. Single cell suspensions were prepared from the two liver lobes and analysis of the IHL population was performed by flow cytometry, exactly as described previously (references 23 and 24). The cells were surface stained with FITC- or phycoerythrin-labeled anti-CD4, anti-CD8, anti-CD3, anti-DX-5, anti-NK1.1, anti-CD19, anti-Gr-1, anti-CD11b, and anti-CD11c Abs (BD PharMingen) for the detection of NK1.1+/CD3− and DX5+ cells (NK cells), CD3+/NK1.1− cells (T cells), CD3+/NK1.1+ cells (NKT cells), CD8+ cells (mostly CTLs), CD4+ (mostly Th cells), CD11b−/CD11c+ (mostly lymphoid dendritic cells), CD11b+/CD11c+ cells (mostly myeloid dendritic cells), CD11b+/CD11c− (mostly macrophages), CD19+ (B cells), and Gr-1+ cells (mostly granulocytes). To detect transferred HBsAg-specific CD8+ CTL clones (6C2) in IHLs, cells were stained with anti-CD8-FITC and H-2(Ld)/HBs28–39 tetramer-PE (provided by John D. Altman, Emory University School of Medicine, Atlanta, GA). Samples were acquired on a FACSCalibur™ flow cytometer, and the data was analyzed using CELLQuest™ software (Becton Dickinson).
Quantitative Analyses of the Intrahepatic Content of the Transferred CTLs.
Male CTLs were injected into female transgenic mice and the animals were killed by 4 h after injection. Total liver DNA was extracted as described previously. Primers specific for the Sry gene contained in the Y chromosome (reference 25; sense: 5′-GCAGTTGCCTCAACAAAACTGT-3′; antisense: 5′-AGGTGTGCAGCTCTACTCCAG-3′) and primers specific for GAPDH (sense: 5′-GGAAAGCTGTGGCGTGAT-3′; antisense: 5′-CCAGTGAGCTTCCCGTTCAG-3′) were added to 250 ng of total liver DNA and triplicate of each samples were subjected to quantitative PCR in an iCycler apparatus (Bio-Rad Laboratories) using SYBR Green as fluorescent dye (26).
Results
In Vitro Characteristics of HBsAg-specific CTLs.
An H-2d restricted, CD8+ CTL clone (designed 6C2) derived from nontransgenic B10D2 mice (H-2d) that recognizes an epitope (IPQSLDSWWTSL) located between residues 28–39 of HBsAg was used in this study (9). As shown in Fig. 1, clone 6C2 produced high levels of IFN-γ and TNF-α mRNAs after a 4-h stimulation in vitro with plate-bound anti-CD3 mAbs. It is noteworthy that the in vitro and in vivo (see below) characteristics of clone 6C2 are representative of various numbers of several independently derived CD8+, H-2d-restricted, HBsAg-specific CTL clones, and lines described previously (6, 9, 27, 28). Similar to other independently derived CTL clones (data not shown), clone 6C2 did not express either (Crg2)IP-10 or Mig before or after in vitro activation (Fig. 1). Clone 6C2 was found, however, to express CXCR3, the common receptor for (Crg2)IP-10 and Mig, although its expression was strongly inhibited upon in vitro activation (Fig. 1). While it is formally possible that this pattern of cytokine, chemokine, and chemokine receptor expression may differ after CTL activation in the liver, these results indicate that these CTLs do not express (Crg2)IP-10 nor Mig but they do express IFN-γ, TNF-α, and CXCR3.
Figure 1.
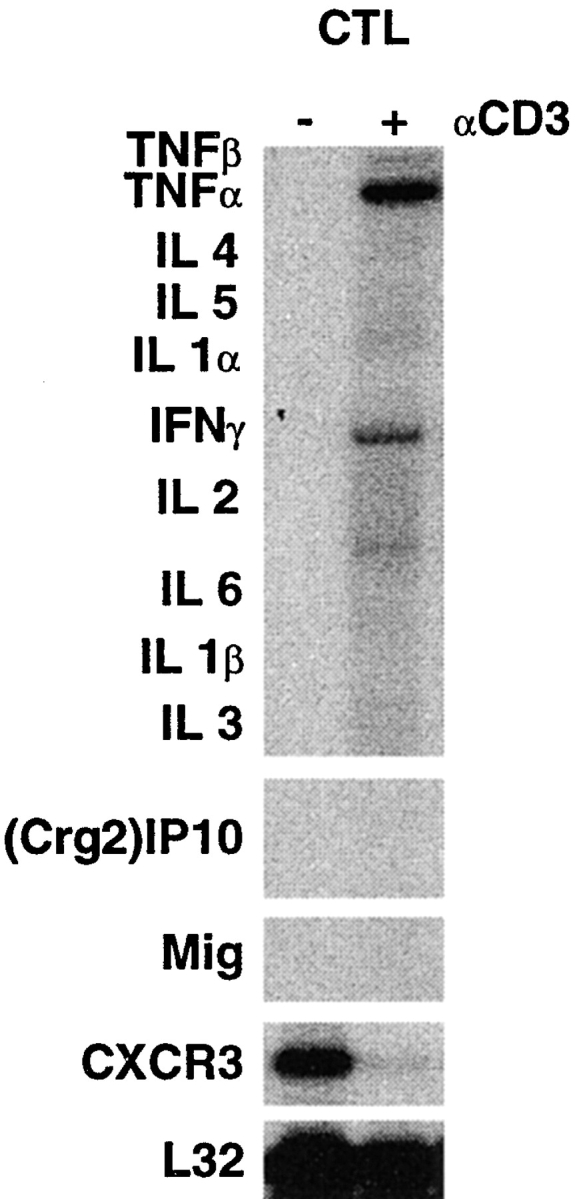
In vitro characteristics of HBsAg-specific CTLs. Total hepatic RNA from clone 6C2 before and after a 4-h stimulation in vitro with plate-bound anti-CD3 mAbs was analyzed by RNase protection assay (RPA) for the expression of various cytokines, (Crg2)IP-10, Mig, and CXCR3 as indicated. The RNA encoding the ribosomal protein L32 was used to normalize the amount of RNA loaded in each lane.
(Crg2)IP-10 and Mig Are Induced in the Liver of CTL-injected HBV Transgenic Mice.
To relate the kinetics of liver disease, antiviral effects, and cytokine expression with the kinetics of chemokine and chemokine receptor induction in the liver of CTL-injected HBV transgenic mice, eight groups (three mice per group) of age-, sex-, and serum HBeAg-matched transgenic mice from lineage 1.3.32 were injected intravenously with 107 HBsAg-specific CTLs (clone 6C2). Mice were bled and killed, and livers were harvested at the indicated time points (Fig. 2). The results were compared with those observed in livers from age-, sex-, and serum HBeAg-matched transgenic littermates injected with saline (NaCl) that were killed 2 d after NaCl injection (Fig. 2).
Figure 2.
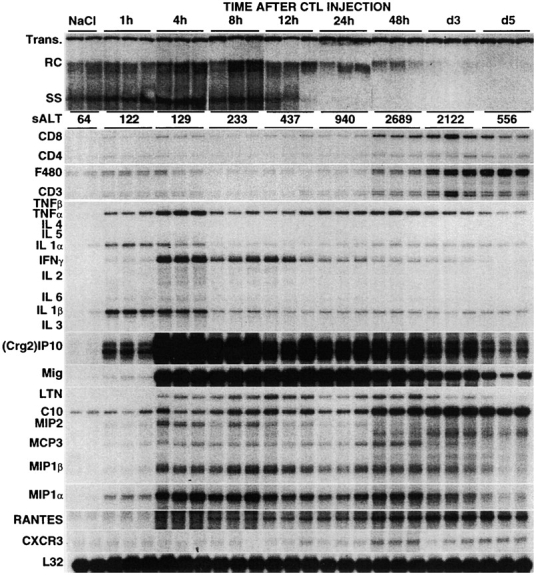
Kinetics of HBV replication, disease, and expression of cytokine, T cell and macrophage markers and chemokines in the liver of CTL-injected HBV transgenic mice. Age- and serum HBeAg–matched male transgenic mice (three mice per group) from lineage 1.3.32 were injected intravenously with 107 CTLs (clone 6C2), killed at the indicated time points, and total hepatic DNA was analyzed for HBV replication by Southern blot analyses. Bands corresponding to the integrated transgene, relaxed-circular (RC) and single-stranded (SS) linear HBV DNA replicative forms are indicated. The integrated transgene can be used to normalize the amount of DNA bound to the membrane. The mean sALT activity, measured at the time of autopsy, is indicated for each group and is expressed in U/l. Total hepatic RNA from the same mice was also analyzed by RPA for the expression of T cell (CD3, CD4, and CD8) and macrophage (F480) markers, various cytokines, chemokines, and CXCR3 as indicated. The RNA encoding the ribosomal protein L32 was used to normalize the amount of RNA loaded in each lane. Results were compared with those observed in livers pooled from 10 age-, sex-, and serum HBeAg–matched transgenic littermates injected with saline (NaCl).
After passive transfer of HBV-specific CTLs into HBV transgenic mice, the transient liver disease (monitored biochemically at multiple time points after injection as sALT activity, a hepatocellular enzyme that is released into the circulation by necrotic hepatocytes) reached maximum severity at days 2–3 (Fig. 2) and completely disappeared within 7 d (data not shown). Viral replication was inhibited within 24 h (Fig. 2). Very low levels of IL-1α and IL-1β (likely produced by Kupffer cells) were the only mRNA species detected in the liver from NaCl-injected controls (Fig. 1). Within 1 h after CTL injection, however, the mRNA levels for these cytokines along with the mRNAs for IFN-γ and TNF-α (likely produced by the CTLs) increased substantially, reaching their peak by 4 h (Fig. 2). The induced transcripts remained elevated for at least 72 h and they decreased toward baseline levels by day 5 (Fig. 2). The kinetics and magnitude of the changes in IFN-γ and TNF-α gene expression reflect the kinetics of CTL entry into the liver (maximal at 4–12 h; reference 5) and the recognition of antigen by these cells. It is also noteworthy that the signal for the macrophage marker F480 increased by 1 h and this was concomitant with the increase of proinflammatory cytokines such as IL-1α, IL-1β, IFN-γ, and TNF-α. This could be due to either the induction of F480 expression on activated resident macrophages (Kupffer cells) or the rapid recruitment of circulating macrophages into the liver.
The intensity of the signal for T cell (CD8, CD4, and CD3) and macrophage (F4–80) markers was virtually maximal at 48–72 h (Fig. 2), indicating that host-derived inflammatory cells were recruited into the liver at these later time points. Since the severity of liver disease was also maximal at 48–72 h (Fig. 2), these results suggest that other cells besides the transferred CTLs contribute to most of the liver disease.
As shown in Fig. 2, the intrahepatic messages for (Crg2)IP-10 and Mig were rapidly (within 4 h) and strongly induced (>200- and 1,000-fold, respectively, when compared by PhosphorImaging analysis with preinjected levels; note that the indicated bands for (Crg2)IP-10 and Mig correspond to an exposure time of the autorad of 16 and 2 h, respectively). Since neither (Crg2)IP-10 nor Mig were produced by HBV-specific CTLs (Fig. 1), these results indicate that other cells in the liver may represent the source of these chemokines in our system. With similar kinetics but lower levels of induction, we also detected several C-C chemokines, including macrophage inflammatory protein (MIP)-1α, MIP-1β, and MIP-2 (chemoattractants for lymphocytes and macrophages), monocyte chemotactic protein (MCP)-1 and MCP-3 (chemoattractants for monocytes/macrophages) and lymphotactin (chemoattractant for lymphocytes and NK cells; Fig. 2).
The C-C chemokines C10 and regulated on activation, normal T cell expressed and secreted (RANTES; chemoattractants for monocytes/macrophages) showed a more delayed peak of induction that was maximal by days 2–5 (Fig. 2), indicating that other factors beside the activation of the transferred CTLs contributed to their induction.
To determine which cells produce (Crg2)IP-10 and Mig, the hepatic content of both chemokine mRNAs was analyzed by in situ hybridization analysis of livers derived from HBV transgenic animals killed at various time points after CTL injection. Interestingly, as shown in Fig. 3 for two representative livers harvested 4 h after CTL injection, (Crg2)IP-10 (Fig. 3 D) and Mig (Fig. 3 B) mRNAs were detected not only in nonparenchymal cells (Kupffer cells and infiltrating inflammatory cells, arrowheads) but, even more abundantly, in parenchymal cells (hepatocytes) of the liver. In keeping with the fact that the intrahepatic levels of Mig mRNA were five times more abundant than those for (Crg2)IP-10 (Fig. 2), it is not surprising that a higher percentage of liver cells were positive for the former chemokine (Fig. 3 B). It is also noteworthy that (Crg2)IP-10 mRNA was more abundant in liver cells (particularly hepatocytes) surrounding small inflammatory foci (Fig. 3 D), while Mig mRNA was more homogeneously distributed throughout the organ (which was more easily observed at shorter exposures, not shown). No signal for (Crg2)IP-10 mRNA was detected in the liver of NaCl-injected control mice that were killed by 4 h after injection (Fig. 3 C). Very low levels of Mig mRNA were observed in similar control animals which were almost exclusively detected within hepatocytes (Fig. 3 A) that surrounded rare, small foci of Mig-mRNA negative inflammatory cells (Fig. 3 A, arrows). No expression of either chemokine was detected in apoptotic hepatocytes (asterisks, Fig. 3 B and D). Finally, a similar induction of (Crg2)IP-10 and Mig was observed at the 24 and 48 h time points (data not shown). All together, these results indicate that the hepatocytes represent a major source of (Crg2)IP-10 and Mig mRNAs in the CTL-injected livers.
Figure 3.
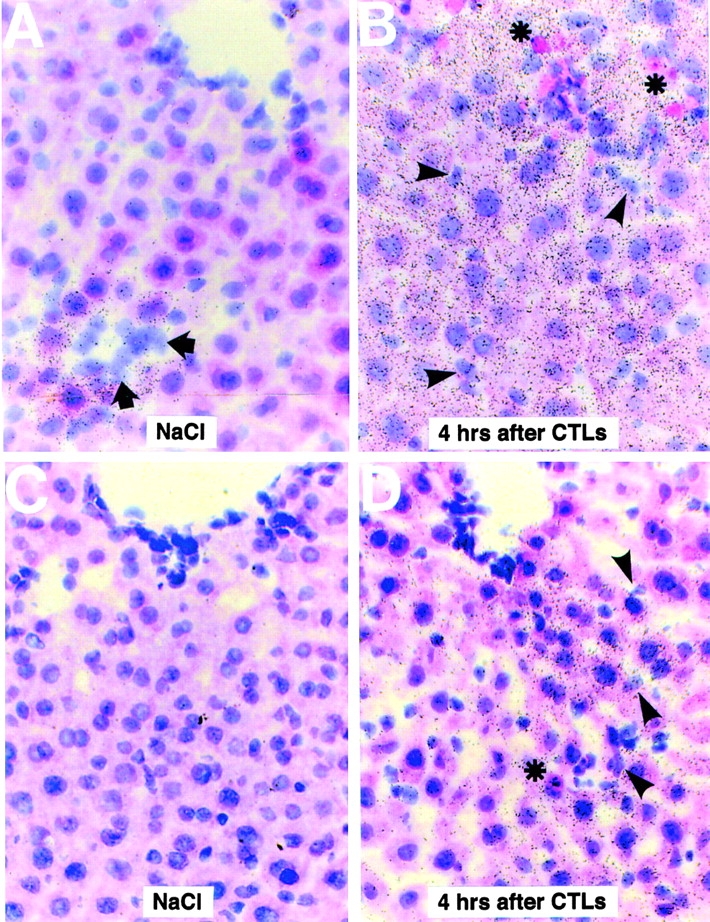
Intrahepatic distribution of (Crg2)IP-10 and Mig mRNA in NaCl- and CTL-injected livers. Livers from HBV transgenic mice (lineage 1.3.32) that were injected 4 h earlier either with saline (NaCl) or HBV-specific CTLs were analyzed for the expression of Mig (A and B) or (Crg2)IP-10 (C and D) mRNA by in situ hybridization using specific 33[P]-labeled riboprobes. Note that most Mig- or (Crg2)IP-10 RNA is contained in the hepatocytes of CTL-injected livers (B and D). Mig- or (Crg2)IP-10–positive nonparenchymal cells (arrowheads, B and D) and Mig- or (Crg2)IP-10–negative apoptotic hepatocytes (asterisks, B and D) and inflammatory cells (arrows, A) are also indicated (H&E; original magnification: ×400).
The Intrahepatic Induction of (Crg2)IP-10 and Mig Is Mediated by IFN-γ Produced by the Transferred CTLs.
Previous studies have shown that IFN-γ is a powerful inducer of both (Crg2)IP-10 and Mig (11). To determine whether the induction of (Crg2)IP-10 and Mig was mediated by IFN-γ in our system as well, we monitored the ability of hamster mAbs specific for IFN-γ to modulate the intrahepatic expression of these two chemokines after CTL injection. Two groups (three mice per group) of age-, sex-, and serum HBeAg–matched transgenic mice (lineage 1.3.32) were injected with either IFN-γ mAb or irrelevant hamster IgG prior transfer of 107 HBsAg-specific CTLs (clone 6C2) and the mice were killed 24 h after CTL administration. As shown in Fig. 4 (note that the indicated bands for (Crg2)IP-10 and Mig correspond to an exposure time of the autorad of 16 and 2 h, respectively), the induction of (Crg2)IP-10 and Mig was almost completely blocked in two representative mice that received Abs to IFN-γ. The levels of (Crg2)IP-10 were induced ∼120-fold (when compared by phosphor imaging analysis with NaCl-injected control mice) and treatment with IFN-γ mAb reduced this increase by 24-fold. These results indicate that the induction of these chemokines depends on IFN-γ.
Figure 4.

The intrahepatic induction of (Crg2)IP-10 and Mig is mediated by IFN-γ produced by the transferred CTLs. (Left panel) Age- and serum HBeAg–matched male transgenic mice (lineage 1.3.32) were intraperitoneally injected with 250 μg of hamster mAbs to IFN-γ and killed 24 h after CTL administration. Control mice (Irr Ab) were simultaneously injected with 250 μg of irrelevant hamster IgG before CTL transfer and killed at the same time after CTL administration. (right panel). Age- and serum HBeAg–matched male transgenic mice (lineage 1.3.46) that were either heterozygous (+/−) or homozygous (−/−) for the IFN-γ null mutation were injected with 107 CTLs (clone 6C2) and killed 24 h later. Total hepatic RNA was analyzed for (Crg2)IP-10, Mig, and CXCR3 as indicated. The RNA encoding the ribosomal protein L32 was used to normalize the amount of RNA loaded in each lane. Results were compared with those observed in livers pooled from 10 age-, sex-, and serum HBeAg–matched transgenic littermates injected with saline (NaCl).
To determine the relative contribution of the transferred CTLs versus host-derived inflammatory cells in the IFN-γ–dependent induction of (Crg2)IP-10 and Mig, we examined the expression levels of these chemokines in CTL-injected livers from HBV transgenic mice that were crossed with mice genetically deficient for IFN-γ. Two groups (three mice per group) of age- (8–10 wk), sex- (male), and serum HBeAg–matched animals that were either heterozygous (+/−) or homozygous (−/−) for the IFN-γ null mutation were injected with 107 IFN-γ–producing HBsAg-specific CTLs (clone 6C2) and killed 24 h later. As shown in Fig. 4 for two representative mice per group, the intrahepatic levels of (Crg2)IP-10 and Mig mRNAs were comparable in both groups of mice, indicating that the amount of IFN-γ produced by passively transferred CTLs mediates the induction of these two chemokines.
(Crg2)IP-10 and Mig only Partially Mediate the Recruitment of HBV-specific CTLs.
To define the role of (Crg2)IP-10 and Mig in the recruitment of transferred HBV-specific CTLs into the liver, we monitored the ability of anti–(Crg2)IP-10 and anti-Mig neutralizing Abs to modulate this process. The recruitment of the passively transferred clone 6C2 (originally produced in male mice; reference 9) was measured by quantifying the amount of Sry-specific sequences (Sry is a gene contained in the Y chromosome (25)) in the liver of HBV transgenic female mice (lineage 1.3.32) using real time PCR. The recruitment of the transferred CTLs was also measured by isolating IHLs and quantifying the number of tetramer-specific CTLs using FACS® analysis. 1 ml of either a cocktail of anti–(Crg2)IP-10 and anti-Mig neutralizing rabbit Ig or NRS was administered intraperitoneally into two groups (three mice per group) of age- (8–10 wk) and serum HBeAg–matched female transgenic mice (lineage 1.3.32) twice, first 16 h before and then simultaneously with the transfer of 5 × 106 HBsAg-specific CTLs (clone 6C2). Mice were killed 4 h after CTL transfer and total liver DNA was extracted.
250 ng of total liver DNA derived from each mouse were subjected to quantitative real time PCR using Sry-specific primers. GAPDH-specific primers were used to normalize the amount of input DNA. Sry-specific amplicons were detected at similar threshold cycles (<1 cycle difference) in mice that received either control or antichemokine Abs (data not shown). Since this assay discriminates differences of twofold or more, these results indicate that the number of CTLs that reached the liver at 4 h after injection was within a twofold difference between the two groups of mice. Similar results were obtained when the number of tetramer-positive CTLs was quantitated in these same livers. Tetramer-positive CTLs accounted for 0.97 and 1.3% of the total IHL recovered from the liver of mice that received either control or antichemokine Abs, respectively. These conclusions are also supported by the finding that the levels of IFN-γ mRNA (a marker of antigen recognition by CTLs) in control mice were induced only about twofold more than those observed in the liver of the mice treated with antichemokine Abs. (Fig. 5). This reiterates the fact that the recruitment of the passively transferred CTLs was only partially dependent on (Crg2)IP-10 and Mig.
Figure 5.

(Crg2)IP-10 and Mig activity do not mediate the antiviral potential of HBV-specific CTLs but mediate part of the accompanying liver disease. 1 ml of either a cocktail of anti–(Crg2)IP-10 and anti-Mig neutralizing rabbit Ig or NRS was administered intraperitoneally into six groups (three mice per group) of age- (8–10 wk), sex-, and serum HBeAg–matched transgenic mice (lineage 1.3.32) twice, first 16 h before and then simultaneously with the transfer of HBsAg-specific CTLs (clone 6C2). Mice were bled and killed, and livers were harvested 4, 24, or 48 h later. Total hepatic DNA was analyzed for HBV replication by Southern blot analyses. Bands corresponding to the integrated transgene, relaxed-circular (RC) and single-stranded (SS) linear HBV DNA replicative forms are indicated. The integrated transgene can be used to normalize the amount of DNA bound to the membrane. The mean sALT activity, measured at the time of autopsy, is indicated for each group and is expressed in U/l. Total hepatic RNA from the same mice was also analyzed by RPA for the expression of various cytokines as indicated. The RNA encoding the ribosomal protein L32 was used to normalize the amount of RNA loaded in each lane. Results were compared with those observed in livers pooled from 10 age-, sex-, and serum HBeAg–matched transgenic littermates injected with saline (NaCl).
(Crg2)IP-10 and Mig Do Not Mediate the Antiviral Potential of HBV-specific CTLs.
To define the role of (Crg2)IP-10 and Mig in the antiviral potential of HBV-specific CTLs into the liver, we monitored the ability of anti–(Crg2)IP-10 and anti-Mig neutralizing Abs to modulate the CTL-dependent antiviral effect.
As before, 1 ml of either a cocktail of anti–(Crg2)IP-10 and anti-Mig neutralizing rabbit Ig or NRS was administered intraperitoneally into four groups (three mice per group) of age- (8–10 wk), sex-, and serum HBeAg-matched transgenic mice (lineage 1.3.32) twice, first 16 h before and then simultaneously with the transfer of 2.5 × 106 HBsAg-specific CTLs (clone 6C2). Mice were bled and killed, and livers were harvested 24 and 48 h later and results were compared with those observed in livers pooled from 10 age-, sex-, and serum HBeAg–matched transgenic littermates injected with saline (NaCl).
As shown in Fig. 5, simultaneous administration of (Crg2)IP-10 and Mig Abs did not block the CTL-induced inhibition of hepatic HBV replication observed at 24 and 48 h that was also detected in control mice that received NRS. These results indicate that (Crg2)IP-10 and Mig do not directly inhibit HBV replication themselves nor do they regulate the antiviral potential of HBV-specific CTLs in our system. The fact that HBV replication was inhibited in this experiment in association with very modest elevations of sALT reiterate the fact that the CTL-dependent antiviral effect is not due to the destruction of hepatocytes (6, 7).
(Crg2)IP-10 and Mig Mediate the Recruitment of Antigen-nonspecific Host-derived Inflammatory Cells that Are Responsible for Most of the Liver Disease.
Importantly, treatment with antichemokine Abs diminished the severity of the liver disease, as measured biochemically or histologically, thus indicating the neutralization ability of these antibodies in our in vivo system. At the time of autopsy, the sALT activity of mice that received antichemokine Abs was reduced two- to threefold when compared with control animals at different time points (Fig. 5). Since in this experiment HBV transgenic mice (those killed at 24 and 48 h) received a lower number of CTLs (2.5 × 10 6) than the mice shown in Fig. 2 (107), it is not surprising that sALT levels (in control animals) were also lower: mean = 479 ± 91 (Fig. 5) versus mean = 940 ± 146 (Fig. 2) at 24 h and mean = 374 ± 83 (Fig. 5) versus mean = 2686 ± 339 (Fig. 2) at 48 h. This is consistent with previous reports showing that in this system sALT elevation is CTL dose dependent (6, 9).
As shown in Fig. 6 (top), histological analysis of the livers from animals treated with control Abs demonstrated necroinflammatory foci scattered throughout the liver parenchyma, containing apoptotic hepatocytes (asterisks), lymphomononuclear (arrows and arrowheads), and polymorphonuclear cells. Antichemokine treatment did not significantly reduce the number of necroinflammatory foci (scored as apoptotic hepatocytes and inflammatory cells) by 24 and 48 h after CTL injection, as measured by quantitative morphometric analysis (79± 7.6 [24 h] and 92 ± 12.6 [48 h] in Ab-treated mice versus 91± 11.2 [24 h] and 109 ± 13.2 [48 h] in control animals: these numbers represent the mean ± SEM per 100 high power fields, corresponding to ∼4 mm2 of liver tissue). This again indicates that similar numbers of CTLs reached the liver in both groups of animals. Importantly, however, antichemokine treatment reduced the size of necroinflammatory foci, both by 24 (data not shown) and 48 h (Fig. 6, right panels) after CTL injection. The average area of these foci (2.2 ± 1.3 × 10−3 mm2) was about twofold smaller than that observed in control mice (4.0 ± 2.2 × 10−3 mm2) as measured by morphometric analysis. Many fewer lymphomononuclear cells (Fig. 6, top right panel, arrows and arrowheads) were detectable in the foci, while the number of polymorphonuclear cells was little or not reduced (Fig. 6, top right panel).
Figure 6.
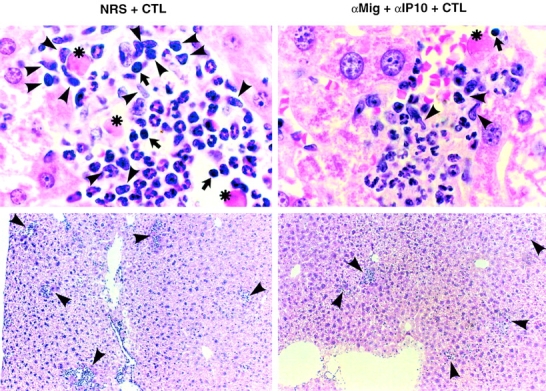
Passive neutralization of (Crg2)IP-10 and Mig reduced the recruitment of lymphomononuclear cells and the size of the intrahepatic inflammatory foci. (Top) Histological analysis of the necroinflammatory foci detected in the livers from animals treated either with control Abs (NRS, left) or antichemokine Abs (αMig + αIP-10, right) that were killed 48 h after CTL transfer. Cells displaying the histological features of apoptotic hepatocytes (asterisks), lymphocytes (arrows), and macrophages (arrowheads) are indicated. Cells displaying the histological features of polymorphonuclear cells are not indicated. Note that fewer apoptotic hepatocytes and lymphomononuclear cells were detectable in the foci of mice treated with antichemokine Abs (right). (Bottom) Histological analysis of the same livers at lower magnification displaying several necroinflammatory foci (arrowheads). Original magnifications: ×1,000 (top) and ×100 (bottom).
To determine the characteristics of the intrahepatic inflammatory infiltrate in the same livers, the absolute number of IHLs recovered was quantitated, and the phenotype of the recruited inflammatory cell subsets was determined by FACS® analysis. Two additional groups of transgenic control mice (three mice per group) were injected with NaCl alone to produce a baseline for the analysis of the intrahepatic infiltrate by flow cytometry.
When compared with NaCl-injected controls (Fig. 7, gray bars), the total number of IHLs as well as most cell subsets significantly increased in the liver of CTL-treated mice that received control Abs (Fig. 7, white bars). The total number of IHLs increased over fivefold at 24 and 48 h after CTL injection, corresponding with a commensurate increase in: (i) NK1.1+/CD3− and DX5+ cells (NK cells) (seven- to eightfold and 10–12-fold at 24 and 48 h, respectively); (ii) CD3+/NK1.1− cells (T cells) (about eightfold at both time points); (iii) CD8+ cells (mostly CTLs) (about five- and eightfold at 24 and 48 h, respectively); (iv) CD4+ (mostly Th cells) (∼5 and 3.5-fold at 24 and 48 h, respectively); (v) CD11b−/CD11c+ (mostly lymphoid dendritic cells) (about threefold at both time points); (vi) CD11b+/CD11c+ cells (mostly myeloid dendritic cells) (∼10- and 16-fold at 24 and 48 h, respectively); (vii) CD11b+/CD11c− (mostly macrophages) (about eight at both time points); (viii) CD19+ (B cells) (almost twofold at both time points) and (ix) Gr-1+ cells (mostly granulocytes) (∼10-fold at both time points). It is also noteworthy that the number of NKT cells was reduced two- to threefold in CTL-injected livers (with and without antichemokine treatment) when compared with the number of NKT cells isolated from NaCl-injected control animals (data not shown). This presumably reflects activation induced cell death of NKT cells, as previously shown in other systems (29, 30). As also shown in Fig. 7, antichemokine treatment reduced the number of total IHLs recruited (black bars) by ∼2.5 and 1.7 fold at 24 and 48 h, respectively. Along with this, the number of all cell subsets recruited was lower in mice that were killed at 24 h and received antichemokine Abs (Fig. 7, black bars). The most pronounced decreases were observed for NK cells, myeloid dendritic cells, and CTLs (all of which are known to express CXCR3; references 31–33). The number of Th cells, macrophages, lymphoid dendritic cells, B cells, and granulocytes also decreased, but to a lesser extent (Fig. 7). By 48 h, very little difference in the number of Th cells and granulocytes was observed between the two groups, while a substantial difference remained for all other subsets (Fig. 7). All together, these results indicate that in association with the reduction of sALT elevation (Fig. 5) and the size of the inflammatory foci (Fig. 6), passive neutralization of (Crg2)IP-10 and Mig reduced the recruitment of most cell subsets and particularly those (NK cells, myeloid dendritic cells, and CTLs) known to express CXCR3.
Figure 7.
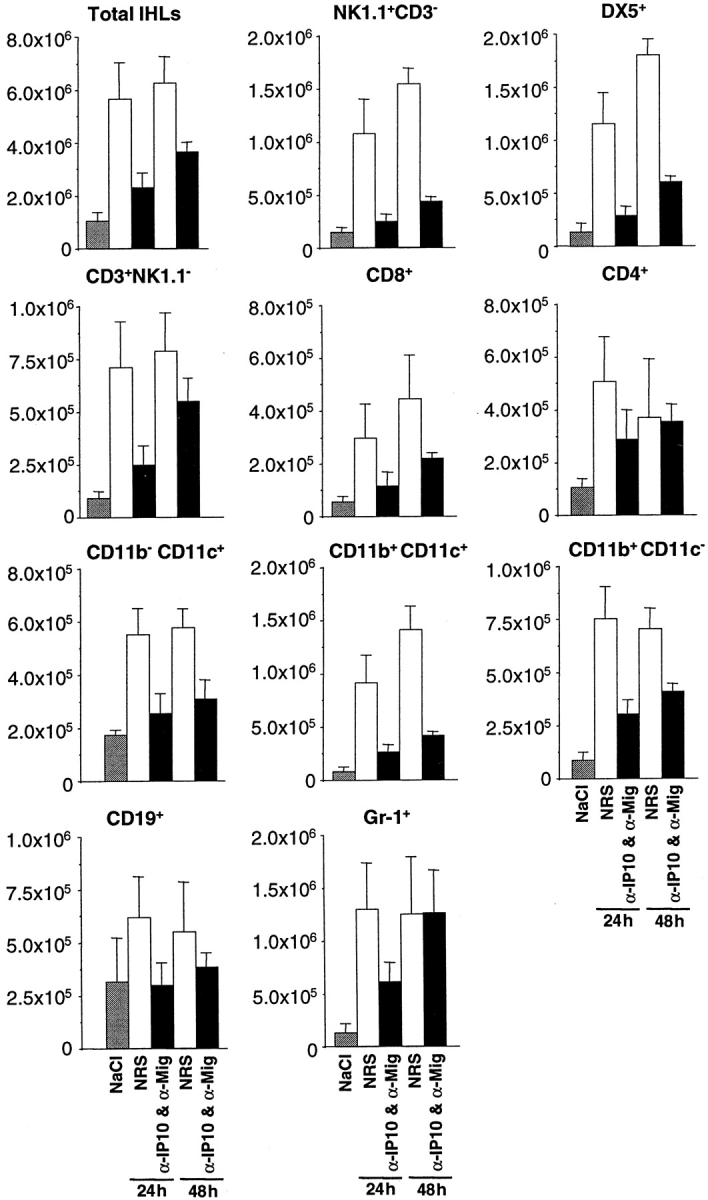
Passive neutralization of (Crg2)IP-10 and Mig reduced the recruitment of most cell subsets and particularly lymphomononuclear cells such as NK cells, myeloid dendritic cells, and CTLs. IHLs analysis in the same animals described in the legend to Fig. 5. Livers were weighed at the time of autopsy. IHLs were isolated from two liver lobes of a known weight and analyzed by flow cytometry. The indicated numbers of total IHLs and different cell subsets represent the numbers detected in the whole liver.
Discussion
In this study we showed that after the passive transfer of HBV-specific CTLs into transgenic mice that replicate HBV at high levels in their hepatocytes, chemokines such as (Crg2)IP-10 and Mig were rapidly and strongly induced in the liver. Interestingly, the transferred CTLs did not produce either chemokine. Rather, upon antigen recognition, they secreted IFN-γ, which in turn activated both nonparenchymal and, especially, parenchymal cells of the liver to express high levels of (Crg2)IP-10 and Mig. The notion that hepatocytes represent a major source of these chemokines in the inflamed liver is supported by a recent study that showed IFN-γ–dependent expression of (Crg2)IP-10 and Mig in primary hepatocytes derived from IL-2/IL-12 treated mice (13). These observations may also shed some light into the pathogenesis of other liver conditions (i.e., ischemia/reperfusion, biliary atresia, liver cancer, protozoan infections) where the hepatic expression of IP-10 and Mig has been detected (14, 34–37). All together, these results suggest that the hepatocyte (the cell type that sustains viral replication during natural infection; reference 38) may play an important role in the recruitment of inflammatory cells into the liver and, perhaps, in its own antiviral defense. Given that during natural infection the peak influx of T cells into the organ is delayed by several weeks after the peak of HBV replication (39), it is also tempting to speculate that the virus may interfere with the production of these chemokines in order to counteract the antiviral effector functions of immune cells. Future studies aimed at monitoring the intrahepatic profiles of chemokine expression in infected human or chimp livers will address this interesting hypothesis.
Passive neutralization of (Crg2)IP-10 and Mig in vivo did not significantly reduce the recruitment of the transferred CTLs and the intrahepatic induction of IFN-γ was reduced by about twofold at the 4-h time point. The notion that the early recruitment of the transferred CTLs was not particularly affected by neutralization of (Crg2)IP-10 and Mig may be due to the fact that other chemokine–chemokine receptor interactions were still operative in our system. In support of this, we found that in addition to CXCR3 (the receptor specific for (Crg2)IP-10 and Mig, Fig. 1) the transferred CTLs expressed CCR1, CCR2, CCR4, CCR5, and CCR7 (data not shown) and ligands specific for these receptors (MIP-1α, MIP-1β, RANTES, MCP-1, and MCP3) were induced in the CTL-injected livers (Fig. 2). It is also noteworthy that the expression of CXCR3 by the HBV-specific CTLs was strongly inhibited upon in vitro activation of (Fig. 1). This effect may explain the lack of intrahepatic detection of CXCR3 mRNA into the liver at the early time points (4–12 h) after CTL transfer (Fig. 2), when CTL entry into the liver and the recognition of antigen by these cell is maximal. Inhibition of chemokine receptor mRNA expression after in vitro activation has also been reported for influenza-virus specific CTLs (40) and it is thought to play an important role in the regulation of the chemokine system (41). Lack of detection of intrahepatic CXCR3 mRNA at the early time points could also be due to the relative lower abundance of this message in the total hepatic RNA population as compared with its higher abundance in the CTL-derived RNA population described in Fig. 1. At any rate, it is worth mentioning that the message for CXCR3 increased at later time points (24–72 h), probably reflecting the recruitment of host-derived, antigen-nonspecific CXCR3+-inflammatory cells.
The extent to which the recruitment of the transferred CTLs was affected by the passive neutralization of (Crg2)IP-10 and Mig at later time points (24 and 48 h) was not directly measured. However, no difference in the intrahepatic levels of IFN-γ and TNF-α mRNAs was observed between antichemokine Ab- and NaCl-treated mice (Fig. 5). Again, since the intrahepatic expression of these cytokines likely reflect the entry of the IFN-γ- and TNF-α–expressing transferred CTLs into liver and the recognition of antigen by these cells, these results suggest that (Crg2)IP-10 and Mig did not mediate the recruitment of the transferred CTLs at these time points. In keeping with this, the number of hepatic inflammatory foci was not significantly reduced and HBV replication was still strongly inhibited in the animals that received antichemokine Abs (Fig. 5). These results also indicate that (Crg2)IP-10 and Mig do not have a direct antiviral activity against HBV and they do not mediate the antiviral potential of HBV-specific CTLs in our system. Pertinent to this, it is worth mentioning that while recent studies in vivo have demonstrated a role for (Crg2)IP-10 and Mig in controlling the replication of viruses such as vaccinia virus (15) mouse hepatitis virus (16, 17) and MCMV (18), they didn't demonstrate that those chemokines directly inhibited viral replication, since these viruses can reinfect potentially cured cells and obscure the observations. In our studies, however, the lack of a direct antiviral potential of (Crg2)IP-10 and Mig against HBV could be demonstrated because HBV is not infectious for mice and it is produced by each hepatocyte without spreading from cell to cell (3).
Importantly, passive neutralization of (Crg2)IP-10 and Mig significantly reduced the recruitment of lymphomononuclear cells and the size of the intrahepatic inflammatory foci (Fig. 6) as well as the severity of the CTL-dependent liver disease (Fig. 5). The notion that viral replication was strongly inhibited in antichemokine Ab-treated animals in which very little liver disease was observed (Fig. 5) reiterates the fact that the antiviral potential of the CTLs is mainly mediated by cytokine-dependent, noncytolytic mechanisms, as demonstrated previously (6–8, 42).
The recruitment of most cell subsets and particularly those (NK cells, myeloid dendritic cells, and CTLs) known to express CXCR3 (31–33) was inhibited in antichemokine Ab-treated animals by 5–10-fold at the 24 and 48 h time points (Fig. 7). The recruitment of Th cells was also reduced, albeit to a lesser extent (less than twofold by 24 h and virtually no reduction by 48 h, Fig. 7). Since Th cells are known to express CXCR3 (43, 44), these results may suggest that other chemoattractant factors play a more important role in the recruitment of these cells into the liver. Similar reasons may explain the partially reduced recruitment of cells not known to express CXCR3 (macrophages and granulocytes) which was mostly observed by 24 h (Fig. 7).
The association of reduced liver disease with reduced recruitment of host-derived, antigen-nonspecific inflammatory cells into the liver implies that these cells can amplify the liver damage initiated by the antigen-specific CTLs. This conclusion is also supported by the fact that the kinetics and magnitude of liver disease (maximal at 48 h, Fig. 2) were delayed compared with the peak of intrahepatic expression of IFN-γ and TNF-α (maximal at 4 h, Fig. 2), which, as suggested before, are likely to mirror the entry into the liver of antigen-specific CTLs and the recognition of antigen by these cells. The pathogenetic mechanisms whereby antigen-nonspecific inflammatory cells may induce liver damage are not understood. Future studies will attempt to address this important issue. It is also noteworthy that the HBV transgenic mice used in this study are profoundly immunologically tolerant to the virus, particularly at the T cell level (4). This notion coupled with the short term nature of the experiments herein described makes extremely unlikely that host-derived antigen-specific T cells contributed to the pathogenesis of liver disease in our system.
In conclusion, we found that blocking (Crg2)IP-10 and Mig in vivo reduces the recruitment of host-derived inflammatory cells (particularly lymphomononuclear cells) into the liver and the severity of the liver disease without affecting the IFN-γ–dependent antiviral potential of antigen-specific CTLs. We conclude that target organ production of (Crg2)IP-10 and Mig leads to an exaggerated recruitment of host-derived inflammatory cells to the liver which play an important role in the pathogenesis of liver disease in our system. Similar mechanisms may contribute to the pathogenesis of viral hepatitis in man, where, like in our system, the number of HBV-specific T cells detected in the liver is outnumbered by recruited nonvirus-specific T cells (2, 45) and other inflammatory cells (46). Since the transgenic mice used in this study are not infected by HBV, it is possible that during natural infection CTL-independent mechanisms (i.e., intrahepatic activation of NK and/or NK T cells) could induce IFN-γ and initiate the cascade of pathogenetic events that results in the recruitment of antigen nonspecific inflammatory cells to the liver. However, it must be noted that the IFN-γ–dependent secondary processes that lead to the intrahepatic recruitment of inflammatory cells during natural infection are likely to be quite similar to those described in this study, irrespective of the nature of the cell type that originally produces this cytokine. In keeping with this, it is worth mentioning that the intrahepatic expression of IP-10 and Mig is induced during chronic HCV infection along with an increased number of liver infiltrating CXCR3-expressing T cells (20). Detection of HCV-specific CXCR3+ CTLs has been associated with phases of enhanced liver disease in HCV-infected patients (47). Based on the studies herein described, the notion that neutralization of IP-10 and Mig is associated with maintenance of antiviral effects but diminished tissue damage may help the design of potential immunotherapeutic approaches for the treatment of HBV infection in chronically infected patients.
Acknowledgments
We thank Timothy Stewart for providing IFN-γ−/− mice; Robert Schreiber for providing anti–IFN-γ antibodies; John D. Altman for providing H-2(Ld)/HBs28-39 tetramer-PE; Monte Hobbs for providing the cytokine gene and T cell marker probe sets used in the RNase protection assays; and Heike Mendez, Rick Koch, and Margie Chadwell for excellent technical assistance.
This work was supported by grants AI40696 (to L.G. Guidotti), CA40489 (to F.V. Chisari), MH50426 (to I.L. Campbell), and NS37336 (to T.E. Lane) from the National Institutes of Health and grant RG30393A1/T (to T.E. Lane) from the National Multiple Sclerosis Society. V.C. Asensio was a postdoctoral fellow of the National Multiple Sclerosis Society. This is manuscript number 14075-MEM from the Scripps Research Institute.
Footnotes
Abbreviations used in this paper: (Crg2)IP-10, chemokine responsive to γ–2/IFN-γ inducible protein; HBV, hepatitis B virus; HBeAg, hepatitis Be antigen; HCV, hepatitis C virus; IHL, intrahepatic lymphocyte; MCP, monocyte chemotactic protein; Mig, monokine induced by IFN-γ; MIP, macrophage inflammatory protein; NRS, normal rabbit preimmune serum; RPA, RNase protection assay; sALT, serum alanine aminotransferase.
References
- 1.Chisari, F.V., and C. Ferrari. 1995. Hepatitis B virus immunopathogenesis. Annu. Rev. Immunol. 13:29–60. [DOI] [PubMed] [Google Scholar]
- 2.Maini, M.K., C. Boni, C.K. Lee, J.R. Larrubia, S. Reignat, G.S. Ogg, A.S. King, J. Herberg, R. Gilson, A. Alisa, et al. 2000. The role of virus-specific CD8+ cells in liver damage and viral control during persistent hepatitis B virus infection. J. Exp. Med. 191:1269–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guidotti, L.G., B. Matzke, H. Schaller, and F.V. Chisari. 1995. High level hepatitis B virus replication in transgenic mice. J. Virol. 69:6158–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shimizu, Y., L.G. Guidotti, P. Fowler, and F.V. Chisari. 1998. Dendritic cell immunization breaks cytotoxic T lymphocyte tolerance in hepatitis B virus transgenic mice. J. Immunol. 161:4520–4529. [PubMed] [Google Scholar]
- 5.Ando, K., L.G. Guidotti, S. Wirth, T. Ishikawa, G. Missale, T. Moriyama, R.D. Schreiber, H.J. Schlicht, S. Huang, and F.V. Chisari. 1994. Class I restricted cytotoxic T lymphocytes are directly cytopathic for their target cells in vivo. J. Immunol. 152:3245–3253. [PubMed] [Google Scholar]
- 6.Guidotti, L.G., T. Ishikawa, M.V. Hobbs, B. Matzke, R. Schreiber, and F.V. Chisari. 1996. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 4:25–36. [DOI] [PubMed] [Google Scholar]
- 7.Guidotti, L.G., and F.V. Chisari. 2001. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu. Rev. Immunol. 19:65–91. [DOI] [PubMed] [Google Scholar]
- 8.McClary, H., R. Koch, F.V. Chisari, and L.G. Guidotti. 2000. Relative sensitivity of hepatitis B virus and other hepatotropic viruses to the antiviral effects of cytokines. J. Virol. 74:2255–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ando, K., T. Moriyama, L.G. Guidotti, S. Wirth, R.D. Schreiber, H.J. Schlicht, S. Huang, and F.V. Chisari. 1993. Mechanisms of class I restricted immunopathology. A transgenic mouse model of fulminant hepatitis. J. Exp. Med. 178:1541–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zlotnik, A., and O. Yoshie. 2000. Chemokines: a new classification system and their role in immunity. Immunity. 12:121–127. [DOI] [PubMed] [Google Scholar]
- 11.Rossi, D., and A. Zlotnik. 2000. The biology of chemokines and their receptors. Annu. Rev. Immunol. 18:217–242. [DOI] [PubMed] [Google Scholar]
- 12.Price, D.A., P. Klenerman, B.L. Booth, R.E. Phillips, and A.K. Sewell. 1999. Cytotoxic T lymphocytes, chemokines and antiviral immunity. Immunol. Today. 20:212–216. [DOI] [PubMed] [Google Scholar]
- 13.Park, J.W., M.E. Gruys, K. McCormick, J.K. Lee, J. Subleski, J.M. Wigginton, R.G. Fenton, J.M. Wang, and R.H. Wiltrout. 2001. Primary hepatocytes from mice treated with IL-2/IL-12 produce T cell chemoattractant activity that is dependent on monokine induced by IFN-γ (Mig) and chemokine responsive to γ-2 (Crg-2). J. Immunol. 166:3763–3770. [DOI] [PubMed] [Google Scholar]
- 14.Amichay, D., R.T. Gazzinelli, G. Karupiah, T.R. Moench, A. Sher, and J.M. Farber. 1996. Genes for chemokines MuMig and Crg-2 are induced in protozoan and viral infections in response to IFN-gamma with patterns of tissue expression that suggest nonredundant roles in vivo. J. Immunol. 157:4511–4520. [PubMed] [Google Scholar]
- 15.Mahalingam, S., J.M. Farber, and G. Karupiah. 1999. The interferon-inducible chemokines MuMig and Crg-2 exhibit antiviral activity In vivo. J. Virol. 73:1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu, M.T., B.P. Chen, P. Oertel, M.J. Buchmeier, D. Armstrong, T.A. Hamilton, and T.E. Lane. 2000. The T cell chemoattractant IFN-inducible protein 10 is essential in host defense against viral-induced neurologic disease. J. Immunol. 165:2327–2330. [DOI] [PubMed] [Google Scholar]
- 17.Liu, M.T., D. Armstrong, T.A. Hamilton, and T.E. Lane. 2001. Expression of Mig (monokine induced by interferon-γ) is important in T lymphocyte recruitment and host defense following viral infection of the central nervous system. J. Immunol. 166:1790–1795. [DOI] [PubMed] [Google Scholar]
- 18.Salazar-Mather, T.P., T.A. Hamilton, and C.A. Biron. 2000. A chemokine-to-cytokine-to-chemokine cascade critical in antiviral defense. J. Clin. Invest. 105:985–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narumi, S., Y. Tominaga, M. Tamaru, S. Shimai, H. Okumura, K. Nishioji, Y. Itoh, and T. Okanoue. 1997. Expression of IFN-inducible protein-10 in chronic hepatitis. J. Immunol. 158:5536–5544. [PubMed] [Google Scholar]
- 20.Shields, P.L., C.M. Morland, M. Salmon, S. Qin, S.G. Hubscher, and D.H. Adams. 1999. Chemokine and chemokine receptor interactions provide a mechanism for selective T cell recruitment to specific liver compartments within hepatitis C-infected liver. J. Immunol. 163:6236-6243. [PubMed] [Google Scholar]
- 21.Schreiber, R.D., L.J. Hicks, A. Celada, N.A. Buchmeier, and P.W. Gray. 1985. Monoclonal antibodies to murine γ-interferon which differentially modulate macrophage activation and antiviral activities. J. Immunol. 134:1609–1618. [PubMed] [Google Scholar]
- 22.Asensio, V.C., and I.L. Campbell. 1997. Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J. Virol. 71:7832–7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kakimi, K., L.G. Guidotti, Y. Koezuka, and F.V. Chisari. 2000. Natural killer T cell activation inhibits hepatitis B virus replication In vivo. J. Exp. Med. 192:921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fulcher, D.A., A.B. Lyons, S.L. Korn, M.C. Cook, C. Koleda, C. Parish, B. Fazekas de St. Groth, and A. Basten. 1996. The fate of self-reactive B cells depends primarily on the degree of antigen receptor engagement and availability of T cell help. J. Exp. Med. 183:2313-2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gubbay, J., N. Vivian, A. Economou, D. Jackson, P. Goodfellow, and R. Lovell-Badge. 1992. Inverted repeat structure of the Sry locus in mice. Proc. Natl. Acad. Sci. USA. 89:7953–7957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bustin, S.A. 2000. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 25:169–193. [DOI] [PubMed] [Google Scholar]
- 27.Nakamoto, Y., L.G. Guidotti, V. Pasquetto, R.D. Schreiber, and F.V. Chisari. 1997. Differential target cell sensitivity to CTL-activated death pathways in hepatitis B virus transgenic mice. J. Immunol. 158:5692–5697. [PubMed] [Google Scholar]
- 28.Ishikawa, T., D. Kono, J. Chung, P. Fowler, A. Theofilopoulos, S. Kakumu, and F.V. Chisari. 1998. Polyclonality and multispecificity of the CTL response to a single viral epitope. J. Immunol. 161:5842–5850. [PubMed] [Google Scholar]
- 29.Eberl, G., and H.R. MacDonald. 1998. Rapid death and regeneration of NKT cells in anti-CD3ε- or IL-12-treated mice: a major role for bone marrow in NKT cell homeostasis. Immunity. 9:345–353. [DOI] [PubMed] [Google Scholar]
- 30.Eberl, G., and H.R. MacDonald. 2000. Selective induction of NK cell proliferation and cytotoxicity by activated NKT cells. Eur. J. Immunol. 30:985–992. [DOI] [PubMed] [Google Scholar]
- 31.Inngjerdingen, M., B. Damaj, and A.A. Maghazachi. 2001. Expression and regulation of chemokine receptors in human natural killer cells. Blood. 97:367–375. [DOI] [PubMed] [Google Scholar]
- 32.Garcia-Lopez, M.A., F. Sanchez-Madrid, J.M. Rodriguez-Frade, M. Mellado, A. Acevedo, M.I. Garcia, J.P. Albar, C. Martinez, and M. Marazuela. 2001. CXCR3 chemokine receptor distribution in normal and inflamed tissues: expression on activated lymphocytes, endothelial cells, and dendritic cells. Lab. Invest. 81:409–418. [DOI] [PubMed] [Google Scholar]
- 33.Jones, D., R.J. Benjamin, A. Shahsafaei, and D.M. Dorfman. 2000. The chemokine receptor CXCR3 is expressed in a subset of B-cell lymphomas and is a marker of B-cell chronic lymphocytic leukemia. Blood. 95:627–632. [PubMed] [Google Scholar]
- 34.Colletti, L.M., M.E. Green, M.D. Burdick, and R.M. Strieter. 2000. The ratio of ELR+ to ELR− CXC chemokines affects the lung and liver injury following hepatic ischemia/reperfusion in the rat. Hepatology. 31:435–445. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi, H., S. Narumi, T. Tamatani, G.J. Lane, and T. Miyano. 1999. Serum IFN-inducible protein-10: a new clinical prognostic predictor of hepatocyte death in biliary atresia. J. Pediatr. Surg. 34:308–311. [DOI] [PubMed] [Google Scholar]
- 36.Yoong, K.F., S.C. Afford, R. Jones, P. Aujla, S. Qin, K. Price, S.G. Hubscher, and D.H. Adams. 1999. Expression and function of CXC and CC chemokines in human malignant liver tumors: a role for human monokine induced by γ-interferon in lymphocyte recruitment to hepatocellular carcinoma. Hepatology. 30:100–111. [DOI] [PubMed] [Google Scholar]
- 37.Bone-Larson, C.L., K.J. Simpson, L.M. Colletti, N.W. Lukacs, S.C. Chen, S. Lira, S.L. Kunkel, and C.M. Hogaboam. 2000. The role of chemokines in the immunopathology of the liver. Immunol. Rev. 177:8–20. [DOI] [PubMed] [Google Scholar]
- 38.Seeger, C., and W.S. Mason. 2000. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64:51–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guidotti, L.G., R. Rochford, L. Chung, M. Shapiro, R. Purcell, and F.V. Chisari. 1999. Viral clearance without destruction of infected cells during acute HBV infection. Science. 284:825–829. [DOI] [PubMed] [Google Scholar]
- 40.Cerwenka, A., T.M. Morgan, A.G. Harmsen, and R.W. Dutton. 1999. Migration kinetics and final destination of type 1 and type 2 CD8 effector cells predict protection against pulmonary virus infection. J. Exp. Med. 189:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mantovani, A., P. Allavena, and S. Sozzani. 1998. Regulation of chemokine receptors in mononuclear phagocytes versus dendritic cells and in the amplification of Th1 versus Th2 responses. Eur. Cytokine Netw. 9:76–80. [PubMed] [Google Scholar]
- 42.Guidotti, L.G., K. Ando, M.V. Hobbs, T. Ishikawa, L. Runkel, R.D. Schreiber, and F.V. Chisari. 1994. Cytotoxic T lymphocytes inhibit hepatitis B virus gene expression by a noncytolytic mechanism in transgenic mice. Proc. Natl. Acad. Sci. USA. 91:3764–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bonecchi, R., G. Bianchi, P.P. Bordignon, D. D'Ambrosio, R. Lang, A. Borsatti, S. Sozzani, P. Allavena, P.A. Gray, A. Mantovani, and F. Sinigaglia. 1998. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J. Exp. Med. 187:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Syrbe, U., J. Siveke, and A. Hamann. 1999. Th1/Th2 subsets: distinct differences in homing and chemokine receptor expression? Springer Semin. Immunopathol. 21:263–285. [DOI] [PubMed] [Google Scholar]
- 45.Bertoletti, A., and M.K. Maini. 2000. Protection or damage: a dual role for the virus-specific cytotoxic T lymphocyte response in hepatitis B and C infection? Curr. Opin. Immunol. 12:403–408. [DOI] [PubMed] [Google Scholar]
- 46.Ishak, K.G. 1976. Light microscopic morphology of viral hepatitis. Am. J. Clin. Pathol. 65:787–827. [PubMed] [Google Scholar]
- 47.Prezzi, C., M.A. Casciaro, V. Francavilla, E. Schiaffella, L. Finocchi, L.V. Chircu, G. Bruno, A. Sette, S. Abrignani, and V. Barnaba. 2001. Virus-specific CD8+ T cells with type 1 or type 2 cytokine profile are related to different disease activity in chronic hepatitis C virus infection. Eur. J. Immunol. 31:894–906. [DOI] [PubMed] [Google Scholar]