

Abstract
Examining the rate of in vivo T cell turnover (proliferation) in aged mice revealed a marked reduction in turnover at the level of memory-phenotype CD44hi CD8+ cells relative to young mice. Based on adoptive transfer experiments, the reduced turnover of aged CD44hi CD8+ cells reflected an inhibitory influence of the aged host environment. Aged CD44hi CD8+ cells also showed poor in vivo responses to IL-15 and IL-15–inducing agents, but responded well to IL-15 in vitro. Two mechanisms could account for the reduced turnover of aged CD44hi CD8+ cells in vivo. First, aging was associated with a prominent and selective increase in Bcl-2 expression in CD44hi CD8+ cells. Hence, the reduced turnover of aged CD44hi CD8+ cells may in part reflect the antiproliferative effect of enhanced Bcl-2 expression. Second, the impaired in vivo response of aged CD44hi CD8+ cells to IL-15 correlated with increased serum levels of type I interferons (IFN-I) and was largely reversed by injection of anti–IFN-I antibody. Hence the selective reduction in the turnover of aged CD44hi CD8+ cells in vivo may reflect the combined inhibitory effects of enhanced Bcl-2 expression and high IFN-I levels.
Keywords: aging, CD8+ cells, IFN-I, IL-15, T cell turnover
Introduction
It is well-established that aging is associated with immunological defects, especially at the level of T cells (1–4). The mechanisms governing hyporesponsiveness in aged T cells are poorly understood. A priori, there could be defects at the level of T cells per se or in APCs or both.
With regard to APCs, there are several reports that macrophages/monocytes from old donors can be inhibitory for T proliferative responses (4–6). Such inhibition could involve multiple mechanisms, including increased production of PGE2 (7, 8) and TGF-β (9). Nevertheless, defects in APC function are not an invariable finding in old age (10–12). Moreover, defects in T cell function are apparent in APC-free systems (2) and in situations where defined cell lines are used as APCs (3).
For T cells, studies with purified populations of APC-depleted murine T cell subsets have demonstrated that old T cells display a number of early activation defects after CD3 ligation in vitro (for a review, see reference 2). To a large extent, these changes in signal transduction are attributable to the marked increased frequency of memory-phenotype cells in old T cell populations (3, 13–16). Nevertheless, at least some of the signal transduction defects found in old T cells are apparent at the level of purified naive phenotype CD4+ cells (2). The defects in these cells include decreased proliferative responses and IL-2 production and correlate with increased expression of P-glycoprotein (2, 15). These data refer to CD4+ cells. Whether aging is associated with defects at the level of CD8+ cells is less clear.
In considering the defective function of old T cells, the lifespan of these cells is an important issue. Since de novo production of T cells by the thymus declines rapidly in old age (17), most of the T cells found in old age are probably the long-term progeny of T cells generated in young life. Indeed, parking studies in mice suggest that, at a population level, T cells in this species have an almost indefinite lifespan (>2 y) (18). Since total T cell numbers remain relatively constant from puberty to old age, T cells are presumably subject to homeostatic mechanisms which balance the rate of cell death and proliferation (19–21). In this respect there is a paucity of firm information on the normal in vivo physiology of T cells in aged hosts, especially with regard to T cell turnover and lifespan. Previous work from this laboratory on T cell turnover in normal young mice showed that, although both naive- and memory-phenotype cells are long lived at a population level, the background rate of proliferation (turnover) of these cells is much faster for memory- than for naive-phenotype cells (22).
For memory-phenotype (CD44hi) CD8+ cells there is increasing evidence that the survival and turnover of these cells is under the control of IL-15 (23–28). In particular, numbers of CD44hi CD8+ cells are selectively reduced in IL-15–/– mice (26) and the normal turnover of CD44hi CD8+ cells in vivo can be partly blocked by antibodies to IL-2Rβ (CD122), an important component of the receptor for IL-15 (and IL-2; reference 27). In this paper, we show that aging is associated with a selective reduction in the turnover of CD44hi CD8+ cells and an impaired response of these cells to IL-15 in vivo (though not in vitro). The reduced turnover of CD44hi CD8+ cells in vivo was not seen after transfer to young mice. Evidence is presented that the slow turnover of CD44hi CD8+ cells in aged mice may reflect the combined inhibitory effects of Bcl-2 overexpression and increased levels of type I IFN-I.
Materials and Methods
Mice.
C57BL/6 (B6) mice together with IL-2Rβ–/– (29) and Bcl-2 transgenic (30) mice, both on a B6 background, were obtained from The Jackson Laboratory; IL-2Rβ–/– mice were derived from heterozygous IL-2Rβ+/– mice purchased from The Jackson Laboratory. Mice were aged by maintaining young mice in our animal facility for 2 y or more under specific pathogen-free conditions; routine serological testing of mice for a variety of pathogens were negative. For studies on long-term T cell turnover in vivo, adult thymectomized (ATx)*mice (22) were used. For aged mice, ATx was performed at 1 y and the mice were used 1 y later; sham thymectomized (STx) mice were used as controls. For young mice, mice were subjected to ATx or STx at 4–6 wk and used 3–4 wk later.
Flow Cytometry.
As described elsewhere (22, 23), surface markers on T cell subsets were defined by staining spleen and/or LN cells with Cy5-coupled anti-CD4 and anti-CD8 mAbs followed by biotin- or phycoerythrin-coupled mAbs specific for CD44, B7.1, B7.2 CD69, CD25, CD122, CD44, Ly6C, and intracellular adhesion molecule 1; these mAbs were purchased from BD PharMingen. Biotinylated mAbs were detected with Red-670 streptavidin (GIBCO BRL). T cells were stained internally for Bcl-2 and Bcl–XL as described previously (31). In brief, cells were stained for surface markers, then fixed with paraformaldehyde, treated with saponin and then stained with a hamster anti–mouse Bcl-2 mAb (BD PharMingen; 3F11) or with a polyclonal rabbit antiserum specific for Bcl–XL (provided by John Reed, Burnham Institute, La Jolla, CA) followed by FITC-conjugated anti–rabbit antibody. Stained cells were analyzed on a FACSort™ flow cytometer.
T Cell Turnover In Vivo.
As described previously (22), T cell proliferation in vivo was measured by maintaining mice continuously on bromodeoxyuridine (BrdU) (Sigma-Aldrich) dissolved in the drinking water. To detect BrdU incorporation, spleen and LN cells were surface stained for CD4 or CD8 and CD44 or IL-2Rβ, fixed and then stained internally with an anti-BrdU mAb. For measuring BrdU incorporation on adoptive transfer, the donor and hosts differed with respect to Ly5.1 versus Ly5.2 or Thy 1.1 versus Thy 1.2. Donor and host cells were then defined by staining cells with allele-specific anti-Ly5 and anti-Thy 1 mAbs (32). In some studies, T cell turnover on adoptive transfer was measured by labeling T cells in vitro before injection with the intracellular fluorescent dye CFSE (32). As measured by flow cytometry, each division of CFSE-labeled cells causes a twofold reduction in mean fluorescence intensity (MFI) in the daughter cells, with the result that extensive cell proliferation results in a progressive step-wise reduction in MFI. For examining T proliferative responses to IL-15 and Poly I:C in vivo, graded concentrations of mouse rIL-15 (e-Bioscience) were injected intravenously and Poly I:C (100 μg/mouse) was injected intraperitoneally; in some mice, polyvalent neutralizing antiserum specific for IFN-β (Access Biomedical) was injected intravenously at 36 and 48 h. Mice were placed on BrdU water, preceded by an intraperitoneal injection of BrdU on day 0. BrdU incorporation was measured in spleen and LNs on day 2 for IL-15 and day 3 for Poly I:C.
T Cell Proliferation In Vitro.
Purified subsets of CD44hi CD8+ cells were prepared from LNs by FACS® sorting and cultured at 0.5 × 106 cells per milliliter with graded concentrations of rIL-15 ± IFN-β (Access Biomedical). 3[H]TdR (1 μCi/ml) was added during the last 8 h of culture. 3[H]TdR incorporation was measured on days 2–4.
Apoptosis.
To measure spontaneous apoptosis, nylon-wool-purified LN T cells were cultured at 106 cells per milliliter in 1 ml 24-well plates for 1 or 2 d in standard RPMI 1640 tissue culture medium supplemented with 10% FCS, glutamine, antibiotics, and 2-ME (33). Apoptosis was measured by TUNEL staining (33) after surface staining for CD4 or CD8 and CD44.
Effect of Cytokines on Bcl-2 Expression.
FACS®-sorted CD44hi CD8+ cells were cultured at 106 cells per milliliter with various cytokines (purchased from R&D Systems) in standard tissue culture medium for 18 h and then stained internally for Bcl-2 expression.
Cytokine Concentrations.
Levels of cytokines in serum and cell supernatants were measured by ELISA. In brief, serum or culture supernatants, together with known concentrations of recombinant TNF-α, IFN-β, IL-12, and IFN-γ as controls, were diluted serially in saline and added to plastic plates coated with antibodies specific for IFN-αβ (polyclonal rabbit antibody purchased from Access Biomedical, San Diego, CA), IFN-γ, TNF-α, or IL-12 (BD PharMingen). After incubation at 37°C for 2 h, the plates were washed, supplemented with biotinylated antibodies, washed again, followed by addition of streptavidin-peroxidase. After 1 h, the substrate O-phenyldiamine was added. The plates were analyzed with an ELISA reader after stopping the reaction with 25% H2SO4. Cytokine induction by Poly I:C (Sigma-Aldrich) was elicited in vivo by injecting mice intraperitoneally with 100 μg Poly I:C and in tissue culture by adding graded concentrations of Poly I:C to thioglycollate-induced macrophages in vitro.
RT-PCR.
Total RNA was isolated from whole spleen. First strand cDNA synthesis was performed using a Superscript Preamplification System (GIBCO BRL). PCR reactions were primed either with pairs of oligonucleotide primers corresponding to the 3′ region of mouse IL-15 (23) or IFN-β (Stratagene) or to the housekeeping gene G3PDH (23). PCR were performed for 30 cycles, with each cycle being 1 min at 94°C, 2 min at 54°C, and 3 min at 72°C. PCR samples were resolved on 1.5% agarose gels.
Results
T Cells Were Characterized in Young versus Aged C57BL/6 (B6) Mice.
For aged mice, young B6 mice were purchased and then maintained in our animal colony for up to 2 y. To prevent de novo production of T cells, ATx mice were used in some experiments. For old mice, the thymus was removed at 1 y and the mice were then tested 1 y later; as controls STx mice were used. In general, there was little difference in the results obtained for ATx, STx, and normal (unoperated) old mice. Unless stated otherwise, the age of the mice used in the experiments discussed below was ∼2 y (22–26 mo) for aged mice and 2–3 mo for young mice. Mice with tumors or lymphadenopathy were discarded.
T Cell Turnover.
To examine T cell turnover, young versus old B6 mice were given BrdU continuously in the drinking water. Groups of the mice were killed at intervals and suspensions of spleen and LN cells were stained for surface markers followed by fixation and intracellular staining for BrdU using an anti-BrdU mAb. Naive- and memory-phenotype T cells were subsetted on the basis of CD44 and IL-2Rβ (CD122) expression (illustrated for BrdU-labeled aged T cells in the right of Fig. 1) . For IL-2Rβ, it should be noted that expression of IL-2Rβ is much higher on CD8+ than CD4+ cells (23). Hence, naive- and memory-phenotype cells were defined as being IL-2Rβint and IL-2Rβhi, respectively, for CD8+ cells and IL-2Rβlo and IL-2Rβint for CD4+ cells.
Figure 1.
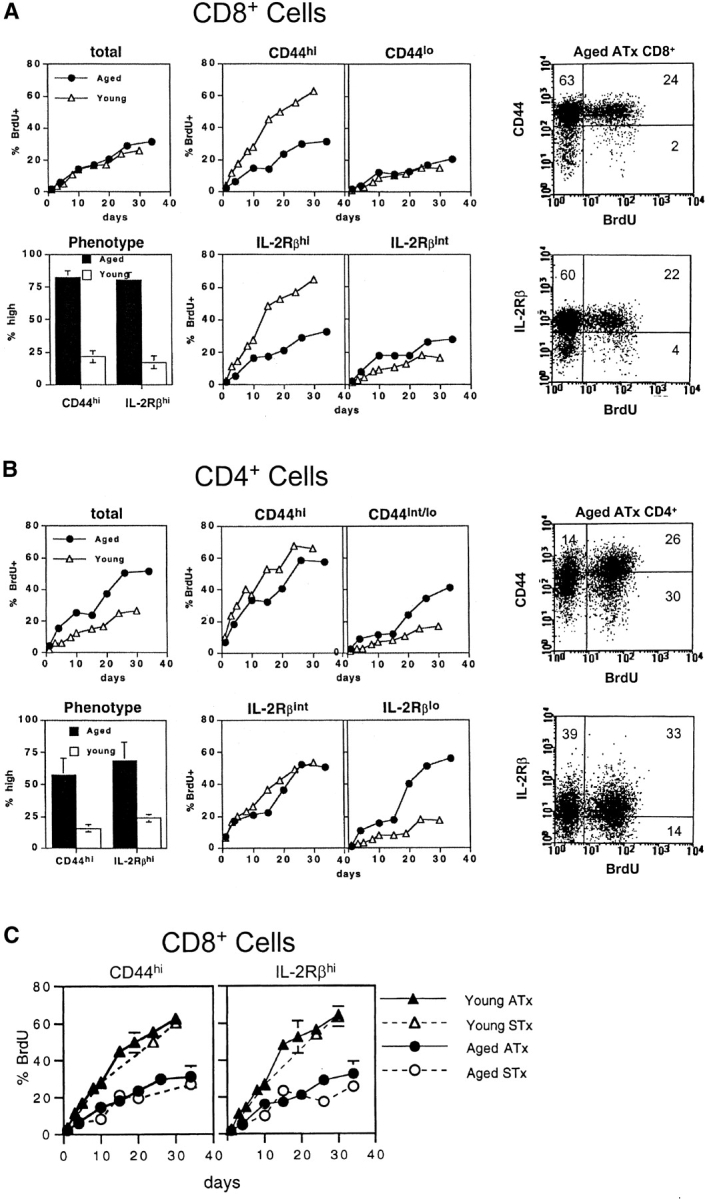
Turnover of T cell subsets in old and young mice. In A and B, groups of ATx old (2 y) and ATx young (3 mo) mice were placed on BrdU water for various periods. Spleen and LN suspensions were surface stained for CD4 and CD8 expression and also for expression of CD44 and IL-2Rβ. After fixation, the cells were then stained internally for BrdU incorporation using an anti-BrdU mAb and FACS® analyzed. The data show the mean percentage of BrdU labeling of pooled LN cells from two mice per group; labeling in spleen was quite similar. The bar graphs on the left show the percentage of T cells that display a memory phenotype in young versus old mice; dot plots for defining CD44hi versus CD44lo and IL-2Rβhi versus IL-2Rβint (for CD8+ cells) and IL-2Rβint versus IL-2Rβlo (for CD4+ cells) on aged ATx CD8+ cells from a representative mouse given BrdU for 26 d are shown on the right; quadrants were set on the basis of parallel staining of young T cells (data not shown). Note that for total (unseparated) CD8+ cells, the lack of a difference in turnover between young and old cells reflected the dominance of naive T cells (slow turnover) in young mice. In C, data on CD8+ cells from ATx mice (the same as in A) are compared with data from STx (euthymic) mice. The data show mean values (± SD) for two mice per point; for several of the points, SDs were too small to display.
In agreement with previous reports (1–3), the proportion of memory-phenotype CD8+ (CD44hi IL-2Rβhi) and CD4+ (CD44hi IL-2Rβint) cells was far higher in old mice than in young mice (Fig. 1). Data for BrdU incorporation by LN T cell subsets for ATx mice are shown in Fig. 1 A and B; data for spleen T cells were very similar. The data make several points.
For total unseparated CD8+ and CD4+ cells, the turnover (BrdU incorporation) of CD8+ cells in young and old mice was very similar (see Fig. 1 legend for explanation), whereas the turnover of CD4+ cells was faster in old mice than young mice.
Confirming previous findings (22), the turnover of T cell subsets in young mice was slow for naive-phenotype cells and rapid for memory-phenotype cells, both for CD8+ and CD4+ cells. The turnover of T cell subsets in old mice was distinctly different in three respects. First, in marked contrast to young mice, the difference in the turnover of naive- and memory-phenotype cells in old mice was mild or nonexistent. Second, for CD4+ cells (but not CD8+ cells) naive-phenotype cells paradoxically had a faster turnover in old mice than in young mice; a likely explanation for this finding is that, for CD4+ cells, many of the naive-phenotype cells in old mice are not truly naive but represent revertants from memory-phenotype cells (22). Third, and most importantly, the turnover of memory-phenotype CD8+ cells was much slower in old than young mice. Slow turnover of memory-phenotype cells in old mice also applied to CD4+ cells but was less prominent than for CD8+ cells.
The data in Fig. 1 A and B are representative of three different experiments on ATx mice. Data on euthymic (STx) mice were comparable and are illustrated in Fig. 1 C for CD8+ cells. For the experiments discussed hereafter, nonoperated mice were used.
T Cell Turnover on Adoptive Transfer to Young versus Old Hosts.
The key finding in the above experiments is that the turnover of memory-phenotype CD8+ cells was much slower in old mice than in young mice. A priori, this finding could reflect an intrinsic defect in old T cells per se or in the nonT microenvironment of old mice. To distinguish between these two possibilities, we examined whether the reduced turnover of old memory-phenotype CD8+ cells could be overcome by adoptively transferring old T cells to young hosts.
In the experiment shown in Fig. 2 A, purified LN T cells from old mice were transferred to Thy-1 different young hosts; 3 d later the T cell recipients plus control old and young mice were placed on BrdU water for 3 d. The notable finding was that BrdU incorporation by old CD44hi CD8+ cells transferred to young hosts was almost as high as for the population of young host CD44hi CD8+ cells (Fig. 2 A, top). These findings thus indicated that the slow turnover of CD44hi CD8+ cells in intact old mice could be substantially reversed by transferring the cells to young hosts. Similar findings applied when a mixture of Thy 1–marked old and young T cells were CFSE labeled (32) and transferred to young hosts. In this situation, proliferation of donor-derived old CD44hi CD8+ cells was almost as high as for donor-derived young CD44hi CD8+ (Fig. 2 A, bottom).
Figure 2.




Turnover of old and young T cells on adoptive transfer to old versus young hosts. (A, top) Doses of 107 nylon-wool-purified LN T cells from old Thy 1.2 donors were transferred intravenously into young Thy 1.1 hosts. After 3 d, the mice were placed on BrdU water for 3 d. Together with cells from normal old and young mice, LN cells from young hosts given old T cells were surface stained for CD8, Thy 1.2, Thy 1.1, and CD44 followed by internal staining for BrdU. The data (mean of three mice per group) show the percentage of BrdU+ cells for gated CD44hi CD8+ cells. In the adoptive transfer system, donor and host cells were detected on the basis of Thy 1.1 versus Thy 1.2 expression. (A, bottom) Purified populations of old Thy 1.2 T cells and young Thy 1.1 T cells were labeled with CFSE and transferred intravenously at a dose of 107 of each into young Thy 1.1 hosts. LN cells were removed from groups of these mice at the intervals shown and stained for CD8, CD44, Thy 1.1, and Thy 1.2. Gating on young (Thy 1.1+) versus old (Thy 1.2+) CD44hi CD8+ cells, the cells were analyzed for the intensity of CFSE labeling. The data show the percentage of CD44hi CD8+ cells that divided one or more times after transfer; the percentage of proliferation was calculated from the percentage of cells in each CFSE peak, followed by correction for cells that divided more than once. (B) Doses of 2 × 106 CFSE-labeled purified LN T cells from young Thy 1.1 mice were transferred intravenously into young versus old hosts (Thy 1.2) pretreated with light irradiation (600 cGy). The data show CFSE labeling of donor (Thy 1.1+) CD8+ cells harvested from host LN on day 8. (C) A mixed population of CFSE-labeled young and old T cells (106 of each) was transferred intravenously to 600 cGy young versus old hosts; different Ly 5 and Thy 1 markers were used to define the two populations of donor cells and distinguish these cells from the host cells. The data show CFSE labeling of the donor cells recovered from host LN on day 8. (D) Doses of 2 × 107 LN T cells from young Bcl-2 transgenic mice (Ly5.1) were transferred intravenously to young normal B6 Ly5.2 mice. At 1 mo after transfer the mice were given BrdU in the drinking water; lymphoid cells were then stained for surface markers and BrdU incorporation. The data (mean of three mice per group) show the percentage of BrdU incorporation by CD44hi CD8+ donor versus host LN cells after 3 d or 10 d on BrdU. Similar findings were seen in a second experiment in which the time between cell transfer and BrdU administration was reduced to 3 d (not shown). Note that for each experiment shown in A–D, the data are representative of 2–6 separate experiments.
These findings implied that the reduced turnover of old T cells in old mice did not reflect an intrinsic defect in T cells but in the host environment. If so, it would follow that young T cells would show a reduction in turnover after transfer to old hosts. This was indeed the case. Thus, after transfer of CFSE-labeled young T cells to young versus old hosts, proliferation was extensive in young hosts but quite limited in old hosts (Fig. 2 B).
A corollary of the above data is that if a mixture of old and young T cells was transferred to young versus old hosts, the young and old T cells would both proliferate well in young hosts but poorly in old hosts. Experiments with allotype-marked CFSE-labeled cells verified this prediction (Fig. 2 C). It should be noted that in Fig. 2 B and C (but not 2 A) the host mice were pretreated with light irradiation to facilitate detection of the donor T cells. Host irradiation enhances “homeostatic” proliferation of donor T cells (32) but the results in irradiated and nonirradiated mice were qualitatively similar (compare Fig. 2 A, bottom, with Fig. 2 C, top).
Collectively, the above data indicate that, both in normal and irradiated hosts, the impaired turnover of memory-phenotype CD8+ cells in old mice reflects the particular milieu of the host environment. It should be mentioned that higher turnover of old CD8+ cells in young hosts than in old hosts has recently been reported by Ku et al. (34). Our finding that this difference applies to both irradiated and nonirradiated hosts argues against the suggestion of Ku et al. that reduced turnover in old hosts reflects increased competition for cytokines because of the increase in memory-phenotype T cells.
Surface Markers.
To examine whether the reduced turnover of old CD44hi CD8+ cells correlated with surface marker changes, old versus young T cells were stained for expression of various cell-surface molecules. Gating on CD44hi CD8+ cells showed no obvious difference in the expression of CD62L, CD45RB, CD69, CD25, B7.1, B7.2, Ly6C, and IL-2Rβ on old versus young T cells (data not shown). In particular, CD44hi CD8+ cells in old mice closely resembled young T cells in expressing a high density of IL-2Rβ; as discussed earlier, IL-2Rβ is an important component of the receptor for IL-15 (and IL-2). The only noticeable difference for this marker was that, for CD8+ cells, the proportion of CD44hi CD8+ cells that were IL-2Rβhi was higher in old mice (90–95%) than in young mice (60–70%) (data not shown).
Bcl-2 and Bcl–XL.
Since overexpression of Bcl-2 or Bcl–XL is reported to inhibit T cell proliferation (30, 35, 36), the reduced turnover of memory-phenotype CD8+ cells in old mice could be a reflection of enhanced expression of Bcl-2 and/or Bcl–XL. In young mice, it was shown previously that Bcl-2 expression is higher in CD44hi CD8+ cells than in CD44lo CD8+ cells but equivalent in CD44hi and CD44lo CD4+ cells (37); Bcl–XL expression was not examined. Intracellular staining for Bcl–2 and Bcl–XL expression in young (2 mo) versus old (14 and 27 mo) T cells is shown in Fig. 3 A.
Figure 3.

Bcl-2 and Bcl–XL expression in young versus old T cells. (A) LN cells from B6 mice aged 2, 14, and 27 mo were surface stained for CD4, CD8 and CD44 and then, after fixation and permeabilization, stained internally for Bcl-2 or Bcl-XL. The data show representative staining for Bcl-2 (top panel) and mean MFI values (± SD) for Bcl-2 and Bcl-XL staining (middle and lower panels) for 3–6 mice per group; in the middle and lower panels, ratios ± SD of MFI staining for Bcl-2 and Bcl–XL versus MFI staining with an irrelevant control mAb are inserted. (B) Effect of cytokines on Bcl-2 expression in young CD44hi CD8+ cells. (Left) CD44hi CD8+ cells were purified by FACS® sorting and cultured in 200 ng/ml of the cytokines shown for 18 h and then stained internally for Bcl-2 expression after surface staining for CD8 and CD44. MFI values relative to cells cultured without cytokines are shown. (Right) Bcl-2 expression in fresh CD44hi CD8+ cells from normal B6 versus B6.IL-2Rβ–/– mice. MFI values relative to staining of B6 cells are shown.
In young mice, confirming the above report, Bcl-2 expression was clearly higher in CD44hi CD8+ cells than CD44lo CD8+ cells but was not elevated in CD44hi CD4+ cells relative to CD44lo CD4+ cells. For Bcl–XL expression, the results were different. As for Bcl-2, Bcl–XL expression in young T cells was appreciably higher in CD44hi cells than CD44lo cells. In contrast to Bcl-2, however, upregulation of Bcl–XL in CD44hi cells applied to both CD4+ and CD8+ cells. Thus, Bcl-2 was selectively elevated in CD44hi CD8+ cells but not CD44hi CD4+ cells whereas Bcl–XL was elevated in both CD44hi CD4+ cells and CD44hi CD8+ cells.
In old mice, the notable finding was that elevation of Bcl-2 expression was much more pronounced than in young mice. Thus, Bcl-2 expression was 2–3-fold higher in old CD44hi CD8+ cells than in young CD44hi CD8+ cells; this finding applied both to direct MFI values for Bcl-2 staining and also to the ratio of Bcl-2:background staining (shown numerically within each middle and lower panel of Fig. 3 A). Bcl-2 expression also increased with age in CD44lo CD8+ cells, but this increase applied only to Bcl-2:background staining ratios and not to direct MFI values for Bcl-2 staining. In addition to Bcl-2 upregulation, old CD44hi CD8+ cells also showed a mild (30–50%) increase in Bcl–XL expression relative to young cells.
These findings indicated that aging was associated with a selective increase in the expression of Bcl-2, and to a lesser extent Bcl–XL, in CD44hi CD8+ cells. By contrast, aging caused little or no change in Bcl-2 or Bcl–XL expression in CD44hi CD4+ cells or in naive-phenotype cells.
The increased Bcl-2/Bcl–XL expression found in old CD44hi CD8+ cells might reflect altered exposure to cytokines. To examine this possibility, purified CD44hi CD8+ cells from young mice were cultured overnight in vitro with eight different cytokines and then stained for Bcl-2 expression. Of the cytokines tested, only IL-15 caused a prominent (twofold) increase in Bcl-2 expression (Fig. 3 B, left). Two other γc-controlled cytokines, IL-4 and IL-7, caused a mild increase in Bcl-2 expression, whereas IFN-I (IFN-β), IFN-γ, and TNF-α had no effect (see below). For IL-15, upregulation of Bcl-2 was restricted to CD44hi CD8+ cells and did not apply to CD44hi CD4+ cells (data not shown), which is consistent with the much lower expression of IL-2Rβ on the latter cells (see above). Significantly, unmanipulated CD44hi CD8+ cells from IL-2Rβ–/– mice, i.e., cells that are unresponsive to IL-15 (and IL-2), showed a prominent decrease in Bcl-2 expression relative to normal cells (Fig. 3 B, right). It should be noted that, unlike IL-15–/– mice, IL-2Rβ–/– mice contain large numbers of CD44hi CD8+ cells, presumably reflecting unrestrained MHC-driven IL-15–independent responses to environmental antigens in the absence of inhibition by IL-2 (27, 29).
Apoptosis In Vitro.
Since Bcl-2 and Bcl–XL are antiapoptotic molecules (30, 35, 36), the elevation of these molecules in old T cells would be expected to retard “background” death of these cells. To assess this possibility, purified young versus old B6 T cells were cultured in standard tissue culture medium and then TUNEL stained to assess apoptosis. The notable finding was that spontaneous apoptosis in culture was far lower with old T cells than with young T cells (Fig. 4) . The resistance of old T cells to spontaneous apoptosis was more prominent for CD8+ cells than CD4+ cells and, especially on day 2 of culture, was more pronounced for CD44hi cells than for CD44int/lo cells. On day 2 of culture, relative to young T cells, apoptosis of old T cell subsets was reduced by 3.7-fold for CD44hi CD8+ cells, 2.4-fold for CD44int/lo CD8+ cells, 1.9-fold for CD44hi CD4+ cells, and 1.4-fold for CD44int/lo CD4+ cells. Thus, resistance of old T cells to apoptosis was most prominent in CD44hi CD8+ cells and correlated with the selective elevation of Bcl-2 in these cells. For the other T cell subsets, the significant but less prominent decrease in apoptosis of old T cells may have reflected minor upregulation of Bcl-2, Bcl–XL, and/or other Bcl-2 family members (which were not studied).
Figure 4.

Spontaneous apoptosis of old versus young T cells after culture in vitro in medium alone. Nylon-wool-purified LN T cells from old versus young mice were cultured in standard tissue culture medium (Materials and Methods). After 1 or 2 d, the cells were surface stained for CD4, CD8, and CD44 followed by fixation and internal staining by TUNEL for apoptosis. The data show TUNEL staining on gated CD8+ and CD4+ subsets of cells. Numbers refer to mean data (± SD) for triplicate cultures. Two other experiments gave comparable results.
Bcl-2 Upregulation and T Cell Turnover.
If the reduced turnover of CD44hi CD8+ cells in old mice reflected elevated Bcl-2 expression, T cell turnover would be expected to be reduced in Bcl-2 transgenic mice. Consistent with this prediction it was found that, after transfer of young Bcl-2 transgenic T cells to young normal hosts, the turnover (BrdU incorporation) of CD44hi CD8+ cells was considerably lower for donor Bcl-2 transgenic cells than for normal host cells (Fig. 2 D). Because of their propensity to develop lymphadenopathy, old Bcl-2 transgenic mice were not studied.
Cytokine Levels.
The capacity of IL-15 to upregulate Bcl-2 expression on CD44hi CD8+ cells raised the possibility that the overexpression of Bcl-2 in aged CD44hi CD8+ cells reflected increased levels of IL-15 in the aged microenvironment. Because IL-15 is difficult to detect at the protein level, we measured IL-15 at the level of mRNA. Since a spectrum of different cell types, including stromal cells, synthesize IL-15 (25, 28), IL-15 mRNA was measured in whole spleen rather than in cell suspensions. As shown in Table I, relative to G3PDH as a “housekeeping” control, IL-15 mRNA levels in spleens of young and old mice were quite similar.
Table I.
Whole Spleen RT-PCR: IL-15 and IFN-β versus G3PDH (± SD)
| IFN-β
|
IL-15
|
|
|---|---|---|
| G3PDH | G3PDH | |
| Young B6 | 1.43 (0.22) | 1.02 (0.11) |
| Old B6 | 3.71 (0.62) | 1.25 (0.24) |
Cytokines in young versus old mice. Expression of IL-15 and IFN-β mRNA in spleens of young versus old mice detected by RT-PCR (Materials and Methods). The data show mean values (three mice per group ± SD) of ratios of IL-15 and IFN-β mRNA relative to G3PDH mRNA.
Levels of IFN-I (IFN-β) mRNA were used as a control in the above experiment. Interestingly, IFN-I mRNA levels were appreciably higher (by 2.6-fold) in old versus young spleens. To confirm this finding at the protein level, we measured levels of IFN-I in the serum of old versus young mice before and after injection of Poly I:C, a powerful activator of IFN-I synthesis by macrophages and other APC (38). As shown in Fig. 5 A, IFN-I levels before and after Poly I:C were ∼2–3-fold higher in old mice than young mice. Old mice also showed elevated baseline levels of IFN-γ and IL-12 but decreased levels of TNF-α; for IL-12 and TNF-α (but not IFN-γ), levels of these cytokines increased after Poly I:C injection, both for young and old mice. For IFN-I and TNF-α, the fold-increase in cytokine levels in old mice relative to young mice is shown in Fig. 5 B. As in serum, IFN-I synthesis by macrophages in response to Poly I:C in vitro was higher for old than young cells (Fig. 5 C).
Figure 5.

(A) Levels of cytokines in serum. To prepare serum, blood was drawn from groups of old and young mice before and after injection of Poly I:C (100 μg intraperitoneally); the mice were killed at each time point shown. Serum concentrations of IFN-αβ (IFN-I), IFN-γ, TNF-α, and IL-12 were measured by ELISA; note that the antibody used to detect IFN-I did not distinguish between IFN-α and IFN-β. For IFN-αβ and IL-12, serum from individual mice was tested, and the data show mean values (± SD) for 4–6 mice for each time point; where not shown, SDs were too small to display. For IFN-γ and TNF-α, cytokine levels were measured on serum pooled from 4–6 mice. (B) For IFN-αβ and TNF-α, the data in A are shown as fold-increases for old mice relative to young mice. (C) To measure cytokine production by macrophages, thioglycollate-induced macrophages from old and young mice were cultured in vitro at 106 cells per milliliter, either alone or in the presence of graded concentrations of Poly I:C. Supernatants were taken from the cultures on day 3 and concentrations of IFN-αβ were measured by ELISA. The data show means (± SD) of cultures from individual mice (four mice per group).
The increased level of IFN-I in old mice is of interest because, in young mice, IFN-I injection augments the background rate of proliferation of CD44hi CD8+ cells (but not CD44hi CD4+ cells) (38), apparently by inducing production of IL-15 (23). As discussed earlier, at least in young mice in vivo proliferation of CD44hi CD8+ cells is largely controlled by IL-15. Hence the increased levels of IFN-I in old mice would be expected to augment IL-15 production and thereby enhance CD44hi CD8+ cell proliferation. Yet, this was not the case, old mice showed near-normal levels of IL-15 and a decrease rather than an increase in in vivo proliferation of CD44hi CD8+ cells. Therefore, we considered an alternative explanation for the data.
In addition to stimulating IL-15 production, IFN-I is known to exert an antiproliferative effect on T cells (39–41). Consistent with this observation we have found that, in young mice, bystander proliferation of CD44hi CD8+ cells induced by injection of Poly I:C falls to low levels when the dose of Poly I:C is raised above a certain level (data not shown). It is conceivable therefore that the reduced turnover of CD44hi CD8+ cells in old mice is due in part to a direct antiproliferative effect on T cells mediated by the increased levels of IFN-I. If so, it would follow that old mice would show decreased responsiveness to exogenous IL-15 and IL-15-inducing agents such as Poly I:C (which induces IL-15 synthesis via production of IFN-I). In line with this prediction, “bystander” proliferation of CD44hi (IL-2Rβhi) CD8+ cells elicited by injection of IL-15 (Fig. 6 A) and Poly I:C (Fig. 6 B) was substantially lower in old than young mice. Significantly, for Poly I:C, this reduced responsiveness in old mice could be largely reversed by injection of anti–IFN-I antibody (Fig. 6 B).
Figure 6.




Capacity of IFN-I to inhibit responses of CD44hi CD8+ cells to IL-15. (A) Response of young versus old mice to IL-15. Various doses of mouse rIL-15 were injected intravenously into young and old B6 mice. The mice were immediately injected with BrdU and then given BrdU in the drinking water. On day 2 after IL-15 injection, LN cells pooled from two mice per group were stained internally for BrdU incorporation after surface staining for CD4, CD8, and CD44. The data show the percentage of BrdU+ cells for gated CD44hi and CD44lo CD8+ cells. (B) Response of young and old mice to Poly I:C (PI:C). (Left) young versus old mice were injected with PI:C (100 μg/mouse intraperitoneally) and then given BrdU as in A, followed by staining on day 3 for BrdU and surface markers. Gating on CD44hi CD8+ cells, the data show the percentage BrdU+ cells for LN pooled from two mice. (Right) Old mice were injected with saline (PBS), PI:C, or PI:C plus anti–IFN-β antiserum (32,000 U/mouse given in two injections at 36 h and 48 h after PI:C) and given BrdU from day 0–3 followed by staining for BrdU and surface markers. Gating on IL-2Rβhi CD8+ cells, the data show mean percentage of BrdU+ cells (± SD) for LN cells from three mice; where not shown SDs were too small to display. Gating on CD44hi CD8+ cells (>90% IL-2Rβhi) gave similar results (data not shown). (C) Response of young versus old CD44hi CD8+ cells to IL-15 in vitro. FACS®-purified CD44hi CD8+ cells were cultured with the indicated concentrations of rIL-15 for 2 d; 3[H]TdR was added during the last 8 h of culture. The data show mean levels of 3[H]TdR incorporation for triplicate cultures (± SD). (D) Response of old CD44hi CD8+ cells to IL-15 in the presence of IFN-I. Purified CD44hi CD8+ cells from old mice were cultured with IL-15 (20 ng/ml) plus various concentrations of IFN-β (doubling dilutions down to 20 U/ml). 3[H]TdR was added on day 2. The data show mean levels of 3[H]TdR incorporation for triplicate cultures on day 2. The arrow marks the response with IFN-β at 40 U/ml. For each experiment shown in the figure, the data are representative of two to three separate experiments.
Tests on purified CD44hi CD8+ cells showed that old and young T cells gave comparable proliferative responses to IL-15 in vitro (Fig. 6 C). Under these conditions, it would follow from the above in vivo data that adding even low concentrations of IFN-I would decrease the in vitro response to IL-15. This was indeed the case. Thus, the response of old CD44hi CD8+ cells to IL-15 in vitro was reduced by IFN-I concentrations as low as 40 U/ml (Fig. 6 D, see arrow), comparable to the levels found in the serum of old mice; very similar data applied to young T cells (data not shown). These in vitro data thus support the notion that the limited in vivo response of aged CD44hi CD8+ cells to IL-15 reflects inhibition mediated by increased levels of IFN-I.
Discussion
The main finding in this paper is that aging is associated with a selective decrease in the turnover of memory-phenotype CD44hi CD8+ cells. Based on adoptive transfer experiments, the decreased turnover of CD44hi CD8+ cells is controlled by the milieu of the aged host environment. Two distinct mechanisms, i.e., (i) overexpression of Bcl-2 and (ii) inhibition by IFN-I, could account for this observation.
Bcl-2 Overexpression.
For young mice, the data confirm the recent report (37) that Bcl-2 expression is higher in memory-phenotype CD8+ cells than in other T cell subsets. In extending this finding, we observed that Bcl-2 expression in CD44hi CD8+ cells was even higher in old mice but was not elevated in CD44hi CD4+ cells. We also found that, like Bcl-2, Bcl–XL expression was elevated in CD44hi cells, both in young and old mice. Unlike Bcl-2, however, Bcl–XL expression was upregulated in both CD44hi CD8+ and CD44hi CD4+ cells and, for CD44hi CD8+ cells, was only slightly increased with aging. In agreement with the known anti-apoptotic function of Bcl-2 (35), selective overexpression of Bcl-2 in old CD44hi CD8+ cells was associated with a prominent decrease in spontaneous apoptosis of these cells in vitro.
Why Bcl-2 expression is selectively upregulated in CD44hi CD8+ cells and is enhanced by aging is unclear. Based on the effects of adding cytokines to purified T cells in vitro, Bcl-2 upregulation may reflect contact with cytokines, especially IL-15. Consistent with this finding, Bcl-2 expression was markedly reduced in IL-2Rβ–/– CD44hi CD8+ cells, i.e., in T cells that lack a key receptor for IL-15 (and IL-2). Hence continuous contact with IL-15 in vivo may explain why Bcl-2 expression is much higher in CD44hi CD8+ cells (which are IL-2Rβhi and therefore IL-15 responsive) than in CD44hi CD4+ cells (IL-2Rβint). However, it is more difficult to account for the further elevation in Bcl-2 expression found in old CD44hi CD8+ cells. Here, the simplest idea is that IL-15 levels are increased in old age. However, at least at the level of mRNA in whole spleen, this was not the case, IL-15 levels being only fractionally higher in old spleen than young spleen. Nevertheless, the possibility that local concentrations of IL-15 in the T cell areas, e.g., on dendritic cells (42), are higher in old mice cannot be discounted. An alternative possibility is that enhanced Bcl-2 expression in old CD44hi CD8+ cells reflects enhanced contact with other cytokines, e.g., IL-4 or IL-7. Thus, like IL-15, these two cytokines enhanced Bcl-2 expression in young T cells in vitro. As yet, however, we have not measured levels of IL-4 and IL-7 in old versus young mice.
Whatever the mechanisms controlling Bcl-2 expression, it is notable that overexpression of Bcl-2 in old CD44hi CD8+ cells correlated with a selective decrease in the background turnover of these cells in vivo. Hence, in view of the evidence that Bcl-2 overexpression impairs entry into cell cycle (30, 35, 36), the enhanced expression of Bcl-2 in old CD44hi CD8+ cells could be at least partly responsible for the slow turnover of these cells in vivo. Support for this idea is provided by the finding that, in young mice, the turnover of CD44hi CD8+ cells was much slower for Bcl-2 transgenic T cells than for normal T cells. For CD44hi CD4+ cells, aging was associated with little change in either Bcl-2 or Bcl–XL expression and only a mild decrease in T cell turnover (see below). Interestingly, others have reported that, relative to young mice, CD44hi CD4+ cells from older lupus-prone BXSB mice show reduced T cell turnover and apoptosis and increased Bcl–XL (but not Bcl-2) expression (43). Hence, as for Bcl-2 in CD8+ cells, enhanced Bcl–XL in CD4+ cells may have an antiproliferative effect on T cell turnover. It should be mentioned that slow turnover of lymphoid cells in old mice has also been reported for B cells (44). Whether this finding is related to Bcl-2/Bcl–XL expression or increased IFN-I levels (see below) has yet to be studied.
Inhibition by IFN-I.
Of the limited number of cytokines studied, levels of three cytokines, IFN-I, IFN-γ, and IL-12 were about threefold higher in the serum of old mice than in young mice. Elevation of IFN-I (which was also found at the mRNA level) is of particular interest because this cytokine can have two diametrically opposite effects on CD44hi CD8+ cells, i.e., (i) increased proliferation induced by stimulation of IL-15 synthesis (23) and (ii) inhibition of proliferation as the result of a direct inhibitory influence on T cells (39–41). Since IL-15 levels were not noticeably enhanced in old mice, we considered the possibility that the reduced turnover of old CD44hi CD8+ cells in vivo reflected a direct antiproliferative response mediated by the increased concentration of IFN-I found in old mice. In accordance with this idea, in vivo proliferation of CD44hi CD8+ cells after injection of IL-15 and IL-15-inducing agents (Poly I:C) was clearly lower in old mice than in young mice. Significantly, for Poly I:C, the reduction in T cell turnover was reversed by injection of anti–IFN-I antibody. Likewise, preliminary work has shown that (a) anti–IFN-I antibody injection partly reverses the slow tempo of CD44hi CD8+ cell turnover in old mice and (b) the slow turnover of these cells is much less apparent in old IFN-IR–/– mice (unpublished data).
If the reduced response of old T cells to IL-15 in vivo reflected inhibition by IFN-I, it would follow that old CD44hi CD8+ cells would respond well to IL-15 in vitro in the absence of IFN-I but would be highly sensitive to inhibition by addition of exogenous IFN-I. This was indeed the case. In fact, concentrations of IFN-I as low as 40 U/ml caused an appreciable reduction in the in vitro response to IL-15; this concentration of IFN-I was close to the level found in the serum of old mice. It is important to note that in vitro responses to IL-15 were highly sensitive to even moderate (twofold) alterations in the concentration of IFN-I in vitro. This finding supports the view that the approximately threefold increase in IFN-I levels found in old mice is sufficient to exert an antiproliferative effect on T cells.
Since the antiproliferative effect of IFN-I is not restricted to CD44hi CD8+ cells, enhanced exposure to IFN-I in old mice would be expected to retard the turnover of CD44hi CD4+ cells as well as CD44hi CD8+ cells. On this point it should be noted that the decrease in T cell turnover in old mice did affect CD44hi CD4+ cells, though to a lesser extent than CD44hi CD8+ cells. Nevertheless, the fact that slow turnover of old T cells was more prominent in CD44hi CD8+ cells than CD44hi CD4+ cells implies that inhibition by IFN-I is not the sole explanation. For this reason, we favor the idea that the selective decrease in the turnover of CD44hi CD8+ cells in old mice reflects the combined inhibitory effects of two different mechanisms working together, i.e., enhanced IFN-I levels plus Bcl-2 overexpression.
The significance of reduced T cell turnover in aging is a matter for speculation. Since thymic function is poor after middle age, maintaining T cell numbers in old age presumably hinges on continuous contact with factors that promote long-term T cell survival. For CD44hi CD8+ cells, at least in young mice exposure to IL-15 is crucial for survival. How IL-15 protects CD44hi CD8+ cells is unclear but, based on the present data, upregulation of Bcl-2 and/or other antiapoptotic molecules is a likely possibility. Placing the survival of T cells under the control of life-sustaining cytokines, however, has the potential danger that even transient increases in these growth factors could cause T cell numbers to rise to excessive levels. Hence to maintain normal homeostasis, one can envisage a delicate equilibrium where factors controlling T cell survival/proliferation are balanced by other factors that either promote cell death or inhibit proliferation. Hence, the increase in Bcl-2 expression and IFN-I levels in aging could be a manifestation of a general need to regulate T cell homeostasis in senescence. It is worth noting, however, that Bcl-2 and IFN-I do not act solely as negative regulators. Thus, countering their inhibitory effect on proliferation, Bcl-2 (30) and IFN-I (45) may augment cell survival via their antiapoptotic properties (and via IL-15 production by IFN-I). Hence, the control of T cell homeostasis in aging is undoubtedly highly complex and presumably reflects the interplay of many different factors.
Acknowledgments
We thank Ms. Barbara Marchand for typing the manuscript.
This work was supported by grants CA38355, CA25803, AI21487, AI32068, AI45809, AI41079, and AG01743 from the United States Public Health Service. C.D. Surh is a Scholar of The Leukemia & Lymphoma Society. Publication no. 13459-IMM from the Scripps Research Institute.
Footnotes
Abbreviations used in this paper: ATx, adult thymectomized; BrdU, bromodeoxyuridine; MFI, mean fluorescence intensity; STx, sham thymectomized.
References
- 1.Hodes, R. 1997. Aging and the immune system. Immunol. Rev. 160:5–8. [DOI] [PubMed] [Google Scholar]
- 2.Miller, R.A., G. Garcia, C.J. Kirk, and J.M. Witkowski. 1997. Early activation defects in T lymphocytes from aged mice. Immunol. Rev. 160:79–90. [DOI] [PubMed] [Google Scholar]
- 3.Linton, P.J., L. Haynes, L. Tsui, X. Zhang, and S. Swain. 1997. From naive to effector—alterations with aging. Immunol. Rev. 160:9–18. [DOI] [PubMed] [Google Scholar]
- 4.Chakravarti, B., and G.N. Abraham. 1999. Aging and T-cell-mediated immunity. Mech. Ageing Dev. 108:183–206. [DOI] [PubMed] [Google Scholar]
- 5.Rosenstein, M.M., and H.R. Strausser. 1980. Macrophage-induced T cell mitogen suppression with age. J. Reticuloendothel. Soc. 27:159–166. [PubMed] [Google Scholar]
- 6.Franklin, R.A., S. Arkins, Y.M. Li, and K.W. Kelley. 1993. Macrophages suppress lectin-induced proliferation of lymphocytes from aged rats. Mech. Ageing Dev. 67:33–46. [DOI] [PubMed] [Google Scholar]
- 7.Vercammen, C., and J.L. Ceuppens. 1987. Prostaglandin E2 inhibits human T-cell proliferation after crosslinking of the CD3-Ti complex by directly affecting T cells at an early step of the activation process. Cell. Immunol. 104:24–36. [DOI] [PubMed] [Google Scholar]
- 8.Hayek, M.G., S.N. Meydani, M. Meydani, and J.B. Blumberg. 1994. Age differences in eicosanoid production of mouse splenocytes: effects on mitogen-induced T-cell proliferation. J. Gerontol. 49:B197–B207. [DOI] [PubMed] [Google Scholar]
- 9.Zhou, D., F.J. Chrest, W. Adler, A. Munster, and R.A. Winchurch. 1993. Increased production of TGF-beta and IL-6 by aged spleen cells. Immunol. Lett. 36:7–11. [DOI] [PubMed] [Google Scholar]
- 10.Thoman, M.L., and W.O. Weigle. 1981. Lymphokines and aging: interleukin-2 production and activity in aged animals. J. Immunol. 127:2102–2106. [PubMed] [Google Scholar]
- 11.Daynes, R.A., B.A. Araneo, W.B. Ershler, C. Maloney, G.Z. Li, and S.Y. Ryu. 1993. Altered regulation of IL-6 production with normal aging. Possible linkage to the age-associated decline in dehydroepiandrosterone and its sulfated derivative. J. Immunol. 150:5219–5230. [PubMed] [Google Scholar]
- 12.Rich, E.A., M.A. Mincek, K.B. Armitage, E.G. Duffy, D.C. Owen, J.D. Fayen, D.L. Hom, and J.J. Ellner. 1993. Accessory function and properties of monocytes from healthy elderly humans for T lymphocyte responses to mitogen and antigen. Gerontol. 39:93–108. [DOI] [PubMed] [Google Scholar]
- 13.Ernst, D.N., M.V. Hobbs, B.E. Torbett, A.L. Glasebrook, M.A. Rehse, K. Bottomly, K. Hayakawa, R.R. Hardy, and W.O. Weigle. 1990. Differences in the expression profiles of CD45RB, Pgp-1, and 3G11 membrane antigens and in the patterns of lymphokine secretion by splenic CD4+ T cells from young and aged mice. J. Immunol. 145:1295–1302. [PubMed] [Google Scholar]
- 14.Nagelkerken, L., A. Hertogh-Huijbregts, R. Dobber, and A. Drager. 1991. Age-related changes in lymphokine production related to a decreased number of CD45RBhi CD4+ T cells. Eur. J. Immunol. 21:273–281. [DOI] [PubMed] [Google Scholar]
- 15.Linton, P.-L., L. Haynes, N.R. Klinman, and S.L. Swain. 1996. Antigen-independent changes in naive CD4+ T cells with age. J. Exp. Med. 184:1891–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swain, S.L., L.M. Bradley, M. Croft, S. Tonkonogy, G. Atkins, A.D. Weinberg, D.D. Duncan, S.M. Hedrick, R.W. Dutton, and G. Huston. 1991. Helper T-cell subsets: phenotype, function and the role of lymphokines in regulating their development. Immunol. Rev. 123:115–144. [DOI] [PubMed] [Google Scholar]
- 17.Mackall, C.L., and R.E. Gress. 1997. Thymic aging and T-cell regeneration. Immunol. Rev. 160:91–102. [DOI] [PubMed] [Google Scholar]
- 18.Sprent, J., M. Schaefer, M. Hurd, C.D. Surh, and Y. Ron. 1991. Mature murine B and T cells transferred to SCID mice can survive indefinitely and many maintain a virgin phenotype. J. Exp. Med. 174:717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sprent, J. 1993. Lifespans of naive, memory and effector lymphocytes. Cur. Opin. Immunol. 5:433–438. [DOI] [PubMed] [Google Scholar]
- 20.Mackall, C.L., F.T. Hakim, and R.E. Gress. 1997. Restoration of T-cell homeostasis after T-cell depletion. Semin. Immunol. 9:339–346. [DOI] [PubMed] [Google Scholar]
- 21.Tanchot, C., M.M. Rosado, F. Agenes, A.A. Freitas, and B. Rocha. 1997. Lymphocyte homeostasis. Semin. Immunol. 9:331–337. [DOI] [PubMed] [Google Scholar]
- 22.Tough, D.F., and J. Sprent. 1994. Turnover of naive- and memory-phenotype T cells. J. Exp. Med. 179:1127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang, X., S. Sun, I. Hwang, D.F. Tough, and J. Sprent. 1998. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity. 8:591–599. [DOI] [PubMed] [Google Scholar]
- 24.Lodolce, J.P., D.L. Boone, S. Chai, R.E. Swain, T. Dassopoulos, S. Trettin, and A. Ma. 1998. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. 9:669–676. [DOI] [PubMed] [Google Scholar]
- 25.Waldmann, T., Y. Tagaya, and R. Bamford. 1998. Interleukin-2, interleukin-15, and their receptors. Int. Rev. Immunol. 16:205–226. [DOI] [PubMed] [Google Scholar]
- 26.Kennedy, M.K., M. Glaccum, S.N. Brown, E.A. Butz, J.L. Viney, M. Embers, N. Matsuki, K. Charrier, L. Sedger, C.R. Willis, et al. 2000. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J. Exp. Med. 191:771–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ku, C.C., M. Murakami, A. Sakamoto, J. Kappler, and P. Marrack. 2000. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science. 288:675–678. [DOI] [PubMed] [Google Scholar]
- 28.Fehniger, T.A., and M.A. Caligiuri. 2001. Interleukin 15: biology and relevance to human disease. Blood. 99:14–32. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki, H., G.S. Duncan, H. Takimoto, and T.W. Mak. 1997. Abnormal development of intestinal intraepithelial lymphocytes and peripheral natural killer cells in mice lacking the IL-2 receptor β chain. J. Exp. Med. 185:499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strasser, A., A.W. Harris, and S. Cory. 1991. Bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell. 67:889–899. [DOI] [PubMed] [Google Scholar]
- 31.Kishimoto, H., Z. Cai, A. Brunmark, M.R. Jackson, P.A. Peterson, and J. Sprent. 1996. Differing roles for B7 and ICAM-1 in negative selection of thymocytes. J. Exp. Med. 184:531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ernst, B., D.-S. Lee, J. Chang, J. Sprent, and C.D. Surh. 1999. The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity. 11:173–181. [DOI] [PubMed] [Google Scholar]
- 33.Kishimoto, H., C.D. Surh, and J. Sprent. 1995. Upregulation of surface markers on dying thymocytes. J. Exp. Med. 181:649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ku, C.C., J. Kappler, and P. Marrack. 2001. The growth of the very large CD8+ T cell clones in older mice is controlled by cytokines. J. Immunol. 166:2186–2193. [DOI] [PubMed] [Google Scholar]
- 35.Cory, S. 1995. Regulation of lymphocyte survival by the bcl-2 gene family. Annu. Rev. Immunol. 13:513–544. [DOI] [PubMed] [Google Scholar]
- 36.O'Reilly, L.A., D.C. Huang, and A. Strasser. 1996. The cell death inhibitor Bcl-2 and its homologues influence control of cell cycle entry. EMBO J. 15:6979–6990. [PMC free article] [PubMed] [Google Scholar]
- 37.Grayson, J.M., A.J. Zagac, J.D. Altman, and R. Ahmed. 2000. Cutting edge: increased expression of Bcl-2 in antigen-specific memory CD8+ T cells. J. Immunol. 164:3950–3954. [DOI] [PubMed] [Google Scholar]
- 38.Tough, D.F., P. Borrow, and J. Sprent. 1996. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science. 272:1947–1950. [DOI] [PubMed] [Google Scholar]
- 39.Krishnaswamy, G., J.K. Smith, S. Srikanth, D.S. Chi, J.H. Kalbfleisch, and S.K. Huang. 1996. Lymphoblastoid interferon-α inhibits T cell proliferation and expression of eosinophil-activating cytokines. J. Interferon Cytokine Res. 16:819–827. [DOI] [PubMed] [Google Scholar]
- 40.Petricoin, E.F., III, S. Ito, B.L. Williams, S. Audet, L.F. Stancato, A. Gamero, K. Clouse, P. Grimley, A. Weiss, J. Beeler, et al. 1997. Antiproliferative action of interferon-α requires components of T-cell-receptor signalling. Nature. 390:629–632. [DOI] [PubMed] [Google Scholar]
- 41.Sun, S., X. Zhang, D.F. Tough, and J. Sprent. 1998. Type I interferon-mediated stimulation of T cells by CpG DNA. J. Exp. Med. 188:2335–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cella, M., D. Jarrossay, F. Facchetti, O. Alebardi, H. Nakajima, A. Lanzavecchia, and M. Colonna. 1999. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 5:919–923. [DOI] [PubMed] [Google Scholar]
- 43.Sabzevari, H., S. Propp, D.H. Kono, and A.N. Theofilopoulos. 1997. G1 arrest and high expression of cyclin kinase and apoptosis inhibitors in accumulated activated/memory phenotype CD4+ cells of older lupus mice. Eur. J. Immunol. 27:1901–1910. [DOI] [PubMed] [Google Scholar]
- 44.Kline, G.H., T.A. Hayden, and N.R. Klinman. 1999. B cell maintenance in aged mice reflects both increased B cell longevity and decreased B cell generation. J. Immunol. 162:3342–3349. [PubMed] [Google Scholar]
- 45.Marrack, P., J. Kappler, and T. Mitchell. 1999. Type I interferons keep activated T cells alive. J. Exp. Med. 189:521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]