

Abstract
Antigen recognition by clonotypic B cell receptor (BcR) is the first step of B lymphocytes differentiation into plasmocytes. This B cell function is dependent on efficient major histocompatibility complex (MHC) class II–restricted presentation of BcR-bound antigens. In this work, we analyzed the subcellular mechanisms underlying antigen presentation after BcR engagement on B cells. In quiescent B cells, we found that MHC class II molecules mostly accumulated at the cell surface and in an intracellular pool of tubulovesicular structures, whereas H2-M molecules were mostly detected in distinct lysosomal compartments devoid of MHC class II. BcR stimulation induced the transient intracellular accumulation of MHC class II molecules in newly formed multivesicular bodies (MVBs), to which H2-M was recruited. The reversible downregulation of cathepsin S activity led to the transient accumulation of invariant chain–MHC class II complexes in MVBs. A few hours after BcR engagement, cathepsin S activity increased, the p10 invariant chain disappeared, and MHC class II–peptide complexes arrived at the plasma membrane. Thus, BcR engagement induced the transient formation of antigen-processing compartments, enabling antigen-specific B cells to become effective antigen-presenting cells.
Keywords: antigen presentation, multivesicular body, lysosome, B lymphocyte, antigen receptor
Introduction
The interaction of antigens with specific B cell antigen receptors (BcRs)* initiates the antibody response by inducing the proliferation and differentiation of B cells and the interaction of B cells with antigen-specific CD4+ T cells. The expression of clonotypic BcR is an important feature of B cells because the binding of antigen to this receptor triggers antigen presentation at very low antigen concentrations whereas the fluid phase uptake of similar low concentrations of antigen does not (1). The components of BcR account for the specificity of antigen presentation in B cells. Surface Ig (sIg) binds antigen, and a noncovalently associated heterodimer, Ig-α/Ig-β, couples the receptor to cytosolic effectors that trigger the uptake of antigen and a signaling cascade leading to B cell activation (2). Thus, BcR facilitates the endocytosis of soluble antigens and their transport to endosomal/lysosomal compartments (3). The antigens are then degraded into peptides, which associate with MHC class II molecules for presentation to T lymphocytes.
The loading of MHC class II molecules with antigenic peptides occurs in distinct compartments of the endocytic pathway, which can be identified based on ultrastructural morphology and protein content. Historically, MHC class II compartments (MIICs) were first described in human B cell lines as multilaminar (late MIICs) or multivesicular prelysosomal compartments (early MIICs; references 4 and 5) containing lysosomal proteins (β hexosaminidase, cathepsin D, lysosomal-associated membrane protein [Lamp] 1, and CD63). Subcellular fractionation studies then identified specialized compartments, such as MHC class II vesicules (CIIV) in A20 mouse B lymphoma cells (6). CIIVs are more closely related to endosomes than to lysosomes because they contain transferrin receptors and are devoid of lysosomal markers. Recent data have reconciled these observations by showing that the accumulation of MHC class II in these two different types of processing compartment is differentially regulated during dendritic cell (DC) maturation (7). In immature DCs, MHC class II accumulates in MIICs, together with H2-M and invariant chain fragments. In LPS-induced mature DCs, mature MHC class II molecules have been detected on their way to the cell surface, in CIIV devoid of H2-M and invariant chain, but rich in costimulatory molecules (7). The low level of invariant chain fragments in mature DCs was attributed to the upregulation of cathepsin S activity during DC maturation (8), suggesting that the content of endolysosomal compartments may be regulated by cell activation.
Recent data have suggested that class II compartments in B cells may also be modified after BcR engagement (9). Indeed, antigen-bound BcR are redistributed at the plasma membrane with lipid microdomains (rafts), which remain associated with BcR during its transport to antigen-processing compartments containing MHC class II molecules (10). BcR stimulation also modifies the structural features of MIICs by enlarging class II- and Lamp 1-positive vesicles in B cells (11). Thus, the binding of antigen to BcR may have a major effect on the membrane microenvironment surrounding BcR in the endocytic pathway and may change the nature and composition of MHC class II–containing compartments.
These data raise questions about how BcR engagement leads to efficient MHC class II–restricted antigen presentation in B cells. We addressed this issue using the mouse B lymphoma cell lines IIA1.6 and A20. Unlike EBV-transformed human B cells, which are constitutively activated by viral proteins, these cells have the phenotype of nonactivated mature B cells, in which MHC class II molecules mostly accumulate at the cell surface and in a tubulovesicular network of endosomal-like vesicles similar to CIIV, as previously described in IIA1.6 and A20 B cells (6, 12).
We found that BCR stimulation induced the downregulation of cathepsin S activity and the intracellular accumulation of class II–invariant chain complexes. MHC class II molecules were thus redistributed — together with Ii, antigen-bound sIg, and H2-M — to newly formed late endosomal multivesicular compartments similar to MIIC. These dynamic changes in the endocytic MHC class II pathway were transient and reversible because, 24 h later, MHC class II were mostly detected in CIIV-like compartments and cathepsin S activity had returned to its initial level. Thus, BcR seem to integrate signaling and antigen-targeting functions, thereby modulating the composition and nature of the endocytic compartments of B cells. Dynamic changes in MIICs are therefore a major consequence of BcR engagement and constitute an original mechanism leading to the selective and efficient presentation of BcR-bound antigens.
Materials and Methods
Cells, Chemical Reagents, and Antibodies.
IIA1.6 cells, A20 cells expressing anti-DNP IgM, and 24.4 Hybidoma T cells were cultured in RPMI 1640, 10% FCS, 1% penicillin-streptomycin, 0.1% β-mercaptoethanol, and 2% sodium pyruvate. The IIA1.6 B lymphoma cell line is an FcγR-deficient variant of A20 B cells (13). G418 (1 mg/ml) was added in the culture medium of A20 cells. A20 IgM anti-DNP cells are A20 B lymphoma cells that have been transfected with genomic clones encoding the light chain and the heavy μ chain specific for the hapten, DNP (14). 24.4 T cells are specific for a λ repressor peptide (12–24) presented by IAd MHC class II molecules (15). IIA1.6 cells expressing IAκ or IAβ were obtained by transfection of cells with the corresponding cDNAs encoding the IA α and β chains (gifts from R. Germain, National Institutes of Health, Bethesda, MD), which were inserted into SR-driven expression vectors bearing neomycin and hygromycin resistance genes, respectively (15). Cells were cultured with 500 μg/ml hygromycin and 1 mg/ml G418, in 96-well plates, and positive clones were identified by cytofluorometry with Y3P or 11.5.2 mAbs specific for the b and k IA alleles, respectively. RPMI 1640, FCS, PBS, penicillin, streptomycin, sodium pyruvate, and l-glutamine were purchased from GIBCO BRL. All other chemicals used in this study were obtained from Sigma-Aldrich.
The antibodies used were a rabbit anti-serum specific for the cytoplasmic domain of the IA α chain and a rabbit anti-serum specific for the cytoplasmic tail of the Ii chain (IiNH2) (a gift from J. Davoust, Centre D'Immunologie de Marseille Luminy, Marseille, France). Anti–H2-M antiserum was obtained by immunizing rabbits with a synthetic peptide corresponding to the cytoplasmic tail of the β chain. Igs were then obtained by affinity purification. For immunogold labeling, MHC class II molecules were detected with the rat anti–class II mAb M5/114 (16). In some experiments, the M5114 mAb was conjugated to biotin. The other antibodies used were anti-Lamp1 mAb (BD PharMingen), anti-β tubulin mAb (Sigma-Aldrich), rabbit anti-biotin antiserum, rabbit anti-fluorescein antiserum (Molecular Probes), F(ab′)2 fragments of goat anti–mouse IgG anti-serum (Cappel), and F(ab′)2 fragments of donkey anti–goat IgG antiserum (Jackson ImmunoResearch Laboratories). Horseradish peroxidase–coupled and AP-coupled goat anti–rabbit IgG antiserum were obtained from Jackson ImmunoResearch Laboratories. Rabbit anti-cathepsin S antiserum was a gift from H. Chapman, Harvard University, Boston, MA; rabbit anti-Ig-α antiserum was a gift from J. Cambier (National Jewish Hospital, Denver, CO) and rat anti–H2-M mAb (2C3A) was a gift from L. Karlssonn (La Jolla, San Diego, CA). The rabbit anti-rab5 and anti-rab7 antisera were gifts from P. Chavrier (Institut Curie, Paris, France). OVA or λ repressor was coupled to DNP (10 molecules of DNP for 1 molecule of protein) as described previously (15).
BcR Stimulation of IIA1.6 Cells.
IIA1.6 cells and A20 IgM anti-DNP cells were stimulated with multivalent ligands of BcR. F(ab′)2 fragments of goat anti–mouse IgG antiserum (10 μg/ml) and F(ab′)2 fragments of donkey anti–goat IgG antiserum (20 μg/ml) were mixed in RPMI media and incubated at 37°C for 30 min to facilitate the formation of multivalent anti–mouse IgG complexes. FACS® analysis showed that these complexes bound specifically to the mouse mIgG of IIA1.6 cells or the IgM of A20 cells (data not shown). IIA1.6 cells or A 20 cells were stimulated by incubation with preformed multivalent ligands at 37°C for 30 min in culture medium and were then washed three times in PBS. A20 IgM anti-DNP cells were stimulated in similar conditions with 10 μg/ml of DNP-coupled OVA or λ repressor. The cells were either analyzed directly or subjected to further chase periods for various lengths of time in complete culture medium. For electron microscopy, IIA1.6 cells were stimulated with F(ab′)2 fragments of goat anti–mouse IgG antiserum complexed with FITC-coupled F(ab′)2 fragments of donkey anti–goat IgG antiserum, whereas A20 IgM anti-DNP cells were stimulated with DNP-coupled OVA.
Cell Fractionation and Immunoblotting.
Cells were fractionated on Percoll gradients as described previously (17). Briefly, 3 × 107 IIA1.6 cells were washed once with PBS, collected by centrifugation and resuspended in homogenization buffer (10 mM triethanolamine, 10 mM acetic acid, 1 mM EDTA, and 250 mM sucrose, pH 7.4) at a density of 3 × 107 cells per milliliter and passed through a ball-bearing homogenizer 8–10 times. Intact cells and nuclei were removed by centrifugation at 3,000 g for 10 min. The postnuclear supernatant (500 μl) was mixed with homogenization buffer and Percoll to give 5 ml of a 22% Percoll solution, which was then centrifuged at 33,000 rpm for 30 min in a Beckman ultracentrifuge, using a TLA-100.4 rotor. Fractions were collected from the bottom of the gradient. β-hexosaminidase and alkaline phosphodiesterase (APDE) enzymological assays were performed as described previously (18) to identify the subcellular fractions containing lysosomes and plasma membranes, respectively. Briefly, 75 μl of each fraction was incubated with 100 μl of the APDE substrate for 1 h at 37°C; a colorimetric assay was then performed in which absorbance was measured at 405 nm. For the β hexosaminidase assay, 5 μl of each fraction was incubated for 30 min with 50 μl of the enzyme substrate buffer. The reaction was stopped by adding 2 ml of stop buffer and the amount of enzyme was determined by fluorimetry (Hoefer) at an excitation wavelength of 365 nm and an emission wavelength of 450 nm. The content of each fraction was also determined by Western blotting with specific anti-rab5, anti-rab7, anti–H2-M, and anti-Lamp1 antibodies. We quantified MHC class II and invariant chain in experiments investigating the redistribution MHC class II–invariant chain complexes by pooling the fractions with β hexosaminidase or APDE activity and subjecting them to SDS-PAGE. The protein bands were blotted onto membranes, which were probed with rabbit anti-IA α chain or anti–Ii-NH2 antibodies, then with alkaline phosphatase-coupled antisera. Binding was detected by incubation at room temperature in buffer containing AP substrate (Boehringer Mannheim). Signals were detected in a Storm 860 apparatus (Molecular Dynamics) and quantified with ImageQuant software.
Antigen Presentation Assay.
In experiments assessing the stimulation of T cells by Percoll fractions, 108 A20 IgM anti-DNP cells were incubated for 30 min at 4°C with 10 μg/ml DNP-coupled λ repressor in RPMI 1640. The cells were washed and incubated at a density of 2 × 106 cells per milliliter for 30 min or 3 h at 37°C in complete medium. Cells were fractionated as described below and pools of the four fractions with β hexosaminidase or APDE activity and containing equivalent amounts of protein, as determined by colorimetric assay, were transferred to 96-well plate DVPP Multiscreen membranes (Millipore) with a 96-well vacuum transfer apparatus (Bio-Rad Laboratories). T cell stimulation was evaluated by adding 100 μl of a cell suspension containing 2 × 104 24.4 T cell hybridoma cells in complete medium to each well. Plates were incubated for 18 h at 37°C, then centrifuged for 10 min at 1,200 g. Supernatants were removed, frozen at –80°C for 2 h, and IL-2 content was then determined by CTLL-2 assay as described previously (15). Percoll fractions from A20 cells, incubated with DNP-coupled OVA, did not stimulate 24.4 T cells previously (data not shown), demonstrating the specificity of 24.4 stimulation in experiments with λ repressor (see Fig. 1).
Figure 1.

Redistribution of MHC class II–invariant chain complexes to lysosomes after BcR stimulation. (A) Percoll gradient fractionation of IIA1.6 cells and A20 cells expressing anti-DNP IgM. Cells were homogenized and the postnuclear supernatant (PNS) was fractionated on a 22% Percoll gradient. β hexosaminidase activity and alkaline phosphodiesterase activities were measured in each fraction and protein content was determined by Western blotting with anti-rab5, anti-rab7, anti-Lamp1, anti-H2-M, and anti-IAα chain antibodies. (B) Fractionation of MHC class II-containing compartments. Unstimulated and BcR-stimulated IIA1.6 cells (cells were incubated for 30 min at 37°C with anti–mouse IgG antibodies) were fractionated and β hexosaminidase and alkaline phosphodiesterase activities determined as in A. Pools of light (black bars) or dense (white bars) fractions were subjected to SDS-PAGE and MHC class II and invariant chain were quantified as described in Materials and Methods. The results shown are the means of three experiments. (C) Immunogold labeling of dense fractions containing MHC class II molecules after BCR stimulation. Membrane fractions were processed, immunogold-labeled, and contrast stained as described in Materials and Methods. (Top) The dense fractions are enriched in electron-dense compartments strongly labeled with an antibody directed against the cytoplasmic domain of IA. Internal membranes can be seen in the lumen of the electron-dense compartments. (Bottom) In the electron-dense fractions, small vesicles, 60–80 nm in diameter, are often observed. These vesicles, labeled with the M5114 antibody, probably correspond to the internal vesicles of multivesicular class II compartments. Their presence in these fractions is almost certainly due to the disruption of multivesicular bodies during fractionation. Scale bars, 100 nm. (D) DNP-coupled λ repressor was bound to A20 IgM anti-DNP cells at 4°C and the cells were incubated for 0 min, 30 min or 3 h at 37°C in complete medium. Cells were fractionated as below and we analyzed 24.4 T cell stimulation for the pools of fractions with β hexosaminidase or APDE activity, but similar amounts of proteins, by determining the IL- 2 content of the supernatant with a CTLL-2 assay.
Western Blot Analysis.
Cells were lysed by incubation in lysis buffer: 0.5% Triton, 300 mM NaCl, 50 mM Tris, pH 7.4, and 10 μg/ml leupeptin, chemostatin, aprotinin, pepstatin, and N-ethyl maleimide. Cell lysates were diluted in reducing sample buffer and boiled for 5 min before electrophoresis in 12% polyacrylamide gels containing SDS. Proteins were transferred to polyvinylidene fluoride membranes (Millipore). The membranes were incubated with blocking solution, followed by primary antibody and then horseradish peroxidase–labeled species-specific antibody. Chemiluminescence was detected with a Boehringer kit. For quantification of the p10 fragment of the invariant chain after BcR stimulation, the membranes were probed with rabbit anti–Ii-NH2 antibody, then with alkaline phosphase-coupled antiserum and binding was detected by incubation at room temperature in a buffer containing AP substrate (Boehringer Mannheim). The filters were sequentially probed with rabbit anti-IA α chain antiserum and an anti-tubulin mAb. Blots were visualized in a Storm 860 machine (Molecular Dynamics), and p10 signals were quantified (ImageQuant). Each lane was normalized on the basis of the corresponding IA or tubulin signals. Similar results were obtained with IA and tubulin normalization.
Immunoelectron Microscopy.
Cells were fixed by incubation for 2 h at room temperature in a mixture of 2% paraformaldehyde in 0.2 M phosphate buffer, pH 7.4, (PB) and 0.125% glutaraldehyde. Fixed cells were processed for ultrathin cryosectioning and immunogold labeling as described previously (5). In brief, cells were washed with PB and 50 mM glycine in PB and were then embedded in 7.5% gelatin. Small blocks were infiltrated with 2.3 M sucrose at 4°C for 2 h and then frozen in liquid nitrogen. Ultrathin cryosections were cut with a Leica ultracut FCS (Vienna, Austria) and retrieved in a mixture of 2% methylcellulose and 2.3 M sucrose (vol/vol). The sections were indirectly immunogold-labeled with the rat mAb anti-I-A M5114, followed by a rabbit anti–rat antibody (Dako) and various rabbit polyclonal antisera. Bound antibodies were detected with protein A coupled to 10- and 15-nm gold particles (purchased from J.W. Slot, Utrecht University, Utrecht, The Netherlands). The sections were contrast stained, embedded in a mixture of methylcellulose and uranyl acetate and viewed with a CM120 Twin Phillips electron microscope (Eindhoven) (19). The distribution of MHC class II in the various compartments of IIA1.6 cells and A20 cells before and after stimulation was determined semiquantitatively on immunogold-labeled ultrathin cryosections by counting the number of gold particles labeling class II molecules in each of the defined compartments in 30 randomly selected cell profiles. For electron microscopy analysis, subcellular fractions were washed in PBS and loaded on formvar-carbon coated grids by incubation for 20 min. The fractions were immunogold-labeled with anti-class II antibodies (polyclonal anti-IA directed against the cytoplasmic domain of class II) or with the M5114 mAb and protein A-gold particles (10 nm diameter).
Immunofluorescence Staining and Confocal Microscopy.
Cells were washed in PBS and collected on glass coverslips coated with poly-L lysine (Sigma-Aldrich), to which they were allowed to adhere for 30 min. Cells were then fixed in 3% paraformaldehyde for 10 min at room temperature and incubated with 100 mM glycine in PBS. Cells were permeabilized by incubation in 0.05% saponin, 0.2% BSA in PBS, and incubated with the rat anti–H2-M mAb (gift from L. Karlsonn). The cells were washed three times and the binding of specific antibodies was detected using F(ab′)2 donkey anti–rat or anti–rabbit antibodies conjugated to FITC or Texas Red. The coverslips were then mounted in Mowiol. Confocal laser scanning microscopy was performed with a Leica TCS microscope, consisting of a DM microscope interfaced with a mixed gas argon/krypton laser (Leica Laser Technik) as described previously (20).
Fluorometric Test for Cathepsin Activity.
IIA1.6 cells were prepared as described above. Equal amounts of membranes (as determined by the Bradford test) in 1 M sodium acetate, 0.2 M EDTA, 0.5 M DTT, 5% CHAPS (pH 5.5 or 7.0) were incubated for 1 h at 37°C, in 96-well plates, with substrates of cathepsins: 200 mM Z-Val-Val-Arg-NHMec (a gift from P. Morton, USF, Chesterfield, MO) for cathepsin S, in the presence or absence of 1 nM N-morpholinurea–leucine–homophenylalanine vinylsulfone-phenyl (LHVS), and 80 mM Z-Phe-Arg-NHMec (Sigma-Aldrich) for cathepsins B/L. Reactions were quantified at λexc. = 355 nm and λem. = 460 nm, by fluorimetry (1420 VICTOR2™ multilabel counter; EG&G Wallac) (21). Same samples were tested for β hexosaminidase activity, as described previously (17). LHVS was provided by H. Ploegh (Harvard University, Boston, MA).
Labeling of the Active Site of Cysteine Proteases.
We detected active cysteine proteases with Mu–[125I]–Tyr–Ala–CH2F, as described elsewhere (22). Cells (107 cells per sample) were incubated with or without the inhibitor LHVS (50 nM) at 37°C for 1 h before labeling and then with 50 nM Mu–[125I]–Tyr–Ala–CH2F for 2 h at 37°C. Cells were washed twice with 1% FCS in PBS and cell lysates were prepared as above. Cell lysates containing the same amount of radioactivity were boiled for 5 min and then subjected to electrophoresis in a 12% acrylamide gel containing SDS.
Results
The mouse B lymphoma cell lines A20 and IIA1.6 were used for two major reasons: (i) they have the phenotype of quiescent mature B cells expressing sIgG, or anti-DNP IgM for A20 cells, and the intracellular machinery triggering BcR-mediated cell activation (15); and (ii) they constitute a homogeneous population of B cells, ideal for functional studies of antigen presentation and cell biological analysis of MHC class II–containing compartments (6). We aimed to analyze in detail the various cellular events occurring during BcR-induced stimulation, focusing on changes in the trafficking of MHC class II molecules and their partners leading to efficient antigen presentation.
BcR Stimulation Induced the Redistribution of MHC Class II into Dense Fractions of the Percoll Gradient.
We began by analyzing the compartmentalization of MHC class II–invariant chain complexes in IIA1.6 cells during BcR-mediated B cell activation. Late endosomes and lysosomes were purified by ultracentrifugation on a Percoll gradient. β hexosaminidase activity and Lamp1 were detected in heavy fractions corresponding to lysosomal and prelysosomal compartments whereas APDE activity, rab5, rab7, and Lamp1 were detected in light fractions corresponding to the other cell membranes (Fig. 1 A). H2-M was detected principally in the heavy fraction, but was also found in other fractions. As previously described in these B lymphoma cells, MHC class II molecules were mostly present in the light fraction (Fig. 1 A), which contained the plasma membrane, ER, and Golgi apparatus, along with endosomes and CIIV (12). In contrast, 30 min after BcR cross-linking with multivalent ligands, MHC class II and the p31 invariant chain were present not only in the light fraction, but also in the heavy lysosome-containing fractions of the Percoll gradient (Fig. 1 B), whereas no change in the intracellular distributions of rab7, rab5, H2-M, and Lamp1 was detected (data not shown). β hexosaminidase-positive fractions were pooled and their MHC class II and invariant chain contents determined: these dense fractions contained 10–12% of total class II or entire invariant chain (Fig. 1 B). A similar distribution of MHC class II was obtained with A20 anti-DNP cells stimulated with soluble DNP-coupled OVA (data not shown). Then we analyzed, by electron microscopy, the morphology of the MIICs that sedimented in the dense fractions of the Percoll gradient after the BcR stimulation of IIA1.6 cells. The pooled dense fractions consisted mostly of large vesicular structures (200–350 nm diameter) with internal membrane sheets and vesicles (Fig. 1 C). Most of the isolated compartments were labeled with an antiserum directed against the cytoplasmic domain of the IA α chain (Fig. 1 C, top). In dense fractions, we also observed small vesicular structures strongly labeled with M5/114, which recognizes the luminal domain of the IA β chain (Fig. 1 C, bottom). These vesicles probably correspond to internal vesicles of the class II-positive MVBs disrupted during washing, after fractionation. Thus, BcR stimulation caused changes in the intracellular distributions of MHC class II molecules and the invariant chain. We then determined whether MHC class II–peptide complexes were present in these MIICs after the internalization of BcR-bound antigens. A20 cells expressing anti-DNP IgM were incubated for various times with DNP-coupled λ-repressor and then fractionated on a Percoll gradient. Pools of dense fractions and of light fractions containing similar amounts of protein were lysed in 96-well plates and bound to nylon membrane. Presentation of the λ repressor was detected with the IAd-restricted T cell hybridoma: 24.4. T cell stimulation was detectable in the dense fraction after 30 min, with weaker stimulation observed in the light fraction (containing plasma membrane) at this time point (Fig. 1 D), even though most of the MHC class II molecules were found in light fractions (Fig. 1 B). After 3 h, T cell stimulation increased in both fractions (Fig. 1 D). MHC class II–peptide complexes were therefore generated in MIICs that were redistributed to dense fractions during BcR stimulation.
BcR Stimulation Induced p10 Accumulation and Downregulated Cathepsin S Activity.
Although MHC class II–peptide complexes were detected in heavy fractions of the Percoll gradient, due to the high sensitivity of T cell hybridoma most MHC class II molecules were associated with the invariant chain in lysosome-related compartments. Indeed, immunoprecipitation of heavy fractions with the anti-class II antibody, M5114, demonstrated that MHC class II were associated with entire invariant chain or with a p10 degradation fragment 3 h after BcR engagement (data not shown). Analysis of B cell lysates, at various times after BcR stimulation, showed an increase in the amount of p10 invariant chain fragment detected with a rabbit antiserum specific for the cytosolic domain of the invariant chain, whereas MHC class II and β tubulin levels remained constant (Fig. 2 A). The effect of BcR stimulation on invariant chain degradation depended on the MHC class II allele; the effect was strongest in IIA1.6 cells–expressing IAκ. In parental IIA1.6 cells expressing IAd, p10 levels doubled (data not shown), whereas they increased by a factor of 5 in IAk-transfected IIA1.6 cells (Fig. 2 A) and in IIA1.6 cells expressing IAb (data not shown). However, the changes in p10 levels over time were similar for both alleles: the p10 fragment was first detected after 30 min, increased to reach a maximum at 3 h and then returned to its initial level after 6 h of BcR stimulation (Fig. 2 A). No change in de novo MHC class II synthesis was observed in pulse-chase experiments (data not shown). Although MHC class II–peptide complexes were detected in dense fractions after BcR stimulation, the increase in p10 invariant chain fragment levels and the detection of invariant chain, together with MHC class II, in late endosomal compartments after BcR cross-linking suggests that the proteolytic degradation of invariant chain was inhibited during B cell activation.
Figure 2.

BcR stimulation downregulates cathepsin S activity in IIA1.6 cells. (A) Accumulation of the p10 invariant chain fragment in BcR-stimulated IIA1.6 cells. IIA1.6 cells were incubated for the times indicated with anti–mouse IgG antibodies, as described in Materials and Methods. The p10 fragment was detected with a rabbit antiserum specific for the cytoplasmic tail of the invariant chain. The same blot was incubated with an anti-IAα chain antiserum and then with an anti–β tubulin antibody (left). In the right panel, the p10 signal is normalized with the MHC class II signal obtained for the same lane and these p10 signals are expressed in arbitrary units. These results are representative of three independent experiments. (B) IIA1.6 cells were left unstimulated or were stimulated for various times with anti–mouse IgG antibodies and membranes were prepared to assess cathepsin S (top) β hexosaminidase (bottom right), and cathepsin B/L (bottom left) activities in fluorometric assays. Cathepsin S activity was tested at pH 7.0, because cathepsin S is functional at neutral pH, with (white bars) or without (back bars) LHVS, a cathepsin S inhibitor. Error bars correspond to the mean of three experiments. (C) Detection of cathepsin S with an iodinated peptide that binds to the active site of cathepsin S. Stimulated or unstimulated cells were incubated for 1 h with 125I-Mu-Tyr-Ala-CH2F, with or without LHVS, and cell lysates were then subjected to electrophoresis in 12% polyacrylamide gels. Cathepsin S was detected as a 26-kD band. The labeling of this protein was inhibited by adding LHVS. LHVS did not inhibit the labeling of an higher molecular weight band in the gel at ∼32 kD, which was identified as cathepsin L. Rat basophilic leukemia cell line cells, which do not produce cathepsin S, were used as a negative control. In the bottom panel, each band was quantified with a PhosphorImager™. Cathepsin S labeling was normalized against labeling of the 32-kD band for each lane and cathepsin S labeling was quantified in arbitrary units.
Therefore, we assessed various proteolytic activities during BcR-induced cell activation. IIA1.6 cells were stimulated for various times as described above, and the activities of cathepsin S, cathepsin L/B, and β hexosaminidase were assessed on the same membrane preparations by means of fluorometric assays (23). Cathepsin S activity was measured at pH 7.0 (Fig. 2 B) in membrane preparations of B cells because cathepsin S is active at this pH, whereas cathepsin B is not. We demonstrated that the activity measured corresponded to cathepsin S by adding low concentrations of LHVS. Such low concentrations (1 nM) of LHVS inhibit cathepsin S, but not cathepsin B. Higher concentrations of LHVS (30 nM) are required to inhibit cathepsin B (24). Cathepsin S activity decreased after BcR cross-linking: maximal inhibition (75%) occurred at 3 h, with cathepsin S activity progressively increasing thereafter and returning to initial values after 6 h; 18 h after BcR cross-linking, 15% inhibition of cathepsin activity was still observed. No change in β hexosaminidase activity was detected in the same samples, whereas cathepsin L/B activity decreased slightly (Fig. 2 B). We directly measured cathepsin S activity in B cells by labeling stimulated and unstimulated IIA1.6 cells with an iodinated peptide that bound specifically to the active site of cathepsin S (22). A 26-kD protein, presumably cathepsin S, was specifically labeled in these conditions and this labeling was inhibited in the presence of LHVS peptide. This assay gave similar results to the original assay, as 80% inhibition of cathepsin S labeling was observed after 3 h and this inhibition was partially reversible after 18 h (Fig. 2 C). No changes in the amounts of cathepsin S and cystatin C proteins present were detected by Western blotting (data not shown). These results demonstrate that BcR-induced B cell activation downregulated cathepsin S activity, accounting for the transient accumulation of MHC class II–invariant chain complexes in lysosomes. The changes in MIIC composition induced by B cell activation suggest that BcR stimulation may trigger morphological changes in processing compartments.
Redistribution of MHC Class II to Multivesicular Compartments after BcR Stimulation.
We analyzed, after BcR stimulation, changes in the intracellular compartmentalization of IIA1.6 cells and A20 anti-DNP cells, by performing immunogold labeling with the rat monoclonal anti-class II antibody, M5/114, on ultrathin cryosections of unstimulated and BCR-stimulated cells. As previously described for unstimulated IIA1.6 and A20 cells, only a small proportion of MHC class II molecules were detected within cells (12) (and Fig. 3 A and E). Most of the intracellular class II molecules were present in small vesicles, and in tubulovesicular structures beneath the plasma membrane (Fig. 3 A and E, and Fig. 4 B). We identified these compartments, which are probably related to CIIV (6), as endosomes because they were labelled with anti–transferrin receptor antibody but not with anti-Lamp1 antibody (12, 20). BcR stimulation of IIA1.6 cells or A20 anti-DNP cells for 30 min to 3 h radically changed the distribution of MHC class II molecules and the morphology of the class II–containing compartments. First, the amount of intracellular class II molecules increased considerably, by factor 3 in IIA1.6 cells after 3 h and by factor 6 in A20 cells after 30 min of IgM stimulation (Fig. 4 A). Second, this intracellular pool of molecules accumulated mainly in multivesicular MIICs, which were rarely observed in unstimulated IIA1.6 and A20 cells. All these modifications of MIICs were specifically related to BcR engagement because the incubation of cells with similar concentrations of irrelevant IgG complexes did not modify the nature of compartments containing MHC class II (data not shown). In these compartments, intense labeling for MHC class II was observed in the internal membrane vesicles (Figs. 3 B, C, F, and 4 B). An important feature of MHC class II redistribution to multivesicular compartments was the kinetics of this structural change in the endocytic pathway. These changes occurred rapidly, as they were detectable 10 min after BcR cross-linking (data not shown). Class II molecules were still detected in multivesicular compartments of IIA1.6 cells after 3 h (Fig. 3 C), with increasing numbers of intracellular class II molecules (Fig. 4 A and B) and an increasing number of compartments. However, MHC class II accumulation in MVBs seemed to be reversible because the number of intracellular class II molecules in IIA1.6 cells had decreased by 18 h (Fig. 4 A). As in unstimulated cells, most of the class II molecules were present in numerous tubulovesicular membrane structures distributed around the microtubule organizing center (data not shown) and beneath the plasma membrane (Figs. 3 D and 4 B). As expected (Figs. 1 and 2), the invariant chain was mostly detected together with MHC class II in the MVBs of BcR-stimulated IIA1.6 cells (Fig. 5 A), whereas the invariant chain was present mostly in the endoplasmic reticulum and Golgi apparatus before stimulation (data not shown). Similar observations were made for A20 anti-DNP cells (data not shown). These data suggest that B cell activation may modify the intracellular transport of MHC class II–invariant chain complexes, leading to the transient accumulation of these complexes in late endosomes or prelysosomal compartments.
Figure 3.
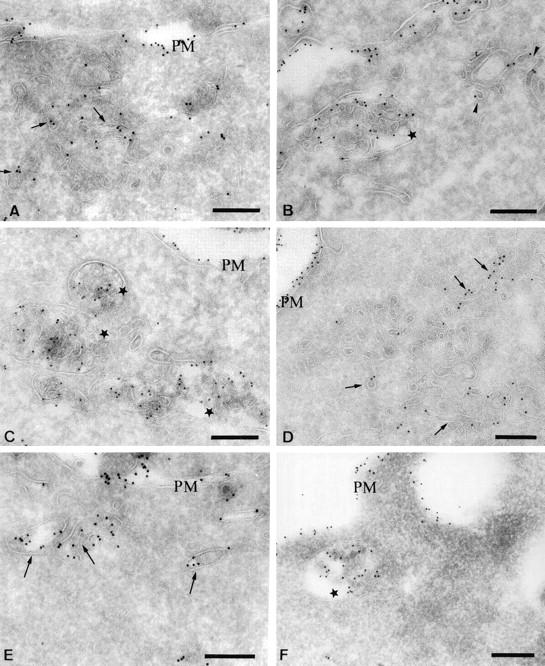
Redistribution of MHC class II molecules after BcR stimulation. Ultrathin cryosections of unstimulated IIA.6 cells (A) and A20 IgM anti-DNP cells (E) or IIA1.6 cells stimulated for 30 min (B), 3 h (C), or 18 h (D), and A20 IgM anti-DNP cells stimulated for 30 min (F) were single immunogold-labeled with the rat monoclonal M5114 antibody and protein A coupled to 10 nm gold particles (PAG 10). (A) In unstimulated cells, the majority of MHC class II molecules were present on the plasma membrane (PM) and in small tubulovesicular structures beneath the plasma membrane (arrows). (B) After 30 min of stimulation, MHC class II molecules were detected on the plasma membrane (PM), in small tubular structures (arrows) and in multivesicular bodies (stars). (C) After 3 h, note the accumulation of MHC class II molecules in numerous multivesicular bodies (stars). The plasma membrane was also labeled (PM) (D) In cells stimulated for 18 h, intracellular MHC class II molecules were present mostly in small vesicles and tubules (arrows) and on the plasma membrane (PM). (E) In unstimulated A20 IgM anti-DNP cells, MHC class II molecules were detected at the plasma membrane and in tubulovesicular structures (arrows). (F) In A20 cells stimulated with DNP-OVA for 30 min, MHC class II molecules were visualized at the plasma membrane and in multivesicular bodies (star). Bars, 200 nm.
Figure 4.

Quantitative analysis of the distribution of MHC class II molecules during BCR stimulation. (A) We counted the number of gold particles labeling intracellular class II molecules, under the electron microscope, on 30 randomly selected cell profiles. Note the increase in labeling of intracellular class II molecules after 30 min and 3 h of BcR stimulation and the slight decrease after 18 h. (B) We counted the number of gold particles labeling MHC class II molecules in various intracellular compartments (tubulovesicular structures, preMVBs, MVBs, and lysosomes), under the electron microscope, on 30 cell profiles. Results are presented as the percentage of gold particles in the various compartments.
Figure 5.
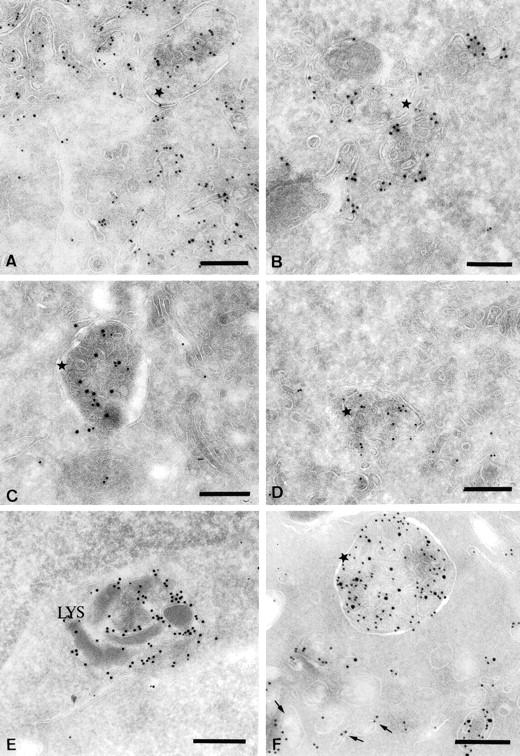
Characterization of the multivesicular compartments induced after BCR stimulation. Ultrathin cryosections of IIA1.6 cells stimulated for 30 min were double immunogold-labeled with the rat monoclonal M5.114 antibody and a polyclonal anti-invariant chain antibody (A), a polyclonal anti-FITC antibody (B) or a rat monoclonal anti-Lamp1 antibody (D). The same cells were also single immunogold-labeled with a rabbit polyclonal anti-Igα antibody (C). In D, a biotinylated M5.114 antibody was used to avoid cross-reactions. In E and F, ultrathin cryosections of unstimulated IIA1.6 cells (E) or cells stimulated for 30 min (F) were double immunogold-labeled with the M5.114 antibody (PAG 10) and a biotinylated anti–H2-M antibody detected with an anti-biotin antibody (PAG 15). (A) Note the presence of the invariant chain (PAG 15) in the class II-rich multivesicular compartments (stars). (B) The FITC-coupled Igs (PAG 15) clearly accumulate in the class II molecule-containing multivesicular compartments (PAG 10). (C) The multivesicular MIICs also contain Ig α (PAG 10) (star). (D) Both Lamp1 (PAG 15) and MHC class II (PAG 10) are present in the multivesicular MIICs. (E) In unstimulated cells, most of the H2-M was present in compartments containing internal electron-dense membranes (Lys). (F) In cells stimulated for 30 min, H2-M was detected in characteristic class II molecule-containing multivesicular compartments (star). Bars, 200 nm.
To determine the precise nature and position in the endocytic pathway of the BcR-induced MIICs, we labeled ultrathin cryosections of stimulated-IIA1.6 cells. We clearly identified these multivesicular compartments as a site of antigen processing, based on the accumulation of multivalent antigens and the BcR subunit, together with MHC class II molecules recognized by the M5/114 antibody. Thus, 30 min after BcR stimulation with FITC-coupled F(ab′)2 fragments of anti–mouse IgG antibodies, FITC was mostly detected, with a specific anti-FITC antibody, in multivesicular class II compartments (Fig. 5 B). We also showed, using a rabbit anti–Ig-α antiserum, that Ig-α subunits accumulated in large amounts in the same compartments (Fig. 5 C). These data indicated that BcR-bound antigens entered newly formed MVBs containing MHC class II molecules. To determine the identity of these MVBs within the late endocytic pathway, ultrathin cryosections of cells stimulated for 30 min or 3 h were double immunogold-labeled with anti-class II and anti-Lamp1 antibodies (Fig. 5 D) or with anti-class II and anti-MPR antibodies (data not shown). The class II-rich MVBs contained Lamp1 (Fig. 5 D). The lower intensity of labeling for MHC class II observed in some of the double immunogold-labeling experiments results from the use of a biotinylated-M5114 antibody to prevent artifactual colabeling with other mAbs. These data demonstrate that BcR stimulation induced the formation in mouse B cells of multivesicular MIICs with the structural and biochemical features of the MIIC, previously described in human B cells.
H2-M Redistribution in BcR-induced MIIC.
We further investigated the composition of BcR-induced MIIC by analyzing the distribution of molecules involved in antigen presentation. First, we investigated the intracellular distribution of nonpolymorphic H2-M during B cell activation by BcR. Unstimulated and stimulated IIA1.6 cells were labeled with antibodies specific for H2-M and MHC class II molecules. Bound antibodies were then detected with FITC-coupled and Texas Red–coupled secondary antibodies, respectively. The immunolabeling of H2-M and MHC class II was analyzed by confocal microscopy. Before activation, the bulk of MHC class II was located at the plasma membrane, with only a minor fraction detected in intracellular compartments (Fig. 6 A). In cells with faint intracellular class II labeling, MHC class II molecules showed only partial colocalization with H2-M, as some H2-M-positive compartments were MHC class II-negative (Fig. 6 A). In a large proportion of cells, MHC class II molecules mostly accumulated at the plasma membrane whereas H2-M was mostly found in intracellular compartments previously identified as lysosomes in these cells (12). As expected from the data shown in Fig. 3, BcR engagement rapidly induced the intracellular accumulation of MHC class II in compartments labeled with specific anti–H2-M antibodies (Fig. 6 A).
Figure 6.


BcR stimulation modifies H2-M intracellular localization. (A) Intracellular accumulation of H2-M and MHC class II after BcR stimulation. IIA1.6 cells were not stimulated (top) or were stimulated (bottom) with anti–mouse IgG antibodies for 60 min. They were then fixed and processed for immunofluorescence staining. Cells were labeled with the biotinylated anti-IAd antibody MKD6 and a rabbit anti–H2-M antiserum. Bound antibody was detected with FITC-conjugated streptavidin and Texas Red–conjugated secondary antibodies, respectively. (B) Quantitative analysis of the distribution of H2-M during BCR stimulation was performed as in Fig. 4. Results are presented as the percentage of gold particles in the various compartments.
This suggests that BcR engagement modified the intracellular distribution of H2-M, inducing their redistribution from lysosomal compartments to newly formed MHC class II multivesicular compartments. No modification of H2-M expression was observed by Western blot analysis (data not shown). We further investigated the nature of the H2-M–containing compartments by immunogold labeling of ultrathin cryosections of stimulated and unstimulated IIA1.6 cells. In unstimulated cells, most H2-M was present in compartments morphologically different from MVBs (Fig. 5 E). These compartments, which we classified as lysosomes based on their morphology and high Lamp1 content, contained very few MHC class II molecules, and H2-M molecules were occasionally detected in small vesicles at the periphery of the cell (Fig. 5 F). BcR cross-linking had a major effect on the distribution of H2-M. 30 min after the start of B cell antigen stimulation, a large proportion of the intracellular H2-M accumulated in multivesicular compartments containing MHC class II (Figs. 5 F and 6 B). Thus, BcR simultaneously induced antigen targeting and the redistribution of H2-M to the MHC class II–containing compartments.
Thus, our data show that BcR stimulation induced the redistribution of MHC class II to newly formed multivesicular compartments, together with antigen-bound BcR and functional H2-M. In addition, the initial downregulation of cathepsin S activity and the dynamic changes in MIICs showed that BcR stimulation induced major changes in the endocytic pathway that could account for the unique function of BcR in antigen presentation by B cells.
Discussion
In B cells, the endosomal and lysosomal compartments are the intracellular sites of antigen degradation and peptide loading onto MHC class II (3, 6). In this study, we demonstrated that the binding of antigens to BcR profoundly changed the distribution of MHC class II molecules in the endocytic pathway of B cells. BcR cross-linking induced the de novo formation of multivesicular compartments, with morphological and biochemical features similar to those of late endosomal MIICs, containing MHC class II, H2-M, BcR, and antigens. Thus, BcR, which is known to induce B cell activation and the uptake and targeting of antigens to class II compartments, induced dynamic and reversible changes in the endocytic pathway, switching antigen presentation on B cells on and off.
How does BcR induce antigen presentation? BcR functions similarly to many other cell surface receptors. The binding of multivalent antigens to BcR results in the cross-linking of the receptor and its recruitment to clathrin-coated pits, increasing the uptake of BcR-bound antigens (25). Thus, the primary function of BcR in antigen presentation is facilitating the entry of antigens into the endocytic pathway, in which proteases degrade proteins into peptides. This proteolytic degradation of antigens is the first step in antigen processing and is necessary but insufficient to induce the efficient presentation of a large spectrum of peptides. Thus, in addition to its quantitative effect on antigen processing, BcR-mediated antigen uptake also induces the presentation of a selective set of peptides (15). In contrast, fluid-phase endocytosis of the same antigen induces the presentation of only a few such peptides. Our data also clearly show that BcR stimulation regulates the composition and nature of the endocytic compartments containing MHC class II molecules.
Before antigen stimulation, class II molecules were mainly present at the cell surface and in small tubulovesicular structures likely to correspond to the previously described CIIVs identified in the same B cell line (IIA1.6) (6). Previous data have shown that BcR engagement may modify MIICs in B cells. Several authors have reported that CIIVs and MIICs may coexist in murine B cells after BcR-mediated antigen uptake (26, 27). Other authors have shown that BCR stimulation affects the nature of MIICs (11). We showed in this study that BcR stimulation induced the formation of compartments corresponding to MVBs, containing internal membrane vesicles and Lamp1. The newly formed MIICs have two features that are important for the antigen presentation functions of B cells. First, they accumulate BCR-bound antigen, as shown previously (27). Second, these newly formed MIICs accumulate H2-M. H2-M play a key role in antigen presentation, displacing the CLIP peptide or p10 fragment, as recently proposed by H. Ploegh et al. (28), and enhancing the loading of MHC class II with antigenic peptides (29). In contrast, the detection of invariant chain in these compartments suggests that the regulation of invariant chain degradation may be an original mechanism for inducing the accumulation of MHC class II and the formation of MVBs after BcR stimulation, as the invariant chain is known to retain MHC class II in the late endocytic pathway (30).
How does BcR stimulation regulate invariant chain degradation? The intracellular retention of MHC class II–invariant chain complexes is potentially important because it induces the concentration of MHC class II, together with antigen-bound BcR, in processing compartments. The downregulation of cathepsin S activity after BCR stimulation may be essential for coordination of the transport of MHC class II and BCR-bound antigen to the same processing compartment. During the initial phase of B cell stimulation, the downregulation of cathepsin S inhibited the dissociation of invariant chain MHC class II complexes, which were retained in lysosomal compartments. MHC class II progressively concentrated together with BcR-bound antigens in MIIC. Cathepsin S activity later returned to basal levels, Ii was degraded, mature MHC class II molecules were transported to the cell surface, and the newly formed MIICs disappeared. Two lines of evidence support this model. First, higher levels of transient intracellular accumulation of MHC class II and p10 fragment were observed in IIA1.6 cells expressing class II alleles with a strong affinity for invariant chain such IAk (Fig. 2 A and data not shown). Second, no modification of class II compartments was observed in transgenic anti-hen egg lysozyme/Ii deficient mice after BcR engagement (31). In addition, we observed that MIIC formation and cathepsin S downregulation occurred simultaneously after BcR stimulation. Such modifications to the endocytic pathway and MHC class II maturation and transport have previously been observed during DC maturation (8). In immature DCs, MHC class II molecules are found with the invariant chain and H2-M in lysosomal MIICs (7). Upon inflammatory stimuli, such as LPS, the DCs mature and MHC class II, devoid of invariant chain is detected in CIIVs. The presence of invariant chain in class II compartments is related to cathepsin S activity (22). Cathepsin S activity is inhibited by cystatin C in immature DCs and maturation signals, such as LPS, downregulate cystatin C production, leading to an increase in cathepsin S activity and degradation of the invariant chain (8). In addition, there is a clear relationship between maturation status and the ability of DCs to process exogenous antigens. Immature DCs are very efficient at capturing antigens and processing them to give peptides that associate with MHC class II in MIIC-related compartments. In contrast, mature DCs express higher levels of MHC class II at the cell surface but inefficiently capture and process antigens. Therefore, the cell biology of antigen presentation seems to be very similar in B cells and DCs. In both cases, efficient antigen processing is associated with the accumulation of MHC class II–invariant chain together with H2-M and antigens in MIIC-related compartments and with the downregulation of cathepsin S activity, with no change in cystatin C production during BcR-mediated B cell activation (data not shown). Another difference from DCs is the expression of a clonotypic receptor, which initiates, in antigen-specific B cells, both cell activation and antigen processing.
This major feature of B cells raises a new question: how does BcR coordinate antigen transport and MHC class II redistribution in the endocytic pathway? Two lines of evidence suggest that the coordination of these two processes is important in BcR-induced antigen presentation. First, we found that BcR stimulation induced the redistribution of H2-M, with MHC class II, to the same multivesicular compartments as BCR-bound antigens. Second, BcR-mediated antigen presentation in A20 cells, the downregulation of cathepsin S activity and MIIC formation showed similar kinetics. MHC class II–peptide complexes were detected in newly formed MVBs after 30 min (Fig. 1 D), probably due to the loading of empty MHC class II in endosomes. However, we previously reported that cell surface antigen presentation began 2–3 h after the binding of antigen to BcR, and reached a maximum after 6–18 h (15, 20) (data not shown), when cathepsin S activity returned to basal levels. Therefore, there is a lag time of a few hours between the entry of antigens into B cells and the expression at the cell surface of MHC class II–peptide complexes. This corresponds to the time required for MIIC formation (30 min–3 h), and the disappearance of these compartments that we observed 18 h after BCR stimulation.
In the current working model for antigen targeting, BcR captures antigens at the surface of B cells, targeting BcR-bound antigen to early endosomes. In these compartments, the receptors are sorted towards preexisting late compartments of the endocytic pathway, which contain MHC class II molecules. BcR cross-linking is thought to induce the concomitant recruitment of cytosolic effectors, which turn on B cell activation, leading to dramatic changes in the form and functions of B cells. Recent data have suggested that the signaling and targeting functions of BcR are related (10, 15). An alternative model may be thus put forward. During the transport of BcR in the endocytic pathway, the recruitment of cytosolic effectors by BcR may modify the composition of BcR-linked endosomal compartments. The direct consequences of BcR-induced B cell activation would therefore include modification of the nature and content of endo/lysosomal compartments and induction of the formation of multivesicular bodies containing MHC class II. Identification of the cytosolic effectors responsible for MVB formation will make it possible to validate this model and to extend our understanding of the intracellular mechanisms underlying the dynamic changes in the endocytic pathway that turn on antigen presentation by B cells.
Acknowledgments
The authors thank Sebastian Amigorena and Claire Hivroz for critical reading of the manuscript and Danielle Tenza for her advice with electron microscopy experiments.
This work was supported by grants from Institut National de la Santé et de la Recherche Médicale, Institut Curie, and the Comité de Paris of the Ligue Nationale contre le Cancer. H. Vincent-Schneider holds a fellowship from the Ministère des Universités et de la Recherche.
Footnotes
Abbreviations used in this paper: APDE, alkaline phosphodiesterase; BcR, B cell receptor; CIIV, MHC class II vesicules; DC, dendritic cell; Lamp, lysosomal-associated membrane protein; LHVS, N-morpholinurea-leucine-homophenylalanine vinylsulfone-phenyl; MIIC, MHC class II compartment; MVB, multivesicular body; sIg, surface Ig.
References
- 1.Lanzavecchia, A. 1990. Receptor-mediated antigen uptake and its effect on antigen presentation to class II-restricted T lymphocytes. Annu. Rev. Immunol. 8:773–793. [DOI] [PubMed] [Google Scholar]
- 2.Cambier, J.C., C.M. Pleiman, and M.R. Clark. 1994. Signal transduction by the B cell antigen receptor and its coreceptors. Annu. Rev. Immunol. 12:457–486. [DOI] [PubMed] [Google Scholar]
- 3.West, M.A., J.M. Lucocq, and C. Watts. 1994. Antigen processing and class II MHC peptide-loading compartments in human B-lymphoblastoid cells. Nature. 349:147–151. [DOI] [PubMed] [Google Scholar]
- 4.Peters, P.J., J.J. Neefjes, V. Oorschot, H.L. Ploegh, and H.J. Geuze. 1991. Segregation of MHC class II molecules from MHC class I molecules in the Golgi complex for transport to lysosomal compartments. Nature. 6311:669–676. [DOI] [PubMed] [Google Scholar]
- 5.Kleijmeer, M.J., S. Morkowski, J.M. Griffith, A.Y. Rudensky, and H.J. Geuze. 1997. Major histocompatibility complex class II compartments in human and mouse B lymphoblasts represent conventional endocytic compartments. J. Cell Biol. 3:639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amigorena, S., J.R. Drake, P. Webster, and I. Mellman. 1994. Transient accumulation of new class II MHC molecules in a novel endocytic compartment in B lymphocytes. Nature. 6476:113–120. [DOI] [PubMed] [Google Scholar]
- 7.Pierre, P., S.J. Turley, E. Gatti, M. Hull, J. Meltzer, A. Mirza, K. Inaba, R.M. Steinman, and I. Mellman. 1997. Developmental regulation of MHC class II transport in mouse dendritic cells. Nature. 6644:787–792. [DOI] [PubMed] [Google Scholar]
- 8.Pierre, P., and I. Mellman. 1998. Developmental regulation of invariant chain proteolysis controls MHC class II trafficking in mouse dendritic cells. Cell. 7:1135–1145. [DOI] [PubMed] [Google Scholar]
- 9.Barois, N., F. Forquet, and J. Davoust. 1997. Selective modulation of the major histocompatibility complex class II antigen presentation pathway following B cell receptor ligation and protein kinase C activation. J. Biol. Chem. 6:3441–3447. [DOI] [PubMed] [Google Scholar]
- 10.Cheng, P.C., M.L. Dykstra, R.N. Mitchell, and S.K. Pierce. 1999. A role for lipid rafts in B cell antigen receptor signaling and antigen targeting. J. Exp. Med. 11:1549–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siemasko, K., B.J. Eisfelder, E. Williamson, S. Kabak, and M.R. Clark. 1998. Cutting edge: signals from the B lymphocyte antigen receptor regulate MHC class II containing late endosomes. J. Immunol. 11:5203–5208. [PubMed] [Google Scholar]
- 12.Brachet, V., G. Raposo, S. Amigorena, and I. Mellman. 1997. Ii chain controls the transport of major histocompatibility complex class II molecules to and from lysosomes. J. Cell Biol. 1:51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones, B., J.P. Tite, and C.A. Janeway, Jr. 1986. Different phenotypic variants of the mouse B cell tumor A20/2J are selected by antigen- and mitogen-triggered cytotoxicity of L3T4-positive, I-A-restricted T cell clones. J. Immunol. 1:348–356. [PubMed] [Google Scholar]
- 14.Watanabe, M., T.H. Watts, J. Gariepy, and N. Hozumi. 1988. Function and behavior of surface immunoglobulin receptors in antigen-specific T cell-B cell interaction. Cell. Immunol. 1:226–235. [DOI] [PubMed] [Google Scholar]
- 15.Lankar, D., V. Briken, K. Adler, P. Weiser, S. Cassard, U. Blank, M. Viguier, and C. Bonnerot. 1998. Syk tyrosine kinase and B cell antigen receptor (BCR) immunoglobulin-α subunit determine BCR-mediated major histocompatibility complex class II-restricted antigen presentation. J. Exp. Med. 188:819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhattacharya, A., M.E. Dorf, and T.A. Springer. 1981. A shared alloantigenic determinant on Ia antigens encoded by the I-A and I-E subregions: evidence for I region gene duplication. J. Immunol. 127:2488–2495. [PubMed] [Google Scholar]
- 17.Bonnerot, C., V. Briken, V. Brachet, D. Lankar, S. Cassard, B. Jabri, and S. Amigorena. 1998. syk protein tyrosine kinase regulates Fc receptor γ-chain-mediated transport to lysosomes. EMBO J. 17:4606–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green, S.A., K.P. Zimmer, G. Griffiths, and I. Mellman. 1987. Kinetics of intracellular transport and sorting of lysosomal membrane and plasma membrane proteins. J. Cell Biol. 105:1227–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raposo, G., M.J. Kleijmeer, G. Posthuma, J.W. Slot, and H.J. Geuze. 1997. Immunogold labeling of ultrathin cryosections: application in immunology. Handbook of Experimental Immunology. 5th edition. Blackwell Science, editor. Elsevier, Cambridge, MA. pp. 1–11.
- 20.Bonnerot, C., D. Lankar, D. Hanau, D. Spehner, J. Davoust, J. Salamero, and W.H. Fridman. 1995. Role of B cell receptor Ig α and Ig β subunits in MHC class II-restricted antigen presentation. Immunity. 3:335–347. [DOI] [PubMed] [Google Scholar]
- 21.Manoury, B., E.W. Hewitt, N. Morrice, P.M. Dando, A.J. Barrett, and C. Watts. 1998. An asparaginyl endopeptidase processes a microbial antigen for class II MHC presentation. Nature. 396:695–699. [DOI] [PubMed] [Google Scholar]
- 22.Driessen, C., R.A. Bryant, A.M. Lennon-Dumenil, J.A. Villadangos, P.W. Bryant, G.P. Shi, H.A. Chapman, and H.L. Ploegh. 1999. Cathepsin S controls the trafficking and maturation of MHC class II molecules in dendritic cells. J. Cell Biol. 147:775–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xing, R., A.K. Addington, and R.W. Mason. 1998. Quantification of cathepsins B and L in cells. Biochem. J. 332:499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xing, R., and R.W. Mason. 1998. Design of a transferrin-proteinase inhibitor conjugate to probe for active cysteine proteinases in endosomes. Biochem. J. 334:667–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watts, C., P.A. Reid, M.A. West, and H.W. Davidson. 1990. The antigen processing pathway in B lymphocytes. Semin. Immunol. 2:247–253. [PubMed] [Google Scholar]
- 26.Geuze, H.J. 1998. The role of endosomes and lysosomes in MHC class II functioning. Immunol. Today. 19:282–287. [DOI] [PubMed] [Google Scholar]
- 27.Drake, J.R., T.A. Lewis, K.B. Condon, R.N. Mitchell, and P. Webster. 1999. Involvement of MIIC-like late endosomes in B cell receptor-mediated antigen processing in murine B cells. J. Immunol. 162:1150–1155. [PubMed] [Google Scholar]
- 28.Villadangos, J.A., R.A. Bryant, J. Deussing, C. Driessen, A.M. Lennon-Dumenil, R.J. Riese, W. Roth, P. Saftig, G.P. Shi, H.A. Chapman, et al. 1999. Proteases involved in MHC class II antigen presentation. Immunol. Rev. 172:109–120. [DOI] [PubMed] [Google Scholar]
- 29.Alfonso, C., and L. Karlsson. 2000. Nonclassical MHC class II molecules. Annu. Rev. Immunol. 18:113–142. [DOI] [PubMed] [Google Scholar]
- 30.Cresswell, P. 1994. Assembly, transport, and function of MHC class II molecules. Annu. Rev. Immunol. 12:259–293. [DOI] [PubMed] [Google Scholar]
- 31.Zimmermann, V., P. Rovere, J. Trucy, K. Serre, P. Machy, F. Forquet, L. Leserman, and J. Davoust. 1999. Engagement of B cell receptor regulates the invariant chain-dependent MHC class II presentation pathway. J. Immunol. 162:2495–2502. [PubMed] [Google Scholar]