

Abstract
Although considerable evidence implicates the cytokine interferon (IFN)-γ in atherogenesis, the proximal inducers and the range of sources of its expression remain unknown. This study tested the hypothesis that interleukin (IL)-18 regulates IFN-γ expression during atherogenesis. Indeed, human atheroma in situ expressed IL-18 and elevated levels of its receptor subunits, IL-18Rα/β, compared with nondiseased arterial tissue. IL-18 occurred predominantly as the mature, 18-kD form and colocalized with mononuclear phagocytes (MØ), while endothelial cells (ECs), smooth muscle cells (SMCs), and MØ all expressed IL-18Rα/β. Correspondingly in vitro, only MØ expressed IL-18, while all three cell types displayed the IL-18Rα/β complex constitutively, exhibiting enhanced expression upon stimulation with LPS, IL-1β, or tumor necrosis factor (TNF)-α. IL-18 signaling evoked effectors involved in atherogenesis, e.g., cytokines (IL-6), chemokines (IL-8), intracellular adhesion molecules (ICAM)-1, and matrix metalloproteinases (MMP-1/-9/-13), demonstrating functionality of the receptor on ECs, SMCs, and MØ. Finally, IL-18, particularly in combination with IL-12, induced the expression of IFN-γ in cultured MØ and, surprisingly, in SMCs (but not in ECs). The expression of functional IL-18 and IL-18 receptor on human atheroma-associated ECs, SMCs, and MØ, and its unexpected ability to induce IFN-γ expression in SMCs, suggests a novel paracrine proinflammatory pathway operating during atherogenesis.
Keywords: atherosclerosis, inflammation, IL-18/IL-18R, interferon-γ, cytokines
Introduction
Recent evidence accords a prominent role of pathways of innate and acquired immunity in the pathogenesis of atherosclerosis (1, 2). Cytokines have garnered considerable interest as potential mediators of this process (1–3). In particular, recent in vitro and in vivo studies demonstrated that the cytokine IFN-γ can promote atherosclerosis (4–7). However, the proximal inducers as well as the spectrum of cell types capable of expressing this pleiotropic cytokine have remained poorly understood. Originally, expression of IFN-γ was considered restricted to T lymphocytes and NK cells (8, 9). However, previous reports suggest a broader panel of cell types as the source of this potent immunomodulator, including B cell and certain tumor cell lines (10, 11). Moreover, mononuclear phagocytes (MØ)*, a cell type central to atherogenesis, may elaborate IFN-γ, although these findings have aroused controversy (12, 13).
In addition to antigenic stimulation, proposed inducers of IFN-γ expression in T lymphocytes within atheroma include proinflammatory cytokines, such as IL-1β and TNF-α, microbial components, such as endotoxin (LPS), as well as IFN-γ itself, probably via paracrine and/or autocrine pathways (8–10, 12). Of note, recent studies identified IL-18, a member of the IL-1 cytokine family, as a potent IFN-γ–inducing factor, originally termed IGIF (14–16). Initially identified in Kupffer cells and MØ (14) IL-18 is further expressed by osteoblasts (17), chondrocytes (18), and epidermal keratinocytes (19). Proinflammatory cytokines including IL-1β, TNF-α, or IL-6, as well as LPS, induce IL-18 gene expression (20, 21), directing the synthesis of an inactive 24-kD precursor. Similar to its analogue IL-1β, biological activity of IL-18 requires processing of the zymogen by the IL-1β–converting enzyme, caspase-1, to the 18-kD mature form (22, 23). Although IL-18 induces IFN-γ expression in T lymphocytes, synergism with IL-12 markedly enhances this function (14, 24). Consistent with a key role for mature IL-18 in the regulation of IFN-γ synthesis, IL-18–, or caspase-1–deficient mice showed a markedly diminished expression of IFN-γ (25, 26). Beyond induction of the central proatherogenic factor IFN-γ (4–6, 27, 28), IL-18 also augments the production of various other mediators implicated in atherogenesis, such as the proinflammatory cytokines IL-1β, IL-6, or TNF-α; the chemokines IL-8, MCP-1, and MIP-1α; the intracellular adhesion molecule (ICAM)-1; the growth factor GM-CSF; inducible nitric oxide synthase (the high capacity isoform); or inducible cyclooxygenase (Cox-2) (15, 16, 18, 20, 21, 24, 29). Furthermore, IL-18 promotes Th1 responses (24, 25, 30, 31), which dominate during human atherogenesis (3). Notably, the Th1 lymphocyte subpopulation, but not Th2 lymphocytes, express the receptor for IL-18 (30). IL-18 acts by binding to the heterodimeric IL-18 receptor complex, comprised of the IL-1 receptor-related protein (IL-1Rrp), termed IL-18Rα, and the IL-1 receptor accessory protein-like (IL-1RacPL), termed IL-18Rβ (32–34). While IL-18Rα represents the ligand-binding chain, IL-18Rβ appears to assist in the formation of a high affinity complex and mediates intracellular signals (20). Inflammatory cytokines overexpressed in human atheroma, such as IL-1β, TNF-α, and IL-12 (2, 35), enhance the expression of both receptor chains on NK cells and T lymphocytes, thereby increasing their responsiveness to IL-18 (20). IL-18–mediated processes may contribute to chronic inflammatory disorders such as rheumatoid arthritis (36, 37). Yet the expression and function of this cytokine and its receptor within human atherosclerotic lesions or atheroma-associated cells types remained poorly understood. We report here overexpression of both IL-18 and IL-18Rα/β within human atheroma in situ compared with nondiseased arterial tissue, and demonstrate that ligation of the receptor on cultured endothelial cells (ECs), smooth muscle cells (SMCs), and MØ triggers atheroma-associated processes in vitro, including the unexpected finding that human vascular SMCs themselves can express IFN-γ in response to IL-18.
Materials and Methods
Materials.
Human recombinant mature IL-1β, TNF-α, and IFN-γ were obtained from Endogen. Human recombinant mature IL-12 and IL-18 were purchased from R&D Systems. Escherichia coli endotoxin (LPS) and Polymyxin B were provided by Sigma-Aldrich.
Cell Isolation and Culture.
Cultured human vascular ECs and SMCs were isolated from saphenous veins by collagenase treatment and explant outgrowth, respectively, and used through passage 3, as described previously (38, 39). MØ were isolated from freshly prepared leukocyte concentrates by density gradient centrifugation using Lymphocyte Separation Medium (Organon-Teknika) and subsequent adherence to plastic culture flasks. MØ were cultured for 10 d in RPMI 1640 containing 2% human serum (Sigma-Aldrich; as described previously in references 38 and 39). All three cell types were cultured before (24 h) and during the experiment in media lacking FBS-serum; ECs in M199 supplemented with 0.1% bovine serum albumin, SMCs in IT (Insulin/Transferrin) medium, and MØ in RPMI 1640 lacking serum (38, 39). The purity of monocytes/MØ was ≥92%, as determined by FACS® analysis (anti–human CD68 mAb FITC; BD PharMingen). Culture media and FBS contained <40 pg endotoxin/ml as determined by the chromogenic Limulus amoebocyte assay (QLC-1000; BioWhittaker).
RT-PCR.
Total RNA isolated from cultures of ECs, SMCs, or MØ using RNazol (Tel-Test) was assessed for purity and yield spectrophotometrically (2100 Bioanalyzer; Agilent Technologies), was DNase-treated (DNase I, 15 min, 25°C; Life Technologies), and was finally reverse transcribed (2 μg total RNA, 50 min, 42°C) in 20 μl total reaction mix (200 U Superscript II Reverse Transcriptase; 25 μg/ml Oligo [dT]12–18 primers; 10 mM DTT; 0.5 mM dNTPs; 4 μl First Strand Buffer; all final concentrations; all Life Technologies). RT reaction products (2 μl) were mixed with either the IL-18Rα (sense: 5′-CTTCACATTCTTGCCCCAAT-3′; antisense: 5′-GCAGCTGCATCCAGTTATGA-3′), IL-18Rβ (sense: 5′-GAAGAACACTTGGCCCTGAG-3′; antisense: 5′-TTTCACAGGCATGTGGTAGC-3′), or IFN-γ (sense: 5′-TTTAGCTCTGCATCGTTTTG-3′; antisense: 5′-CATGTATTGCTTTGCGTTGG-3′) primer pair (0.2 μM each) in 50 μl total reaction mix (1.5 mM MgCl2, 0.2 mM dNTPs, 2.5 U Platinum Taq DNA Polymerase, and 5 μl 10× PCR buffer; all Life Technologies). The PCR reaction mix was applied to (i) 35 cycles at 94°C (60 s), 60°C (60 s), and 72°C (90 s) for IL-18Rα and IFN-γ, or (ii) 40 cycles at 94°C (60 s), 55°C (60 s), and 72°C (90 s) for IL-18Rβ. Aliquots (10 μl) of the PCR products were run on 1.5% agarose gels and visualized by UV-transillumination. The expected size for the RT-PCR product was 450 bp for IL-18Rα, 399 bp for IL-18Rβ, and 375 bp for IFN-γ. Loading of equal amounts of template was verified by RT-PCR for GAPDH yielding similar band intensities for all samples (data not shown). Mock RT reaction products, obtained in the absence of reverse transcriptase, or H2O served as templates for negative control studies (data not shown).
Western Blot Analysis.
To extract proteins, tissue of nonatherosclerotic (n = 5) and atheromatous human carotids and aorta (n = 7) as well as abdominal aortic aneurysm (n = 3) were snap frozen and homogenized under liquid nitrogen using mortar and piston. Pulverized specimens were incubated with ice-cold lysis buffer (0.3 g tissue/ml lysis buffer: 10 mM NaH2PO4, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, 0.2% NaN3, 5 mM EDTA, 20 μg/ml soybean trypsin inhibitor, 0.1 mM PMSF, 1 μg/ml aprotinin, and 1 μg/ml leupeptin). Tissue extracts as well as cell extracts (both 50 μg total protein per lane) and supernatants of cultured ECs, SMCs, and MØ were separated by SDS-PAGE and applied to Western blot analysis as described previously (38, 39), using the respective primary (goat anti–human IL-18 (0.2 μg/ml; R&D Systems); goat anti–human IL-18Rα (1 μg/ml; R&D Systems); mouse anti–human IL-12p40 (1 μg/ml; Genzyme); mouse anti–human IL-1β (1 μg/ml; Upstate Biotechnologies); rabbit anti–human Caspase-1p20 (Santa Cruz Biotechnology, Inc.; 0.2 μg/ml); all final concentration) and secondary antibody. Immunoreactive proteins were visualized using the Western blot chemiluminescence system (NEN™). To test antibody specificity, Western blots were developed with antibodies preincubated (2 h, 37°C) with the respective recombinant protein (5 μg/ml IL-18 or IL-18Rα; R&D Systems). Moreover, results were validated in additional experiments using antibodies of different origin (rabbit anti–human IL-18 (1 μg/ml; Serotec); mouse anti–human IL-18Rα (1 μg/ml; R&D Systems). Loading of equal amounts of protein was verified by subsequent staining of these blots with Coomassie blue as well as redeveloping blots with an mouse anti–human GAPDH antibody (1 μg/ml; Biodesign) (data not shown). Densitometric analysis of digital images of the respective Western blots employed ImagePro software (Media Cybernetics).
ELISA.
Supernatants of cultured ECs, SMCs, or MØ were added (2 h) to 96-well modules coated with capturing monoclonal mouse anti–human IL-6, IL-8, or IFN-γ antibody (1 μg/ml, 4°C, overnight) and blocked with PBS/2% BSA (2 h, RT). Plates were washed (PBS/0.1% Tween 20) three times and the respective biotin-labeled antibody (0.5 μg/ml) was added (1 h). Finally, wells were incubated for 30 min with alkaline phosphatase (Vectastain ABC kit, AK-500; Vector Laboratories), and were washed four times. Antibody binding was visualized by adding p-nitrophenyl phosphate (1.39 mg/ml; Sigma-Aldrich), and absorbance determined at 405 nm. Cytokine concentrations were calculated using a standard curve prepared with the respective recombinant cytokine. Samples were assayed in triplicates. Error bars represent standard deviation.
Flow Cytometry.
Confluent cultures of human vascular ECs, SMCs, or MØ were washed with ice-cold PBS, harvested by trypsinization, fixed (PBS/4% paraformaldehyde, 15 min), and subsequently washed once with PBS/0.2% BSA before being incubated (1 h, 4°C) with either buffer alone, PE-conjugated control IgG, PE-conjugated mouse anti–human IL-18Rα antibody (4 μg/ml; R&D Systems), or mouse anti–human ICAM-1 antibody (10 μg/ml; provided by M.A. Gimbrone, Jr., Brigham and Women's Hospital, Boston, MA) followed by PE-conjugated rabbit anti–mouse antibody (20 μg/ml). To determine activity of the IFN-γ released by SMCs, supernatants of SMC cultures (1:2 diluted in M199) were applied (48 h) in absence or presence of neutralizing mouse anti–human IFN-γ antibody (5 μg/ml; R&D Systems) to confluent EC cultures. ECs were processed as described above and incubated with PE-conjugated control IgG or mouse anti–human MHC II antibody (1 h, 4°C; Beckman Coulter). Subsequently, cells were washed with PBS/0.2% BSA and analyzed in a Becton Dickinson FACScan™ flow cytometer using CELLQuest™ software (Becton Dickinson). At least 20,000 viable cells per condition were analyzed.
Immunohistochemistry.
5-μm serial cryostat sections of surgical specimens of human carotid atheroma (n = 3), normal carotids from autopsies, and nondiseased aorta from cardiac transplantation donors (n = 3) were cut, air-dried onto microscope slides, fixed in acetone (−20°C, 5 min), and preincubated with PBS containing 0.3% hydrogen peroxide. Subsequently, sections were incubated (30 min) with primary mouse anti–human IL-18 (0.25 μg/ml, final concentration), goat anti–human IL-18Rα (3 μg/ml, final concentration; both R&D Systems), mouse anti–human muscle actin mAb for SMCs (1:35; Enzo Diagnostics), mouse-anti-human CD31 mAb for EC (1: 35; Dako), mouse anti–human CD68 for MØ (1:500; Dako), mouse anti–human CD3 for T cells (1:25; Dako), or control antibody (0.25 μg/ml mouse myeloma protein MOPC-21 or 3 μg/ml goat IgG, both final concentration; both Sigma-Aldrich), diluted in PBS supplemented with 5% appropriate serum, and processed according to the recommendations provided by the supplier (Universal Dako LSAB kit; Dako). For control purposes staining was validated in additional experiments using goat anti–human IL-18 (5 μg/ml) and mouse anti–human IL-18Rα (1.5 μg/ml, both final concentration; both R&D Systems). For colocalization with the respective cell type by immunofluorescence double labeling, goat anti–human IL-18 or IL-18Rα antibody was applied for 90 min followed by biotinylated secondary antibody (45 min) and Texas red-conjugated streptavidin (20 min; Amersham Pharmacia Biotech). Subsequent to application of the avidin/biotin blocking kit (Vector Laboratories), anti-muscle actin mAb for SMCs (1:35; Enzo Diagnostics), anti-CD31 mAb for EC (1:35; Dako), or anti-CD68 mAb for MØ (1:500; Dako) were added (overnight, 4°C). Finally, the appropriate secondary antibodies were applied for 45 min followed by streptavidin-FITC (Amersham Pharmacia Biotech) for 20 min.
In Situ Hybridization.
In situ hybridization was performed accordingly to the instructions of the manufacturer (Innogenex). Briefly, frozen tissue sections as well as cultures of ECs, SMCs, and MØ on four-chamber slides were fixed in cold acetone, air-dried, and incubated (10 min, 65°C; subsequently for 2 h at 37°C) with a mixture of FITC-labeled IL-18Rβ (5′-CCCGTGTTCCTGTCATCACTCCAATAACTTCATTCCTTTG-3′; 5′-TGCAACAAGCCAAAGAAATATCCAGCCCAAACAGAGCATT-3′; 5′-GCCCTGGTGATAGTCATAAACTTC- ATCCACTACGATTCGG-3′; 5′-CTCTGGTAGGTCCGGTACAGCAGCACTATTTCAATCCAGT-3′), and IFN-γ (5′-CATCGTTTCCGAGAGAATTAAGCCAAAGAAGTTGAAATCA-3′; 5′-AAGAGAACCCAAAACGATGCAGAGCTGAAAAGCCAAGATA-3′; 5′-TTTTCTGTCACTCTCCTC- TTTCCAATTCTTCAAAATGCCT-3′) antisense oligomers, or the respective sense (control) oligomers (all 4 μg/ml, final concentration) in hybridization-buffer. Finally, slides were washed three times, incubated with biotinylated mouse anti-FITC antibody (1 h), followed by alkaline phosphatase-conjugated streptavidin (30 min) and NBT/BCIP chromogen solution (1 h). Adjacent sections were analyzed for SMCs, MØ, and T cell content by immunohistochemistry as described above.
Statistical Analysis.
Data are presented as mean ± SD and groups were compared using the Student's t test. A value of P ≤ 0.05 was considered significant.
Results
Human Atherosclerotic Lesions Express IL-18 and IL-18 Receptor (IL-18Rα/β) In Situ.
Non-diseased arterial tissue lacked immunoreactive IL-18, but showed basal expression of IL-18Rα (Fig. 1). In contrast, human atheroma stained strongly for IL-18 and showed enhanced staining for IL-18Rα, compared with nonatherosclerotic tissue. Control IgG showed no signal. Evaluation of IL-18Rβ expression, using in situ hybridization since antibodies were not available, demonstrated faint basal expression of IL-18Rβ transcripts in nondiseased tissue, which was markedly enhanced in atherosclerotic lesions (Fig. 1). Controls using the respective sense probe did not yield a specific signal.
Figure 1.
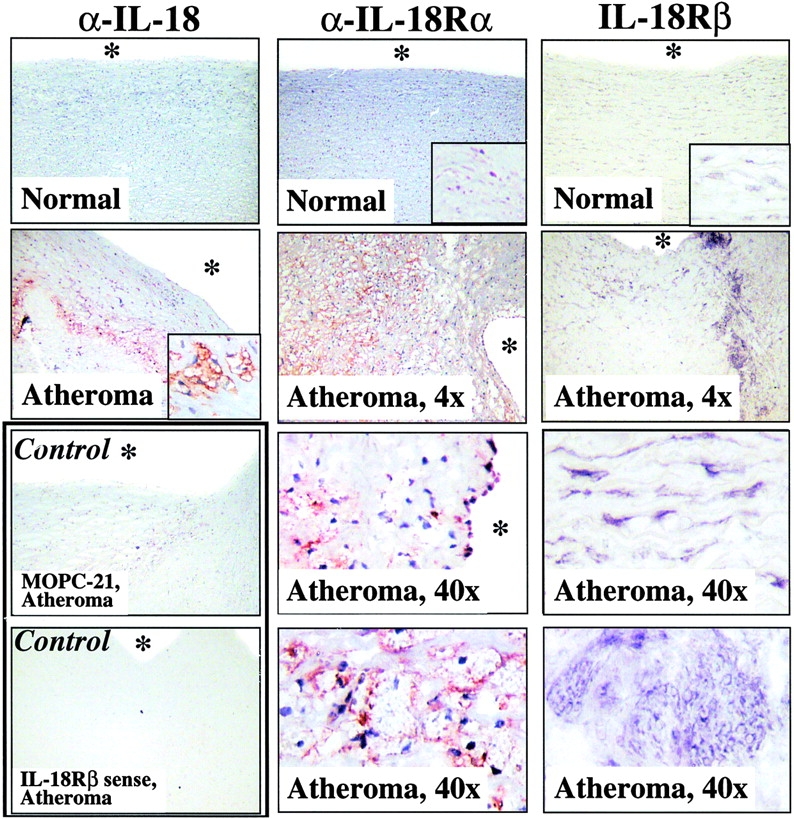
Human atherosclerotic lesions express IL-18, IL-18Rα, and IL-18Rβ. Serial cryostat sections of frozen specimens of nonatherosclerotic aorta (Normal) and carotid atheroma (Atheroma) were analyzed by either immunohistochemistry, using mouse anti–human IL-18 (top panels) or goat anti–human IL-18Rα antibodies (middle column), or by in situ hybridization, using IL-18Rβ antisense oligomers (right column). The corresponding lower panels as well as insets depict higher magnifications (40×) of the overviews (4×) presented in the top panels. Control experiments (bottom panels) demonstrated that application of MOPC-21, goat IgG (data not shown), or IL-18Rβ sense oligomers did not yield specific staining in atherosclerotic specimens. The asterisks indicate the lumen of the vessels. Analysis of tissue obtained from two other nonatherosclerotic as well as atherosclerotic specimens showed similar results.
In accord with these studies, immunofluorescence double-labeling demonstrated that IL-18 localized with lesional MØ, rather than SMCs or ECs, whereas IL-18Rα colocalized with all three atheroma-associated cell types (Fig. 2). Moreover, extracts of atheromatous tissue, but not of nondiseased arterial tissue, contained immunoreactive IL-18 (Fig. 3 A). All plaques analyzed (n = 7) contained immunoreactive 24-kD and 18-kD proteins, corresponding to the predicted molecular weight for precursor and mature IL-18. Typically the 18-kD form predominated in Western blot analysis. Of note, protein extracts obtained from abdominal aortic aneurysm specimens revealed expression of predominantly the 24-kD precursor form (Fig. 3 B), suggesting that the protein extraction procedure employed did not cause artifactual, postharvesting degradation of precursor IL-18, and did not account for the 18-kD band in atherosclerotic tissue. Parallel addition of recombinant human IL-1β precursor, the other substrate of caspase-1, to protein extracts from atherosclerotic tissue yielded recovery of the 33-kD precursor, but not smaller forms (Fig. 3 B), a further indication that posthoc proteolysis did not explain the abundance of mature IL-18 in plaques. Human atheroma furthermore contained significantly higher concentrations of IL-18Rα compared with nondiseased tissue (8.9 ± 1.7-fold; n = 7; P ≤ 0.005) (Fig. 3 A). Preincubation (2 h) of the anti–IL-18 or anti–IL-18Rα antibody with the respective recombinant protein markedly diminished immunoreactive bands in Western blots, supporting specificity of the antibodies employed (data not shown). Furthermore, IL-18 and IL-18Rα antibodies of different origin (rabbit anti–human IL-18 and mouse anti–IL-18Rα, respectively) yielded similar data (data not shown). In view of the involvement of caspase-1 in the processing of IL-18 (22, 23) and the reported synergism of IL-18 with IL-12 (14, 24), we further analyzed atherosclerotic tissue extracts for the expression of caspase-1 and IL-12. In agreement with previous immunohistochemical analysis (40, 41), atherosclerotic but not nondiseased arterial tissue contained the active p20 subunit of Caspase-1 (Fig. 3 A), consistent with the presence of processed, mature IL-18. Moreover, atherosclerotic but not nondiseased specimens expressed IL-12p40 (Fig. 3 A), in accord with the previously reported transcriptional regulation (35).
Figure 2.

Differential expression of IL-18 and IL-18Rα in ECs, SMCs, and MØ in human atherosclerotic lesions. Double-immunofluorescence staining colocalized IL-18 (red, left) or IL-18Rα (red, right) with ECs (anti-CD31), SMCs (anti-α-actin), or MØ (anti-CD68) within atherosclerotic plaques. Analysis of three atheroma from different individuals showed similar results.
Figure 3.

Human atherosclerotic lesions contain immunoreactive IL-18 and IL-18Rα. (A) Extracts (50 μg/lane) of nonatherosclerotic arterial specimens (Normal) as well as atherosclerotic lesions (Atheroma) were applied to SDS-PAGE and subsequent Western blot analysis using either an anti–IL-18, anti–IL-18Rα, anti–caspase-1P20, or anti–IL-12P40 antibody. (B) As control for protein degradation, (top) protein extracts of abdominal aortic aneurysm (AAA) were applied to Western blot analysis for IL-18 or (bottom) exogenous recombinant IL-1β precursor (300 ng/mg tissue) was added to extracts of atherosclerotic tissue. The positions of the molecular weight markers are indicated on the left. Analysis of tissue obtained from a total of five nonatherosclerotic as well as seven atherosclerotic specimens showed similar results.
Differential Expression of IL-18 and IL-18 Receptor (IL-18Rα/β) in Human Vascular ECs, SMCs, and MØ In Vitro.
Further studies tested the hypothesis that atheroma-associated mediators regulate expression of IL-18 and its receptor in ECs, SMCs, and MØ. In accord with the in situ observation of the MØ-restricted expression of IL-18 in atheroma, we did not detect immunoreactive ligand in either unstimulated, IL-1β–, or TNF-α–stimulated ECs or SMCs in vitro (Fig. 4). However, in agreement with previous reports (20, 31), these proinflammatory cytokines as well as endotoxin induced expression and release of IL-18 by MØ, detected by Western blotting as a 24-kD and 18-kD immunoreactive protein, corresponding to the predicted molecular weight for the precursor and mature form, respectively. In contrast to the ligand, all three cell types showed basal IL-18Rα expression that rose after stimulation with IL-1β or TNF-α (Fig. 4). FACS® analysis revealed surface expression of IL-18Rα on these vascular wall cells (Fig. 5), which was enhanced after stimulation with IL-1β or TNF-α. Interestingly, IFN-γ employed as a sole stimulus did not enhance expression of IL-18Rα, whereas in combination with IL-1β or TNF-α it evoked more IL-18Rα cell surface expression on ECs and SMCs than the other stimuli tested. Combinations of cytokines present in atheroma, elicited IL-18Rα expression on ECs and SMCs time- (Fig. 5) and concentration- (data not shown) dependent, requiring a minimum of 12-h stimulation with 0.1 ng IL-1β/0.5 ng TNF-α/10 U IFN-γ/ml. Maximal expression occurred after 48 h of stimulation with 10 ng IL-1β/50 ng TNF-α/1,000 U IFN-γ/ml and did not increase after more prolonged incubation (96 h; data not shown). Analysis of the expression of the second component of the IL-18 receptor complex, IL-18Rβ, employed RT-PCR, since antibodies were not available. Amplification of cDNA prepared from cultures of unstimulated ECs, SMCs, or MØ demonstrated constitutive expression of IL-18Rβ transcripts (Fig. 6). In accordance with the data obtained regarding the regulation of IL-18Rα protein expression, stimulation with proinflammatory cytokines, such as IL-1β (data not shown) or TNF-α, but also IL-18 itself, enhanced the detectable band intensities for both IL-18Rα and IL-18Rβ transcripts in ECs and SMCs. Although RT-PCR studies do not allow for quantitative analysis, these studies suggest that the expression of both IL-18 receptor subunits may be regulated similarly.
Figure 4.

Differential expression of IL-18 and IL-18Rα on cultured ECs, SMCs, and MØ in vitro. Supernatants (50 μl/lane; for IL-18) or lysates (50 μg total protein/lane; for IL-18Rα) of ECs, SMCs, or MØ cultures, incubated for 24 h with serum-free medium alone (None) or human recombinant IL-1β (10 ng/ml), TNF-α (50 ng/ml), combinations thereof, or LPS (100 ng/ml) were analyzed by Western blotting using anti–human IL-18 (left) or IL-18Rα (right) antibody. The positions of the molecular weight markers are indicated on the left. Three independent experiments yielded similar data.
Figure 5.

Proinflammatory cytokines induce the cell surface expression of IL-18Rα in human ECs and SMCs. Human vascular SMCs and ECs were incubated 36 h (stimuli) or for the respective times (time dependency) with either medium alone (None) or medium supplemented with IL-1β (10 ng/ml), IFN-γ (1,000 U/ml), TNF-α (50 ng/ml), or combinations thereof, and were studied for the expression of IL-18Rα by FACS® analysis. Staining for IL-18Rα (solid histograms) is compared with the isotype control (dotted line) and unstained control (solid line). At least 20,000 viable cells were analyzed for each staining. Values of geometric mean fluorescence are indicated in each panel. Similar results were obtained in three independent experiments using cells of different donors.
Figure 6.

Proinflammatory cytokines induce the expression of IL-18Rα and IL-18Rβ transcripts in human ECs, SMCs, and MØ. Expression of IL-18Rα and IL-18Rβ transcripts was analyzed by RT-PCR using RNA obtained from ECs, SMCs, and MØ cultured in medium alone (None), or medium supplemented with TNF-α (50 ng/ml), endotoxin (100 ng/ml LPS), 50 μg/ml IL-12, 10 μg/ml IL-18, or a combination of the latter two. PCR products were applied to 1.5% agarose gels. Positions of the base pair markers are indicated on the left. Comparable data were obtained using cells from at least three different donors.
The IL-18 Receptor Expressed on Human Vascular ECs and SMCs Is Functional.
To test the functionality of the IL-18 receptor complex expressed on ECs and SMCs we incubated either cell type with recombinant IL-18. Stimulation with the ligand indeed induced the expression of the proinflammatory cytokine IL-6 and the chemokine IL-8, in a time- and concentration-dependent fashion (Fig. 7 A), demonstrating functionality of the IL-18 receptor expressed on vascular ECs and SMCs. Maximal activity required stimulation of either cell type with 15 ng recombinant human IL-18/ml for 24 h. Higher concentrations (up to 150 ng IL-18/ml, data not shown) or prolonged stimulation (up to 48 h) did not increase IL-6 or IL-8 release from ECs or SMCs.
Figure 7.


Expression of functional IL-18 receptor on cultured human ECs, SMCs, and MØ. (A) Cultures of ECs or SMCs were stimulated with recombinant human IL-18 (50 ng/ml) for the respective times (left) or with the respective concentrations of IL-18 for 24 h (right). Culture supernatants were analyzed by ELISA for IL-6 or IL-8. Data represent mean ± SD. Similar results were obtained in three independent experiments. (B) Culture supernatants of SMCs or MØ stimulated with IL-1β/TNF-α (10/50 ng/ml), IL-18 (50 ng/ml), IL-12 (10 ng/ml), or a combination of the latter two, were applied to Western blot analysis for MMP-1 or MMP-13, or to gelatin-zymography. Positions of the molecular weight markers are indicated on the left. Comparable data were obtained using cells from at least three different donors.
To study additional proatherogenic functions triggered by IL-18, we further tested whether stimulation with this cytokine induced the expression of matrix metalloproteinase (MMP), enzymes that likely render atheroma prone to rupture and hence promote thrombosis. Indeed, IL-18 induced the expression of MMP in all three cell types, as demonstrated here for the interstitial collagenases MMP-1 and MMP-13 as well as the gelatinase MMP-9 in human vascular SMC and MØ (Fig. 7 B). Although IL-18 as a sole stimulus in several experiments induced both collagenases only weakly, combination with IL-12 enhanced the expression to levels comparable to those obtained with ‘classical’ inducers of these MMP, such as IL-1β and TNF-α. Furthermore, IL-18 induced the expression of ICAM-1 on ECs as determined by FACS® analysis (data not shown).
IL-18 Induces IFN-γ Production in MØ and Unexpectedly by SMCs.
We further investigated whether IL-18 also mediates its originally recognized function, the induction of IFN-γ expression, on human vascular ECs and SMCs as well as MØ. Indeed, IL-18, particularly in synergism with IL-12, elicited IFN-γ protein and transcript in MØ and, even more surprisingly, in human vascular SMCs, a cell type previously not implicated in the synthesis of this cytokine, as shown by ELISA (Fig. 8 A), in situ hybridization, and RT-PCR (Fig. 8 B). Of note, addition of Polymyxin B did not diminish the expression of IFN-γ transcripts or protein, whereas heat inactivation of IL-12/IL-18 abolished this function, excluding endotoxin contamination as the cause of our observation (Fig. 8 A and B). Moreover, other proinflammatory cytokines, such as IL-1β or TNF-α, did not induce expression of IFN-γ in these cell types. The SMC cultures employed for these experiments expressed smooth muscle α-actin, however, did not yield a signal for CD14 by RT-PCR or for CD3, CD4, CD31, CD64, or CD68 by immunohistochemistry (data not shown), suggesting that the passaged SMC cultures employed indeed contained negligible if any T lymphocytes, ECs, or MØ. Of note, within the total of 15 different donors, SMC cultures from five donors did not show induction of IFN-γ (0.8 ± 1.4 U IFN-γ/ml) compared with supernatants of unstimulated cultures (1.4 ± 1.8 U IFN-γ/ml), even when maximal concentrations of IL-18 or IL-12/IL-18 concentrations were applied. Within the group of 10 experiments yielding significantly (P ≤ 0.05) enhanced IFN-γ expression upon stimulation with IL-12/IL-18 combination, low and high “responders” were identified. Five donors yielded an average of 16.5 ± 12.9 U IFN-γ/ml and the remaining five donors of 260 ± 115 U IFN-γ/ml. Although the cause remains unknown, neither gender and age of the cell donor nor passage number of the cells appeared to account for this observed variation. In contrast to SMCs, MØ expressed IFN-γ more uniformly after stimulation with IL-12/IL-18 (220 ± 43 U IFN-γ/ml; n = 4). EC did not express IFN-γ in response to any of the stimuli or combinations thereof tested, supporting the cell type specificity of our finding (Fig. 8 A).
Figure 8.




IL-18 induces expression of IFN-γ on vascular SMCs and MØ. (A) Cultures of SMCs or ECs were stimulated (36 h) with either IL-1β/TNF-α (10/50 ng/ml), IL-12 (10 ng/ml), respective concentrations of IL-18, or combinations of the latter two, and supernatants were analyzed by ELISA for IFN-γ. Polymyxin B and heat treated IL-12/IL-18 (95°C for 10 min) were applied to exclude activation via endotoxin contamination. Data represent mean ± SD. Similar results were obtained in at least three independent experiments. (B, right) Total RNA obtained from cultured SMCs stimulated with medium alone (None), IL-12, IL-18, or combinations thereof, was analyzed by RT-PCR for IFN-γ transcripts. (B, left) MØ or SMCs cultured to confluence on 4-well chamber slides were analyzed for the expression of IFN-γ transcripts by in situ hybridization using the respective antisense or sense oligomers. (C) Supernatants of SMC cultures stimulated with medium alone (SMC-SN, none) or combination of IL-12 (10 ng/ml) and IL-18 (50 ng/ml) (SMC-SN (IL-12/18)) were applied in absence or presence of neutralizing α-IFN-γ antibody (SMC-SN (IL-12/18) + α-IFN-γ) to confluent EC cultures for 48 h and MHC II expression was compared with unstimulated (none) or IFN-γ–stimulated (rIFN-γ, 1,000 U/ml) ECs by FACS® analysis. Staining (solid histograms) was compared with isotype control (dotted line). At least 20,000 viable cells were analyzed for each staining. Values of geometric mean fluorescence are indicated in each panel. Similar results were obtained in three independent experiments using cells of different individuals. (D, left) Serial cryostat sections of frozen specimens of carotid atheroma were analyzed for the expression of IFN-γ transcripts by in situ hybridization using the respective antisense (top) or sense (bottom) oligomers. Low (original magnification: 4×, left) and high (original magnification: 40×, right) magnifications are shown. The asterisks indicate the lumen of the vessels. (D, right) Adjacent sections were stained for SMCs (anti–α-actin), MØ (anti-CD68), or T lymphocytes (anti-CD3). Low (4×) and high (40×, insets) magnifications are shown.
In view of the unexpected expression of IFN-γ by SMCs, we further tested the ability of medium conditioned by IL-12/IL-18–stimulated SMCs to exhibit a characteristic biological activity of IFN-γ. Indeed, incubation with culture supernatants derived from IL-12/IL-18-stimulated SMCs enhanced the expression of MHC II in EC (Fig. 8 C), compared to those cultured with supernatants of nontreated SMCs. Addition of neutralizing anti–IFN-γ antibody to the culture medium abrogated induction of MHC II on ECs, further validating the specificity of our observations.
To demonstrate the potential relevance of the novel observation of IFN-γ expression by SMCs to the pathogenesis of atherosclerosis, we performed in situ hybridization studies on sections of human atheroma. In support of the in vitro observations outlined above, IFN-γ in atherosclerotic lesions localized with T cells, but also within MØ- and SMC-enriched areas, which were T cell deprived (Fig. 8 D). In contrast, nonatherosclerotic tissue did not contain IFN-γ mRNA. Moreover, application of sense oligomers did not yield detectable signals. Unavailability of appropriate antibodies hampered extension of these in situ results to the protein level (see discussion).
Discussion
Recent research points to a prominent role of immunity and inflammation in the pathogenesis of atherosclerosis (1–3). Considerable data support the hypothesis that cytokines orchestrate many of the complex processes underlying this prevalent human disease (1–4, 6, 7, 42). The present study highlights a novel potential pathway for signaling in atherogenesis that involves the IL-18 receptor/ligand dyad. It further reports the unexpected ability of vascular SMCs to produce biologically active IFN-γ after stimulation with IL-18. Together, these findings considerably add to the burgeoning evidence of a local, self-sustaining immune, and inflammatory response within the atheromatous plaque. Within the cytokine network, IFN-γ has garnered considerable interest as a regulator of atherogenesis. Early studies implicated this cytokine in various proatherogenic processes, including loss of fibrillar collagen and modulation of inflammatory processes (27, 28, 43, 44). Indeed, absence of IFN-γ significantly diminishes formation of atherosclerotic lesions in hypercholesterolemic mice. Gupta et al. demonstrated that IFN-γ promotes and modifies atherosclerosis through both local effects in the arterial wall, e.g., lesional lipid accumulation and enhanced collagen content, as well as a systemic effect on plasma lipoproteins, concluding that therapeutic inhibition of IFN-γ signaling may lead to the formation of more lipid-depleted, “stable” atheromata (4). Studies from our own and other groups further demonstrated a causal role for IFN-γ in transplantation-associated arteriopathy (5, 45). Interestingly, IFN-γ can mediate atherosclerosis in the absence of T lymphocytes, the classical source of this cytokine (6). Our surprising finding that IL-18 induced the expression of IFN-γ not only in MØ, but also in vascular SMCs, a previously unsuspected source, points to a novel potential pathogenic pathway at play in both native and transplantation-associated atherosclerosis.
As mentioned above, unavailability of appropriate antibodies hampered immunohistochemical analysis of IFN-γ expression in situ. Indeed, a very recent report has called into question the specificity of various (13 different) anti–IFN-γ antibodies due to “cross-reactivity” with other cell types than T lymphocytes, namely SMCs (46). Our present findings would suggest a reevaluation of this conclusion, as some of the putatively spurious staining might actually be genuine.
Our in situ findings indicate that MØ in human atheroma strongly express IL-18, a finding supported by a report published after submission of this manuscript (47). In contrast, nondiseased arterial tissue, which lacks MØ, did not contain IL-18. The localization of the cytokine agrees with previous reports, demonstrating IL-18 expression by human MØ in vitro (14, 48). Inducers of IL-18 include IL-1β and TNF-α, proinflammatory cytokines expressed in human atheroma, but not in nondiseased arteries (3, 44). Detection of the mature 18-kD form of IL-18 in protein extracts of human atheroma and cultured MØ suggests the presence of the active form of the cytokine, a finding in accordance with the expression of active caspase-1 subunits. Although expression of IL-18 in atheroma appears restricted to one cell type, several cells found in atheroma express the IL-18 receptor, namely ECs, SMCs, MØ, and T lymphocytes, as shown here and demonstrated elsewhere (15, 16, 37, 47). Low level staining for IL-18Rα in nondiseased tissue, supported by Western blot analysis, FACS®, and PCR data, suggested modest basal expression of the receptor on vascular ECs, SMCs, and MØ, a finding consistent with reports of constitutive expression of the IL-18 receptor on hematopoetic cell lines (49). Interestingly, the combination of several cytokines found in plaques, namely IL-1β, TNF-α, and IFN-γ, promoted the expression of both IL-18 receptor chains. Thus, the enhanced expression of the IL-18 receptor in human atheroma in situ corroborates the in vitro observations, indicating the likelihood of IL-18 signaling in early and/or advanced atherosclerotic lesions. In agreement, transfer of an IL-18 binding protein plasmid into ApoE-deficient mice diminishes atherosclerosis in vivo (50). The finding that IFN-γ alone did not enhance, but, when combined with IL-1β and TNF-α, did promote expression of the IL-18 receptor, might reflect distinct pathways separating physiologic from pathologic responses, since IL-1β and TNF-α commonly coexist at sites of chronic inflammation and are not required for physiologic T lymphocyte or NK cell activation. Moreover, the inducibility of IL-18Rα/β by IFN-γ in an inflammatory setting combined with the induction of IFN-γ expression via IL-18 signaling suggests a positive feedback loop in the vasculature, which might contribute to the dysregulated inflammatory response characterizing atheroma. Such a self-reinforcing IL-18/IFN-γ cascade could furthermore contribute to the dominant Th1 response during atherogenesis (3). Notably, IL-18 promotes Th1 immune responses via its well recognized function to evoke IFN-γ expression in lymphocytes (14–16) as well as via IFN-γ–independent pathways, probably in synergy with IL-12, although this function remains controversial (20, 24, 25, 51). Accordingly, the Th1 lymphocyte subpopulation, but not Th2 lymphocytes, express the receptor for IL-18 (30). Our observations extend the implications for the IL-18/IL-18R dyad in atherosclerosis beyond regulation of IFN-γ or Th1-mediated immune responses. Ligation of the IL-18 receptor on ECs, SMCs, MØ, and, as published previously, on T lymphocytes, induced expression of cytokines and chemokines, such as IL-6, IL-8, and as shown by others, of IL-1β and TNF-α, molecules implicated in atherogenesis (1, 2, 16). Moreover, IL-18 induced the expression of adhesion molecules and at least two classes of matrix degrading enzymes, including interstitial collagenases, enzymes considered crucial for the degradation of fibrillar collagen, the major load-bearing molecule of the atheroma's fibrous cap (38). Thus, IL-18 signaling probably promotes processes underlying weakening of the plaque's extracellular matrix, rendering lesions prone to rupture, the proximate cause of most coronary thromboses. In addition, IL-18 might contribute to atherogenesis in other ways, for example promoting production of proinflammatory cytokines and adhesion molecules, as well as growth factors (e.g., GM-CSF), inducible nitric oxide synthase, or inducible cyclooxygenase (15, 16, 18, 20, 21, 24). Aside from mediating proatherogenic functions directly, IL-18 might further augment atherogenesis by inhibiting TGF-β function (18), a pathway that may mitigate this disease (52). In conclusion, our data identify a novel step proximal to IFN-γ in the inflammatory pathways of atherosclerosis. We have uncovered a new source of IFN-γ in atheroma, a finding that should stimulate the evaluation of SMC-derived IFN-γ in other chronic inflammatory diseases and in vasculitides.
Acknowledgments
The authors thank E. Shvartz, A. Papautsky, S. LaClair, Karen E. Williams, and E. Simon-Morrissey (Brigham and Women's Hospital) for their skillful technical assistance and Prof. Jonathan Howard (University of Cologne, Cologne, Germany) for his insightful discussion and support.
This work was supported by a MERIT Award to Dr. Peter Libby (NIH/NHLBI, HL34636).
This work was presented in part at the Scientific Sessions of the American Heart Association, New Orleans on November 12–15, 2000, and published in abstract form (Circulation. 2000. 18:11–66). Further parts of this work were presented at the 11th International Congress of Immunology, Stockholm, Sweden, July 22–27, 2001, and published in abstract form (Scand. J. Immunol. 2001. 54:A5).
Footnotes
Abbreviations used in this paper: EC, endothelial cell; ICAM, intracellular adhesion molecule; MMP, matrix metalloproteinase; MØ, mononuclear phagocytes; SMC, smooth muscle cell.
References
- 1.Libby, P. 1990. Inflammatory and immune mechanisms in atherogenesis. Atherosclerosis Reviews. A. Leaf and P. Weber, editors. Raven Press, New York. 79–89.
- 2.Ross, R. 1999. Atherosclerosis-an inflammatory disease. N. Engl. J. Med. 340:115–126. [DOI] [PubMed] [Google Scholar]
- 3.Frostegard, J., A.K. Ulfgren, P. Nyberg, U. Hedin, J. Swedenborg, U. Andersson, and G.K. Hansson. 1999. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis. 145:33–43. [DOI] [PubMed] [Google Scholar]
- 4.Gupta, S., A.M. Pablo, X. Jiang, N. Wang, A.R. Tall, and C. Schindler. 1997. IFN-γ potentiates atherosclerosis in ApoE knock-out mice. J. Clin. Invest. 99:2752–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagano, H., R.N. Mitchell, M.K. Taylor, S. Hasegawa, N.L. Tilney, and P. Libby. 1997. Interferon-γ deficiency prevents coronary arteriosclerosis but not myocardial rejection in transplanted mouse hearts. J. Clin. Invest. 100:550–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tellides, G., D.A. Tereb, N.C. Kirkiles-Smith, R.W. Kim, J.H. Wilson, J.S. Schechner, M.I. Lorber, and J.S. Pober. 2000. Interferon-γ elicits arteriosclerosis in the absence of leukocytes. Nature. 403:207–211. [DOI] [PubMed] [Google Scholar]
- 7.Whitman, S.C., P. Ravisankar, H. Elam, and A. Daugherty. 2000. Exogenous interferon-γ enhances atherosclerosis in apolipoprotein E−/− mice. Am. J. Pathol. 157:1819–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trinchieri, G., M. Matsumoto-Kobayashi, S.C. Clark, J. Seehra, L. London, and B. Perussia. 1984. Response of resting human peripheral blood natural killer cells to interleukin 2. J. Exp. Med. 160:1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young, H.A., and K.J. Hardy. 1995. Role of interferon-γ in immune cell regulation. J. Leukoc. Biol. 58:373–381. [PubMed] [Google Scholar]
- 10.Pang, Y., Y. Norihisa, D. Benjamin, R.R. Kantor, and H.A. Young. 1992. Interferon-γ gene expression in human B-cell lines: induction by interleukin-2, protein kinase C activators, and possible effect of hypomethylation on gene regulation. Blood. 80:724–732. [PubMed] [Google Scholar]
- 11.Nitta, T., M. Ebato, K. Sato, and K. Okumura. 1994. Expression of tumour necrosis factor-α, -β and interferon-γ genes within human neuroglial tumour cells and brain specimens. Cytokine. 6:171–180. [DOI] [PubMed] [Google Scholar]
- 12.Di Marzio, P., P. Puddu, L. Conti, F. Belardelli, and S. Gessani. 1994. Interferon γ upregulates its own gene expression in mouse peritoneal macrophages. J. Exp. Med. 179:1731–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson, B.W., T.L. McLemore, and R.G. Crystal. 1985. γ interferon is spontaneously released by alveolar macrophages and lung T lymphocytes in patients with pulmonary sarcoidosis. J. Clin. Invest. 75:1488–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okamura, H., H. Tsutsi, T. Komatsu, M. Yutsudo, A. Hakura, T. Tanimoto, K. Torigoe, T. Okura, Y. Nukada, K. Hattori, et al. 1995. Cloning of a new cytokine that induces IFN-γ production by T cells. Nature. 378:88–91. [DOI] [PubMed] [Google Scholar]
- 15.Ushio, S., M. Namba, T. Okura, K. Hattori, Y. Nukada, K. Akita, F. Tanabe, K. Konishi, M. Micallef, M. Fujii, et al. 1996. Cloning of the cDNA for human IFN-γ-inducing factor, expression in Escherichia coli, and studies on the biologic activities of the protein. J. Immunol. 156:4274–4279. [PubMed] [Google Scholar]
- 16.Puren, A.J., G. Fantuzzi, Y. Gu, M.S. Su, and C.A. Dinarello. 1998. Interleukin-18 (IFNγ-inducing factor) induces IL-8 and IL-1β via TNFα production from non-CD14+ human blood mononuclear cells. J. Clin. Invest. 101:711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Udagawa, N., N.J. Horwood, J. Elliott, A. Mackay, J. Owens, H. Okamura, M. Kurimoto, T.J. Chambers, T.J. Martin, and M.T. Gillespie. 1997. Interleukin-18 (interferon-γ-inducing factor) is produced by osteoblasts and acts via granulocyte/macrophage colony-stimulating factor and not via interferon-γ to inhibit osteoclast formation. J. Exp. Med. 185:1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olee, T., S. Hashimoto, J. Quach, and M. Lotz. 1999. IL-18 is produced by articular chondrocytes and induces proinflammatory and catabolic responses. J. Immunol. 162:1096–1100. [PubMed] [Google Scholar]
- 19.Stoll, S., G. Muller, M. Kurimoto, J. Saloga, T. Tanimoto, H. Yamauchi, H. Okamura, J. Knop, and A.H. Enk. 1997. Production of IL-18 (IFN-γ-inducing factor) messenger RNA and functional protein by murine keratinocytes. J. Immunol. 159:298–302. [PubMed] [Google Scholar]
- 20.Nakanishi, K., T. Yoshimoto, H. Tsutsui, and H. Okamura. 2001. Interleukin-18 regulates both th1 and th2 responses. Annu. Rev. Immunol. 19:423–474. [DOI] [PubMed] [Google Scholar]
- 21.Dinarello, C.A. 1999. Interleukin-18. Methods. 19:121–132. [DOI] [PubMed] [Google Scholar]
- 22.Gu, Y., K. Kuida, H. Tsutsui, G. Ku, K. Hsiao, M.A. Fleming, N. Hayashi, K. Higashino, H. Okamura, K. Nakanishi, et al. 1997. Activation of interferon-γ inducing factor mediated by interleukin-1beta converting enzyme. Science. 275:206–209. [DOI] [PubMed] [Google Scholar]
- 23.Ghayur, T., S. Banerjee, M. Hugunin, D. Butler, L. Herzog, A. Carter, L. Quintal, L. Sekut, R. Talanian, M. Paskind, et al. 1997. Caspase-1 processes IFN-γ-inducing factor and regulates LPS-induced IFN-γ production. Nature. 386:619–623. [DOI] [PubMed] [Google Scholar]
- 24.Micallef, M.J., T. Ohtsuki, K. Kohno, F. Tanabe, S. Ushio, M. Namba, T. Tanimoto, K. Torigoe, M. Fujii, M. Ikeda, et al. 1996. Interferon-γ-inducing factor enhances T helper 1 cytokine production by stimulated human T cells: synergism with interleukin-12 for interferon-γ production. Eur. J. Immunol. 26:1647–1651. [DOI] [PubMed] [Google Scholar]
- 25.Takeda, K., H. Tsutsui, T. Yoshimoto, O. Adachi, N. Yoshida, T. Kishimoto, H. Okamura, K. Nakanishi, and S. Akira. 1998. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity. 8:383–390. [DOI] [PubMed] [Google Scholar]
- 26.Fantuzzi, G., A.J. Puren, M.W. Harding, D.J. Livingston, and C.A. Dinarello. 1998. Interleukin-18 regulation of interferon γ production and cell proliferation as shown in interleukin-1β-converting enzyme (caspase-1)-deficient mice. Blood. 91:2118–2125. [PubMed] [Google Scholar]
- 27.Warner, S.J.C., G.B. Friedman, and P. Libby. 1989. Immune interferon inhibits proliferation and induces 2;-5′-Oligoadenylate synthetase gene expression in human vascular smooth muscle cells. J. Clin. Invest. 83:1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansson, G.K., L. Jonasson, J. Holm, M.K. Clowes, and A. Clowes. 1988. γ interferon regulates vascular smooth muscle proliferation and Ia expression in vivo and in vitro. Circ. Research. 63:712–719. [DOI] [PubMed] [Google Scholar]
- 29.Kohka, H., T. Yoshino, H. Iwagaki, I. Sakuma, T. Tanimoto, Y. Matsuo, M. Kurimoto, K. Orita, T. Akagi, and N. Tanaka. 1998. Interleukin-18/interferon-γ-inducing factor, a novel cytokine, up-regulates ICAM-1 (CD54) expression in KG-1 cells. J. Leukoc. Biol. 64:519–527. [DOI] [PubMed] [Google Scholar]
- 30.Xu, D., W.L. Chan, B.P. Leung, D. Hunter, K. Schulz, R.W. Carter, I.B. McInnes, J.H. Robinson, and F.Y. Liew. 1998. Selective expression and functions of interleukin 18 receptor on T helper (Th) type 1 but not Th2 cells. J. Exp. Med. 188:1485–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dinarello, C.A. 1999. IL-18: A TH1-inducing, proinflammatory cytokine and new member of the IL-1 family. J. Allergy Clin. Immunol. 103:11–24. [DOI] [PubMed] [Google Scholar]
- 32.Torigoe, K., S. Ushio, T. Okura, S. Kobayashi, M. Taniai, T. Kunikata, T. Murakami, O. Sanou, H. Kojima, M. Fujii, et al. 1997. Purification and characterization of the human interleukin-18 receptor. J. Biol. Chem. 272:25737–25742. [DOI] [PubMed] [Google Scholar]
- 33.Born, T.L., E. Thomasson, T.A. Bird, and J.E. Sims. 1998. Cloning of a novel receptor subunit, AcPL, required for IL-18 signaling. J. Biol. Chem. 273:29445–29450. [DOI] [PubMed] [Google Scholar]
- 34.Hoshino, K., H. Tsutsui, T. Kawai, K. Takeda, K. Nakanishi, Y. Takeda, and S. Akira. 1999. Generation of IL-18 receptor-deficient mice: evidence for IL-1 receptor-related protein as an essential IL-18 binding receptor. J. Immunol. 162:5041–5044. [PubMed] [Google Scholar]
- 35.Uyemura, K., L.L. Demer, S.C. Castle, D. Jullien, J.A. Berliner, M.K. Gately, R.R. Warrier, N. Pham, A.M. Fogelman, and R.L. Modlin. 1996. Cross-regulatory roles of interleukin (IL)-12 and IL-10 in atherosclerosis. J. Clin. Invest. 97:2130–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gracie, J.A., R.J. Forsey, W.L. Chan, A. Gilmour, B.P. Leung, M.R. Greer, K. Kennedy, R. Carter, X.Q. Wei, D. Xu, et al. 1999. A proinflammatory role for IL-18 in rheumatoid arthritis. J. Clin. Invest. 104:1393–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McInnes, I.B., J.A. Gracie, B.P. Leung, X.Q. Wei, and F.Y. Liew. 2000. Interleukin 18: a pleiotropic participant in chronic inflammation. Immunol. Today. 21:312–315. [DOI] [PubMed] [Google Scholar]
- 38.Sukhova, G.K., U. Schönbeck, E. Rabkin, F.J. Schoen, A.R. Poole, R.C. Billinghurst, and P. Libby. 1999. Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation. 99:2503–2509. [DOI] [PubMed] [Google Scholar]
- 39.Schönbeck, U., F. Mach, G.K. Sukhova, E. Atkinson, E. Levesque, M. Herman, P. Graber, P. Basset, and P. Libby. 1999. Expression of stromelysin-3 in atherosclerotic lesions: regulation via CD40-CD40 ligand signaling in vitro and in vivo. J. Exp. Med. 189:843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geng, Y.J., and P. Libby. 1995. Evidence for apoptosis in advanced human atheroma. Colocalization with interleukin-1β-converting enzyme. Am. J. Pathol. 147:251–266. [PMC free article] [PubMed] [Google Scholar]
- 41.Young, J.L., G.K. Sukhova, D. Foster, W. Kisiel, P. Libby, and U. Schönbeck. 2000. The serpin proteinase inhibitor 9 is an endogenous inhibitor of interleukin 1β-converting enzyme (caspase-1) activity in human vascular smooth muscle cells. J. Exp. Med. 191:1535–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Libby, P., and R. Ross. 1996. Cytokines and growth regulatory molecules. Atherosclerosis and Coronary Artery Disease. V. Fuster, R. Ross, and E. Topol, editors. Lippincott-Raven, New York. 585–594.
- 43.Hansson, G.K., M. Hellstrand, L. Rymo, L. Rubbia, and G. Gabbiani. 1989. Interferon-γ inhibits both proliferation and expression of differentiation-specific α-smooth muscle actin in arterial smooth muscle cells. J. Exp. Med. 170:1595–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Libby, P., H. Loppnow, J.C. Fleet, H. Palmer, H.M. Li, S.J.C. Warner, R.N. Salomon, and S.K. Clinton. 1991. Production of cytokines by vascular wall cells: an update and implications for atherogenesis. Atherosclerosis: cellular and molecular interactions in the artery wall. A.I. Gotlieb, B.L. Langille and S. Federoff, editors. Plenum Press, New York. 161–169.
- 45.Raisanen-Sokolowski, A., T. Glysing-Jensen, J. Koglin, and M.E. Russell. 1998. Reduced transplant arteriosclerosis in murine cardiac allografts placed in interferon-γ knockout recipients. Am. J. Pathol. 152:359–365. [PMC free article] [PubMed] [Google Scholar]
- 46.van Der Loos, C.M., M.A. Houtkamp, O.J. de Boer, P. Teeling, A.C. van Der Wal, and A.E. Becker. 2001. Immunohistochemical detection of interferon-γ. Fake or fact? J. Histochem. Cytochem. 49:699–710. [DOI] [PubMed] [Google Scholar]
- 47.Mallat, Z., A. Corbaz, A. Scoazec, S. Besnard, G. Leseche, Y. Chvatchko, and A. Tedgui. 2001. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation. 104:1598–1603. [DOI] [PubMed] [Google Scholar]
- 48.Puren, A.J., G. Fantuzzi, and C.A. Dinarello. 1999. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1β are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc. Natl. Acad. Sci. USA. 96:2256–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamura, S., T. Otani, R. Okura, Y. Ijiri, R. Motoda, M. Kurimoto, and K. Orita. 2000. Expression and responsiveness of human interleukin-18 receptor (IL-18R) on hematopoietic cell lines. Leukemia. 14:1052–1059. [DOI] [PubMed] [Google Scholar]
- 50.Mallat, Z., A. Corbaz, A. Scoazec, P. Graber, S. Alouani, B. Esposito, Y. Humbert, Y. Chvatchko, and A. Tedgui. 2001. Interleukin-18/interleukin-18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ. Res. 89:E41–45. [DOI] [PubMed] [Google Scholar]
- 51.Robinson, D., K. Shibuya, A. Mui, F. Zonin, E. Murphy, T. Sana, S.B. Hartley, S. Menon, R. Kastelein, F. Bazan, and A. O'Garra. 1997. IGIF does not drive Th1 development but synergizes with IL-12 for interferon-γ production and activates IRAK and NFκB. Immunity. 7:571–581. [DOI] [PubMed] [Google Scholar]
- 52.Pinderski Oslund, L.J., C.C. Hedrick, T. Olvera, A. Hagenbaugh, M. Territo, J.A. Berliner, and A.I. Fyfe. 1999. Interleukin-10 blocks atherosclerotic events in vitro and in vivo. Arter. Thromb. Vasc. Biol. 19:2847–2853. [DOI] [PubMed] [Google Scholar]