

Abstract
To elucidate the mechanism(s) by which Vav3, a new member of the Vav family proteins, participates in B cell antigen receptor (BCR) signaling, we have generated a B cell line deficient in Vav3. Here we report that Vav3 influences phosphoinositide 3-kinase (PI3K) function through Rac1 in that phosphatidylinositol-3,4,5-trisphosphate (PIP3) generation was attenuated by loss of Vav3 or by expression of a dominant negative form of Rac1. The functional interaction between PI3K and Rac1 was also demonstrated by increased PI3K activity in the presence of GTP-bound Rac1. In addition, we show that defects of calcium mobilization and c-Jun NH2-terminal kinase (JNK) activation in Vav3-deficient cells are relieved by deletion of a PIP3 hydrolyzing enzyme, SH2 domain-containing inositol polyphosphate 5′-phosphatase (SHIP). Hence, our results suggest a role for Vav3 in regulating the B cell responses by promoting the sustained production of PIP3 and thereby calcium flux.
Keywords: Akt, calcium, gene targeting, JNK, Rac
Introduction
Stimulation of the B cell antigen receptor (BCR)* leads to the sequential activation of protein tyrosine kinases (PTKs) that phosphorylate numerous cellular proteins including Vav isoforms and phospholipase C (PLC)-γ2 (1–4). The proto-oncogene Vav (herein referred to as Vav1) was first discovered by virtue of a mutation that rendered it able to transform fibroblasts. Vav1 contains a guanine nucleotide exchange factor (GEF) domain for the Rho/Rac/Cdc42 family of small GTPases, a pleckstrin homology (PH) domain, and two src homology (SH)3 domains that flank one SH2 domain (5). In vitro studies show that the GEF activity of Vav1 is regulated by tyrosine phosphorylation (6) and by inositol lipids that interact with the Vav1 PH domain (7). However, whether this mechanism operates in vivo is still unclear.
Since Vav1 discovery, two new Vav members (Vav2 and Vav3) have been identified. Although all Vav family proteins have similar structural features, they display different expression patterns. Vav1 is primarily expressed in hematopoietic lineages (8), while Vav2 and Vav3 have a relatively broad expression profile (9–11).
Vav1 has been shown to be critical for T cell development as well as mast cell activation (12–15). Moreover, the importance of Vav1 and Vav2 proteins for B cell development and activation has been recently underscored by gene targeting and overexpression experiments. The B cell developmental and functional defects are relatively mild in mice lacking either Vav1 or Vav2, while double-deficient mice exhibit more severe defects (12, 13, 16, 17). The small numbers of peripheral B cells which can develop in the absence of both Vav1 and Vav2 display a profound defect in BCR-mediated calcium mobilization. Conversely, overexpression of either Vav1 or Vav2 in Bal-17 B cells results in an increase of BCR-mediated calcium mobilization, thereby leading to enhancement of nuclear factor of activated T cell (NF-AT) activation (18). However, the exact molecular mechanism(s) by which Vav1 and Vav2 participate in BCR signaling still remains to be determined. Moreover, despite the high expression of Vav3 in peripheral lymphocytes and spleen cells (10, 11), the role of Vav3 in BCR signaling has not been addressed.
In this study, we first characterized BCR signaling of DT40 B cells deficient in Vav3. Then we examined how Vav3 exerts its functions. Vav3-deficient B cells exhibited similar defects to those in phosphoinositide 3-kinase (PI3K)-deficient B cells, which led us to consider the functional interaction between Vav3 and PI3K. Here we show that phosphatidylinositol-3,4,5-trisphosphate (PIP3) generation and subsequent Akt activation are attenuated by loss of Vav3 or by introduction of dominant negative Rac1. Supporting the positive effect of Vav3 on PI3K, PI3K enzymatic activity is upregulated by Rac1 in a GTP-dependent manner. Furthermore, the defects in Vav3-deficient cells can be suppressed by deletion of a PIP3 hydrolyzing enzyme, SH2 domain-containing inositol polyphosphate 5′-phosphatase (SHIP). Thus, our data suggest that Vav3 is involved in sustaining PIP3 generation, which in turn regulates downstream events such as calcium flux.
Materials and Methods
Cells, Abs, and Reagents.
Various mutant chicken DT40 B cells were cultured as described previously (19, 20). Human 293T cells were cultured in DME containing 10% FCS. Preparation of murine splenic B cells was described previously (21).
Anti-Vav3 and anti-Vav2 Abs were obtained by immunizing rabbits with bacterially expressed GST fusion protein containing chicken Vav3 (128–193 amino acid region) and chicken Vav2 (311–430 amino acid region), respectively. The anti-chicken IgM mAb (M4), rabbit anti-Lyn Abs, and rabbit anti-PLC-γ2 Abs were described previously (22). The following Abs were purchased: anti-phosphotyrosine mAb (4G10), anti-Rac1 mAb, and rabbit anti-PI3K p85 Abs from Upstate Biotechnology, goat anti-PI3K p110α Abs (sc-1331) and goat anti-Akt1 Abs (sc-1618) from Santa Cruz Biotechnology, Inc., anti-c-Jun NH2-terminal kinase 1 (JNK1) mAb from BD PharMingen, anti-T7•Tag mAb from Novagen, and goat anti-mouse IgM from Jackson ImmunoResearch Laboratories. Recombinant GST-Rac1 proteins were purchased from Cytoskeleton.
cDNA Cloning.
A chicken EST clone (GenBank/EMBL/DDBJ accession no. AJ395373) was found by homology search of the NCBI database using the mouse Vav3 cDNA sequences. A λZAP DT40 cDNA library was screened by using the EST cDNA fragment as a probe, giving rise to isolation of 26 clones. The longest insert that contains the open reading frame encoding 846 amino acids has been deposited into GenBank (GenBank/EMBL/DDBJ accession no. AY046915).
To isolate chicken Vav2 cDNA, a cDNA fragment encoding human Vav2 was used as a probe for screening the λZAP DT40 cDNA library under the low-stringency conditions. 23 positive clones were identified and further characterized. The complete sequence of the longest insert (encoding protein of 839 amino acids) has been deposited into GenBank (GenBank/EMBL/DDBJ accession no. AY046916).
Expression Constructs and Transfection.
The chicken Vav3 GEF mutant construct (altering amino acids 336–342 from LLLQELV to IIIQDAA) (23) was generated by oligonucleotide-directed mutagenesis. The cDNAs encoding wild-type Vav3, its GEF mutant, or wild-type Vav2 were subcloned into the pApuro expression vector (24). T7-tagged Btk cDNA in the pApuro expression vector was described previously (25). These constructs were transfected into Vav3-deficient DT40 cells by electroporation, and selected in the presence of puromycin (0.5 μg/ml). Expression of transfected cDNAs was confirmed by Western blot analysis.
cDNAs encoding bovine p85α subunit and chicken p110α subunit of PI3K were provided by Drs. Michael D. Waterfield (London, UK) and Peter K. Vogt (La Jolla, CA), respectively. A kinase-negative p110α cDNA (R916P) was generated by oligonucleotide-directed mutagenesis. These cDNAs were subcloned into the pAzeo expression vector (26). 293T cells were transfected with p85α cDNA together with either wild-type or kinase-negative p110α cDNA using calcium phosphate precipitation method.
Generation of Vav3- and PI3K p110α-deficient DT40 Cells.
DT40 genomic fragments of Vav3 and PI3K p110α were obtained by PCR. The targeting vectors pVav3-bsr, pVav3-hisD, and pVav3-neo were constructed by replacing the 0.3-kb genomic DNA containing exons that encode a part of Dbl homology region (308–338 amino acids) with bsr, hisD, and neo cassettes, respectively. These cassettes were flanked by 2.5 kb and 6.5 kb of Vav3 genomic sequences on the 5′ and 3′ sides, respectively. To isolate Vav3-deficient cells, DT40 cells transfected sequentially with pVav3-hisD and pVav3-bsr were selected in the presence of proper drugs. DT40 cells deficient for both SHIP and Vav3 were generated by successive transfection with pVav3-hisD and pVav3-neo into SHIP-deficient cells (19).
The neo and hisD targeting vectors of p110α were constructed by replacing the 0.5-kb genomic fragment containing exons corresponding to a part of kinase domain (831–888 amino acids) with neo and hisD cassettes, respectively. These constructs spanned 1.7 kb (5′ side) and 0.8 kb (3′ side) of p110α sequences. These targeting vectors were sequentially transfected into DT40 cells to obtain a null mutant.
Selection for drug-resistance clones was performed by using either alone or combinations of histidiol (1 mg/ml), blastcidin S (50 μg/ml), and G418 (2 mg/ml).
Biochemical Analyses.
For immunoprecipitation, cells were solubilized in lysis buffer (10 mM Tris-HCl [pH7.5], 150 mM NaCl, and 1% Nonidet P-40) supplemented with protease and phosphatase inhibitors as described previously (24). Precleared lysates were incubated with proper Abs and protein A-agarose. For Western blot analysis, immunoprecipitates, affinity precipitates, or whole-cell lysates were resolved on SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membrane (Bio-Rad Laboratories), and detected by the indicated Abs and enhanced chemiluminescence (ECL) system (Amersham Pharmacia Biotech). In vitro kinase assays of Akt and JNK1 were performed as described previously (27). The reaction products were resolved on SDS-PAGE, dried, and subjected to quantification of radioactivity by using a Fuji FLA2000 bioimaging analyzer (Fuji Photo Film). Based on these analyses, the stimulation folds are shown in Figs. 3 E, 4 B, 5 C, 5 D, 9 B, 9 C, and 10 C. Phospholipase hydrolysis activity of PLC-γ2 was measured by quantitating inositol 1,4,5-trisphosphate (IP3) production as described previously (28).
Figure 3.
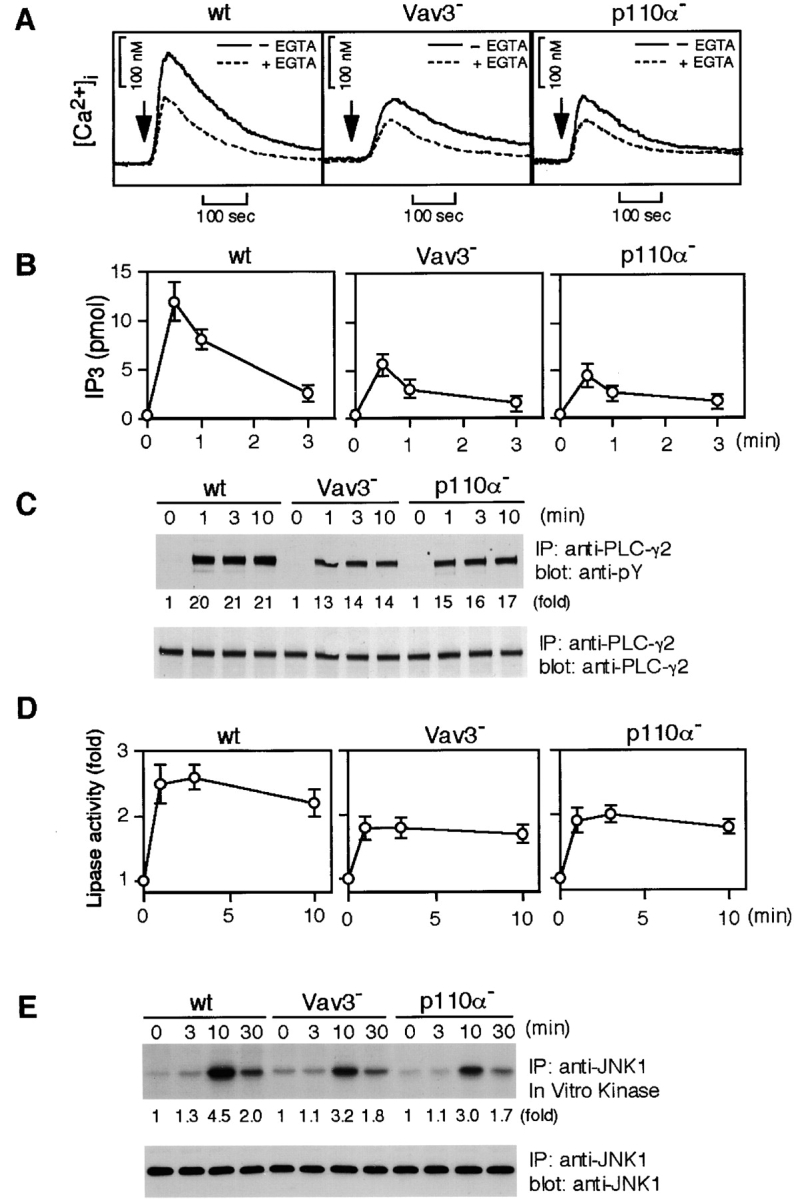
Characterization of Vav3- and PI3K p110α-deficient DT40 B cells. Cells were stimulated with M4 (4 μg/ml) for indicated time periods and then subjected to the following analyses. (A) Calcium mobilization. Intracellular free calcium levels in Fura-2-loaded cells were monitored by a fluorescence spectrophotometer. Calcium release from intracellular calcium stores was measured in the presence of 1 mM EGTA (dotted line). wt, wild-type. (B) IP3 generation. Soluble IP3 was extracted from 2 × 106 cells and subjected to a Biotrak competitive binding assay system. The results (2 × 106 cell equivalents per point) were shown by mean ± standard error of three independent experiments. (C) Tyrosine phosphorylation of PLC-γ2. Immunoprecipitation (IP) was performed with anti-PLC-γ2 Abs. The blots (5 × 106 cell equivalents per lane) were stained with 4G10 mAb (top). The blots were stripped and reprobed with anti-PLC-γ2 Abs (bottom). (D) BCR-induced PLC-γ2 activation. PLC-γ2 was immunoprecipitated from lysates (2 × 107 cell equivalents per point), and in vitro phospholipase activity was measured as described in Materials and Methods. The results were shown by mean ± standard error of three independent experiments. (E) BCR-induced JNK1 activation. Lysates from 5 × 106 cells were immunoprecipitated with anti-JNK1 mAb, and the resulting immunoprecipitates were divided. Half of them was used for Western blot analysis using anti-JNK1 mAb (bottom). The remaining half was used for in vitro kinase assay using GST-c-Jun as an exogenous substrate. The kinase reaction products were resolved on 12.5% SDS-PAGE, and their phosphorylation was quantified by autoradiography (top).
For affinity precipitation assays using GST-CRIB (Cdc42/Rac interactive binding domain [CRIB] of rat PAK1 [amino acid 1–125] fused to GST), bacterially expressed GST-CRIB, prebound glutathione-sepharose beads (20 μl packed beads, 40 μg of protein) were prepared as described (29). Cell lysates in lysis buffer (25 mM Hepes [pH 7.3], 150 mM NaCl, 5 mM EGTA, 20 mM β-glycerophosphate, 10 mM NaF, 2 mM sodium vanadate, 0.5% Triton X-100, 4% glycerol, 5 mM dithiothreitol, 0.5 mM PMSF, and 5 μg/ml each leupeptin and pepstatin) were incubated with the beads for 10 min at 4°C. After quick wash in the lysis buffer, bound proteins were eluted from the beads with SDS-PAGE sample buffer. Eluted samples were resolved on 12.5% SDS-PAGE and subsequently subjected to Western blot analysis using anti-Rac1 mAb.
Glycolipid-enriched microdomains (GEMs) were isolated from cell lysates in Triton X-100 and floatation on sucrose density gradients as described previously (30).
The amount of proteins detected by Western blotting was determined by scanning the autoradiography followed by processing of the data with NIH-image program. Based on these analyses, the stimulation folds are shown in Figs. 3 C, 4 D, 5 B, and 10 B.
Measurements of [Ca2+]i, Inositol Phosphates, and Phosphoinositides.
Calcium mobilization was measured using fura-2 in a bulk spectrofluorimeter as described (30). Production of IP3 was measured using a commercial IP3 assay system (Biotrak TRK 1000; Amersham Pharmacia Biotech) as described previously (26).
Determination of phosphatidylinositol-4,5,-bisphosphate (PIP2) was also performed by using the Biotrak TRK 1000 assay kit according to the manufacturer's instructions. Briefly, PIP2 extracted from the cells using methanol/chloroform/12 M HCl (80:40:1 vol/vol) were hydrolyzed into IP3 by 1 M KOH at 100°C for 15 min. Samples were neutralized and then assayed as described above.
Thin-layer chromatography (TLC) analyses of phosphoinositides were performed by 32P labeling of cells, extraction of lipids, and TLC analysis as described previously (27, 31). The radioactivity of PIP3 spots was quantified by using a Fuji FLA2000 bioimaging analyzer and was normalized to the radioactivity in phospholipids of the samples. Then, the resulting values were normalized to the value at time zero of BCR stimulation for each of three independent experiments and averaged.
PI3K Assay.
PI3K activity was essentially measured as described previously with minor modifications (32). 293T cells transfected together with PI3K p85α and p110α cDNAs were solubilized in lysis buffer (20 mM Tris-HCl [pH8.0], 100 mM NaCl, 1 mM EDTA, 0.3 mM dithiothreitol, 0.5 mM PMSF, 1% Triton X-100, and 10 μg/ml each aprotinin and leupeptin), and PI3K was immunoprecipitated with anti-p85 Abs. Immunocomplexes were collected on protein A-agarose beads, washed twice with wash buffer (20 mM Tris-HCl [pH8.0], 100 mM NaCl, and 10 mM MgCl2), and washed once with kinase assay buffer (10 mM Tris-HCl [pH7.4] and 10 mM MgCl2). Wild-type or mutant GST-Rac1 recombinant proteins (2 μg) prebound to different guanine nucleotides were added to the washed immunoprecipitates, and PI3K activity was assayed by the addition of 5 μg of sonicated phosphatidylinositol and phosphatidylserine (1:1) and 20 μM [γ-32P]ATP (200 μCi/ml) in 45 μl of kinase assay buffer. The reactions were terminated after 10 min at 25°C by the addition of 100 μl of 1 M HCl. The lipids were then immediately extracted with 200 μl of chloroform/methanol (1:1 vol/vol), and the organic phase was washed once with 80 μl of methanol/1 M HCl (1:1 vol/vol). Extracted products were submitted to TLC on potassium oxalate-coated silica gel 60 plates (Merck) in a chloroform/methanol/4 M ammonium hydroxyside (9:7:2 vol/vol) developing solvent. Phosphorylated products were visualized and quantified by a Fuji FLA2000 bioimaging analyzer.
To determine BCR-induced PI3K activity, DT40 cells were solubilized in lysis buffer (20 mM Tris-HCl [pH8.0], 137 mM NaCl, 1% Triton X-100, 10% glycerol, 2 mM EDTA, 1 mM sodium vanadate, 1 mM PMSF, 1 μg/ml leupeptin, and 0.1 μg/ml aprotin) (33), and solubilized proteins were immunoprecipitated by using anti-p85 Abs and protein A-agarose beads. Beads were washed twice with lysis buffer and washed three times with wash buffer. After wash in the kinase assay buffer, immune complexes were then subjected to PI3K assay as described above.
Results
Vav3 Is Tyrosine Phosphorylated by BCR Cross-linking.
Vav3-specific antibodies were used to establish whether Vav3 is tyrosine phosphorylated after BCR engagement. Increased Vav3 phosphorylation in isolated murine B cells, as measured with anti-phosphotyrosine mAb 4G10, was apparent after 1 min of BCR stimulation, with a peak level detected 3 min after BCR cross-linking, after which phosphorylation declined (Fig. 1) . These data demonstrate that Vav3, like Vav1 and Vav2 (18), is a substrate for PTKs downstream of the BCR in murine primary B cells.
Figure 1.

Vav3 is tyrosine phosphorylated after antigen-receptor ligation in B lymphocytes. Primary B cells purified from mouse spleen (107 cells per sample, left panels) and chicken DT40 B cells (5 × 106 cells per sample, right panels) were incubated with 15 μg/ml polyclonal F(ab′)2 anti–mouse IgM and 4 μg/ml monoclonal anti–chicken IgM, M4, respectively. After stimulation, immunoprecipitates with anti-Vav3 Abs were separated on 7.5% SDS-PAGE and analyzed by Western blotting with 4G10 mAb (top) and anti-Vav3 Abs (bottom). IP, immunoprecipitation.
Chicken DT40 B cells are a useful model for studies of BCR-mediated signal transduction; therefore, we examined whether chicken Vav3 also underwent tyrosine phosphorylation in DT40 B cells. As shown in Fig. 1, BCR stimulation of DT40 B cells resulted in a rapid increase in Vav3 phosphorylation, although the kinetics differed from those of murine primary B cells. Vav3 tyrosine phosphorylation was already maximal at 1 min, after which phosphorylation gradually declined to a lower level.
Role of Vav3 in BCR Signaling.
To elucidate the function of Vav3, we established Vav3-deficient DT40 lines by the gene-targeting method (see Materials and Methods). Lack of Vav3 expression was verified by Western blot analysis (Fig. 2 A). The level of cell surface expression of BCR on Vav3-deficient DT40 cells was essentially the same as that on parental DT40 cells (Fig. 2 B). Little change of the BCR-induced overall tyrosine phosphorylation was detected between wild-type and Vav3-deficient DT40 cells (data not shown), suggesting that the BCR-associated PTKs such as Lyn and Syk are activated normally in the absence of Vav3.
Figure 2.
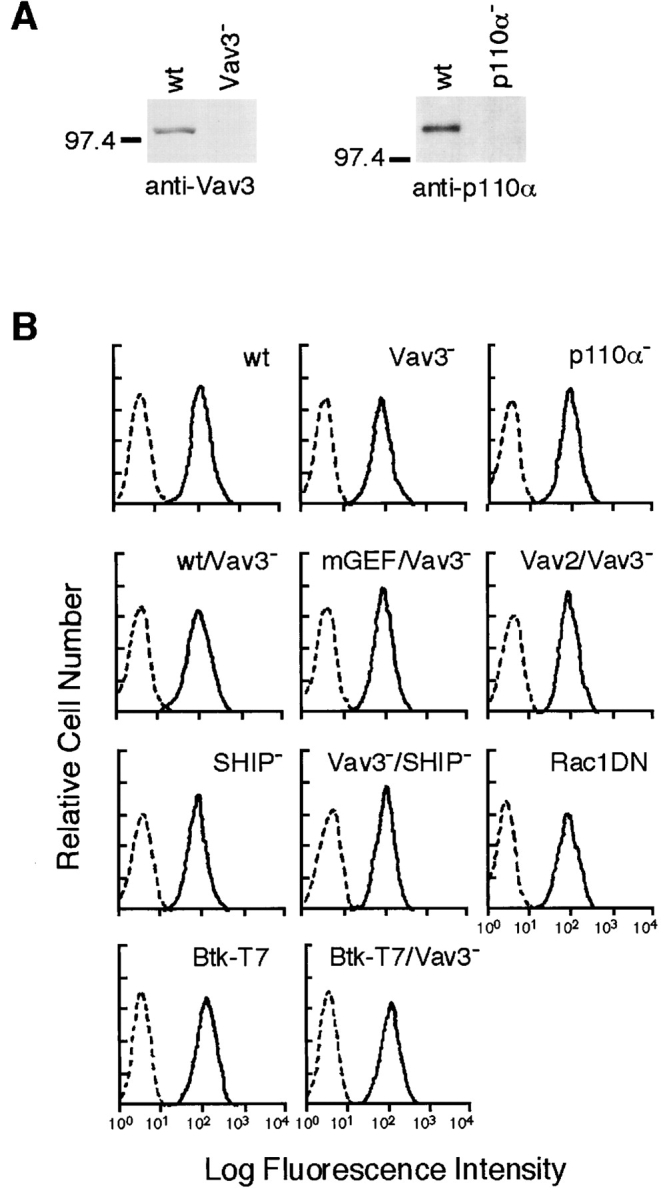
Generation of Vav3- and PI3K p110α-deficient DT40 B cells. (A) Cell lysates were separated on 7.5% SDS-PAGE and analyzed by Western blotting with anti-Vav3 Abs (left) or anti-p110α Abs (right). (B) Cell surface expression of BCR on wild-type and mutant DT40 cells used in this study: wt, parental DT40 cells; Vav3−, Vav3-deficient cells; p110α−, p110α-deficient cells; wt/Vav3−, Vav3-deficient cells expressing wild-type Vav3; mGEF/Vav3−, Vav3-deficient cells expressing GEF mutant Vav3; Vav2/Vav3−, Vav3-deficient cells overexpressing Vav2; SHIP−, SHIP-deficient cells; Vav3−/SHIP−, Vav3/SHIP double-deficient cells; Rac1DN, cells expressing dominant negative Rac1 (Rac1N17); Btk-T7, cells expressing Btk-T7; Btk-T7/Vav3−, Vav3-deficient cells expressing Btk-T7. Unstained cells were used as negative controls (dotted line).
One of the hallmarks of BCR-mediated signaling is the mobilization of intracellular calcium ([Ca2+]i). Indeed, calcium flux has been implicated in B cell maturation and immune responses (21, 34). Therefore, we measured BCR-induced calcium mobilization in wild-type and Vav3-deficient B cells. As shown in Fig. 3 A, the peak height of the [Ca2+]i increase upon receptor stimulation was decreased by ∼50% by loss of Vav3. This decrease in Vav3-deficient cells, compared with wild-type cells, was still observed when extracellular Ca2+ was chelated with EGTA, suggesting the impairment of calcium release from intracellular stores. In addition, these mutant cells exhibited reduced BCR-mediated IP3 generation (Fig. 3 B). Together, we conclude that the calcium defect in Vav3-deficient DT40 B cells is, at least partly, due to insufficient PLC-γ2 activation. Normal calcium mobilization as well as IP3 generation was restored by transfection of wild-type Vav3 cDNA (data not shown).
Previous studies including ours showed that B cells, when treated with PI3K inhibitors such as wortmannin, manifested the partial defects in calcium mobilization and IP3 generation (31, 35–37). To formally demonstrate the resemblance of the defects between Vav3- and PI3K-deficient B cells, we took a genetic approach, as the PI3K inhibitors are known to inhibit other lipid metabolizing enzymes, in addition to PI3K (38). Heterodimer-type (class Ia) PI3Ks consist of a regulatory subunit (p85) and a catalytic subunit (p110) encoded by three distinct genes (p110α, p110β, and p110δ; reference 39). Among three p110 isoforms, p110α was dominantly expressed in DT40 B cells, as determined by Northern blot analysis (data not shown). Thus, we established p110α-deficient DT40 B cells (Fig. 2, A and B) and characterized them. In fact, both calcium mobilization and IP3 generation were partially inhibited in p110α-deficient cells (Fig. 3, A and B). Moreover, in contrast to wild-type cells, p110α- and Vav3-deficient B cells exhibited a reduction in both PLC-γ2 tyrosine phosphorylation and its in vitro lipase activity upon receptor cross-linking (Fig. 3, C and D). Further resemblance was observed in BCR-mediated JNK1 response; in vitro kinase assay revealed attenuated JNK1 activation in both p110α- and Vav3-deficient cells (Fig. 3 E).
Vav3 Regulates PI3K Activation through Rac1.
The above results suggest that Vav3 and PI3K p110α influence each other's functions or that these two molecules function independently, thereby contributing to downstream responses. To test these possibilities, we first determined whether Vav3 modulated PI3K activation in BCR signaling. As demonstrated in Fig. 4 A, BCR-mediated PIP3 generation was attenuated by ∼25% in Vav3-deficient cells, while this attenuation was ∼30% in p110α-deficient cells. As expected, inhibition extents of Akt activation in Vav3- and p110α-deficient B cells correlated well with those of PIP3 generation (Fig. 4 B). A previous study proposed the possibility that Vav1 is involved in PIP2 synthesis by stimulating PIP 5-kinase (40). As PIP2 is a substrate for PI3K, attenuation of PIP3 generation in Vav3-deficient cells might be similarly accounted for by insufficient replenishment of PIP2 rather than by insufficient PIP3 synthesis. However, this possibility is unlikely in DT40 B cells, because we could not detect significant differences of PIP2 levels between wild-type and Vav3-deficient cells during the course of BCR stimulation (Fig. 4 C). Therefore, we conclude that Vav3 regulates PI3K activation in DT40 B cells. PIP3 is thought to be involved in PLC-γ2 activation through its binding to the Btk PH domain and its subsequent activation (31, 37). Hence, our conclusion is further supported by the observation that BCR-mediated tyrosine phosphorylation of Btk was significantly reduced in Vav3-deficient cells (Fig. 4 D).
Figure 4.
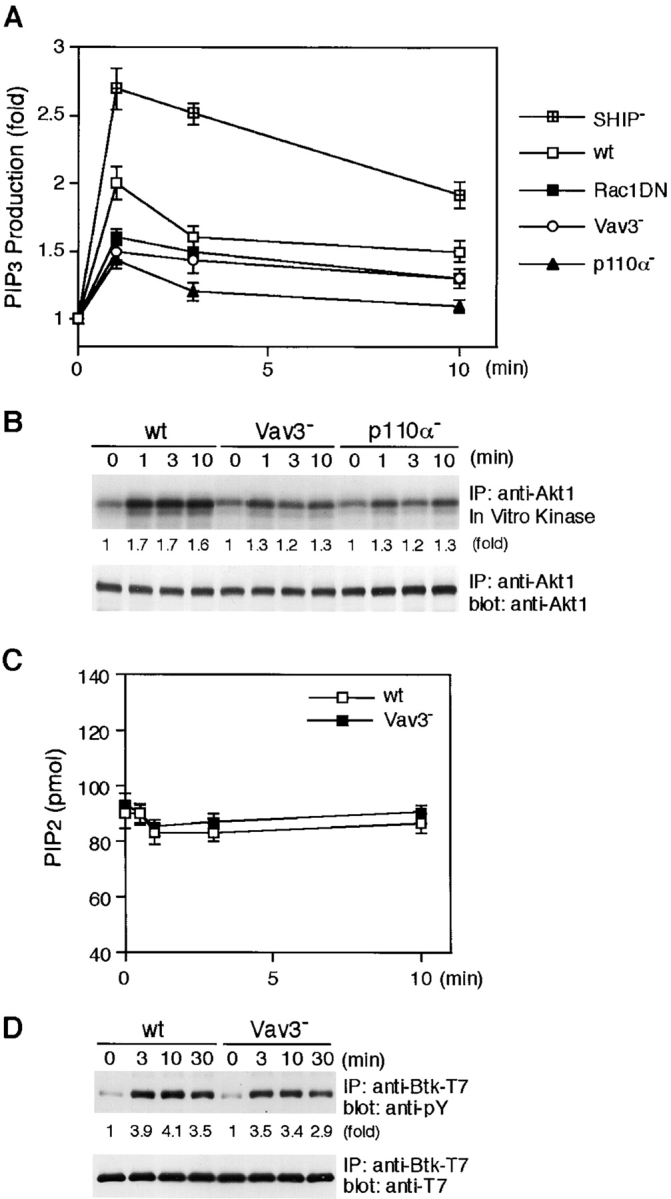
Impairment of BCR-induced PI3K signaling in the absence of Vav3. (A) PIP3 generation. Cells loaded with [32P]orthophosphate (107 cells per condition) were stimulated with M4 (4 μg/ml) for indicated time periods. PIP 3 was extracted and analyzed as described in Materials and Methods. Fold increases of PIP3 level normalized to total phospholipids after M4 stimulation were shown. These results were shown by mean ± standard error of three independent experiments. wt, wild-type. (B) BCR-induced Akt activation. Lysates from 5 × 106 cells were immunoprecipitated with anti-Akt1 Abs, and the resulting immunoprecipitates were divided. Half of them was used for Western blot analysis using anti-Akt1 Abs (bottom). The remaining half was used for in vitro kinase assay using histone H2B as an exogenous substrate. The kinase reaction products were resolved on 15% SDS-PAGE, and their phosphorylation was quantified by autoradiography (top). IP, immunoprecipitation. (C) PIP2 levels. Wild-type and Vav3-deficient DT40 cells (2 × 106) were stimulated with M4 (4 μg/ml) for indicated times and then processed to determine the levels of PIP2 as described in Materials and Methods. The results (2 × 106 cell equivalents per point) were shown by mean ± standard error of three independent experiments. (D) Tyrosine phosphorylation of Btk. Lysates from wild-type and Vav3-deficient DT40 cells expressing Btk-T7 were immunoprecipitated with anti-T7 mA. The blots (5 × 106 cell equivalents per lane) were stained with 4G10 (top). The blots were stripped and reprobed with anti-T7 mAb (bottom).
As predicted, BCR-mediated Rac1 activation, as measured by a PAK-binding assay, was attenuated by ∼50% in Vav3-deficient DT40 cells (Fig. 5 B). Introduction of wild-type Vav3, but not its GEF mutant, into Vav3-deficient cells restored Rac1 activation. Moreover, BCR-mediated Akt activation was restored only by wild-type Vav3 (Fig. 5 C). Then, to formally demonstrate that the effect of Vav3 on PI3K is indeed mediated by Rac1, we examined DT40 cells expressing a dominant-negative form of Rac1, Rac1N17 (Fig. 5 A; reference 20). The transfected cells exhibited attenuation of both PIP3 generation (Fig. 4 A) and Akt activation upon BCR engagement (Fig. 5 D).
Figure 5.
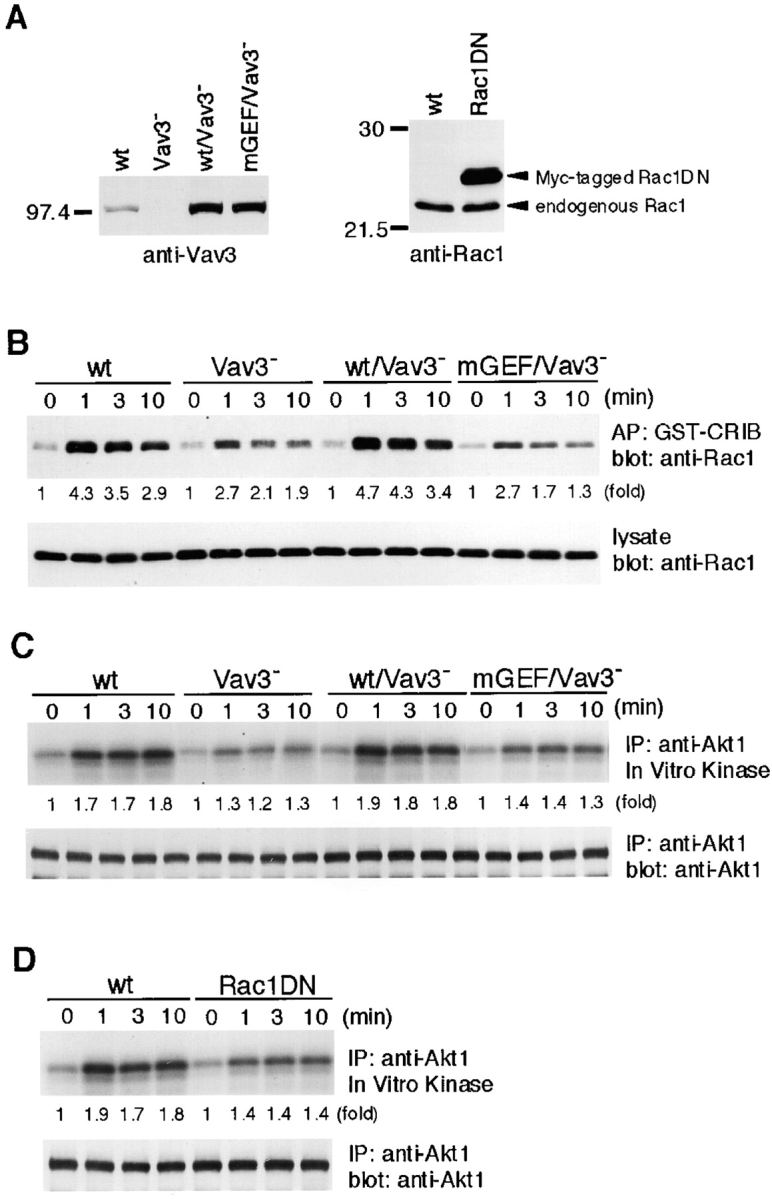
Vav3 regulates BCR-mediated Akt response through Rac1. (A) Expression levels of wild-type (wt) or GEF mutant Vav3 in Vav3-deficient DT40 cells (left) or of Myc-tagged dominant-negative Rac1 in wild-type DT40 cells (right). Whole-cell lysates were analyzed by Western blotting using anti-Vav3 Abs (left) or anti-Rac1 mAb (right). (B) Rac1 activation in response to BCR cross-linking. Cells (5 × 106) were stimulated with M4 (4 μg/ml), and cell lysates were mixed with beads containing the CRIB of PAK. Elutes of the beads (top) or cell lysates (bottom) were resolved on 12.5% SDS-PAGE and subjected to Western blot analysis with anti-Rac1 mAb. AP, affinity precipitation. (C and D) Effects of mutations of Vav3 and Rac1 on Akt responses. Cells were stimulated with M4 (4 μg/ml) for indicated times. Cell lysates were subsequently subjected to in vitro kinase assay of Akt, as described in the legend to Fig. 4 B. IP, immunoprecipitation.
Rac1 Stimulates PI3K Activity in a GTP-dependent Manner.
Optimal PI3K activation is thought to require at least two events in the context of antigen receptor signaling; (a) recruitment of PI3K to the plasma membrane, particularly to GEMs (41–43); (b) an increase in the PI3K enzymatic activity by its conformational change (44). To obtain more insight into how Vav3 affects PI3K activation in BCR signaling, we studied whether these two events were modulated by Vav3-dependent Rac1 activation. First, we compared BCR-mediated recruitment of PI3K p85 subunit to GEMs in wild-type and Vav3-deficient cells, revealing no differences (Fig. 6) .
Figure 6.

Inactivation of Vav3 does not affect PI3K recruitment to glycolipid-enriched microdomains (GEMs). Wild-type (wt) and Vav3-deficient DT40 cells (108 cells per condition), unstimulated (minus symbol) or stimulated with M4 (4 μg/ml) for 1 min (plus symbol), were lysed and then subjected to discontinuous sucrose density gradient centrifugation. Fractions (30 μl/lane, numbered from low to high density) were resolved on 7.5% SDS-PAGE and immunoblotted with anti-p85 Abs or anti-Lyn Abs. Lyn was used as a positive control for GEM fraction.
In vitro kinase experiments were employed to examine the second possibility. Anti-p85 immunoprecipitates from 293T cells transfected with both PI3K p85α and p110α subunits were mixed with different guanidine nucleotide-bound forms of GST-Rac1 and subjected to a PI3K enzymatic assay. Consistent with a previous report (45), the GTP-bound GST-Rac1 stimulated PI3K activity about threefold, while GDP-bound GST-Rac1 stimulated the PI3K activity by 1.2-fold (Fig. 7 A). As a negative control, immunoprecipitates from 293T cells expressing wild-type p85α and kinase-negative p110α were used, showing undetectable levels of in vitro PI3K activity in our assay conditions. Proper expression and association of these two subunits were verified by Western blot analysis (Fig. 7 B). The biological relevance of the above findings with 293T cells was examined by precipitating PI3K from resting and stimulated DT40 B cells and measuring its in vitro kinase activity. As shown in Fig. 7 C, Vav3-deficient B cells exhibited reduced PI3K activation, and this defect was restored by wild-type Vav3, but not its GEF mutant. Taken together, these findings suggest that Rac1, after being activated by Vav3, contributes to enzymatic activation of PI3K rather than to its recruitment to GEMs.
Figure 7.

PI3K enzymatic activity is upregulated by Rac1 in a GTP-dependent manner. (A and B) Effect of Rac1 on PI3K activity in vitro. (A) p85 was immunoprecipitated from lysates of 293T cells transfected with plasmids encoding either wild-type (wt; lane 1–4) or kinase-negative (KN; lane 5–8) p110α, together with p85α. Immunoprecipitates were mixed with GST/GTPγS (lane 1 and 5), GST-Rac1/GDPβS (lane 2 and 6), GST-Rac1/GTPγS (lane 3 and 7), or GST-Rac1N17/GTPγS (lane 4 and 8), and PI3K activity was assayed as described in Materials and Methods. In the top panel, one of representative results is shown. The bottom panel shows quantitative analysis of PI3K activity. The intensity of the PIP spots revealed in autoradiogram was measured by scanning densitometry. The activity in the presence of GST/GTPγS was used to normalize individual activities in each experiment. The results were shown by mean ± standard error of three independent experiments. (B) Aliquots of immunoprecipitates with anti-p85 Abs were subjected to Western blot analysis using anti-p85 Abs (left) or anti-p110α Abs (right). (C) BCR-induced PI3K activity. Lysates from 5 × 106 cells were immunoprecipitated with anti-p85 Abs, and the immunoprecipitates were subjected to PI3K assay. One of the representative results is shown in the top panel. Aliquots of immunoprecipitates were used for Western blot analysis with anti-p85 Abs (bottom). The doublet bands shown in the immunoblot are presumably due to p85α and p85β isoforms, as we used anti-pan-p85 Abs. IP, immunoprecipitation.
BCR-mediated Tyrosine Phosphorylation of Vav3 Occurs Normally in the Absence of PI3K p110α.
Previous in vitro studies place Vav1 downstream of PI3K; PIP3 binds to the Vav1 PH domain and promotes tyrosine phosphorylation-mediated activation of the enzymatic activity of Vav1 (7). To determine whether PI3K similarly modulates Vav3 activation, we assessed Vav3 tyrosine phosphorylation and subsequent Rac1 activation using p110α-deficient DT40 B cells. As shown in Fig. 8 , significant changes in both parameters in wild-type versus mutant DT40 cells could not be detected, except a small decrease of Vav3 tyrosine phosphorylation after 10 min stimulation in p110α-deficient cells. These readouts could not be affected in DT40 B cells, even when treated with 100 nM wortmannin.
Figure 8.

Neither disruption of PI3K p110α nor treatment with wortmannin affects Vav3 activation. Treatment with 100 nM wortmannin was conducted 20 min before stimulation. Wild-type (wt) and p110α-deficient DT40 cells were stimulated with M4 (4 μg/ml) and then subjected to the following analyses. (A) Tyrosine phosphorylation of Vav3, as described in the legend to Fig. 1. IP, immunoprecipitation. (B) Rac1 activation, as described in the legend to Fig. 5 B. AP, affinity precipitation.
Deletion of SHIP Suppresses Defects in Vav3-deficient Cells.
Collectively, the above data suggest that Vav3 functions upstream of PI3K activation, thereby contributing to calcium mobilization and JNK1 activation. This hypothesis predicts that defects in Vav3-deficient DT40 cells should be suppressible by manipulations which enhance PIP3 accumulation. To test this prediction, we deleted a PIP3 hydrolyzing enzyme, SHIP, in a Vav3-deficient background. As shown in Fig. 9 , both of these defects, in addition to downmodulation of Akt in Vav3-deficient cells, were relieved by deletion of SHIP. Hence, reduction in PIP3 levels likely results in inhibition of calcium mobilization as well as JNK1 activation in Vav3-deficient cells.
Figure 9.

Inactivation of SHIP relieves defects of BCR-induced calcium, JNK1, and Akt responses in Vav3-deficient cells. Vav3-, SHIP-, and Vav3/SHIP double-deficient DT40 cells were stimulated with M4 (4 μg/ml) and then subjected to following analyses. (A) Calcium mobilization, as described in the legend to Fig. 3 A. (B) Activation of JNK1, as described in the legend to Fig. 3 E. IP, immunoprecipitation. (C) Activation of Akt, as described in the legend to Fig. 4 B.
Overexpression of Vav2 Rescues Defects in Vav3-deficient Cells.
Although we cloned chicken homologs of Vav2 and Vav3, Vav1 isoform could not be detected in spite of our several efforts including low-stringency cross-hybridization, degenerative-oligonucleotide reverse transcription (RT)-PCR, and EST database search. Given the evidence that Vav2, like Vav3, undergoes tyrosine phosphorylation after BCR cross-linking in DT40 B cells (data not shown), Vav2 could have overlapping functions with those of Vav3. If so, we speculate that Vav2, once overexpressed in a Vav3-deficient background, can compensate some functional defects of Vav3-deficient DT40 B cells. To examine this possibility, we established Vav3-deficient cells that expressed fourfold higher level of Vav2 than that in wild-type cells. As shown in Fig. 10 , defects in Rac1, Akt, and calcium responses were compensated by overexpression of Vav2, suggesting the existence of some functional redundancy between Vav2 and Vav3.
Figure 10.

Overexpression of Vav2 restores BCR-induced Rac1, Akt, calcium responses in Vav3-deficient cells. (A) Expression levels of Vav2. Whole-cell lysates prepared from wild-type (wt), Vav3-deficient, and Vav3-deficient overexpressing Vav2 DT40 cells were analyzed by Western blotting with anti-Vav2 Abs. Cells were stimulated with M4 (4 μg/ml) and then subjected to following analyses. (B) Rac1 activation, as described in the legend to Fig. 5 B. AP, affinity precipitation. (C) Akt activation, as described in the legend to Fig. 4 B. IP, immunoprecipitation. (D) Calcium mobilization, as described in the legend to Fig. 3 A.
Discussion
Vav1 and Vav2 play an important role in coupling BCR to calcium flux (16, 17). Here we extended this concept to show that Vav3, like Vav1 and Vav2, participates in calcium mobilization. Then, we addressed the mechanisms by which Vav3 influences the calcium flux. Many aspects of PLC-γ2/calcium defects are similar in Vav3-deficient and PI3K p110α-deficient DT40 B cells. For instance, BCR-mediated PLC-γ2 tyrosine phosphorylation and subsequent IP3 generation are similarly attenuated in both backgrounds (Fig. 3, B and C), thereby promoting us to examine the relationship between Vav3 and PI3K. Previous in vitro studies favor the model that PI3K functions upstream of Vav as regulators of GEF activity (7). Our data instead support a different mechanism, in which Vav3 functions as a positive regulator of PI3K in BCR signaling.
We demonstrate that BCR-mediated PIP3 generation and subsequent Akt activation are inhibited by loss of Vav3 (Fig. 4, A and B) or by introduction of Rac1N17 into DT40 B cells (Figs. 4 A and 5 D). Strengthening the idea that Vav3-mediated Rac1 activation is involved in PIP3 generation, a Vav3 GEF mutant fails to restore Rac1 activation as well as PI3K activation (Figs. 5 B and 7 C). Hence, the results presented here are consistent with reports that, in Jurkat T cells and RBL mast cells, Akt activation is blocked by overexpression of dominant negative Rac1 and by treatment of Clostridium sordellii lethal toxin, a Rac-inactivating toxin, respectively (46, 47). The inhibition of PIP3 generation in Vav3-deficient cells could be accounted for by the decreased availability of a PI3K substrate, PIP2, and/or by the decreased PIP3 synthesis from PIP2. Two lines of our evidence suggest that the latter mechanism is dominantly operating in DT40 B cells. First, overall PIP2 levels are not significantly changed between wild-type and Vav3-deficient DT40 B cells (Fig. 4 C). Second, the enzymatic activity of PI3K, a responsible kinase for PIP3 synthesis, is indeed increased by Rac1 in a GTP-dependent manner (Fig. 7 A). However, the former mechanism cannot be completely excluded by our data. Recent studies showed that PI3K activation occurs in subdomains of the plasma membrane, called GEMs or lipid rafts (42), suggesting that PIP2 localized specifically in GEMs serves as a PI3K substrate in BCR signaling context. In this regard, our overall PIP2 assay system might not have sufficed enough to detect small changes in the level of PIP2 which reside in GEMs.
Based on the in vitro data that PIP3 binds to the Vav1 PH domain and promotes PTK-mediated activation of its GEF activity, PI3K has been proposed to function as an upstream regulator for Vav family members (7). Hence, our finding that Vav3 is involved in PI3K activation implies two possibilities; (a) in addition to regulation of PI3K by Vav3, PI3K could reciprocally influence Vav3 function, thereby making a positive feed-forward regulatory loop; (b) PI3K-mediated Vav3 activation mechanism does not operate significantly in the context of BCR signaling. Our data support the second possibility. Deletion of PI3K p110α affects neither Vav3 tyrosine phosphorylation nor Rac1 activation. In addition, as reported previously in FcεRI signaling (47), these readouts could not be affected in DT40 B cells, even when PIP3 generation was almost completely inhibited by wortmannin (Fig. 8, A and B). However, given the evidence that PIP3 cannot activate nonphosphorylated Vav1 (7), one interpretation for our results is that a role of PIP3 is the fine modulation of the enzyme activity of Vav3 only after being phosphorylated, rather than acts as a critical Vav3 regulator. This fine modulation by PIP3 might have escaped detection in our experimental conditions.
Activation of PI3K upon BCR engagement is thought to involve recruiting PI3K to GEMs as well as increasing the specific activity of the enzyme. BCR-induced translocation of PI3K to GEMs occurs normally in Vav3-deficient DT40 cells (Fig. 6), which suggests that other molecules play a major role in PI3K recruitment to GEMs, whereas Vav3 participates in upregulation of the PI3K enzymatic activity. Like PI3K, Vav3 is also targeted to GEMs after receptor cross-linking (data not shown), thereby being able to gain access to its substrate Rac1, as Rac1 is known to be concentrated in GEMs (48). Subsequently, PI3K is activated by GTP-bound Rac1. In regard to recruitment of PI3K to GEMs, docking proteins such as CD19, Gab, and BCAP could participate in this process, directly or indirectly, because these proteins, after being tyrosine phosphorylated, associate with the SH2 domains of the PI3K p85 subunit (27, 49–52). Colocalization of Vav3 and PI3K in GEMs appears to be necessary, but their mere colocalization in GEMs might not be sufficient for PIP3 generation. For instance, when CD19 is tyrosine phosphorylated upon BCR ligation alone or coligation of BCR and CD19, the phosphorylated CD19 provides binding sites for both Vav1 and PI3K (51, 53). Assuming that Vav3, like Vav1, is recruited to the phosphorylated CD19, CD19 can bring Vav3 and PI3K into the close proximity with each other, which in turn, could facilitate the efficient coupling between Vav3 and PI3K in B cells. Thus, an organized multiprotein complex including Vav3 and PI3K in GEMs may be required for efficient PIP3 generation.
The reduced PLC-γ2 activation in Vav3-deficient DT40 B cells after BCR engagement could be accounted for by the decreased PI3K activation. In support of this explanation, the PLC-γ2/calcium defects in Vav3-deficient B cells are relieved by deletion of a PIP3 hydrolyzing enzyme, SHIP. In fact, consistent with a previous report using SHIP−/− primary B cells (54), PIP3 levels upon BCR activation are increased in SHIP-deficient DT40 B cells (Fig. 4 A). PIP3 is thought to function as a positive regulator of PLC-γ2 via two distinct mechanisms. One mechanism is that PIP3 is involved in PLC-γ2 activation through its binding to the Btk PH domain and its subsequent activation (31, 37), as Btk is essential for PLC-γ2 activation upon BCR engagement. In this regard, we observed reduced BCR-mediated tyrosine phosphorylation of Btk in Vav3-deficient DT40 cells, compared with wild-type cells (Fig. 4 D). The second mechanism is that the PLC-γ2 PH domain binds to PIP3, which in turn contributes to PLC-γ2 activation by its targeting to GEMs and/or by its conformational changes (55–57). Although it is clear that PIP3-mediated PLC-γ2 activation contributes to calcium mobilization, this mechanism may not entirely account for the effect of Vav3 on calcium mobilization. For example, activated Rac1 could directly modulate a calcium influx channel on the plasma membrane (58), in addition to activating PI3K.
Given our previous evidence that calcium mobilization, in addition to Rac1 activation, is required for coupling BCR to JNK1 (20, 22), the reduced activation of JNK1 in Vav3-deficient cells could be explained, at least in part, by the insufficient calcium mobilization. Indeed, Vav3/SHIP double-deficient DT40 B cells exhibit augmentation in both calcium mobilization and JNK1 activation (Fig. 9, A and B). Thus, it is reasonable to anticipate that Rac1 regulates MAP3K as well as PLC-γ2, both of which contribute to the optimal JNK1 response in the BCR signaling context.
Although this study has not addressed the functional differences among Vav isoforms, our results demonstrate that Vav2, when overexpressed, can compensate for Vav3-regulated Rac1 activation and calcium flux in B cells. Thus, given the previous evidence that both Vav1 and Vav2 are able to augment calcium flux and NF-AT activation in B cell lines (18), we suggest that all three Vav isoforms possess overlapping functions in BCR signaling. Consistent with this, mice deficient in Vav1 or Vav2 show a modest defect in calcium mobilization in response to BCR cross-linking, whereas this defect is exaggerated in mice lacking both Vav1 and Vav2 (16, 17). Hence, the mechanism we have described for Vav3 in chicken B cells is likely to be applicable to other Vav isoforms in the B cell context. Indeed, in agreement with our conclusion, recent works from other groups have shown that Vav1 could also regulate antigen receptor-induced PI3K activation in murine mast cells (15) and thymocytes (unpublished data).
Our implications, however, do not invalidate the previous contention that each Vav isoform has an unique function in other cell types. For instance, T cell studies demonstrate that Vav1 and Vav3 are not simply interchangeable in that overexpression of Vav1, but not Vav3, potentiates TCR-mediated NF-AT activation (59). Considering that Vav family proteins must associate with adaptor molecules for exerting their functions, the inability of Vav3 to induce calcium signaling in T cells may be due to its insufficient interaction with adaptor molecules such as SLP-76 (60). In contrast, BLNK and CD19 in B cells may interact with Vav3 and Vav1 with the same efficiency (40, 51, 53, 61, 62). Differences in the substrate specificity of the Rho family GTPases between Vav1 and Vav3 could be another possibility. High preference of Vav1 toward Rac1 might be required for T cells to activate calcium signaling. Thus, each Vav isoform, like Vav1, has GEF-dependent and -independent functions (23), both of which coordinately contribute to signal transduction pathways, depending upon differential cellular contexts.
Acknowledgments
We would like to thank Drs. P.K. Vogt, M.D. Waterfield, A. Weiss, and R. Goitsuka for providing chicken p110α cDNA, bovine p85α cDNA, human Vav1 and Vav2 cDNAs, and DT40 cDNA library, respectively. We also thank Drs. V.L.J. Tybulewicz and L.F. Reynolds for advice on the PIP2 determination, Drs. A. Weiss and G.M. Ku for advice on the Rac1/PAK pull-down assay, Dr. D. Watanabe for advice on the phospholipid hydrolyzing assay, Dr. M. Turner for helpful discussions, and M. Kurosaki, M. Takezaki, and N. Narumai for technical assistance.
This work was supported by grants from the Ministry of Education, Science, Sports, and Culture of Japan (to K. Inabe, M. Ishiai, and T. Kurosaki), the Uehara Memorial Foundation (to T. Kurosaki), the Toray Science Foundation (to T. Kurosaki), and the Human Frontier Science Program (to T. Kurosaki).
Footnotes
Abbreviations used in this paper: BCR, B cell antigen receptor; CRIB, Cdc42/Rac interactive binding domain; GEF, guanine nucleotide exchange factor; GEM, glycolipid-enriched microdomain; IP3, inositol 1,4,5-trisphosphate; PH, pleckstrin homology; PI3K, phosphoinositide 3-kinase; PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol-3,4,5-trisphosphate; PLC, phospholipase C; PTK, protein tyrosine kinase; SH, src homology; SHIP, SH2 domain-containing inositol polyphosphate 5′-phosphatase.
References
- 1.DeFranco, A.L. 1997. The complexity of signaling pathways activated by the BCR. Curr. Opin. Immunol. 9:296–308. [DOI] [PubMed] [Google Scholar]
- 2.Reth, M., and J. Wienands. 1997. Initiation and processing of signals from the B cell antigen receptor. Annu. Rev. Immunol. 15:453–479. [DOI] [PubMed] [Google Scholar]
- 3.Tamir, I., and J.C. Cambier. 1998. Antigen receptor signaling: integration of protein tyrosine kinase functions. Oncogene. 17:1353–1364. [DOI] [PubMed] [Google Scholar]
- 4.Kurosaki, T. 1999. Genetic analysis of B cell antigen receptor signaling. Annu. Rev. Immunol. 17:555–592. [DOI] [PubMed] [Google Scholar]
- 5.Bustelo, X.R. 2000. Regulatory and signaling properties of the Vav family. Mol. Cell. Biol. 20:1461–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crespo, P., K.E. Schuebel, A.A. Ostrom, J.S. Gutkind, and X.R. Bustelo. 1997. Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature. 385:169–172. [DOI] [PubMed] [Google Scholar]
- 7.Han, J., K. Luby-Phelps, B. Das, X. Shu, Y. Xia, R.D. Mosteller, U.M. Krishna, J.R. Falck, M.A. White, and D. Broek. 1998. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science. 279:558–560. [DOI] [PubMed] [Google Scholar]
- 8.Bustelo, X.R., S.D. Rubin, K.L. Suen, D. Carrasco, and M. Barbacid. 1993. Developmental expression of the vav protooncogene. Cell. Growth Differ. 4:297–308. [PubMed] [Google Scholar]
- 9.Schuebel, K.E., X.R. Bustelo, D.A. Nielsen, B.J. Song, M. Barbacid, D. Goldman, and I.J. Lee. 1996. Isolation and characterization of murine vav2, a member of the vav family of proto-oncogenes. Oncogene. 13:363–371. [PubMed] [Google Scholar]
- 10.Movilla, N., and X.R. Bustelo. 1999. Biological and regulatory properties of Vav-3, a new member of the Vav family of oncoproteins. Mol. Cell. Biol. 19:7870–7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeng, L., P. Sachdev, L. Yan, J.L. Chan, T. Trenkle, M. McClelland, J. Welsh, and L.H. Wang. 2000. Vav3 mediates receptor protein tyrosine kinase signaling, regulates GTPase activity, modulates cell morphology, and induces cell transformation. Mol. Cell. Biol. 20:9212–9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tarakhovsky, A., M. Turner, S. Schaal, P.J. Mee, L.P. Duddy, K. Rajewsky, and V.L. Tybulewicz. 1995. Defective antigen receptor-mediated proliferation of B and T cells in the absence of Vav. Nature. 374:467–470. [DOI] [PubMed] [Google Scholar]
- 13.Zhang, R., F.W. Alt, L. Davidson, S.H. Orkin, and W. Swat. 1995. Defective signalling through the T- and B-cell antigen receptors in lymphoid cells lacking the vav proto-oncogene. Nature. 374:470–473. [DOI] [PubMed] [Google Scholar]
- 14.Fischer, K.D., A. Zmuldzinas, S. Gardner, M. Barbacid, A. Bernstein, and C. Guidos. 1995. Defective T-cell receptor signalling and positive selection of Vav-deficient CD4+ CD8+ thymocytes. Nature. 374:474–477. [DOI] [PubMed] [Google Scholar]
- 15.Manetz, T.S., C. Gonzalez-Espinosa, R. Arudchandran, S. Xirasagar, V. Tybulewicz, and J. Rivera. 2001. Vav1 regulates phospholipase Cγ activation and calcium responses in mast cells. Mol. Cell. Biol. 21:3763–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doody, G.M., S.E. Bell, E. Vigorito, E. Clayton, S. McAdam, R. Tooze, C. Fernandez, I.J. Lee, and M. Turner. 2001. Signal transduction through Vav-2 participates in humoral immune responses and B cell maturation. Nat. Immunol. 2:542–547. [DOI] [PubMed] [Google Scholar]
- 17.Tedford, K., L. Nitschke, I. Girkontaite, A. Charlesworth, G. Chan, V. Sakk, M. Barbacid, and K.D. Fischer. 2001. Compensation between Vav-1 and Vav-2 in B cell development and antigen receptor signaling. Nat. Immunol. 2:548–555. [DOI] [PubMed] [Google Scholar]
- 18.Doody, G.M., D.D. Billadeau, E. Clayton, A. Hutchings, R. Berland, S. McAdam, P.J. Leibson, and M. Turner. 2000. Vav-2 controls NFAT-dependent transcription in B- but not T-lymphocytes. EMBO J. 19:6173–6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okada, H., S. Bolland, A. Hashimoto, M. Kurosaki, Y. Kabuyama, M. Iino, J.V. Ravetch, and T. Kurosaki. 1998. Role of the inositol phosphatase SHIP in B cell receptor-induced Ca2+ oscillatory response. J. Immunol. 161:5129–5132. [PubMed] [Google Scholar]
- 20.Hashimoto, A., H. Okada, A. Jiang, M. Kurosaki, S. Greenberg, E.A. Clark, and T. Kurosaki. 1998. Involvement of guanosine triphosphatases and phospholipase C-γ2 in extracellular signal-regulated kinase, c-Jun NH2-terminal kinase, and p38 mitogen-activated protein kinase activation by the B cell antigen receptor. J. Exp. Med. 188:1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hashimoto, A., K. Takeda, M. Inaba, M. Sekimata, T. Kaisho, S. Ikehara, Y. Homma, S. Akira, and T. Kurosaki. 2000. Cutting edge: essential role of phospholipase C-γ2 in B cell development and function. J. Immunol. 165:1738–1742. [DOI] [PubMed] [Google Scholar]
- 22.Ishiai, M., M. Kurosaki, R. Pappu, K. Okawa, I. Ronko, C. Fu, M. Shibata, A. Iwamatsu, A.C. Chan, and T. Kurosaki. 1999. BLNK required for coupling Syk to PLCγ2 and Rac1-JNK in B cells. Immunity. 10:117–125. [DOI] [PubMed] [Google Scholar]
- 23.Kuhne, M.R., G. Ku, and A. Weiss. 2000. A guanine nucleotide exchange factor-independent function of Vav1 in transcriptional activation. J. Biol. Chem. 275:2185–2190. [DOI] [PubMed] [Google Scholar]
- 24.Takata, M., H. Sabe, A. Hata, T. Inazu, Y. Homma, T. Nukada, H. Yamamura, and T. Kurosaki. 1994. Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+ mobilization through distinct pathways. EMBO J. 13:1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurosaki, T., and M. Kurosaki. 1997. Transphosphorylation of Bruton's tyrosine kinase on tyrosine 551 is critical for B cell antigen receptor function. J. Biol. Chem. 272:15595–15598. [DOI] [PubMed] [Google Scholar]
- 26.Yasuda, T., A. Maeda, M. Kurosaki, T. Tezuka, K. Hironaka, T. Yamamoto, and T. Kurosaki. 2000. Cbl suppresses B cell receptor-mediated phospholipase C (PLC)-γ2 activation by regulating B cell linker protein-PLC-γ2 binding. J. Exp. Med. 191:641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okada, T., A. Maeda, A. Iwamatsu, K. Gotoh, and T. Kurosaki. 2000. BCAP: the tyrosine kinase substrate that connects B cell receptor to phosphoinositide 3-kinase activation. Immunity. 13:817–827. [DOI] [PubMed] [Google Scholar]
- 28.Watanabe, D., S. Hashimoto, M. Ishiai, M. Matsushita, Y. Baba, T. Kishimoto, T. Kurosaki, and S. Tsukada. 2001. Four tyrosine residues in phospholipase C-γ2, identified as Btk-dependent phosphorylation sites, are required for B cell antigen receptor-coupled calcium signaling. J. Biol. Chem. 276:38595–38601. [DOI] [PubMed] [Google Scholar]
- 29.Manser, E., T.H. Loo, C.G. Koh, Z.S. Zhao, X.Q. Chen, L. Tan, I. Tan, T. Leung, and L. Lim. 1998. PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol. Cell. 1:183–192. [DOI] [PubMed] [Google Scholar]
- 30.Ishiai, M., M. Kurosaki, K. Inabe, A.C. Chan, K. Sugamura, and T. Kurosaki. 2000. Involvement of LAT, Gads, and Grb2 in compartmentation of SLP-76 to the plasma membrane. J. Exp. Med. 192:847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scharenberg, A.M., O. El-Hillal, D.A. Fruman, L.O. Beitz, Z. Li, S. Lin, I. Gout, L.C. Cantley, D.J. Rawlings, and J.P. Kinet. 1998. Phosphatidylinositol-3,4,5-trisphosphate (PtdIns-3,4,5-P3)/Tec kinase- dependent calcium signaling pathway: a target for SHIP-mediated inhibitory signals. EMBO J. 17:1961–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng, Y., S. Bagrodia, and R.A. Cerione. 1994. Activation of phosphoinositide 3-kinase activity by Cdc42Hs binding to p85. J. Biol. Chem. 269:18727–18730. [PubMed] [Google Scholar]
- 33.Gold, M.R., V.W. Chan, C.W. Turck, and A.L. DeFranco. 1992. Membrane Ig cross-linking regulates phosphatidylinositol 3-kinase in B lymphocytes. J. Immunol. 148:2012–2022. [PubMed] [Google Scholar]
- 34.Wang, D., J. Feng, R. Wen, J.C. Marine, M.Y. Sangster, E. Parganas, A. Hoffmeyer, C.W. Jackson, J.L. Cleveland, P.J. Murray, and J.N. Ihle. 2000. Phospholipase C-γ2 is essential in the functions of B cell and several Fc receptors. Immunity. 13:25–35. [DOI] [PubMed] [Google Scholar]
- 35.Buhl, A.M., C.M. Pleiman, R.C. Rickert, and J.C. Cambier. 1997. Qualitative regulation of B cell antigen receptor signaling by CD19: selective requirement for PI3-kinase activation, inositol-1,4,5-trisphosphate production and Ca2+ mobilization. J. Exp. Med. 186:1897–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hippen, K.L., A.M. Buhl, D. D'Ambrosio, K. Nakamura, C. Persin, and J.C. Cambier. 1997. FcγRIIB1 inhibition of BCR-mediated phosphoinositide hydrolysis and Ca2+ mobilization is integrated by CD19 dephosphorylation. Immunity. 7:49–58. [DOI] [PubMed] [Google Scholar]
- 37.Bolland, S., R.N. Pearse, T. Kurosaki, and J.V. Ravetch. 1998. SHIP modulates immune receptor responses by regulating membrane association of Btk. Immunity. 8:509–516. [DOI] [PubMed] [Google Scholar]
- 38.Balla, T., G.J. Downing, H. Jaffe, S. Kim, A. Zolyomi, and K.J. Catt. 1997. Isolation and molecular cloning of wortmannin-sensitive bovine type III phosphatidylinositol 4-kinases. J. Biol. Chem. 272:18358–18366. [DOI] [PubMed] [Google Scholar]
- 39.Vanhaesebroeck, B., S.J. Leevers, G. Panayotou, and M.D. Waterfield. 1997. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem. Sci. 22:267–272. [DOI] [PubMed] [Google Scholar]
- 40.O'Rourke, L.M., R. Tooze, M. Turner, D.M. Sandoval, R.H. Carter, V.L. Tybulewicz, and D.T. Fearon. 1998. CD19 as a membrane-anchored adaptor protein of B lymphocytes: costimulation of lipid and protein kinases by recruitment of Vav. Immunity. 8:635–645. [DOI] [PubMed] [Google Scholar]
- 41.Klippel, A., C. Reinhard, W.M. Kavanaugh, G. Apell, M.A. Escobedo, and L.T. Williams. 1996. Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Mol. Cell. Biol. 16:4117–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gillham, H., M.C. Golding, R. Pepperkok, and W.J. Gullick. 1999. Intracellular movement of green fluorescent protein-tagged phosphatidylinositol 3-kinase in response to growth factor receptor signaling. J. Cell Biol. 146:869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xavier, R., T. Brennan, Q. Li, C. McCormack, and B. Seed. 1998. Membrane compartmentation is required for efficient T cell activation. Immunity. 8:723–732. [DOI] [PubMed] [Google Scholar]
- 44.Shoelson, S.E., M. Sivaraja, K.P. Williams, P. Hu, J. Schlessinger, and M.A. Weiss. 1993. Specific phosphopeptide binding regulates a conformational change in the PI 3-kinase SH2 domain associated with enzyme activation. EMBO J. 12:795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bokoch, G.M., C.J. Vlahos, Y. Wang, U.G. Knaus, and A.E. Traynor-Kaplan. 1996. Rac GTPase interacts specifically with phosphatidylinositol 3-kinase. Biochem. J. 315:775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Genot, E.M., C. Arrieumerlou, G. Ku, B.M. Burgering, A. Weiss, and I.M. Kramer. 2000. The T-cell receptor regulates Akt (protein kinase B) via a pathway involving Rac1 and phosphatidylinositide 3-kinase. Mol. Cell. Biol. 20:5469–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Djouder, N., G. Schmidt, M. Frings, A. Cavalie, M. Thelen, and K. Aktories. 2001. Rac and phosphatidylinositol 3-kinase regulate the protein kinase B in FcεRI signaling in RBL 2H3 mast cells. J. Immunol. 166:1627–1634. [DOI] [PubMed] [Google Scholar]
- 48.Arudchandran, R., M.J. Brown, M.J. Peirce, J.S. Song, J. Zhang, R.P. Siraganian, U. Blank, and J. Rivera. 2000. The Src homology 2 domain of Vav is required for its compartmentation to the plasma membrane and activation of c-Jun NH2-terminal kinase 1. J. Exp. Med. 191:47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tuveson, D.A., R.H. Carter, S.P. Soltoff, and D.T. Fearon. 1993. CD19 of B cells as a surrogate kinase insert region to bind phosphatidylinositol 3-kinase. Science. 260:986–989. [DOI] [PubMed] [Google Scholar]
- 50.Buhl, A.M., and J.C. Cambier. 1999. Phosphorylation of CD19 Y484 and Y515, and linked activation of phosphatidylinositol 3-kinase, are required for B cell antigen receptor-mediated activation of Bruton's tyrosine kinase. J. Immunol. 162:4438–4446. [PubMed] [Google Scholar]
- 51.Fujimoto, M., Y. Fujimoto, J.C. Poe, P.J. Jansen, C.A. Lowell, A.L. DeFranco, and T.F. Tedder. 2000. CD19 regulates Src family protein tyrosine kinase activation in B lymphocytes through processive amplification. Immunity. 13:47–57. [DOI] [PubMed] [Google Scholar]
- 52.Gold, M.R. 2000. Intermediary signaling effectors coupling the B-cell receptor to the nucleus. Curr. Top. Microbiol. Immunol. 245:77–134. [DOI] [PubMed] [Google Scholar]
- 53.Weng, W.K., L. Jarvis, and T.W. LeBien. 1994. Signaling through CD19 activates Vav/mitogen-activated protein kinase pathway and induces formation of a CD19/Vav/phosphatidylinositol 3-kinase complex in human B cell precursors. J. Biol. Chem. 269:32514–32521. [PubMed] [Google Scholar]
- 54.Brauweiler, A., I. Tamir, J. Dal Porto, R.J. Benschop, C.D. Helgason, R.K. Humphries, J.H. Freed, and J.C. Cambier. 2000. Differential regulation of B cell development, activation, and death by the src homology 2 domain-containing 5′ inositol phosphatase (SHIP). J. Exp. Med. 191:1545–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rhee, S.G., and Y.S. Bae. 1997. Regulation of phosphoinositide-specific phospholipase C isozymes. J. Biol. Chem. 272:15045–15048. [DOI] [PubMed] [Google Scholar]
- 56.Falasca, M., S.K. Logan, V.P. Lehto, G. Baccante, M.A. Lemmon, and J. Schlessinger. 1998. Activation of phospholipase Cγ by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J. 17:414-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bae, Y.S., L.G. Cantley, C.S. Chen, S.R. Kim, K.S. Kwon, and S.G. Rhee. 1998. Activation of phospholipase C-γ by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273:4465–4469. [DOI] [PubMed] [Google Scholar]
- 58.Djouder, N., U. Prepens, K. Aktories, and A. Cavalie. 2000. Inhibition of calcium release-activated calcium current by Rac/Cdc42-inactivating clostridial cytotoxins in RBL cells. J. Biol. Chem. 275:18732–18738. [DOI] [PubMed] [Google Scholar]
- 59.Moores, S.L., L.M. Selfors, J. Fredericks, T. Breit, K. Fujikawa, F.W. Alt, J.S. Brugge, and W. Swat. 2000. Vav family proteins couple to diverse cell surface receptors. Mol. Cell. Biol. 20:6364–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu, J., D.G. Motto, G.A. Koretzky, and A. Weiss. 1996. Vav and SLP-76 interact and functionally cooperate in IL-2 gene activation. Immunity. 4:593–602. [DOI] [PubMed] [Google Scholar]
- 61.Fu, C., C.W. Turck, T. Kurosaki, and A.C. Chan. 1998. BLNK: a central linker protein in B cell activation. Immunity. 9:93–103. [DOI] [PubMed] [Google Scholar]
- 62.Wienands, J., J. Schweikert, B. Wollscheid, H. Jumaa, P.J. Nielsen, and M. Reth. 1998. SLP-65: a new signaling component in B lymphocytes which requires expression of the antigen receptor for phosphorylation. J. Exp. Med. 188:791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]