

Abstract
It is widely accepted that immunoglobulin (Ig)E triggers immediate hypersensitivity responses by activating a cognate high-affinity receptor, FcεRI, leading to mast cell degranulation with release of vasoactive and proinflammatory mediators. This apparent specificity, however, is complicated by the ability of IgE to bind with low affinity to Fc receptors for IgG, FcγRII and III. We have addressed the in vivo significance of this interaction by studying IgE-mediated passive systemic anaphylaxis in FcγR-deficient mice. Mice deficient in the inhibitory receptor for IgG, FcγRIIB, display enhanced IgE-mediated anaphylactic responses, whereas mice deficient in an IgG activation receptor, FcγRIII, display a corresponding attenuation of IgE-mediated responses. Thus, in addition to modulating IgG-triggered hypersensitivity responses, FcγRII and III on mast cells are potent regulators of IgE-mediated responses and reveal the existence of a regulatory pathway for IgE triggering of effector cells through IgG Fc receptors that could contribute to the etiology of the atopic response.
Keywords: systemic anaphylaxis, Fc receptor, immunoglobulin E, mast cell, gene targeting
The anaphylaxis reaction in mice has been considered to be a typical immediate hypersensitivity response determined primarily by the activation of mast cells via antigen-induced aggregation of an IgE-sensitized high-affinity receptor for IgE (FcεRI),1 causing the release of potent systemic mediators (1, 2). The central role of FcεRI in mediating the response was demonstrated by observations that mice deficient in this receptor fail to undergo IgE-dependent, passive cutaneous (3) and passive systemic anaphylaxis (4). These results were interpreted as indicating a necessary and sufficient role for FcεRI in mediating the IgE-dependent anaphylactic response, excluding the possibility for involvement of other potential receptors for IgE (5). However, earlier observations indicated that the low-affinity Fc receptors for IgG (FcγRIIB and FcγRIII) on mouse mast cells, macrophages, and the rat mucosal type mast cell RBL-2H3 can bind IgE immune complexes in vitro (6, 7), and the engagement of FcγRIIB/III with IgE immune complexes triggers C57.1 mast cells to release serotonin (6), suggesting a greater potential complexity to the IgE-mediated anaphylactic response.
Studies on active anaphylaxis in gene-targeted mice further challenged the simple model of IgE and FcεRI as the sole initiators of anaphylaxis and revealed a critical role for IgG and FcγR in this response. Induction of active anaphylaxis in mice deficient in IgE indicated that IgE antibodies were not essential for the expression of systemic anaphylaxis (8). In addition, mice deficient in FcεRI mounted an undiminished active systemic anaphylactic response, whereas active sensitization and challenge of animals deficient in the common γ chain (FcRγ−/−) resulted in protection (9, 10). Further support for the conclusion that type I immediate hypersensitivity has a significant dependence on IgG1 and FcγRs came from studies demonstrating that FcγRIIB-deficient (FcγRIIB−/−) mice exhibited an enhanced reaction in IgG1-mediated passive cutaneous anaphylaxis, thereby establishing the importance of FcγRIIB as an inhibitory receptor under physiologic conditions (11), as suggested previously in extensive in vitro studies by Daëron and colleagues (12, 13; for review see reference 14).
Although the evidence supporting a direct role for IgG and FcγRs in the anaphylaxis reaction is compelling, the contribution of these receptors to the canonical IgE-mediated response is generally considered to be minimal. To directly analyze the roles of FcγRIIB and FcγRIII in the IgE-dependent component of the systemic anaphylaxis reaction, we compared the responses elicited in FcγRIIB−/− and FcγRIII−/− mice upon passive transfer of either anti-TNP IgE or IgG followed by intravenous challenge with TNP-OVA. As expected, FcγRIIB−/− and FcγRIII−/− mice displayed enhanced or attenuated systemic anaphylaxis to IgG1 sensitization, respectively. However, contrary to the accepted dogma, intense modulation of IgE-dependent systemic anaphylaxis was also observed in these FcγR−/− mice as a result of the low-affinity interactions of IgE–antigen complexes with these receptors. These studies demonstrate the in vivo physiological significance of low-affinity IgE interactions with FcγRs and represent a novel regulatory pathway for classical type I hypersensitivity responses.
Materials and Methods
Antibodies.
Rat anti–mouse FcγRIIB/III (2.4G2; PharMingen) and mouse anti-TNP IgE (IGELa2; American Type Culture Collection) and anti-TNP IgG1 (G1; 15) were purified from the ascites of hybridomas by ion exchange chromatography on DEAE– cellulose (Merck) (16) and by affinity isolation with protein G column (17), followed by removal of aggregated materials by ultracentrifugation at 130,000 g for 90 min at 20°C.
Animals.
All experiments were performed on 6–12-wk-old mice. Male and female FcγRIIB−/− (11) or FcγRIII−/− mice (Y. Ishikawa, J.V. Ravetch, and T. Takai, unpublished results) were generated by breeding the F2 offspring of crosses between chimeras and C57BL/6 mice, and the wild-type mice generated by the same breeding protocol were used as wild-type animals. FcγR−/− mice were generated as described previously (3) and back-crossed to C57BL/6 background over six generations. FcγRIII−/− mice were generated using RW4 embryonic stem cells (GenomeSystems Inc.) as described previously (3, 11). Mice were housed in cages in cabinets supplied with high efficiency particulate-free air and were monitored monthly as specific pathogen free.
Induction of Passive Systemic Anaphylaxis.
Mouse IgG1 or IgE anti-TNP mAbs were administered intravenously through the tail vein in volumes of ∼200 μl/mouse. 30 min after injection of anti-TNP IgG1 or 24 h after injection of IgE, mice were injected with 1.0 mg i.v. TNP4-OVA in PBS. Control mice received OVA in PBS instead. The concentration of IgG1 and IgE mAbs used for passive sensitization and the amount of TNP-OVA used for challenge was determined based on preliminary dose–response experiments required to produce significant drops in body temperature in wild-type and FcγRIIB−/− or FcγRIII−/− mice. Alternatively, systemic anaphylaxis was induced by the intravenous injection of 10 μg 2.4G2 in 200 μl PBS. The amount was determined based on the preliminary dose–response experiment in the same way described above. In a blocking experiment in FcγRIII−/− mice, 100 μg 2.4G2 was administered.
Monitoring of Rectal Temperature and Heart Rate.
Changes in core body temperature associated with systemic anaphylaxis were monitored by measuring changes in rectal temperature using a rectal probe coupled to a digital thermometer (Natsume Seisakusyo Co.) as described (4, 9, 10). Heart rate was recorded as electrocardiograms (Nihon Kohden) of mice under 2,2,2-tribromoethanol (0.25 mg/g body weight, i.p.) anesthesia.
Flow Cytometric Analysis.
Bone marrow–derived cultured mast cells (BMMC) were prepared as described previously (3). For monitoring of upregulation of FcεRI protein on BMMC membrane, cells were cultured in the presence of 0.1 or 5 μg/ml biotinylated IgE or 5 μg/ml biotinylated 2.4G2 for 4 d before final staining with biotinylated IgE (5 μg/ml) plus PE-conjugated streptavidin. Peritoneal resident cells were collected by washing with Tyrode's buffered solution and incubated with 5 μg/ml IgE for 20 min at 4°C to saturate IgE binding to FcεRI, followed by staining with FITC-conjugated rat anti–mouse IgE (Serotec Ltd.) for 20 min at 4°C. Flow cytometric analyses were performed with FACSCalibur™ (Becton Dickinson), and peritoneal mast cells were sorted as c-kit and IgE-positive cells as described (18).
ELISA Determinations for Blood Histamine.
Blood was collected from subocular plexus of mice into microcentrifuge tubes containing EDTA on ice at 5 min after antigen challenge, and plasma was prepared. Histamine in the plasma samples was quantified using ELISA plates (ICN Pharmaceuticals, Inc.) according to the manufacturer's instructions.
Histological Study.
Mice were killed by cervical dislocation. Their tissues were removed and fixed in 10% (vol/vol) neutral buffered formalin and then embedded in paraffin. The specimens were sectioned at 3 μm and stained with toluidine blue at pH 4.0. The number of mast cells/mm2 was determined under a light microscope. A ‘degranulated' mast cell was defined as a cell showing extrusion of >10% cell granules.
Statistical Analysis.
Statistical differences were calculated using Student's t test or Fisher's test. P < 0.05 was considered significant.
Results and Discussion
Modulation of IgG1-mediated Systemic Anaphylaxis in FcγRIIB−/− or FcγRIII−/− Mice.
Bocek et al. (7) reported that coclustering of FcγRIIB and FcγRIII on RBL-2H3 cells did not lead to stimulation of the cells, suggesting a possible inhibitory role of FcγRIIB in this process. In addition, in vitro observations by Daëron et al. (12) demonstrated that mast cell secretory responses triggered by FcεRI may be controlled by FcγRIIB/III. Moreover, the regulatory role of FcγRIIB was also observed in the cellular activation process via B cell receptors (19–21) and T cell receptors (13; for review see reference 14). Our previous studies using gene-targeted mice had demonstrated the role of FcγRIIB in modulating IgG1-mediated passive cutaneous anaphylaxis (11). To establish the generality of those in vivo observations, we investigated IgG1-mediated passive systemic anaphylaxis in FcγRIIB−/− and FcγRIII−/− mice. We chose to evaluate a passive rather than active model in our studies because FcγRIIB− /− mice display enhanced humoral immune responses (11) that could complicate the comparison and interpretation of the anaphylactic responses. To elicit the anaphylactic response, mice were injected intravenously with IgG1 specific for TNP, followed by intravenous administration of TNP-OVA 30 min later. Fig. 1 A shows that FcγRIIB− /− mice developed an enhanced IgG1-dependent passive systemic anaphylactic response as compared with passively sensitized wild-type controls challenged with TNP-OVA. In wild-type mice, the decrease in core temperature was also transient, reaching a nadir ∼15 min after induction, whereas the drop in temperature of FcγRIIB− /− mice persisted for more than 30 min without returning to baseline.
Figure 1.
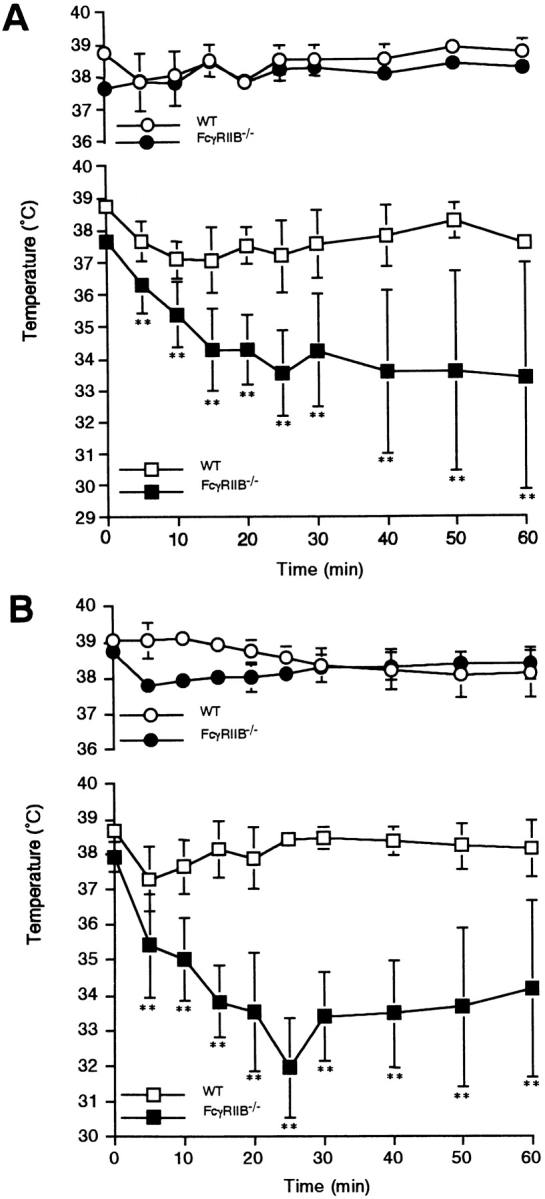

IgG1-mediated or 2.4G2-induced systemic anaphylaxis in FcγRIIB−/− or FcγRIII−/− mice. (A) Changes in rectal temperature of mice during IgG1-induced systemic anaphylaxis. 10 wild-type (□) and 8 FcγRIIB−/− animals (▪) received 200 μg i.v. anti-TNP IgG1. All of the animals received 1.0 mg i.v. TNP4-OVA 30 min later. Six additional wild-type (○) as well as FcγRIIB−/− mice (•) received 200 μg IgG1 and then 1.0 mg OVA as controls. The monitoring of rectal temperature was started at the time of antigen injection. Data are shown as mean ± SD. **P < 0.01. (B) Changes in rectal temperature in response to intravenous injection of 10 μg rat mAb 2.4G2 in 14 wild-type mice (□) and 8 FcγRIIB−/− mice (▪). As controls, five wild-type (○) as well as four FcγRIIB−/− mice (•) received 10 μg normal rat IgG. Data are shown as mean ± SD. **P < 0.01. (C) Changes in rectal temperature during IgG1- induced systemic anaphylaxis in three FcRγ−/− (•), five FcγRIII−/− (▪), or three wild-type mice (□). For the induction, mice received 400 μg i.v. anti-TNP IgG1 and then received 4.0 mg i.v. TNP4-OVA 30 min later. Data are shown as mean ± SD. *P < 0.05; **P < 0.01, compared with wild-type mice.
The mAb 2.4G2 is specific for the extracellular domains of murine FcγRIIB and FcγRIII (22). 2.4G2 induces a degranulative response in BMMC, which is enhanced in cells derived from FcγRIIB−/− mice (11). This enhancement is apparent in vivo as well as shown in Fig. 1 B, where the decrease in core temperature after administration of 2.4G2 was more pronounced in FcγRIIB−/− mice than in control mice. These results indicate that FcγRIIB on effector cells, such as mast cells, inhibits the systemic anaphylaxis elicited via FcγRIII. In contrast to the enhanced responses in FcγRIIB−/− mice described above (Fig. 1, A and B), both FcγRIII−/− mice and FcRγ−/− mice failed to develop IgG1-mediated passive systemic anaphylaxis (Fig. 1 C), directly establishing that IgG1-mediated anaphylaxis is triggered through FcγRIII, as was indirectly suggested by others (9, 10).
Enhancement of IgE-mediated Anaphylaxis in FcγRIIB−/− Mice.
As IgE immune complexes can bind with low affinity to FcγRII and III in vitro, we next induced passive systemic anaphylaxis upon anti-TNP IgE adoptive transfer and TNP-OVA administration into FcγRIIB−/− mice. IgE-mediated systemic anaphylaxis was significantly enhanced in FcγRIIB−/− mice, as assessed by changes in core temperature (Fig. 2 A), heart rate (Fig. 2 B), and augmented hemorrhage in the ileum villi (Fig. 2 C). These results indicate that IgE/FcεRI-mediated anaphylaxis is facilitated by the deletion of FcγRIIB in vivo without any apparent involvement of IgG-immune complexes.
Figure 2.
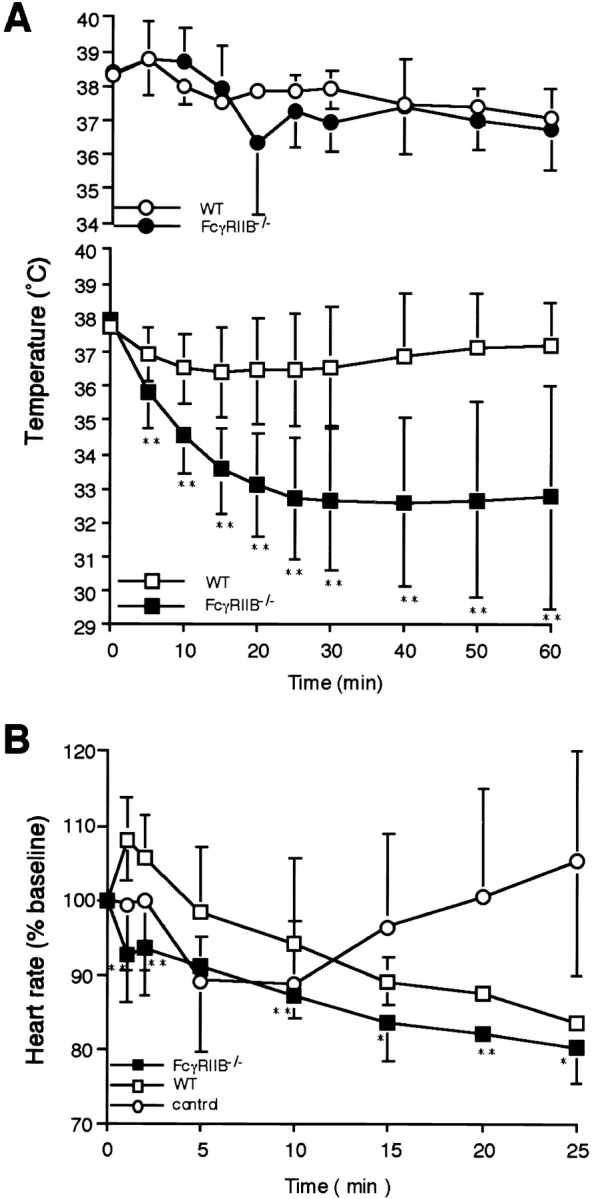

IgE-mediated systemic anaphylaxis in FcγRIIB−/− mice. (A) Changes in rectal temperature during IgE-mediated systemic anaphylaxis. 29 wild-type (WT; □) and 24 FcγRIIB−/− animals (▪) received 20 μg i.v. anti-TNP IgE. All of the animals received 1.0 mg i.v. TNP4-OVA 24 h later. Three additional wild-type (○) as well as FcγRIIB−/− mice (•) received IgE and then OVA. The monitoring of rectal temperature was started at the time of antigen injection. Data are shown as mean ± SD. **P < 0.01. (B) Changes in heart rate during IgE-mediated systemic anaphylaxis in three FcγRIIB−/− (▪) and three wild-type (□) mice. The induction protocols were the same as in A. As controls, three wild-type mice (○) received OVA. Note the transient rise maximizing ∼1 min after TNP-OVA injection in wild-type mice and the gradual decrease during the 25 min after induction, in contrast to the changes in FcγRIIB−/− mice, showing no transient rise and continuous decrease in heart rate. *P < 0.05; **P < 0.01, compared with wild-type mice. (C) Increased hemorrhage in ileum villi during IgE-mediated systemic anaphylaxis in FcγRIIB−/− mice. 1 h after the anaphylaxis induction, mice were killed, and ileum samples were observed under light microscopy. Hemorrhage in tips of microvilli was evident in FcγRIIB−/− mice. Magnification 40.
Systemic anaphylaxis can result in a fatal outcome. In mice, this mortality has been shown to be associated with IgG1 and FcγRIII (9). As shown in Table I, we observed mortality as a consequence of the anaphylactic response only in FcγRIIB−/− mice upon administration of either IgG1 or IgE and the corresponding antigen, or 2.4G2. These results confirm that either IgE- or IgG-induced systemic anaphylaxis is indeed augmented in FcγRIIB−/− mice, as assessed by mortality during anaphylaxis.
Table I.
Mortality During Systemic Anaphylaxis
| Inductiona | Death ratesb | Times until death (min) | ||||||
|---|---|---|---|---|---|---|---|---|
| Wild type | FcγRIIB−/− | Wild type | FcγRIIB−/− | |||||
| IgE | 0/29 (0%) | 5/29 (17%)* | N.A. | 5, 20, 25, 40, 40 | ||||
| IgG1 | 0/10 (0%) | 2/10 (20%)§ | N.A. | 20, 30 | ||||
| 2.4G2 | 0/14 (0%) | 6/14 (43%)‡ | N.A. | 10, 25, 25, 30, 30, 30 | ||||
a Systemic anaphylaxis was induced with anti-TNP IgG1 transfer and TNP-OVA injection, 2.4G2 administration, or anti-TNP IgE transfer and TNP-OVA injection. b Statistical analyses were performed between wild-type and FcγRIIB−/− mice using Fisher's test:
P < 0.05;
P < 0.01;
NS. N.A., not applicable.
Neither FcεRI Expression Level nor Mast Cell Density Is Upregulated in FcγRIIB−/− Mice.
These unexpected observations for IgE-mediated anaphylaxis prompted us to examine whether deletion of FcγRIIB influenced FcεRI expression levels on effector cells. We confirmed by flow cytometric analysis that the expression level of FcεRI on BMMC from FcγRIIB− /− mice was comparable to the level on wild-type BMMC (data not shown). In addition, we could not demonstrate any significant difference in the expression levels of FcεRI on mast cells after IgE-induced upregulation in vitro or in vivo (Fig. 3, A and B). As shown in Fig. 3 A, BMMC derived from either from FcγRIIB− /− or wild-type mice displayed the same level of upregulation of FcεRI in response to IgE (18). Similarly, peritoneal mast cells isolated from FcγRIIB− /− and wild-type mice 24 h after intravenous administration of 20 μg IgE had equivalent levels of FcεRI (Fig. 3 B). Histopathological examinations indicated that the density and morphology of mast cells in ear, abdominal skin, and trachea from the mutant mice were not significantly different from those in wild-type mice (data not shown).
Figure 3.

Expression levels of FcεRI on BMMC and peritoneal mast cells after induction with IgE. (A) FcεRI upregulation in vitro. BMMC were cultured for 4 d in the presence of 0.1 or 5 μg/ml IgE or 5 μg/ml 2.4G2. Mean values of FcεRI expression levels were assessed by flow cytometric measurement of IgE binding. (B) FcεRI levels in in vivo mast cells. FcγRIIB−/− (•) and wild-type (○) mice received 20 μg i.v. IgE, and 24 h later, mean fluorescence intensities of IgE binding on peritoneal mast cells were compared by flow cytometry as described in Materials and Methods.
Increases in the Number of Degranulated Mast Cells and in Blood Histamine Levels after IgE-mediated Anaphylaxis Induction.
The mechanism by which FcγRIIB−/− mice augmented IgE-mediated anaphylaxis was examined by determining the activation of effector cells in these animals as compared with their wild-type counterparts. Blood histamine levels were measured after the induction of anaphylaxis in FcγRIIB−/− and wild-type mice. As shown in Fig. 4 A, blood obtained both from wild-type or FcγRIIB−/−-sensitized animals 5 min after challenge with antigen or 2.4G2 revealed increased histamine concentrations. The histamine levels seen in FcγRIIB−/−-challenged mice were consistently higher in response to IgE, IgG1, or 2.4G2 stimulation than in control mice, suggesting that the enhanced anaphylaxis in FcγRIIB−/− mice could be interpreted in part by accelerated activation of mast cells in the mutant animals. To directly demonstrate enhanced degranulation, lung samples from FcγRIIB−/− or wild-type mice were removed before and 30 min after the induction of IgG-mediated passive systemic anaphylaxis and examined histopathologically. As shown in Fig. 4 B and E, mast cells around bronchi in FcγRIIB−/− mice displayed quantitatively more degranulation than comparable samples taken from wild-type mice subjected to similar treatment.
Figure 4.

Enhanced mast cell activation in FcγRIIB−/− mice during systemic anaphylaxis. (A) Elevated plasma histamine in FcγRIIB−/− mice during IgE- or IgG1-mediated or 2.4G2-induced systemic anaphylaxis. Plasma histamine 5 min after antigen challenge in each wild-type (+/+) and mutant (−/−) mouse is presented as μM. Horizontal bars, mean values. (B) Enhanced degranulation of lung mast cells in FcγRIIB−/− mice during IgE-mediated systemic anaphylaxis. Densities of lung mast cells were calculated by counting the cells in four different sections derived from two mice under light microscopy. The results are expressed as mean ± SD. The densities of control (before induction), wild-type (WT), and FcγRIIB−/− mice were not significantly different. However, the number of degranulated mast cells (closed columns) was significantly higher in FcγRIIB−/− mice (P < 0.005, Fisher's test). (C–E) Photographs of lung mast cells in wild-type mice before anaphylaxis induction (C), and in wild-type (D) or FcγRIIB−/− (E) mice after induction. Toluidine blue staining. Magnification 1,000.
Conclusions.
Although Takizawa et al. (6) demonstrated that FcγRIIB and FcγRIII act as low-affinity receptors for IgE on cultured mast cells and macrophages in vitro, the physiological significance of this interaction between IgE and FcγRIIB/III has not been established. The consequence of a low-affinity interaction between IgE and FcγRs in vivo would result in IgE immune complexes binding not only to FcεRI but also to FcγRIIB/III on those cells and potentially modulating mediator release. Dombrowicz et al. (4) have shown that although BMMC from FcεRI−/− mice can bind IgE immune complexes via FcγRIIB/III in vitro, the abrogation of IgE-mediated systemic anaphylaxis in vivo by deletion of FcεRI would indicate that the interaction of IgE with FcγRs is not significant. However, an alternative explanation for their data is suggested by the present studies, as the FcεRI−/− strain retains FcγRIIB as well as FcγRIII on its mast cells (4). Based on our data, we propose that the IgE immune complex–mediated response would represent the sum of three components, i.e., an FcεRI-mediated major positive factor, an FcγRIIB negative response, and an FcγRIII-mediated positive component, respectively. When the FcεRI component had been lost, the sum of the remaining FcγRIIB and FcγRIII components would be negligible. Our present results predict that a sum of the components of FcεRI and FcγRIIB would be a positive, although diminished, response. This prediction is supported by the IgE-mediated anaphylactic response in FcγRIII−/− mice. As shown in Fig. 5 A, FcγRIII−/− mice indeed show a decreased response in IgE-mediated systemic anaphylaxis. Moreover, we found that blocking of FcγRIIB by preadministration of 2.4G2 resulted in an enhanced response in IgE-mediated systemic anaphylaxis in FcγRIII−/− mice (Fig. 5 B). Taken together, these results support the conclusion that FcγRIIB attenuates IgE-mediated anaphylactic responses triggered by FcεRI or FcγRIII.
Figure 5.
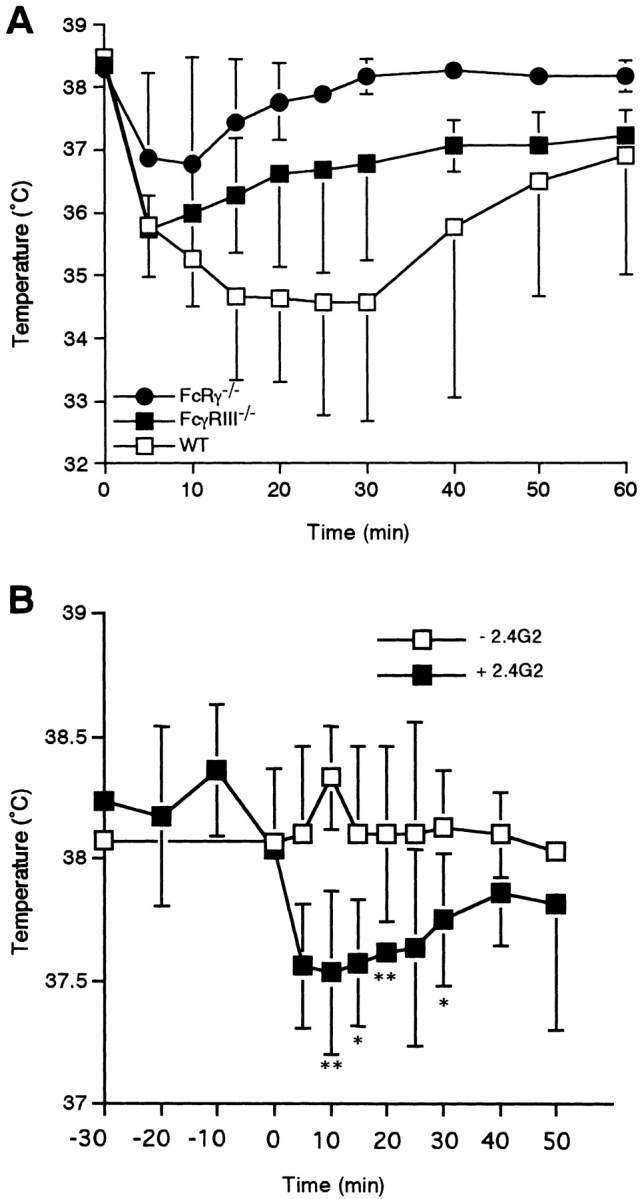
IgE-mediated systemic anaphylaxis in FcγRIII−/− mice. (A) Changes in rectal temperature of mice during IgE-induced systemic anaphylaxis. Three wild-type (WT; □) and three FcγRIII−/− (▪) animals as well as three FcRγ−/− mice (•) received 20 μg i.v. anti-TNP IgE. All of the animals received 1.0 mg i.v. TNP-OVA 24 h later. Data are shown as mean ± SD. (B) Effect of preadministration of 2.4G2 on changes in rectal temperature in IgE-mediated systemic anaphylaxis. At time −24 h, FcγRIII−/− mice received 20 μg IgE; they were administered 100 μg 2.4G2 (▪) or vehicle alone (□) at time −30 min and then received TNP-OVA at time 0. Data are shown as mean ± SD. *P < 0.05; **P < 0.01.
Further support for the role of FcγRIIB in modulating the IgE-mediated response comes from studies in Src homology 2–containing inositol phosphatase (SHIP)-deficient mice (23). This inositol polyphosphate phosphatase is recruited to FcγRIIB upon cross-linking with an immunoreceptor tyrosine-based activation motif (ITAM)-containing activation receptor through its SH2 (Src homology 2) domain and leads to the hydrolysis of phosphatidylinositol 3,4,5-trisphosphate, with release of Bruton's tyrosine kinase and phospholipase Cγ from the inner leaflet of the cell membrane (24). The net result of this pathway is the termination of calcium influx, with subsequent inhibition of activation responses (20, 21, 25). Mast cells derived from SHIP-deficient mice display a hyperresponsive IgE phenotype similar to the response seen in FcγRIIB−/− mice (26). Thus, functional uncoupling of FcγRIIB from its signaling pathway results in similar phenotype deletion of the receptor itself.
The observations presented here support the hypothesis that IgE-mediated activation is modulated by inhibitory receptors like FcγRIIB. Perturbation of an inhibitory pathway would be predicted to render mast cells more sensitive to IgE activation and could account for some atopic phenotypes. Upregulation of FcγRIIB or its constitutive engagement would result in desensitization of mast cells to IgE triggering and reversal of the atopic state.
Acknowledgments
The authors wish to thank Dr. Yutaka Kagaya, Department of Internal Medicine, Tohoku University, Japan, for his assistance in the measurement of heart rate and for helpful discussions.
This work was supported by research grants from the Ministry of Education, Science, Sports, and Culture of Japan; Core Research for Evolutional Science and Technology (CREST), Japan Science and Technology Corporation (JST) (to T. Takai); and from the National Institutes of Health and the Juvenile Diabetes Foundation (to J.V. Ravetch).
Abbreviations used in this paper
- BMMC
bone marrow–derived cultured mast cells
- FcεRI
high-affinity receptor for IgE
- FcγR
Fc receptor for IgG
- FcRγ
Fc receptor γ subunit
- FcγRIIB and FcγRIII
type IIB and type III low-affinity receptors for IgG, respectively
References
- 1.Ando, A., Martin, T.R., and S.J. Galli. 1993. Effects of chronic treatment with the c-kit ligand, stem cell factor, on immunoglobulin E–dependent anaphylaxis in mice. J. Clin. Invest. 92:1639–1649. [DOI] [PMC free article] [PubMed]
- 2.Martin TR, Galli SJ, Katona IM, Drazen JM. Role of mast cells in anaphylaxis. Evidence for the importance of mast cells in the cardiopulmonary alterations and death induced by anti-IgE in mice. J Clin Invest. 1989;83:1375–1383. doi: 10.1172/JCI114025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR γ chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 4.Dombrowicz D, Flamand V, Brigman KK, Koller BH, Kinet J-P. Abolition of anaphylaxis by targeted disruption of the high affinity immunoglobulin E receptor α chain gene. Cell. 1993;75:969–975. doi: 10.1016/0092-8674(93)90540-7. [DOI] [PubMed] [Google Scholar]
- 5.Segal DM, Sharrow SO, Jones JF, Siraganian RP. Fc (IgG) receptors on rat basophilic leukemia cells. J Immunol. 1981;126:138–145. [PubMed] [Google Scholar]
- 6.Takizawa F, Adamczewski M, Kinet J-P. Identification of the low affinity receptor for immunoglobulin E on mouse mast cells and macrophages as FcγRII and FcγRIII. J Exp Med. 1992;176:469–476. doi: 10.1084/jem.176.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bocek P, Jr, Dráberová L, Dráber P, Pecht I. Characterization of Fcγ receptors on rat mucosal mast cells using a mutant FcεRI-deficient rat basophilic leukemia line. Eur J Immunol. 1995;25:2948–2955. doi: 10.1002/eji.1830251035. [DOI] [PubMed] [Google Scholar]
- 8.Oettgen HC, Martin TR, Wynshaw-Boris A, Deng C, Drazen JM, Leder P. Active anaphylaxis in IgE-deficient mice. Nature. 1994;370:367–370. doi: 10.1038/370367a0. [DOI] [PubMed] [Google Scholar]
- 9.Miyajima I, Dombrowicz D, Martin TR, Ravetch JV, Kinet J-P, Galli SJ. Systemic anaphylaxis in the mouse can be mediated largely through IgG1 and FcγRIII. Assessment of the cardiopulmonary changes, mast cell degranulation, and death associated with active or IgE- or IgG1-dependent passive anaphylaxis. J Clin Invest. 1997;99:901–914. doi: 10.1172/JCI119255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dombrowicz D, Flamand V, Miyajima I, Ravetch JV, Galli SJ, Kinet JP. Absence of FcεRI α chain results in upregulation of FcγRIII-dependent mast cell degranulation and anaphylaxis. Evidence of competition between FcεRI and FcγRIII for limiting amounts of FcR β and γ chain. J Clin Invest. 1997;99:915–925. doi: 10.1172/JCI119256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in FcγRII-deficient mice. Nature. 1996;379:346–349. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- 12.Daëron M, Malbec O, Latour S, Arock M, Fridman WH. Regulation of high-affinity IgE receptor– mediated mast cell activation by murine low-affinity IgG receptors. J Clin Invest. 1995;95:577–585. doi: 10.1172/JCI117701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daëron M, Latour S, Malbec O, Espinosa E, Pina P, Pasmans S, Fridman WH. The same tyrosine-based inhibition motif, in the intracytoplasmic domain of FcγRIIB, regulates negatively BCR-, TCR-, and FcR-dependent cell activation. Immunity. 1995;3:635–646. doi: 10.1016/1074-7613(95)90134-5. [DOI] [PubMed] [Google Scholar]
- 14.Daëron M. Fc receptor biology. Annu Rev Immunol. 1997;15:203–234. doi: 10.1146/annurev.immunol.15.1.203. [DOI] [PubMed] [Google Scholar]
- 15.Ohmori H, Hase N, Hikida M, Takai T, Endo N. Enhancement of antigen-induced interleukin 4 and IgE production by specific IgG1 in murine lymphocytes. Cell Immunol. 1992;145:299–310. doi: 10.1016/0008-8749(92)90333-k. [DOI] [PubMed] [Google Scholar]
- 16.Kleine-Tebbe J, Hamilton RG, Roebber M, Lichtenstein LM, MacDonald SM. Purification of immunoglobulin E (IgE) antibodies from sera with high IgE titers. J Immunol Methods. 1995;179:153–164. doi: 10.1016/0022-1759(94)00279-6. [DOI] [PubMed] [Google Scholar]
- 17.Peng Z, Becker AB, Simons FE. Binding properties of protein A and protein G for human IgE. Int Arch Allergy Immunol. 1994;104:204–206. doi: 10.1159/000236731. [DOI] [PubMed] [Google Scholar]
- 18.Yamaguchi M, Lantz CS, Oettgen HC, Katona IM, Fleming T, Miyajima I, Kinet J-P, Galli SJ. IgE enhances mouse mast cell FcεRI expression in vitro and in vivo: evidence for a novel amplification mechanism in IgE-dependent reactions. J Exp Med. 1997;185:663–672. doi: 10.1084/jem.185.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amigorena S, Bonnerot C, Drake JR, Choquet D, Hunziker W, Guillet JG, Webster P, Sautes C, Mellman I, Fridman WH. Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B-lymphocytes. Science. 1992;256:1808–1812. doi: 10.1126/science.1535455. [DOI] [PubMed] [Google Scholar]
- 20.Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig MC, Ravetch JV. A 13-amino-acid motif in the cytoplasmic domain of FcγRIIB modulates B-cell receptor signalling. Nature. 1994;368:70–73. doi: 10.1038/368070a0. [DOI] [PubMed] [Google Scholar]
- 21.Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor FcγRIIB. Nature. 1996;383:263–266. doi: 10.1038/383263a0. [DOI] [PubMed] [Google Scholar]
- 22.Unkeless JC. Characterization of a monoclonal antibody directed against mouse macrophage and lymphocyte Fc receptors. J Exp Med. 1979;150:580–596. doi: 10.1084/jem.150.3.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helgason CD, Damen JE, Rosten P, Grewal R, Sorensen P, Chappel SM, Borowski A, Jirik F, Krystal G, Humphries RK. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12:1610–1620. doi: 10.1101/gad.12.11.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bolland S, Pearse RN, Kurosaki T, Ravetch JV. SHIP modulates immune receptor responses by regulating membrane association of Btk. Immunity. 1998;8:509–516. doi: 10.1016/s1074-7613(00)80555-5. [DOI] [PubMed] [Google Scholar]
- 25.Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch JV. Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell. 1997;90:293–301. doi: 10.1016/s0092-8674(00)80337-2. [DOI] [PubMed] [Google Scholar]
- 26.Huber M, Helgason CD, Damen JE, Liu L, Humphries RK, Krystal G. The src homology 2-containing inositol phosphatase (SHIP) is the gatekeeper of mast cell degranulation. Proc Natl Acad Sci USA. 1998;95:11330–11335. doi: 10.1073/pnas.95.19.11330. [DOI] [PMC free article] [PubMed] [Google Scholar]