

Abstract
Natural killer (NK) cells and interferon (IFN)-γ have been implicated in immune surveillance against tumor development. Here we show that tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) plays a critical role in the NK cell–mediated and IFN-γ–dependent tumor surveillance. Administration of neutralizing monoclonal antibody against TRAIL promoted tumor development in mice subcutaneously inoculated with a chemical carcinogen methylcholanthrene (MCA). This protective effect of TRAIL was at least partly mediated by NK cells and totally dependent on IFN-γ. In the absence of TRAIL, NK cells, or IFN-γ, TRAIL-sensitive sarcomas preferentially emerged in MCA-inoculated mice. Moreover, development of spontaneous tumors in p53+/− mice was also promoted by neutralization of TRAIL. These results indicated a substantial role of TRAIL as an effector molecule that eliminates developing tumors.
Keywords: NK cells, IFN-γ, methylcholanthrene-induced fibrosarcoma, p53, innate immune response
Introduction
Immune surveillance against tumors is mediated by both innate and adaptive components of the cellular immunity (1). The adaptive component mainly consists of CD8+ CTLs that recognize tumor antigens presented by MHC class I molecules on tumor cells. Natural killer (NK) cells have long been implicated in innate immunity against tumors, especially MHC class I–deficient variants (2, 3). Our recent studies have substantiated a pivotal role of NK cells in natural protection from primary tumor development induced by a chemical carcinogen methylcholanthrene (MCA)*and tumor metastasis (4–6). NK cells exert antitumor effects by direct cytotoxicity and by producing IFN-γ (7). We recently demonstrated that perforin-mediated cytotoxicity and IFN-γ act independently, yet together mostly account for the NK cell–mediated protection from MCA-induced sarcoma development and experimental tumor metastasis (8). However, the final effector mechanism by which IFN-γ inhibits the tumor development and metastasis remains unclear.
IFN-γ is a pleiotropic cytokine that can act on both tumor cells and host immunity (9, 10). IFN-γ directly inhibits proliferation of some tumor cells in vitro (11) and indirectly inhibits tumor growth in vivo by suppressing tumor angiogenesis (12, 13). IFN-γ enhances NK cell cytotoxicity by upregulating the expression of adhesion molecules and by increasing the sensitivity of tumor cells to perforin- and Fas ligand (FasL)-mediated cytotoxicity (14, 15). In addition, we have recently revealed a critical contribution of TNF-related apoptosis-inducing ligand (TRAIL) to the IFN-γ–dependent NK cell protection from tumor metastasis (16, 17). TRAIL is a type-II membrane protein belonging to the TNF family, which preferentially induces apoptotic cell death in a wide variety of tumor cells but not in normal cells in vitro (18, 19). Preclinical studies in mice and nonhuman primates have shown that administration of recombinant soluble form of TRAIL could suppress the growth of TRAIL-sensitive tumor xenografts with no apparent systemic toxicity (20, 21). However, a physiological role of TRAIL in tumor surveillance was largely unknown. We recently found that murine liver NK cells constitutively expressed TRAIL in an IFN-γ–dependent manner, which was at least partly responsible for natural antimetastatic function of liver NK cells against TRAIL-sensitive tumor cells (16). We have also demonstrated that IFN-γ–mediated TRAIL induction on NK cells plays some role in IFN-γ–dependent antimetastatic effects of IL-12 and α-galactosylceramide (17). Given that both NK cells and IFN-γ have also been implicated in natural protection from primary tumor development, TRAIL may be responsible for the NK cell–mediated and IFN-γ–dependent mechanism of tumor elimination. To address this possibility, in the present study, we examined the effects of neutralizing anti-TRAIL mAb on tumor development in mice subcutaneously inoculated with MCA. The tumor-promoting effect of anti-TRAIL mAb and the preferential emergence of TRAIL-sensitive MCA-induced sarcomas in anti–TRAIL-treated, NK cell-depleted, or IFN-γ–deficient mice indicated a pivotal role of TRAIL in NK cell– and IFN-γ–mediated immune surveillance against primary tumor development. A substantial contribution of TRAIL to the immune surveillance against spontaneous tumor development caused by p53 mutation was also elucidated.
Materials and Methods
Mice.
Male C57BL/6 (B6) mice at 6–8 wk of age were obtain from Charles River Laboratories and The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia. B6 IFN-γ–deficient mice were provided by Genentech. B6 p53-deficient (p53−/−) mice were obtained from The Jackson Laboratory and were bred to wild-type B6 mice to generate B6 p53 heterozygous (p53+/−) mice. All mice were maintained under specific pathogen-free conditions and used in accordance with the institutional guidelines of Juntendo University and Peter MacCallum Cancer Institute.
Fibrosarcoma Induction by MCA.
Wild-type, p53+/−, and IFN-γ–deficient B6 mice were inoculated subcutaneously in the hind flank with 5, 25, 100, or 400 μg of 3-MCA (Sigma-Aldrich) in 0.1 ml maize oil. Some mice were administered intraperitoneally with anti-TRAIL mAb (N2B2 or N2B1) (300 μg), control rat IgG2a (300 μg) (R35–95; BD PharMingen), or antiasialo GM1 (ASGM1) Ab (200 μg) (Wako) every 5 d starting on the day of MCA inoculation. The neutralizing anti–mouse TRAIL mAbs (N2B2 and N2B1) were prepared as described previously (22). Depletion of NK cells by the anti-ASGM1 Ab treatment was verified by flow cytometry (4, 16). Development of fibrosarcoma was monitored periodically over the course of 100–200 d. Tumors >2 mm in diameter and demonstrating progressive growth were recorded as positive. Tumor size was measured periodically with a caliper as the product of two perpendicular diameters (cm2) and represented as the mean ± SD of 3–10 tumor-bearing mice in each group. All data are representative of 2–3 independent experiments with similar results.
Isolation and Culture of MCA-induced Sarcoma Cell Lines.
When 100 μg MCA-induced sarcomas or spontaneously developing subcutaneous tumors in mice had reached a size of 0.5 cm2, mice were killed and tumors were removed aseptically. Tumors were cut into small pieces and treated with collagenase (Sigma type IV) at 37°C for 1 h, clumps were removed, and single cells were cultured in RPMI1640 medium supplemented with 10% FCS and 2 mM l-glutamine. The cells were split with 0.1% EDTA when they reached confluency. All tumor cell lines were kept in culture for at least 3 mo. The TRAIL-sensitive MCA-III and the TRAIL-resistant MCA-IV fibrosarcoma cell lines had been formerly established from wild-type B6 mice inoculated with 25 μg MCA and treated with anti-ASGM1 antibody.
Subcutaneous Outgrowth of MCA-induced Fibrosarcoma Cell Lines.
The indicated numbers of TRAIL-sensitive MCA-III and the TRAIL-resistant MCA-IV fibrosarcoma cell lines in 0.1 ml PBS were inoculated subcutaneously in the hind flank of B6 mice. Some mice were intraperitoneally administered with anti-TRAIL mAb (N2B2) (250 μg) or control rat IgG2a (250 μg) on day –1, 0, 7, 10, and 14. Tumor size was measured periodically over the course of 40 d with a caliper as described above and represented as the mean ± SD of 4–5 mice in each group. All data are representative of two to three independent experiments with similar results.
Spontaneous Tumor Development in p53+/− Mice.
p53+/− B6 mice were administered intraperitoneally with anti-TRAIL mAb (N2B2) (300 μg) or control rat IgG2a (300 μg) every 5 d starting at the 3 wk of age. Spontaneous development of tumor was monitored periodically by palpation over the course of 24 mo. Tumors >5 mm in diameter and demonstrating progressive growth were recorded as positive, and finally confirmed by postmortem examination. Some mice were killed and the developing tumors were analyzed by histological examination.
Cytotoxicity Assay.
The susceptibility of tumor cells to TRAIL-mediated cytotoxicity was examined using mouse TRAIL-transfected 2PK-3 (mTRAIL/2PK-3) or mock-transfected 2PK-3 as the effector cells at an E/T = 10 by an 8 h 51Cr release assay as described previously (16). In some experiments, tumor cells were precultured with 2,000 U (100 ng)/ml of mouse recombinant IFN-γ (BD PharMingen) for 24 h before the cytotoxic assay.
Statistical Analysis.
Significant differences in incidence at one time point were determined by the Fisher's exact test. Significant differences in progressive incidence and growth were determined by the unpaired Mann-Whitney U test. P values <0.05 were considered significant.
Results
Contribution of TRAIL to Suppression of MCA-induced Sarcoma Development.
To explore the role of TRAIL in natural protection from tumor development, we examined the effect of neutralizing anti-TRAIL mAb on the primary tumor development induced by the chemical carcinogen MCA. Initially, we employed p53+/− B6 mice to facilitate the sarcoma development. Although inoculation of 25 μg or 100 μg MCA eventually induced fibrosarcomas in all mice treated with either anti-TRAIL mAb or control Ig, a significantly accelerated onset of tumor and tumor growth was observed in the anti-TRAIL mAb-treated mice (P < 0.05; Fig. 1 A). At a high dose of 400 μg MCA, however, no significant difference was observed. In the second set of experiments, we also examined the effect of anti-TRAIL mAb on the development of MCA-induced fibrosarcomas in wild-type B6 mice. As shown in Fig. 1 B, wild-type mice were more resistant to low-dose MCA and showed a later onset at high doses of MCA than p53+/− mice. Remarkably, 25 μg MCA induced fibrosarcomas in 8/10 of anti-TRAIL mAb-treated mice but not (0/10) in control Ig-treated mice. Although 100 μg MCA eventually induced fibrosarcomas in 8/10 of either anti-TRAIL mAb- or control Ig-treated mice, tumor onset was significantly accelerated in anti-TRAIL mAb-treated mice (P < 0.05). At a high dose of 400 μg MCA, however, no significant difference was observed. Fibrosarcoma induction by MCA was similarly promoted by the administration of another neutralizing anti–mouse TRAIL mAb, N2B1 (data not shown).
Figure 1.

Effect of anti-TRAIL mAb on development of MCA-induced fibrosarcoma. p53+/− (A) or wild-type (B) B6 mice were inoculated subcutaneously in the hind flank with the indicated amount of MCA. Mice (n = 10 in each group) were administered intraperitoneally with anti-TRAIL mAb (circles) or isotype-matched control rat Ig (squares) every 5 d, and then observed for sarcoma development over the course of 100–200 d. Tumor sizes in p53+/− mice were also recorded over that period and are represented as the mean ± SD of 3–10 mice in each group.
We established fibrosarcoma cell lines from 100 μg MCA-inoculated and anti-TRAIL mAb or control Ig-treated p53 +/− mice or wild-type B6 mice, and analyzed their susceptibility to TRAIL-mediated cytotoxicity. As shown in Fig. 2 A, 5/8 of the fibrosarcoma cell lines derived from anti-TRAIL mAb-treated p53 +/− mice were susceptible to TRAIL-mediated cytotoxicity, while only 1/8 of those from control Ig-treated p53 +/− mice were susceptible. As shown in Fig. 2 B, 3/4 of the fibrosarcoma cell lines derived from anti-TRAIL mAb-treated wild-type mice were susceptible to TRAIL-mediated cytotoxicity, while 1/4 of those from control Ig-treated wild-type mice was susceptible. These results suggested that the TRAIL-sensitive fibrosarcoma cells that preferentially emerged in the anti-TRAIL mAb-treated mice were mostly eliminated in the control Ig-treated mice. Collectively, the tumor-promoting effect of anti-TRAIL mAb (Fig. 1) and the preferential emergence of TRAIL-sensitive tumor cells in anti-TRAIL mAb-treated mice (Fig. 2) indicated a substantial contribution of TRAIL to natural protection from MCA-induced fibrosarcoma development.
Figure 2.


Selection of TRAIL-resistant fibrosarcomas in vivo. Cell lines were originated from MCA-induced fibrosarcomas developed in isotype-matched control rat Ig- or anti-TRAIL mAb-treated p53+/− (A) or wild-type (B) B6 mice. Then, their susceptibility to TRAIL-mediated cytotoxicity was determined by an 8 h 51Cr release assay using mTRAIL-transfected 2PK-3 (mTRAIL-2PK3) and mock-transfected 2PK-3 cells as effector cells. Data are represented as the mean ± SD of triplicate samples at an E/T = 10. The cytotoxic activity of mock-transfected 2PK-3 against all tumor cells was <2% (data not shown).
Contribution of TRAIL to Suppression of Subcutaneous Outgrowth of MCA-induced Fibrosarcoma.
To explore the inhibitory effect of TRAIL on the outgrowth of MCA-induced fibrosarcomas, we next examined the effect of anti-TRAIL mAb on the subcutaneous growth of TRAIL-sensitive or TRAIL-resistant MCA-induced fibrosarcoma cell lines, which we formerly established from anti-ASGM1 Ab-treated (NK cell–depleted) B6 mice. As shown in Fig. 3 A, MCA-III was sensitive and MCA-IV was resistant to TRAIL-mediated cytotoxicity. At the three doses of tumor cells tested, anti-TRAIL mAb treatment consistently promoted the growth of TRAIL-sensitive MCA-III, although the growth of TRAIL-resistant MCA-IV was not affected (Fig. 3 B). These results indicated that the suppressive effect of TRAIL on tumor outgrowth is primarily determined by the susceptibility of tumor cells to TRAIL and that the direct cytotoxic activity of TRAIL on the tumor cells is the primary mechanism of tumor suppressive action.
Figure 3.

Effect of anti-TRAIL mAb on outgrowth of subcutaneously inoculated fibrosarcoma cell lines. (A) Susceptibility of MCA-induced fibrosarcoma cell lines to TRAIL-mediated cytotoxicity. Cytotoxic activity of mTRAIL-transfected 2PK-3 (mTRAIL/2PK-3) and mock-transfected 2PK-3 cells against MCA-induced fibrosarcoma cell lines (MCA-III and MCA-IV) was tested by an 8 h 51Cr release assay. Data are represented as the mean ± SD of triplicate samples at an E/T ratio = 10. (B) The indicated number of MCA-III (white symbols) and MCA-IV (black symbols) cells were inoculated subcutaneously in the hind flank of wild-type B6 mice. Mice were administered intraperitoneally with anti-TRAIL mAb (circles) or isotype-matched control rat Ig (squares) every 5 d. Data are represented as the mean ± SD of five mice in each group.
Contribution of TRAIL to NK Cell–mediated and IFN-γ-dependent Tumor Surveillance.
We previously demonstrated that the TRAIL-mediated protection from tumor metastasis was mostly mediated by NK cells expressing TRAIL in an IFN-γ–dependent manner (16, 17) and that IFN-γ played a substantial role in NK cell–mediated protection from MCA-induced sarcoma development independently of perforin-mediated cytotoxicity (8). To address the possible contribution of TRAIL to the NK cell–mediated and IFN-γ–dependent protection from tumor development, we first examined the effect of NK cell depletion by anti-ASGM1 Ab on the tumor-promoting effect of anti-TRAIL mAb in the MCA model (25 μg). As shown in Fig. 4 A, the tumor-promoting effect of anti-ASGM1 Ab was almost equivalent to that of anti-TRAIL mAb, and the combined treatment with anti-TRAIL mAb and anti-ASGM1Ab showed an additional effect (P < 0.05), suggesting that TRAIL might protect independently of NK cells. However, MCA-induced fibrosarcoma cell lines derived from the NK cell–depleted mice were more frequently sensitive to TRAIL-mediated cytotoxicity than those derived from the control Ig-treated mice (Fig. 5 B), suggesting that TRAIL was involved in the NK cell-mediated surveillance against MCA-induced sarcoma development. Therefore, in addition to NK cells, some other effector cells might participate in the TRAIL-mediated surveillance against MCA-induced sarcoma development.
Figure 4.


Contribution of TRAIL to NK cell–mediated tumor surveillance. (A) p53+/− B6 mice (n = 10 in each group) were subcutaneously inoculated with 25 μg MCA and treated with isotype-matched control rat Ig (○), anti-TRAIL mAb (•), anti-ASGM1 Ab (▵), or anti-ASGM1 Ab and anti-TRAIL mAb (▴). Mice were observed for sarcoma development over the course of 125 d. (B) Cell lines were originated from MCA-induced fibrosarcomas developed in control Ig-treated or anti-ASGM1 Ab-treated wild-type B6 mice, and their susceptibility to TRAIL-mediated cytotoxicity was determined as described in Fig. 2. Data are represented as the mean ± SD of triplicate samples at an E/T = 10. The cytotoxic activity of mock-transfected 2PK-3 cells against all tumor cells was <2% (data not shown).
Figure 5.

Contribution of TRAIL to IFN-γ–mediated tumor surveillance. Groups of 10 wild-type (circles) and IFN-γ-deficient (triangles) B6 mice were inoculated subcutaneously with the indicated amount of MCA, and treated with isotype-matched control rat IgG (white) or anti-TRAIL mAb (black) as described in Fig. 1. Mice were observed weekly for sarcoma development over the course of 180 d.
Next, we examined the contribution of TRAIL to IFN-γ–dependent tumor surveillance by using IFN-γ–deficient mice in the MCA-induced fibrosarcoma development. As shown in Fig. 5, the tumor-promoting effect of IFN-γ deficiency was equivalent to that of anti-TRAIL mAb in wild-type mice, and anti-TRAIL mAb did not further promoted the tumor development in IFN-γ–deficient mice. In addition, 4/4 of MCA-induced fibrosarcoma cell lines derived from IFN-γ–deficient mice were sensitive to TRAIL-mediated cytotoxicity (Fig. 6) . These results indicated that IFN-γ was essential for the TRAIL-mediated tumor surveillance and that TRAIL was mostly responsible for the IFN-γ–mediated tumor surveillance against MCA-induced fibrosarcomas.
Figure 6.
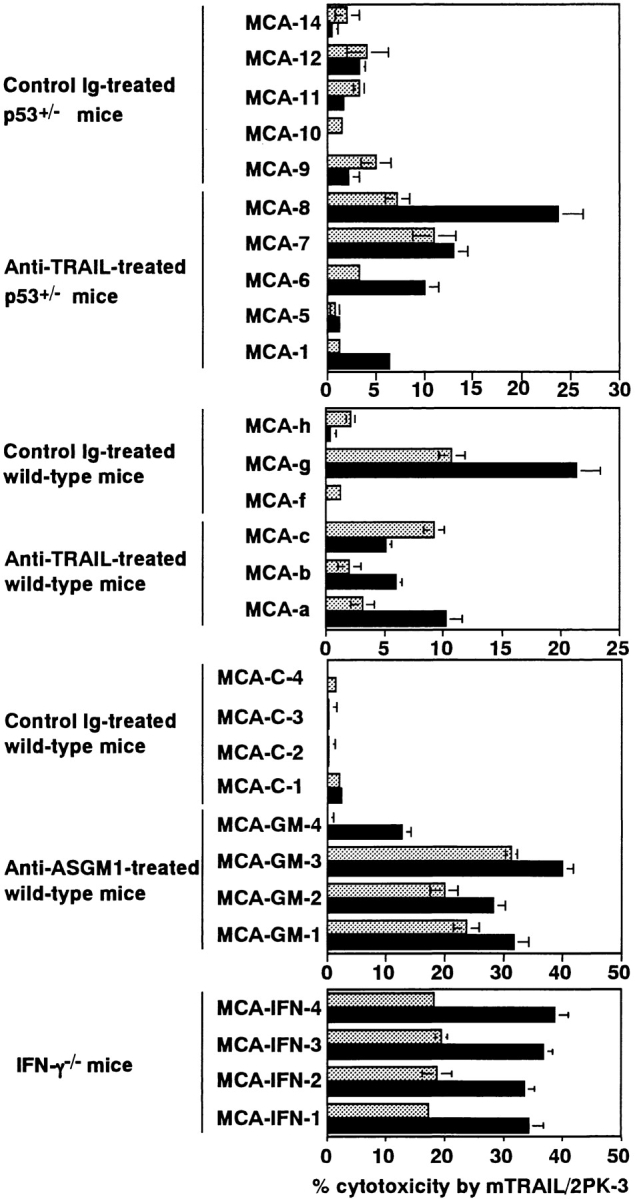
Effect of IFN-γ on susceptibility of MCA-induced fibrosarcoma cells to TRAIL-mediated cytotoxicity. MCA-induced fibrosarcoma cells from control Ig- or anti-TRAIL mAb-treated p53+/− or wild-type mice, control Ig- or anti-ASGM1 Ab-treated wild-type mice, or IFN-γ–deficient mice were preincubated with (solid black bars) or without (gray bars) IFN-γ for 24 h. Then, their susceptibility to TRAIL-mediated cytotoxicity was determined as described in Fig. 2. Data are represented as the mean ± SD of triplicate samples at an E/T = 10. The cytotoxic activity of mock-transfected 2PK-3 cells against all tumor cells was <2% (data not shown).
We further examined whether IFN-γ might affect the susceptibility of tumor cells to TRAIL-mediated cytotoxicity, since the IFN-γ responsiveness of tumor cells has been reported to be critical for the IFN-γ–mediated surveillance against MCA-induced sarcoma (23). As shown in Fig. 6, pretreatment with IFN-γ increased the TRAIL susceptibility of 3/5 of MCA-induced fibrosarcoma cell lines derived from anti-TRAIL mAb-treated p53+/− mice and 2/3 of those from anti-TRAIL mAb-treated wild-type mice. Moreover, IFN-γ enhanced the TRAIL susceptibility of 4/4 of MCA-induced fibrosarcoma cell lines from anti-ASGM1 Ab-treated mice and 4/4 of those from IFN-γ–deficient mice. In contrast, IFN-γ did not substantially increase the TRAIL susceptibility of MCA-induced fibrosarcoma cell lines from control Ig-treated mice, except for MCA-g which was susceptible to TRAIL without IFN-γ treatment. These results suggested that endogenous IFN-γ might control the TRAIL-mediated surveillance against MCA-induced fibrosarcomas not only by upregulating TRAIL expression in effector cells but also by sensitizing tumor cells to TRAIL-mediated cytotoxicity.
Contribution of TRAIL to Suppression of Spontaneous Tumor Development in p53+/− Mice.
To further explore the general role of TRAIL in natural protection from primary tumor development in vivo, we finally examined the effect of neutralizing anti-TRAIL mAb treatment on the spontaneous tumor development in p53+/− mice. No tumor development was observed before 11 mo of age in both control Ig-treated and anti-TRAIL mAb-treated p53+/− mice. At 11 mo of age, development of sarcomas was initially observed in anti-TRAIL mAb-treated p53+/− mice, and then the anti-TRAIL mAb treatment significantly promoted the tumor development as compared with control Ig-treated mice until 24 mo of age (P < 0.05) (Fig. 7 A). At autopsy, the tumors mainly consisted of sarcomas and disseminated lymphomas as previously reported for untreated p53+/− B6 mice (24, 25), and no apparent difference in tumor types was observed between the anti-TRAIL mAb-treated group and the control Ig-treated group (data not shown). We established sarcoma cell lines from the anti-TRAIL mAb- or control Ig-treated p53+/− mice, and analyzed their susceptibility to TRAIL-mediated cytotoxicity. As shown in Fig. 7 B, the tumor cell lines derived from anti-TRAIL mAb-treated p53+/− mice were susceptible to TRAIL-mediated cytotoxicity, which was augmented by pretreatment with IFN-γ, while the tumor cell lines derived from control Ig-treated p53+/− mice were not susceptible to TRAIL even after IFN-γ treatment. These results suggested that the TRAIL-sensitive spontaneously arising tumor cells that preferentially emerged in the anti-TRAIL mAb-treated p53+/− mice were mostly eliminated in the control Ig-treated p53+/− mice, as in the case of MCA-induced fibrosarcomas. These results indicated a substantial contribution of TRAIL to the natural protection from spontaneous tumor development due to the p53 mutation.
Figure 7.


Effect of anti-TRAIL mAb on spontaneous tumor development in p53+/− mice. (A) p53+/− B6 mice were administered intraperitoneally with anti-TRAIL mAb (circles) or isotype-matched control rat Ig (squares) every 5 d starting at 3 wk of age, and then observed for tumor development over the course of 24 mo. Difference between two groups was statistically significant (P < 0.05) as analyzed by unpaired Mann-Whitney U test. (B) Cell lines were originated from spontaneous sarcoma development in isotype-matched control rat Ig- or anti-TRAIL mAb-treated p53+/− mice. Then, their susceptibility to TRAIL-mediated cytotoxicity was determined as described in Fig. 2 after the preincubated with (solid black bars) or without (gray bars) IFN-γ for 24 h. Data are represented as the mean ± SD of triplicate samples at an E/T = 10. The cytotoxic activity of mock-transfected 2PK-3 cells against all tumor cells was <2% (data not shown).
Discussion
In this study, we demonstrated that the administration of neutralizing mAbs against TRAIL promoted MCA-induced tumor development in p53+/− or wild-type B6 mice and spontaneous tumor development in p53+/− mice. These results suggested a substantial role of TRAIL in natural protection from tumor development. Although potent therapeutic effects of soluble recombinant TRAIL against human tumor xenografts in nude and SCID mice have been reported (20, 21), this is the first indication that endogenous TRAIL plays a critical role in host surveillance against primary tumor development in vivo. Consistent with our present results using neutralizing anti-TRAIL mAbs, we have recently observed that MCA-induced sarcoma development was promoted in TRAIL-deficient B6 mice generated by gene targeting to just a similar extent to that was observed in the anti-TRAIL mAb-treated wild-type B6 mice (unpublished data). The contribution of TRAIL to the suppression of spontaneous tumor development in p53 mutant mice is now to be verified by crossing the TRAIL-deficient mice with the p53 mutant mice.
Heterogenous tumor cells with different TRAIL sensitivity are expected to arise during the MCA-induced or spontaneous tumor development in individual mice. However, the preferential emergence of TRAIL-sensitive fibrosarcoma cells in the anti-TRAIL mAb-treated mice, but rarely in the control Ig-treated mice, strongly suggested a selective pressure against TRAIL-sensitive cells during MCA-induced or spontaneous tumor development. Although no obvious TRAIL-mediated suppression of tumor initiation was observed at overwhelming doses of MCA (400 μg), this may be due to an emergence of too many transformed cells to be depleted by endogenous TRAIL. Consistent with a lack of effective selection, fibrosarcoma cell lines derived from 400 μg MCA-inoculated mice were more frequently TRAIL-sensitive than those from 100 μg MCA-inoculated mice (data not shown). Although the presented data suggested that the direct cytotoxic effect of TRAIL on developing tumor cells is the primary mechanism for the tumor suppression, immune responses against tumors might be also affected by the TRAIL blockade, since it has been reported that TRAIL may play a regulatory role in some autoimmune disease models in mice (26, 27).
In the case of MCA-induced sarcoma development, we previously demonstrated a critical contribution of NK cells as the effector cells and that of perforin and IFN-γ as the effector mechanism (4, 8). In the present study, the predominant contribution of TRAIL to IFN-γ–dependent protection from tumor development was illustrated in the MCA-induced fibrosarcoma model. Some dissociation between the NK cell-mediated protection and the TRAIL-mediated protection was observed, which may be explained by the perforin-dependent protection by NK cells and the possible expression of TRAIL on some effector cells other than NK cells. It has been shown that TRAIL was expressed on IFN-γ–stimulated monocytes and dendritic cells in vitro (28, 29). Therefore, these cells may also participate in the IFN-γ–dependent and TRAIL-mediated protection against MCA-induced sarcoma development. Nevertheless, the preferential emergence of TRAIL-susceptible fibrosarcomas in the NK cell–depleted mice and the IFN-γ–deficient mice strongly supports the contribution of TRAIL to NK cell– and IFN-γ–mediated surveillance against MCA-induced fibrosarcomas.
It remains to be determined whether TRAIL also plays a substantial role in natural protection from primary tumors induced by chemical carcinogens other than MCA or spontaneously arising tumors in tumor-prone mice other than p53+/− mice. Perforin was shown to be critical in protection from MCA-induced sarcomas, but not that from 12-O-tetradecanoylphorbol-13-acetate plus 7,12-dimethylbenzanthracene–induced skin papillomas or Moloney murine sarcoma and leukemia virus-induced sarcomas (30). We also recently reported a critical role of perforin in surveillance against spontaneous lymphoma, but not sarcoma, development in p53+/− or p53−/− mice (25). The contribution of TRAIL to IFN-γ–dependent protection suggests that tumor models where IFN-γ controls tumor development are of greatest interest. Notably, IFN-γ has been implicated in surveillance against the development of various nonlymphoid tumors in p53−/− mice (23). However, it should be noted that the expression of TRAIL receptor 2 (TRAIL-R2), which is the only known TRAIL receptor in mice (31), has been reported to be regulated by p53 in human tumor cell lines (32, 33), arguing against the potential utility of the p53−/− model. To address the general role of TRAIL in tumor surveillance, further studies are now underway by using the neutralizing anti-TRAIL mAbs and the TRAIL-deficient mice.
We have previously demonstrated that IFN-γ plays an essential role in inducing TRAIL expression on NK cells in vivo (16, 17). IFN-γ may also induce TRAIL expression on monocytes and dendritic cells (28, 29). Our present results suggested that IFN-γ may regulate TRAIL-mediated tumor surveillance not only by regulating TRAIL expression on effector cells, but also by sensitizing tumor cells to TRAIL-mediated cytotoxicity. The mechanism by which IFN-γ sensitizes tumor cells to TRAIL remains to be determined. Although two death-inducing receptors (TRAIL-R1/DR4 and TRAIL-R2/DR5) and two decoy receptors (TRAIL-R3/DcRI and TRAIL-R4/DcR2) have been identified in humans (34, 35), only one homologous to TRAIL-R2 has been identified in mice (31). IFN-γ may upregulate the expression of TRAIL-R2 on tumor cells. Alternatively, IFN-γ may downregulate the expression of intracellular Fas-associated death domain-like IL-1β-converting enzyme-inhibitory proteins (FLIPs) that protects cells from TRAIL-induced apoptosis (36, 37) or upregulate the expression of caspases that execute TRAIL-induced apoptosis (38). It is noteworthy that human tumor cell lines are more frequently TRAIL-sensitive than murine tumor cell lines. This may be due to the presence of decoy receptors in humans, which are expressed on normal cells and may protect newly transformed cells from TRAIL-mediated surveillance during tumor development. Further studies are needed to explore the possible difference between mice and humans in the role of TRAIL in tumor surveillance.
Recently, Shankaran et al. demonstrated a critical contribution of IFN-γ to immune surveillance against transplanted MCA-induced sarcomas in 129/Sv/Ev mice, that appeared to be mediated by MHC class I–restricted CD8+ CTLs (39). In this model, IFN-γ appeared to act predominantly on tumor cells to upregulate MHC class I and transporter associated protein 1, increasing tumor immunogenicity for adaptive immunity (23, 39). This study is complementary in demonstrating an additional function of IFN-γ in controlling TRAIL-mediated innate immune surveillance against tumors. Since the adaptive immunity against tumors depends on the presence of appropriate tumor antigens, the TRAIL-mediated innate protection may be more versatile as the natural defense against tumors. Further studies on the molecular mechanisms by which IFN-γ and TRAIL contribute to immune surveillance against tumors may provide a novel strategy to prevent tumor development in humans.
Acknowledgments
This work was supported by research grants from the Human Frontier Science Program Organization and the Ministry of Education, Science, and Culture, Japan. M.J. Smyth was supported by the National Health and Medical Research Council of Australia.
Footnotes
Abbreviations used in this paper: ASGM1, asialo GM1; MCA, methylcholanthrene; TRAIL, TNF-related apoptosis-inducing ligand.
References
- 1.Smyth, M.J., D.I. Godfrey, and J.A. Trapani. 2001. A fresh look at tumor immunosurveillance and immunotherapy. Nat. Immunol. 2:293–299. [DOI] [PubMed] [Google Scholar]
- 2.Kärre, K., H.G. Ljunggren, G. Piontek, and R. Kiessling. 1986. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 319:675–678. [DOI] [PubMed] [Google Scholar]
- 3.Trinchieri, G. 1989. Biology of natural killer cells. Adv. Immunol. 47:187–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smyth, M.J., N.Y. Crowe, and D.I. Godfrey. 2001. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int. Immunol. 13:459–463. [DOI] [PubMed] [Google Scholar]
- 5.Smyth, M.J., K.Y. Thia, S.E. Street, E. Cretney, J.A. Trapani, M. Taniguchi, T. Kawano, S.B. Pelikan, N.Y. Crowe, and D.I. Godfrey. 2000. Differential tumor surveillance by natural killer (NK) and NKT cells. J. Exp. Med. 191:661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kodama, T., K. Takeda, O. Shimozato, Y. Hayakawa, M. Atsuta, K. Kobayashi, M. Ito, H. Yagita, and K. Okumura. 1999. Perforin-dependent NK cell cytotoxicity is sufficient for anti-metastatic effect of IL-12. Eur. J. Immunol. 29:1390–1396. [DOI] [PubMed] [Google Scholar]
- 7.Trinchieri, G. 1995. Natural killer cells wear different hats: effector cells of innate resistance and regulatory cells of adaptive immunity and of hematopoiesis. Semin. Immunol. 7:83–88. [DOI] [PubMed] [Google Scholar]
- 8.Street, S.E., E. Cretney, and M.J. Smyth. 2001. Perforin and interferon-γ activities independently control tumor initiation, growth, and metastasis. Blood. 97:192–197. [DOI] [PubMed] [Google Scholar]
- 9.Boehm, U., T. Klamp, M. Groot, and J.C. Howard. 1997. Cellular responses to interferon-γ. Annu. Rev. Immunol. 15:749–795. [DOI] [PubMed] [Google Scholar]
- 10.Dighe, A.S., E. Richards, L.J. Old, and R.D. Schreiber. 1994. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN γ receptors. Immunity. 1:447–456. [DOI] [PubMed] [Google Scholar]
- 11.Hawley, R.G., and L.C. Berger. 1998. Growth control mechanisms in multiple myeloma. Leuk. Lymphoma. 29:465–475. [DOI] [PubMed] [Google Scholar]
- 12.Angiolillo, A.L., C. Sgadari, D.D. Taub, F. Liao, J.M. Farber, S. Maheshwari, H.K. Kleinman, G.H. Reaman, and G. Tosato. 1995. Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J. Exp. Med. 182:155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farber, J.M. 1997. Mig and IP-10: CXC chemokines that target lymphocytes. J. Leukoc. Biol. 61:246–257. [PubMed] [Google Scholar]
- 14.Tamura, T., M. Ishihara, M.S. Lamphier, N. Tanaka, I. Oishi, S. Aizawa, T. Matsuyama, T.W. Mak, S. Taki, and T. Taniguchi. 1995. An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen-activated T lymphocytes. Nature. 376:596–599. [DOI] [PubMed] [Google Scholar]
- 15.Dai, C., and S.B. Krantz. 1999. Interferon γ induces upregulation and activation of caspases 1, 3, and 8 to produce apoptosis in human erythroid progenitor cells. Blood. 93:3309–3316. [PubMed] [Google Scholar]
- 16.Takeda, K., Y. Hayakawa, M.J. Smyth, N. Kayagaki, N. Yamaguchi, S. Kakuta, Y. Iwakura, H. Yagita, and K. Okumura. 2001. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat. Med. 7:94–100. [DOI] [PubMed] [Google Scholar]
- 17.Smyth, M.J., E. Cretney, K. Takeda, R.H. Wiltrout, L.M. Sedger, N. Kayagaki, H. Yagita, and K. Okumura. 2001. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon γ-dependent natural killer cell protection from tumor metastasis. J. Exp. Med. 193:661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiley, S.R., K. Schooley, P.J. Smolak, W.S. Din, C.P. Huang, J.K. Nicholl, G.R. Sutherland, T.D. Smith, C. Rauch, and C.A. Smith. 1995. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 3:673–682. [DOI] [PubMed] [Google Scholar]
- 19.Pitti, R.M., S.A. Marsters, S. Ruppert, C.J. Donahue, A. Moore, and A. Ashkenazi. 1996. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 271:12687–12690. [DOI] [PubMed] [Google Scholar]
- 20.Walczak, H., R.E. Miller, K. Ariail, B. Gliniak, T.S. Griffith, M. Kubin, W. Chin, J. Jones, A. Woodward, T. Le, et al. 1999. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 5:157–163. [DOI] [PubMed] [Google Scholar]
- 21.Ashkenazi, A., R.C. Pai, S. Fong, S. Leung, D.A. Lawrence, S.A. Marsters, C. Blackie, L. Chang, A.E. McMurtrey, A. Hebert, et al. 1999. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Invest. 104:155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kayagaki, N., N. Yamaguchi, M. Nakayama, K. Takeda, H. Akiba, H. Tsutsui, H. Okamura, K. Nakanishi, K. Okumura, and H. Yagita. 1999. Expression and function of TNF-related apoptosis-inducing ligand on murine activated NK cells. J. Immunol. 163:1906–1913. [PubMed] [Google Scholar]
- 23.Kaplan, D.H., V. Shankaran, A.S. Dighe, E. Stockert, M. Aguet, L.J. Old, and R.D. Schreiber. 1998. Demonstration of an interferon γ-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA. 95:7556–7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacks, T., L. Remington, B.O. Williams, E.M. Schmitt, S. Halachmi, R.T. Bronson, and R.A. Weinberg. 1994. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 4:1–7. [DOI] [PubMed] [Google Scholar]
- 25.Smyth, M.J., K.Y. Thia, S.E. Street, D. MacGregor, D.I. Godfrey, and J.A. Trapani. 2000. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J. Exp. Med. 192:755–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song, K., Y. Chen, R. Göke, A. Wilmen, C. Seidel, A. Göke, B. Hilliard, and Y. Chen. 2000. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is an inhibitor of autoimmune inflammation and cell cycle progression. J. Exp. Med. 191:1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hilliard, B., A. Wilmen, C. Seidel, T.S. Liu, R. Goke, and Y. Chen. 2001. Roles of TNF-related apoptosis-inducing ligand in experimental autoimmune encephalomyelitis. J. Immunol. 166:1314–1319. [DOI] [PubMed] [Google Scholar]
- 28.Griffith, T.S., S.R. Wiley, M.Z. Kubin, L.M. Sedger, C.R. Maliszewski, and N.A. Fanger. 1999. Monocyte-mediated tumoricidal activity via the tumor necrosis factor-related cytokine, TRAIL. J. Exp. Med. 189:1343–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fanger, N.A., C.R. Maliszewski, K. Schooley, and T.S. Griffith. 1999. Human dendritic cells mediate cellular apoptosis via tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J. Exp. Med. 190:1155–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van den Broek, M.E., D. Kägi, F. Ossendorp, R. Toes, S. Vamvakas, W.K. Lutz, C.J. Melief, R.M. Zinkernagel, and H. Hengartner. 1996. Decreased tumor surveillance in perforin-deficient mice. J. Exp. Med. 184:1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu, G.S., T.F. Burns, Y. Zhan, E.S. Alnemri, and W.S. El-Deiry. 1999. Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res. 59:2770–2775. [PubMed] [Google Scholar]
- 32.Sheikh, M.S., T.F. Burns, Y. Huang, G.S. Wu, S. Amundson, K.S. Brooks, A.J. Fornace, Jr., and W.S. el-Deiry. 1998. p53-dependent and -independent regulation of the death receptor KILLER/DR5 gene expression in response to genotoxic stress and tumor necrosis factor α. Cancer Res. 58:1593–1598. [PubMed] [Google Scholar]
- 33.Wu, G.S., T.F. Burns, E.R. McDonald III, W. Jiang, R. Meng, I.D. Krantz, G. Kao, D.D. Gan, J.Y. Zhou, R. Muschel, et al. 1997. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 17:141–143. [DOI] [PubMed] [Google Scholar]
- 34.Ashkenazi, A., and V.M. Dixit. 1998. Death receptors: signaling and modulation. Science. 281:1305–1308. [DOI] [PubMed] [Google Scholar]
- 35.Golstein, P. 1997. Cell death: TRAIL and its receptors. Curr. Biol. 7:R750–753. [DOI] [PubMed] [Google Scholar]
- 36.Thome, M., P. Schneider, K. Hofmann, H. Fickenscher, E. Meinl, F. Neipel, C. Mattmann, K. Burns, J.L. Bodmer, M. Schroter, et al. 1997. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 386:517–521. [DOI] [PubMed] [Google Scholar]
- 37.Irmler, M., M. Thome, M. Hahne, P. Schneider, K. Hofmann, V. Steiner, J.L. Bodmer, M. Schroter, K. Burns, C. Mattmann, et al. 1997. Inhibition of death receptor signals by cellular FLIP. Nature. 388:190–195. [DOI] [PubMed] [Google Scholar]
- 38.Griffith, T.S., W.A. Chin, G.C. Jackson, D.H. Lynch, and M.Z. Kubin. 1998. Intracellular regulation of TRAIL-induced apoptosis in human melanoma cells. J. Immunol. 161:2833–2840. [PubMed] [Google Scholar]
- 39.Shankaran, V., H. Ikeda, A.T. Bruce, J.M. White, P.E. Swanson, L.J. Old, and R.D. Schreiber. 2001. IFN-γ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 410:1107–1111. [DOI] [PubMed] [Google Scholar]