

Abstract
An effective type I interferon (IFN-α/β) response is critical for the control of many viral infections. Here we show that in vesicular stomatitis virus (VSV)-infected mouse embryonic fibroblasts (MEFs) the production of IFN-α is dependent on type I IFN receptor (IFNAR) triggering, whereas in infected mice early IFN-α production is IFNAR independent. In VSV-infected mice type I IFN is produced by few cells located in the marginal zone of the spleen. Unlike other dendritic cell (DC) subsets, FACS®-sorted CD11cintCD11b−GR-1+ DCs show high IFN-α expression, irrespective of whether they were isolated from VSV-infected IFNAR-competent or -deficient mice. Thus, VSV preferentially activates a specialized DC subset presumably located in the marginal zone to produce high-level IFN-α largely independent of IFNAR feedback signaling.
Keywords: IFN type I, virus infection, dendritic cell subsets, IFN regulatory factor 7, type I IFN receptor
Introduction
Type I IFNs (IFN-α/β) constitute a family of cytokines comprising in the mouse at least 11 IFN-α isoforms and one IFN-β, which were identified by their ability to protect cells against viral infection (1). For a broad spectrum of viruses a functional type I IFN response is critical for the survival of the infected host (2, 3). Beyond the direct antipathogenic activity, type I IFNs synergize with other proinflammatory stimuli to activate innate effectors such as NK cells, macrophages, and dendritic cells (DCs)* (4) and may modulate antigen presentation and the adaptive immune response (5).
The biologic activity of all IFN-α subtypes and of IFN-β is mediated by binding to the common type I IFN receptor (IFNAR), a heterodimer consisting of the α-chain (IFNAR-1) and the β-chain (IFNAR-2, references 6 and 7). IFNAR signaling activates a multitude of IFN-inducible genes whose products may drastically alter cell homeostasis; in order to impede viral replication, cell proliferation is halted and both transcription and translation are strongly reduced. High-level expression of type I IFN, that can be detrimental to the host (8, 9), is tightly regulated (10) and usually restricted to the state of acute viremia.
The molecular mechanism of type I IFN induction was intensively investigated by analyzing virus-stimulated mouse embryonic fibroblasts (MEFs) derived from gene-targeted mice deficient of factors involved in the type I IFN signaling cascade. MEFs from mice lacking IFN-β, IFNAR-1, signal transducer and activator of transcription 1, or IFN regulatory factor (IRF)-9 showed a defective IFN-α response (11–14). This suggested that after the production of early IFN-β, and possibly IFN-α4, expression of other IFN-α genes was dependent on IFNAR feedback signaling. Two members of the IRF family, IRF-3 and IRF-7, have been shown to play a key role in the sequential induction of type I IFN genes. Virus-mediated serine phosphorylation leads to IRF-3 activation and translocation to the nucleus (15). There, IRF-3 is part of an enhanceosome complex promoting the expression of IFN-β, and presumably of IFN-α4. Early type I IFN is secreted and triggers IFNAR signaling in an autocrine fashion. Among other type I IFN–induced genes, IFNAR signaling strongly upregulates IRF-7 expression (12, 13). Virus infection leads to IRF-7 activation by phosphorylation that drives the expression of the majority of IFN-α subtypes, and hence amplifies the type I IFN response. It is not known, however, which serine kinase(s) is/are responsible for the activation of IRF-3 and IRF-7. It appears that several pathways may lead to IRF-3 and IRF-7 activation. Some viruses, such as type I herpes simplex virus (HSV), induce an IFN response independent of viral replication whereas others, e.g., vesicular stomatitis virus (VSV), lose the ability to induce type I IFN upon inactivation. Furthermore, a variety of nonviral IFN inducers has been described, including prokaryotic DNA motifs (CpG-DNA), synthetic double-stranded RNA (poly[I:C]), lipopolysaccharide, and imiquimod derivatives (16, 17).
In cell culture, virtually any cell type can produce type I IFN in response to appropriate stimulation, yet, depending on the expression of pattern recognition receptors distinct cell types are stimulated by different IFN inducers. For example CpG-DNA but not poly(I:C)–treated human CD11c− DCs produce type I IFN, whereas a reverse correlation was found for CD11c+ DCs (17, 18). Accordingly, CD11c− DCs express the Toll-like receptor (TLR)-9 and CD11c+ DCs express TLR-3 (17, 19), which have been shown to be involved in the recognition of CpG or poly(I:C), respectively (20, 21). So far, the major IFN-producing cell (IPC) has not been defined in the context of infection with specific pathogens. Several studies addressed the nature of the human IPC (22–24), which eventually was defined as the subset of CD11c− plasmacytoid DCs (25, 26). There are indications that upon stimulation of mice certain macrophages (27) or DC subsets can produce type I IFN (28–30).
Considering the above information, in an acutely infected host fibroblasts presumably do not produce the majority of type I IFN. Therefore, we investigated whether the positive feedback regulation of type I IFN induction, as identified in MEFs, also plays a role in viral pathogenesis in vivo. Surprisingly, VSV- and UV-HSV–treated mice mount early IFN-α responses largely independent of IFNAR feedback signaling. We provide evidence that unlike fibroblasts, mouse IPCs can produce early IFN-α independent of IFNAR signaling. These data are discussed in the context of an extended model of IFN-α induction based on the cell type involved in the production of IFN-α.
Materials and Methods
Mice and Viruses.
IFNAR-deficient mice (IFNAR−/−) on the SV129 background (3) and IFN-β2/− mice on the Balb/c background (14) were bred under specific pathogen free (SPF) conditions. Unmutated SV129 mice (referred to as wild-type [WT]) were obtained from the SPF-breeding colony of the European Mutant Mouse Archive (EMMA), Monterotondo, Italy. C57BL/6 mice were purchased at IFFA-Credo. Experimental mouse work was done under SPF conditions in compliance with regulations of the Ministerio della Sanità, Rome, Italy (decreto ministeriale n° 15/2001-b, 3/3.1). VSV-IND (Mudd-Summers isolate) was originally obtained from D. Kolakofsky, University of Geneva, Geneva, Switzerland. VSV was grown on BHK-21 cells (31) and plaqued on Vero cells. Stocks were maintained in MEM containing 2% heat-inactivated FCS. HSV strain F originally obtained by American Type Culture Collection was grown on Vero cells. HSV was purified from cell culture supernatant by PEG 40.000 (Serva) precipitation. After several pelleting steps for 1 h at 43,000 g virus was resuspended in PBS and titrated on Vero cells. After UV irradiation (1.2 Joule/cm2) HSV inactivation was verified by plaquing on Vero cells.
Cell Culture.
WT and IFNAR−/− MEFs were derived from day 13 embryos (32) and maintained in DMEM supplemented with 10% FCS. Cell monolayers were infected with VSV at a multiplicity of infection (MOI) of 10. After 1 h of incubation at 37°C the virus suspension was replaced by DMEM 10% FCS.
Expression Analysis.
Preparation of total RNA was performed using the TRIZOL reagent (GIBCO BRL) according to the manufacturer's instructions. Tissue samples were snap-frozen and homogenized in Trizol using a Polytron (Kinematica). On cultured cell monolayers and FACS®-sorted cells Trizol was applied directly. To eliminate possible DNA contamination total RNA was incubated for 15 min at 37°C with 10 U of DNase (Boehringer Mannheim). cDNA was prepared using Superscript II (GIBCO BRL) according to the manufacturer's instructions. To estimate relative amounts of specific mRNAs, PCRs were performed with serially fivefold diluted cDNA using published primers for IFN-α (consensus primers annealing with all IFN-α subtypes) and IRF-7 (13). The mRNA content was normalized by RT-PCR analysis with GAPDH-specific primers from the same publication. The absence of contaminating DNA was verified by PCR analysis of RNA preparations not treated with reverse transcriptase.
Quantification of IFN Activity.
The IFN bioassay was based on the protection of L929 cells from the cytopathic effect (CPE) of VSV (CPE inhibition assay). To inactivate potential viral contamination, 1:5 prediluted serum samples from VSV-infected mice were UV-irradiated with 1.2 Joule/cm2. Duplicates of serially twofold diluted sera were transferred to 24 h preincubated semiconfluent monolayers of L929 cells in 96-well plates. After 24 h incubation at 37°C supernatants were removed and VSV was added at a MOI of 0.05. After 48 h incubation at 37°C, supernatants were taken away and protected cells were stained with 0.5% crystal violet in 5% formaldehyde, 50% ethanol, and 0.8% sodium chloride. After extensive washing with water and air drying, the dye was extracted from stained cells with 100 μl/well 0.1 M sodium citrate in 50% ethanol, pH 4.2, and the crystal violet content was determined on an ELISA reader at 570 nm. In the CPE inhibition assay an international mouse IFN-α/β reference standard of 100 IU/ml (Gu02–901–511; NIAID Repository) and recombinant IFN-αA standards (PBL Biomedical Laboratories) of 100, 500, 2,500, and 10,000 IU/ml were included. For IFN quantification the 50% protective serum dilution was indicative. A log25 titer corresponded to 1,000 IU IFN-αA. The contribution of IFN-β or IFN-α to the total type I IFN activity was determined by preincubating sera for 1 h with excess amounts of neutralizing anti–IFN-α (clone 4E-A1; Yamasa Shoyu) or anti-IFN-β (clone 7F-D3; Yamasa Shoyu) mAbs. Protection from CPE due to IFN-γ was formally excluded by an IFN-γ Elisa (Promega) indicating that the IFN-γ content of all samples tested was below the detection threshold of the assay.
Immunohistochemistry.
Freshly removed organs were immersed in HBSS and snapped frozen in liquid nitrogen. Tissue sections of 5-μm thickness were cut in a cryostat, placed on siliconized glass slides, air dried, fixed with acetone for 10 min, and stored at –70°C. IFN was stained using a polyclonal sheep anti–mouse IFN-α/β antiserum (PBL Biomedical Labs) or monoclonal rat anti–mouse IFN-α antibody (Serotec). The primary sheep antibody was detected with affinity purified biotinylated donkey anti–sheep Ig antibodies (Jackson ImmunoResearch Laboratories) and alkaline phosphatase labeled avidin/biotin complexes (ABC/AP Dako). The primary rat mAb was detected with affinity purified peroxidase labeled goat anti–rat Ig antibody. For signal amplification by catalyzed reporter deposition, biotinylated thyramine (33) and 3% H2O2 was added for 7 min, and covalently bound biotin was revealed by ABC/AP. Alkaline phosphatase was visualized using naphthol AS-BI (6-bromo-2-hydroxy-3-naphtholic acid-2-methoxy anilide) phosphate and new fuchsin as substrate. Endogenous alkaline phosphatase was blocked by levamisole. The dilutions of secondary antibodies were made in TBS containing 5% normal mouse serum. Incubations were done at room temperature for 30 min; TBS was used for all washing steps. Color reactions were performed at room temperature for 15 min with reagents from Sigma-Aldrich. Sections were counterstained with hemalum and coverslips mounted with glycerol and gelatin.
In Situ Hybridization of IRF-7 mRNA.
Spleens were fixed overnight in 4% paraformaldehyde in PBS and then embedded in paraffin wax. Sections were cut at 5–7-μm and mounted onto Superfrost-Plus slides (BDH). The IRF-7 riboprobe was generated by cloning a 351 bp RT-PCR product into pBluescript followed by Digoxygenin-11-UTP-labeling using the Boehringer RNA labeling kit. In situ hybridization was performed as described previously (34).
DC Subset Isolation by FACS® Sorting.
Single cell suspension of 3–5 spleens was prepared and CD11c+ cells were enriched by magnetic adsorption cell sorting (MACS®; Miltenyi Biotec). Three-color stainings were made using anti-CD11c–biotin–, anti-CD11b–FITC–, and anti-GR-1–PE–conjugated mAbs followed by streptavidin-APC (all from BD PharMingen). 4–10 × 104 cells of each subset were sorted from ∼107 cells on a MoFlo cytometer (Cytomation).
Results
Feedback Signaling Is Required for the Expression of IFN-α mRNA in VSV-infected MEFs but not in VSV-infected Mice.
Tissue culture experiments have shown previously that the expression of all IFN-α genes, except IFN-α4, was dependent on IFNAR feedback signaling in Newcastle disease virus (NDV)-stimulated MEFs (12, 13). To validate this finding under conditions of a productive virus infection, IFNAR competent (WT) and IFNAR-deficient (IFNAR−/−) MEFs were VSV-infected and IFN-α mRNA expression was monitored by RT-PCR analysis. WT fibroblasts showed elevated IFN-α mRNA levels beginning 4 h after VSV infection and peak expression between 9 h and 12 h (Fig. 1 a). Similar to NDV-stimulated MEFs, VSV-infected MEFs predominantly expressed IFN-α4 as determined by subcloning of RT-PCR products and sequence analysis of random clones (Fig. 2 a). In contrast, IFN-α upregulation was not observed in VSV-infected MEFs lacking the IFNAR (Fig. 1 a). Thus, in VSV-infected MEFs the IFN-α production is strictly dependent on IFNAR feedback signaling.
Figure 1.

VSV infection stimulates IFNAR-independent production of IFN-α in mice, but not in MEFs. (a) MEFs derived from WT or IFNAR-deficient mice (IFNAR−/−) were infected with VSV at a MOI of 10 and (b) WT and IFNAR−/− mice were intravenously injected with 2 × 108 PFU VSV. Total RNA of 106 cultured MEFs or of spleen tissue was extracted, and the mRNA content of different samples was normalized by a GAPDH-specific RT-PCR. The expression of IFN-α mRNA was monitored using consensus primers amplifying all IFN-α subtypes. The analysis was performed with serially fivefold diluted cDNA samples starting with undiluted material; spleen derived samples were fivefold prediluted.
Figure 2.
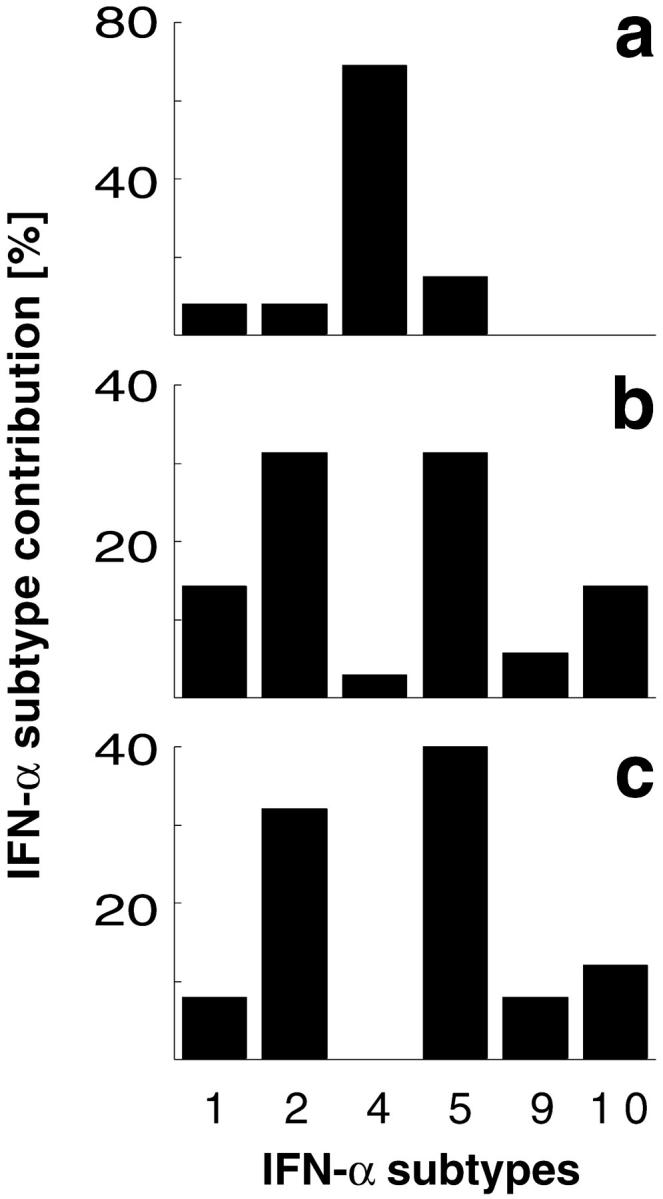
Differential IFN-α expression profiles of VSV infected MEFs and mice. PCR products of the IFN-α expression analysis 6 h after VSV infection of (a) WT MEFs, (b) spleen of WT mice, or (c) spleen of IFNAR−/− mice were subcloned and DNA sequence of 13, 35, and 25 single clones was analyzed, respectively. IFN-α sequences were classified according to EMBL database entries. The sequence termed IFN-α10 corresponds to the IFN-α/β sequence available on the EMBL database (GenBank/EMBL/DDBJ accession no. L38698).
To next examine IFN responses in vivo, IFN-α gene expression was analyzed in spleen from WT and IFNAR−/− mice infected with VSV. Surprisingly, IFN-α mRNA was induced rapidly and to a similar extent in spleen of WT and IFNAR−/− mice. At later time points, IFN-α expression in spleen of IFNAR−/− mice decreased slightly, whereas spleen from WT mice showed a further increase in IFN-α expression (Fig. 1 b). The IFN-α subtype analysis revealed that both VSV-infected IFNAR-competent and -deficient mice showed abundant expression of IFN-α2 and 5, whereas IFN-α4 was found only rarely (Fig. 2 b and c). Thus, different IFN-α patterns found after VSV infection of MEFs and mice suggested that in vivo the majority of IFN-α is produced by a cell type different from fibroblasts. Moreover, similar IFN-α patterns found in WT and IFNAR−/− mice indicated a largely feedback-independent production of early IFN-α in VSV-infected mice.
Early IFNAR-independent and Late IFNAR-dependent IFN-α Expression in VSV- and UV-HSV–treated Mice.
To correlate IFN-α mRNA levels and IFN serum activities, blood serum of VSV-infected mice was taken and analyzed in a CPE inhibition assay. As early as 4 h after VSV infection IFN activity was found in the serum of both WT and IFNAR−/− animals. Specific inhibition of IFN-α and/or IFN-β by neutralizing mAbs in the CPE inhibition assay revealed a major contribution of IFN-α to the serum IFN activity, irrespective of whether IFNAR signaling was functional or not. Serum IFN titers of WT animals increased more rapidly than IFNAR−/− mice eventually leading to ∼30-fold higher IFN titers in WT animals than in IFNAR−/− mice (Fig. 3 a).
Figure 3.

Type I IFN activity in the serum of mice treated with VSV, poly(I:C), or UV-HSV. WT mice (black circles) and IFNAR−/− (white triangles) mice were intravenously stimulated with (a) 2 × 108 PFU VSV, (b) 100 μg poly(I:C), or (c) 2 × 107 PFU UV-HSV. Mice were bled at the indicated time points and serum IFN titers were determined in a CPE protection assay. A log2 of 5 corresponds to 1,000 IU of IFN-α. The contribution of IFN-α or IFN-β to the total IFN activity was determined by specific inhibition with mAbs.
Since VSV replication is not controlled in IFNAR-deficient mice, a highly elevated virus load and increased cell death could have interfered with cytokine production in IFNAR−/− mice. Thus, we next analyzed type I IFN responses after stimulation with nonreplicating virus. Since UV-inactivated VSV does not induce type I IFN responses (data not shown), mice were injected with UV-inactivated HSV. 2 h after the treatment, WT and IFNAR−/− mice showed similar peak IFN activity with ∼50 and 35% IFN-α contribution, respectively. While the IFN response was short-lived in IFNAR−/− mice, IFN titers in WT mice were sustained for several hours, and a switch to a more pronounced IFN-α expression was found (Fig. 3 c). To verify the induction of early IFN-α in the absence of IFNAR-feedback signaling, mice deficient of the IFNAR or IFN-β were intercrossed and IFNAR−/− IFN-β2/− double-knockout mice and IFNAR+/− IFN-β2/− littermates were stimulated with UV-HSV. Even in the absence of IFN-β initial IFN-α titers showed the same magnitude in receptor deficient and competent mice. At later time points IFN-α production was sustained and further enhanced in IFN receptor competent mice as compared with IFNAR−/− mice (Fig. 4). Thus, upon treatment with live VSV- or UV-inactivated HSV early IFNAR-independent IFN-α production is observed. Yet, at later time points IFNAR-feedback signaling is critical to sustain the production of IFN-α.
Figure 4.

IFN-α is produced in mice deficient of IFN-β and IFNAR. IFN-β–deficient mice (black diamonds) and double-deficient mice lacking IFN-β and IFNAR (white diamonds) were intravenously injected with 2 × 107 PFU UV-HSV. Sera were taken at indicated time points and analyzed in a CPE protection assay. Mean IFN activity of three mice per time point is indicated. Results are shown of one out of two independent experiments. Note that the indicated IFN serum activities exclusively derived form IFN-α because the analyzed mice were IFN-β deficient.
Early IFN-α Production in the Absence of IRF-7 Induction.
The transcription factor IRF-7 has been shown to be upregulated upon IFNAR triggering (12, 13) and was proposed to be critically required for the expression of IFN-α subtype genes (12, 35). Therefore, we asked whether early IFNAR-independent IFN-α was produced in the absence of IRF-7 induction. The RT-PCR analysis of IRF-7 mRNA levels in VSV-infected WT and IFNAR−/− mice revealed a prompt IRF-7 upregulation only in WT mice but not in IFNAR-deficient mice (Fig. 5). These experiments indicated that in VSV-infected mice early IFN-α was induced in the absence of detectable IRF-7 upregulation. At later time points IFNAR-independent IRF-7 upregulation was observed (data not shown) which, however, did not suffice to promote a sustained production of IFN-α in IFNAR−/− mice.
Figure 5.

VSV infected IFNAR−/− mice show an early induction of IFN-α before the onset of IRF-7 expression. Total RNA from spleen of IFNAR−/− or WT mice was isolated 3 h after intravenous infection with 2 × 108 PFU VSV. RT-PCR was performed as described in Fig. 1.
Poly(I:C) Treatment of Mice Induces Late IFN-α Production.
To further study the role of feedback signaling with a nonreplicating type I IFN inducer, mice were treated with poly(I:C). Sera of poly(I:C)–treated WT and IFNAR−/− mice displayed early peak IFN activity, followed by a rapid decline in the case of IFNAR−/− mice. In WT mice IFN activity was sustained at a high level for >20 h. Neutralization of IFN-α and/or IFN-β revealed an abundant contribution of IFN-β to early type I IFN activity in both WT and IFNAR−/− mice. At later time points, strong IFN-α production was observed in WT but not in IFNAR-deficient mice (Fig. 3 b).
Preferential production of IFN-β after poly(I:C) treatment, while mainly IFN-α is produced after virus stimulation, indicates that different cell types may be involved in the production of poly(I:C)– versus virus-induced type I IFN. Moreover, these results further support the hypothesis that the sustained production of IFN-α is IFNAR dependent.
VSV Infection but not Poly(I:C) Treatment Stimulates High-Level Type I IFN Production in Cells Located in the Marginal Zone of the Spleen.
To localize IPCs, mice were infected with VSV and 9 h later, spleens were analyzed immunohistochemically with type I IFN-specific antibodies. Consecutive sections were stained with polyclonal antibody directed against type I IFN or with an IFN-α–specific mAb. While staining with the polyclonal serum appeared more sensitive and decorated a structure surrounding follicles, i.e., the marginal zone, stainings with mAb revealed distinct type I IPCs scattered in groups within the marginal zone (Fig. 6 a and b). After treatment with VSV or UV-HSV, spleens of WT and IFNAR−/− mice showed similar marginal zone stainings with polyclonal antibody, albeit staining of spleens from IFNAR-deficient mice was somewhat weaker (Fig. 6 c). Despite comparable IFN levels in the serum, no specific type I IFN staining was detected in the marginal zone of poly(I:C)–treated mice (Fig. 6 c). Thus, after injection of virus particles, irrespective of whether they are replicative or not, type I IFN production is localized to the marginal zone. The soluble inducer poly(I:C) did not induce high enough type I IFN production in the spleen to be detected by immunohistology. Probably under these conditions type I IFN is produced at a low level by numerous cell types and in various tissues.
Figure 6.

VSV and UV-HSV injection, but not poly(I:C) stimulation, leads to high-level production of type I IFN by cells located in the marginal zone of the spleen. (a) WT mice were VSV infected and after 9 h spleens were analyzed immunohistochemically with a polyclonal serum against type I IFN (pAb, top two panels) or with an IFN-α–specific mAb (mAb, bottom two panels). (b) A higher magnification of the marginal zone area of the bottom right panel in (a) stained with mAb is shown. (c) WT and IFNAR−/− mice were VSV, poly (I:C), or UV-HSV injected. After 9 h and 6 h after UV-HSV stimulation, spleens were analyzed immunohistochemically with a polyclonal serum against type I IFN. Sections in (a–c) were counterstained with hemalum (blue) to visualize lymph follicles. (d) In situ hybridization of IRF-7 mRNA (blue) was performed on paraffin sections of spleen from WT and IFNAR−/− mice prepared 9 h after VSV infection.
In situ hybridization of spleen sections of untreated mice, with an IRF-7 probe, did not reveal evidence for constitutive IRF-7 expression, neither in few marginal zone cells nor anywhere else in the spleen (data not shown). After VSV infection of WT mice a strong IRF-7 induction was observed all over the spleen. In contrast, no IRF-7 induction was detected in spleens of VSV-infected IFNAR−/− mice (Fig. 6 d). Together with the above RT-PCR data these observations indicate that virus-induced early IFN-α can be produced independent of IFNAR signaling and IRF-7 upregulation.
Identification of the Major IPC.
The phenotype of the major IPC in VSV pathophysiology is not known. Since the marginal zone is a complex tissue containing specialized macrophages, DCs, endothelial cells, and nonrecirculating B cells, colocalization studies with markers for marginal zone cells could not further resolve the nature of mouse IPCs (data not shown). In analogy to the DC origin of human and mouse IPCs (25, 26, 29, 30) we aimed for the analysis of mouse DC subsets. Mice were VSV-infected and, 9 h later, several CD11c+ DC subsets were FACS®-sorted and analyzed for the IFN-α mRNA content (Fig. 7 a). Surprisingly, only CD11cintCD11b−GR-I+ DCs (fraction D) were strongly positive for IFN-α mRNA, whereas neither lymphoid DCs (CD11c+CD11b−, fraction A), myeloid DCs (CD11c+CD11b+, fraction B), CD11cint CD11b+ DCs (fraction E), nor granulocytes (CD11c+GR-1+, fraction F) showed high IFN-α expression (Fig. 7 b). CD11cint CD11b− DCs (fraction C) showed slightly enhanced IFN-α mRNA levels (which were at least 125-fold lower than fraction D), that might be due to contaminating cells of fraction D. We next asked whether CD11cintCD11b−GR-I+ cells are producers of IFN-α independent of IFNAR feedback signaling. For this purpose DC subsets were isolated from VSV-infected IFNAR−/− mice. Again only CD11cint CD11b−GR-I+ cells showed high-level IFN-α mRNA (Fig. 7 c). These results indicate that although other DC subsets are able to produce IFN-α upon in vitro stimulation (28), they do not play a major role in IFN-α production after VSV infection in vivo.
Figure 7.
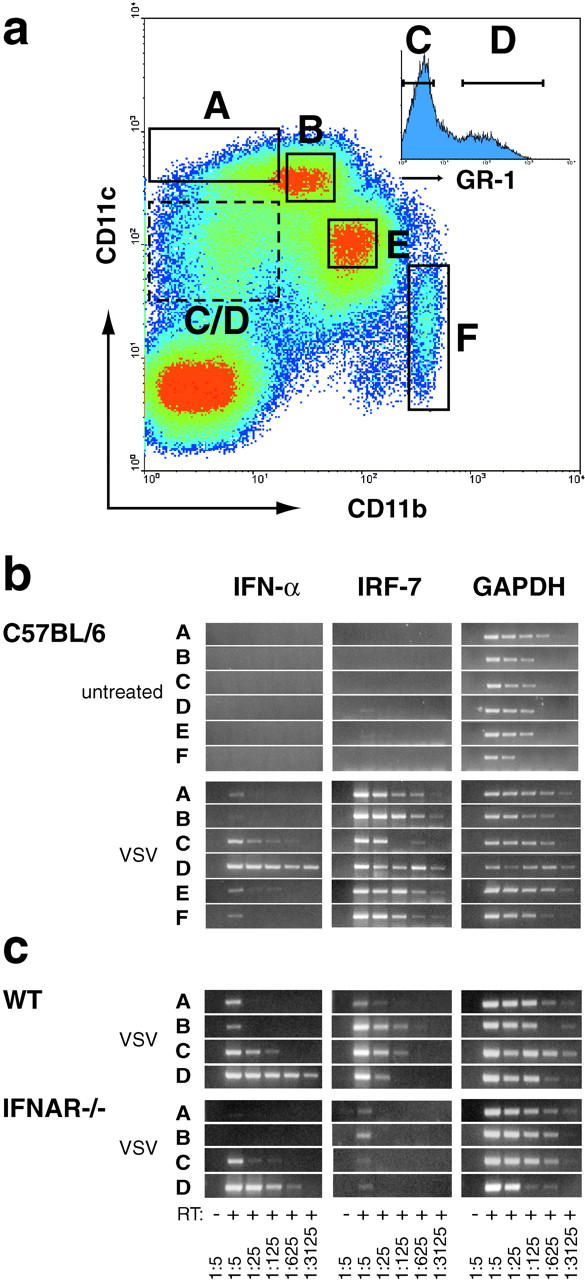
In VSV-infected mice CD11cintCD11b−GR-1+ DCs express high-level IFN-α. (a) Mice were intravenously infected with 2 × 108 PFU VSV and 9 h later spleens were removed to prepare single cell suspensions. MACS®-enriched CD11c+ cells were stained with anti-CD11c-Biot./Str.-APC, anti-CD11b-FITC, and anti-GR-1-PE, and DC subsets were FACS®-sorted using the indicated gates A–F. Cells from gate C/D were further subdivided by separating GR-1− (fraction C) and GR-1+ cells (fraction D). Sorted fractions were derived from spleen of (b) VSV infected or untreated C57BL/6 mice, and of (c) VSV-infected Sv129 (WT) and IFNAR−/− mice. Total RNA of ∼4–10 × 104 sorted cells was prepared and analyzed by RT-PCR as described in Fig. 1.
Discussion
Here we show that after viral infection of mice not only IFN-β but also IFN-α is produced immediately and largely independent of IFNAR feedback signaling. This is in contrast to the current model of IFNAR feedback-dependent expression of IFN-α, that was established based on in vitro data. To reconcile the previous in vitro data with our in vivo observations, we hypothesized that after viral infection of mice a cell type different from fibroblasts, i.e., the murine IPC, produced the majority of type I IFN, and that in these cells IFN-α expression was regulated differently than in fibroblasts. Indeed, we found that after in vivo infection the subset of CD11cint.GR-1+ DCs produced IFN-α at high level, and that early IFN-α production by this cell type was largely independent of IFNAR feedback.
The positive feedback regulation of IFN-α genes was first observed in NDV-infected MEFs that lack IFN-α expression in the absence of a functional IFN signaling cascade (11, 12). One study showed IFNAR-independent IFN-α4 expression in NDV-infected MEFs (13). Interestingly, IFN-α4 was previously found to dominate IFN-α responses of NDV-stimulated L929 fibroblasts (36). However, MEFs infected with VSV (Fig. 1), or vaccinia virus (37) showed a strictly IFNAR-dependent expression of all IFN-αs. Equally, Sendai virus-infected MEFs from IFN-β–deficient mice were found unable to produce any IFN-α, unless stimulated by the addition of exogenous IFN-β (14).
In contrast to these in vitro data, we found that VSV-infected mice mount an early IFNAR-independent IFN-α response. Furthermore, IFN-β–deficient and IFN-β/IFNAR double-deficient mice treated with UV-HSV still expressed substantial IFN-α levels (Fig. 4). While IFN-α production in IFN-β2/− mice could have been promoted by priming with natural type I IFN, produced pathogen independently (38), IFN-α production in IFN-β/IFNAR double-deficient mice indicated that IFNAR triggering was not a prerequisite for early IFN-α production. IFN-α levels reached in IFN-β2/− were sufficient to control intravenous VSV infection (data not shown). In contrast, IFN-β–deficient mice infected peripherally with vaccinia virus showed a markedly increased susceptibility to lethal disease (37): after intranasal infection, IFN-β2/− mice showed up to 105-fold increased virus titers in the lung as compared with WT mice, and eventually succumbed to the infection. Interestingly, in these experiments virus titers in spleen of WT and IFN-β2/− mice were comparably low, indicating that upon local infection vaccinia virus replication was better controlled in the spleen than in the peripherally infected organ. Thus, it appears that depending on the route of infection virus may activate IPCs or other susceptible cell types that differ in their requirement of IFNAR feedback for the IFN-α induction.
Apart from the route of infection also the nature of the stimulus may determine the cell type(s) involved in type I IFN production. Our histology data show that poly(I:C) does not induce marginal zone IPCs to produce high-level type I IFN, suggesting that the observed IFN titers were contributed mainly by nonIPCs. It is possible that in analogy to the human plasmacytoid DCs, mouse IPCs express TLR-9 but not TLR-3 that is involved in poly(I:C) recognition (19, 21), and thus mouse IPCs are not triggered by poly(I:C). IFN titers in poly(I:C)–treated mice showed an early peak of IFN-β expression that only in animals with a functional receptor feedback was followed by sustained IFN-α expression. This indicated that similar to in vitro VSV-infected MEFs, IFN-α expression by nonIPCs in vivo required IFNAR feedback signaling.
In the current model of positive feedback regulation, IFNAR-dependent upregulation of the transcription factor IRF-7 plays a central role. Virus-treated mutant MEFs that failed to induce IRF-7 also lacked expression of IFN-α. The IFN-α response could be restored by ectopic expression of IRF-7 (12). In IFNAR-deficient mice, we observed early IFN-α responses in the absence of detectable IRF-7 upregulation. One possibility is that under these conditions low constitutive IRF-7 levels suffice to drive early IFN-α expression. In this context it is worth noting that in all RT-PCR experiments, unlike other DC subsets, CD11cintCD11b−GR-1+ DCs (fraction D) showed some background IRF-7 expression already in uninfected WT mice (Fig. 7 b). Alternatively, IPCs might be able to utilize an IRF-7–independent pathway for IFN-α expression, as suggested by the recent finding, that ectopic expression of IRF-5 can substitute for IRF-7 to allow expression of certain IFN-α genes (39). Ultimately, only the generation and analysis of IRF-7–deficient mice can define the role of IRF-7 in the expression of virus-induced IFN-α in vivo.
Our immune histological analysis of VSV-infected mice revealed that IPCs are predominantly located in the marginal zone. Similarly, we and others have found the same IPC distribution in UV-HSV–treated mice (27, 40). The importance of the marginal zone for pathogen surveillance was demonstrated by the scavenger function of marginal zone cells, which can retain microscopically small particles, including pathogens, from the blood (41). Mice that were experimentally depleted of macrophages in the marginal zone (42), and osteopetrotic (op) mutant mice that are M-CSF deficient and lack the same macrophage populations, showed an increased sensitivity to pathogen infection (43). Thus, in order to be stimulated early in the course of an infection it appears “reasonable” that IPCs are located in the marginal zone. From histological colocalization studies it had been concluded that MOMA-1+ metallophilic and ERTR-9+ marginal zone macrophages might be involved in type I IFN production (27). Since we could not observe a strict correlation of MOMA-1 and ERTR-9 versus IFN-α/β stainings, and after recent evidence that the human and mouse IPC are plasmacytoid DCs (25, 26, 29, 30), we FACS®-sorted several DC subsets from spleen of VSV-infected mice and analyzed them for IFN-α mRNA expression. These experiments indicated that the subset of CD11cintCD11b−GR-1+ DCs showed high IFN-α mRNA levels. Interestingly, we did not find increased levels of IFN-α message in myeloid or lymphoid DCs (fractions A and B, respectively) isolated from VSV-infected mice, as could have been expected form recent in vitro data (28).
In conclusion, we suggest to extend the current model of IFNAR feedback dependent expression of IFN-α as follows. (i) In vivo, local infections may stimulate the production of mainly IFN-β by nonIPCs to protect surrounding tissues in a paracrine fashion. These cells produce IFN-α IFNAR dependently, i.e., only if sufficient autocrine type I IFN stimulation is provided. (ii) Systemic infection and acute viremia that require a generalized antiviral state may stimulate IPCs to produce IFN-α early and at high-level independent of IFNAR feedback signaling; however, IFNAR feedback defines the magnitude and duration of sustained IFN-α production.
To our knowledge, we present the first set of ex vivo data defining IPCs in viral pathogenesis. Our results support the concept that highly specialized DCs, located in the marginal zone, produce type I IFN upon pathogen contact. The observation that early IFN-α production of mouse IPCs is largely independent of IFNAR signaling indicates a less stringently regulated IFN-α expression in IPCs as compared with other cell types. Further experiments will reveal, whether, depending on the tissue tropism and the stimulation mechanism, different pathogens may activate discrete cell populations to produce type I IFN, and whether mouse IPCs do play a role in antigen processing and presentation. A better understanding of the role of IPCs in viral pathophysiology can have important diagnostic and therapeutic implications, as exemplified by publications showing reduced IPC counts in HIV-infected AIDS patients (44, 45).
Acknowledgments
We thank M. Huth, L. Vlk, N. Wey, and E. Perlas for technical assistance, T. Hayden and H. Kohler for expert cell sorting, S. Weiss for providing IFN-β–deficient mice, G. Schönrich and M. Raftery for providing HSV, M. Ferrantini and F. Belardelli for showing us the method of the CPE inhibition assay, K. Rajewsky for helpful suggestions, and P. Seiler and P. Aichele for critical comments on the manuscript.
This work was supported in part by the Max Planck Research Award to K. Rajewsky. The Basel Institute for Immunology was founded and supported by F. Hoffmann-La Roche AG, Basel, Switzerland.
Footnotes
Abbreviations used in this paper: CPE, cytopathic effect; CpG, prokaryotic DNA motif; DC, dendritic cell; HSV, type I herpes simplex virus; IFNAR, type I IFN receptor; IPC, IFN-producing cell; IRF, IFN regulatory factor; MEF, mouse embryonic fibroblast; MOI, multiplicity of infection; NDV, Newcastle disease virus; poly(I:C), synthetic double-stranded RNA; SPF, specific pathogen free; TLR, Toll-like receptor; VSV, vesicular stomatitis virus; WT, wild-type.
References
- 1.Isaacs, A., and J. Lindenmann. 1957. Virus interference. I. The interferon. Proc. R. Soc. Lond. 147:258–267. [PubMed] [Google Scholar]
- 2.Gresser, I., M.G. Tovey, C. Maury, and M.T. Bandu. 1976. Role of interferon in the pathogenesis of virus diseases as demonstrated by the use of anti-interferon serum. II. Studies with herpes simplex, Moloney sarcoma, vesicular stomatitis, Newcastle disease, and influenza viruses. J. Exp. Med. 144:1316–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muller, U., U. Steinhoff, L.F. Reis, S. Hemmi, J. Pavlovic, R.M. Zinkernagel, and M. Aguet. 1994. Functional role of type I and type II interferons in antiviral defense. Science. 264:1918–1921. [DOI] [PubMed] [Google Scholar]
- 4.Cella, M., M. Salio, Y. Sakakibara, H. Langen, I. Julkunen, and A. Lanzavecchia. 1999. Maturation, activation, and protection of dendritic cells induced by double-stranded RNA. J. Exp. Med. 189:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le Bon, A., G. Schiavoni, G. D'Agostino, I. Gresser, F. Belardelli, and D.F. Tough. 2001. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity. 14:461–470. [DOI] [PubMed] [Google Scholar]
- 6.Uze, G., G. Lutfalla, and I. Gresser. 1990. Genetic transfer of a functional human interferon α receptor into mouse cells: cloning and expression of its cDNA. Cell. 60:225–234. [DOI] [PubMed] [Google Scholar]
- 7.Lutfalla, G., and G. Uze. 1994. Structure of the murine interferon α/β receptor-encoding gene: high-frequency rearrangements in the interferon-resistant L1210 cell line. Gene. 148:343–346. [DOI] [PubMed] [Google Scholar]
- 8.Akwa, Y., D.E. Hassett, M.L. Eloranta, K. Sandberg, E. Masliah, H. Powell, J.L. Whitton, F.E. Bloom, and I.L. Campbell. 1998. Transgenic expression of IFN-α in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J. Immunol. 161:5016–5026. [PubMed] [Google Scholar]
- 9.Gresser, I. 1997. Wherefore interferon? J. Leukoc. Biol. 61:567–574. [DOI] [PubMed] [Google Scholar]
- 10.Wathelet, M.G., C.H. Lin, B.S. Parekh, L.V. Ronco, P.M. Howley, and T. Maniatis. 1998. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-β enhancer in vivo. Mol. Cell. 1:507–518. [DOI] [PubMed] [Google Scholar]
- 11.Harada, H., M. Matsumoto, M. Sato, Y. Kashiwazaki, T. Kimura, M. Kitagawa, T. Yokochi, R.S. Tan, T. Takasugi, Y. Kadokawa, et al. 1996. Regulation of IFN-α/β genes: evidence for a dual function of the transcription factor complex ISGF3 in the production and action of IFN-α/β. Genes Cells. 1:995–1005. [DOI] [PubMed] [Google Scholar]
- 12.Sato, M., N. Hata, M. Asagiri, T. Nakaya, T. Taniguchi, and N. Tanaka. 1998. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 441:106–110. [DOI] [PubMed] [Google Scholar]
- 13.Marie, I., J.E. Durbin, and D.E. Levy. 1998. Differential viral induction of distinct interferon-α genes by positive feedback through interferon regulatory factor-7. EMBO J. 17:6660–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erlandsson, L., R. Blumenthal, M.L. Eloranta, H. Engel, G. Alm, S. Weiss, and T. Leanderson. 1998. Interferon-β is required for interferon-α production in mouse fibroblasts. Curr. Biol. 8:223–226. [DOI] [PubMed] [Google Scholar]
- 15.Sato, M., N. Tanaka, N. Hata, E. Oda, and T. Taniguchi. 1998. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-β gene. FEBS Lett. 425:112–116. [DOI] [PubMed] [Google Scholar]
- 16.Megyeri, K., W.C. Au, I. Rosztoczy, N.B. Raj, R.L. Miller, M.A. Tomai, and P.M. Pitha. 1995. Stimulation of interferon and cytokine gene expression by imiquimod and stimulation by Sendai virus utilize similar signal transduction pathways. Mol. Cell. Biol. 15:2207–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bauer, M., V. Redecke, J.W. Ellwart, B. Scherer, J.P. Kremer, H. Wagner, and G.B. Lipford. 2001. Bacterial CpG-DNA triggers activation and maturation of human CD11c−, CD123+ dendritic cells. J. Immunol. 166:5000–5007. [DOI] [PubMed] [Google Scholar]
- 18.Kadowaki, N., S. Antonenko, and Y.J. Liu. 2001. Distinct CpG DNA and polyinosinic-polycytidylic acid double-stranded RNA, respectively, stimulate CD11c- type 2 dendritic cell precursors and CD11c+ dendritic cells to produce type I IFN. J. Immunol. 166:2291–2295. [DOI] [PubMed] [Google Scholar]
- 19.Kadowaki, N., S. Ho, S. Antonenko, R. de Waal Malefyt, R.A. Kastelein, F. Bazan, and Y.-J. Liu. 2001. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 194:863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hemmi, H., O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsumoto, K. Hoshino, H. Wagner, K. Takeda, and S. Akira. 2000. A Toll-like receptor recognizes bacterial DNA. Nature. 408:740–745. [DOI] [PubMed] [Google Scholar]
- 21.Alexopoulou, L., A.C. Holt, R. Medzhitov, and R.A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature. 413:732–738. [DOI] [PubMed] [Google Scholar]
- 22.Sandberg, K., M.L. Eloranta, A. Johannisson, and G.V. Alm. 1991. Flow cytometric analysis of natural interferon-α producing cells. Scand. J. Immunol. 34:565–576. [DOI] [PubMed] [Google Scholar]
- 23.Ferbas, J.J., J.F. Toso, A.J. Logar, J.S. Navratil, and C.R. Rinaldo, Jr. 1994. CD4+ blood dendritic cells are potent producers of IFN-α in response to in vitro HIV-1 infection. J. Immunol. 152:4649–4662. [PubMed] [Google Scholar]
- 24.Svensson, H., A. Johannisson, T. Nikkila, G.V. Alm, and B. Cederblad. 1996. The cell surface phenotype of human natural interferon-α producing cells as determined by flow cytometry. Scand. J. Immunol. 44:164–172. [DOI] [PubMed] [Google Scholar]
- 25.Siegal, F.P., N. Kadowaki, M. Shodell, P.A. Fitzgerald-Bocarsly, K. Shah, S. Ho, S. Antonenko, and Y.J. Liu. 1999. The nature of the principal type 1 interferon-producing cells in human blood. Science. 284:1835–1837. [DOI] [PubMed] [Google Scholar]
- 26.Cella, M., D. Jarrossay, F. Facchetti, O. Alebardi, H. Nakajima, A. Lanzavecchia, and M. Colonna. 1999. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 5:919–923. [DOI] [PubMed] [Google Scholar]
- 27.Eloranta, M.L., and G.V. Alm. 1999. Splenic marginal metallophilic macrophages and marginal zone macrophages are the major interferon-α/β producers in mice upon intravenous challenge with herpes simplex virus. Scand. J. Immunol. 49:391–394. [DOI] [PubMed] [Google Scholar]
- 28.Hochrein, H., K. Shortman, D. Vremec, B. Scott, P. Hertzog, and M. O'Keeffe. 2001. Differential production of IL-12, IFN-α, and IFN-γ by mouse dendritic cell subsets. J. Immunol. 166:5448–5455. [DOI] [PubMed] [Google Scholar]
- 29.Nakano, H., M. Yanagita, and M.D. Gunn. 2001. Cd11c+b220+gr-1+ cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J. Exp. Med. 194:1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asselin-Paturel, C., A. Boonstra, M. Dalod, I. Durand, N. Yessaad, C. Dezutter-Dambuyant, A. Vicari, A. O'Garra, C. Biron, F. Briere, and G. Trinchieri. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Imm. 2:1144–1150 [DOI] [PubMed] [Google Scholar]
- 31.McCaren, L.C., J.J. Holland, and J.T. Syverton. 1959. The mammalian cell-virus relationship. I. Attachment of poliovirus to cultivated cells of primate and non-primate origin. J. Exp. Med. 109:475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Torres, R.M., and R. Kühn. 1997. Laboratory protocols for conditional gene targeting. Oxford University Press, Oxford. 1–167 pp.
- 33.Bobrow, M.N., T.D. Harris, K.J. Shaughnessy, and G.J. Litt. 1989. Catalyzed reporter deposition, a novel method of signal amplification. Application to immunoassays. J. Immunol. Methods. 125:279–285. [DOI] [PubMed] [Google Scholar]
- 34.Neubuser, A., H. Koseki, and R. Balling. 1995. Characterization and developmental expression of Pax9, a paired-box-containing gene related to Pax1. Dev. Biol. 170:701–716. [DOI] [PubMed] [Google Scholar]
- 35.Yeow, W.S., W.C. Au, Y.T. Juang, C.D. Fields, C.L. Dent, D.R. Gewert, and P.M. Pitha. 2000. Reconstitution of virus-mediated expression of interferon α genes in human fibroblast cells by ectopic interferon regulatory factor-7. J. Biol. Chem. 275:6313–6320. [DOI] [PubMed] [Google Scholar]
- 36.Hoss-Homfeld, A., E.C. Zwarthoff, and R. Zawatzky. 1989. Cell type specific expression and regulation of murine interferon α and β genes. Virology. 173:539–550. [DOI] [PubMed] [Google Scholar]
- 37.Deonarain, R., A. Alcami, M. Alexiou, M.J. Dallman, D.R. Gewert, and A.C. Porter. 2000. Impaired antiviral response and α/β interferon induction in mice lacking β interferon. J. Virol. 74:3404–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taniguchi, T., and A. Takaoka. 2001. A weak signal for strong responses: interferon-α/β revisited. Nat. Rev. Mol. Cell. Biol. 2:378–386. [DOI] [PubMed] [Google Scholar]
- 39.Barnes, B.J., P.A. Moore, and P.M. Pitha. 2001. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon α genes. J. Biol. Chem. 276:23382–23390. [DOI] [PubMed] [Google Scholar]
- 40.Eloranta, M.L., K. Sandberg, and G.V. Alm. 1996. The interferon-α/β responses of mice to herpes simplex virus studied at the blood and tissue level in vitro and in vivo. Scand. J. Immunol. 43:356–360. [DOI] [PubMed] [Google Scholar]
- 41.Matsuno, K., H. Fujii, and M. Kotani. 1986. Splenic marginal-zone macrophages and marginal metallophils in rats and mice. Cell Tissue Res. 246:263–269. [DOI] [PubMed] [Google Scholar]
- 42.Seiler, P., P. Aichele, B. Odermatt, H. Hengartner, R.M. Zinkernagel, and R.A. Schwendener. 1997. Crucial role of marginal zone macrophages and marginal zone metallophils in the clearance of lymphocytic choriomeningitis virus infection. Eur. J. Immunol. 27:2626–2633. [DOI] [PubMed] [Google Scholar]
- 43.Guleria, I., and J.W. Pollard. 2001. Aberrant macrophage and neutrophil population dynamics and impaired Th1 response to Listeria monocytogenes in colony-stimulating factor 1-deficient mice. Infect. Immun. 69:1795–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Howell, D.M., S.B. Feldman, P. Kloser, and P. Fitzgerald-Bocarsly. 1994. Decreased frequency of functional natural interferon-producing cells in peripheral blood of patients with the acquired immune deficiency syndrome. Clin. Immunol. Immunopathol. 71:223–230. [DOI] [PubMed] [Google Scholar]
- 45.Soumelis, V., I. Scott, F. Gheyas, D. Bouhour, G. Cozon, L. Cotte, L. Huang, J.A. Levy, and Y.J. Liu. 2001. Depletion of circulating natural type 1 interferon-producing cells in HIV-infected AIDS patients. Blood. 98:906–912. [DOI] [PubMed] [Google Scholar]