

Abstract
The precise role of B cells in systemic autoimmunity is incompletely understood. Although B cells are necessary for expression of disease (Chan, O., and M.J. Shlomchik. 1998. J. Immunol. 160:51–59, and Shlomchik, M.J., M.P. Madaio, D. Ni, M. Trounstine, and D. Huszar. 1994. J. Exp. Med. 180:1295–1306), it is unclear whether autoantibody production, antigen presentation, and/or other B cell functions are required for the complete pathologic phenotype. To address this issue, two experimental approaches were used. In the first, the individual contributions of circulating antibodies and B cells were analyzed using MRL/MpJ-Faslpr (MRL/lpr) mice that expressed a mutant transgene encoding surface immunoglobulin (Ig), but which did not permit the secretion of circulating Ig. These mice developed nephritis, characterized by cellular infiltration within the kidney, indicating that B cells themselves, without soluble autoantibody production, exert a pathogenic role. The results indicate that, independent of serum autoantibody, functional B cells expressing surface Ig are essential for disease expression, either by serving as antigen-presenting cells for antigen-specific, autoreactive T cells, or by contributing directly to local inflammation.
Keywords: nephritis, transgenic, T cell, vasculitis, antigen presentation
Systemic lupus erythematosus (SLE) is considered to be an immune disease in which the deposition of immune complexes or direct autoantibody deposition leads to the activation of complement, ligation of FcRs, and subsequent inflammation (1). Nevertheless, mononuclear infiltrates along with autoantibodies are usually present in affected tissues, including vessels, salivary glands, skin, and kidneys (2–10). In these situations, infiltrating T cells, macrophages, and other cells reside within destructive lesions (4–6). In particular, interstitial nephritis (IN)1 is relatively common in human SLE; and this finding is associated with a poor prognosis (7, 9, 11, 12). These observations raise questions about the relative roles and connections between humoral and cellular autoimmunity. On the one hand, immune complex formation may lead to cellular recruitment. Alternatively, T cell infiltration may occur directly through antigen-specific events involving either macrophages or parenchymal cells. Deciphering the contributions of these events in tissue injury has both mechanistic and therapeutic implications.
To develop a better understanding of the role of B cells during systemic autoimmunity, we created the JHD-MRL/ MpJ-Faslpr mouse strain, (JHD), a strain of MRL/lpr that lacks B cells (13, 14). The MRL/lpr strain develops a spectrum of disease manifestations that is quite similar to fulminant human SLE, including severe nephritis, vasculitis, sialoadenitis, and skin disease (15). When rendered B cell deficient, these animals lacked not only the classic immune-deposit manifestations of nephritis, such as glomerulonephritis (GN), but also failed to develop cellular infiltrates within organs, including IN, vasculitis (13), and skin disease (Chan, O.T.M., J. McNiff, and M.J. Shlomchik, manuscript in preparation). Furthermore, the absence of B cells substantially blocked the accumulation of activated and memory T cells (16).
Although these results clearly define a critical role for a B cell function(s) in the development of pathogenic lesions, they do not distinguish the precise roles of autoantibodies and B cells (i.e., independent from soluble antibodies). Here, we present a novel system to distinguish these mechanisms by developing an MRL/lpr mouse with B cells but no circulating antibodies. These animals demonstrate T cell activation, cellular infiltration including vasculitis and IN, and increased mortality, indicating that autoreactive B cells promote the development of pathogenic T cells in this disease. The results support a novel and important role for B cells in the pathogenesis of systemic autoimmunity.
Materials and Methods
Transgene Construction.
The mIgM construct: a VDJ region containing the canonical anti-(4-hydroxy-3-nitro-phenyl) acetyl membrane (NP) VH186.2 was cut from plasmid CμB1-8.24, containing the V region of the hybridoma B1-8 (17), using EcoRI and XbaI. It was then ligated to an XbaI-EcoRI fragment containing the IgH intronic enhancer region from plasmid Cμ19.7.1 (17). The BglII-XhoI fragment of pICEM-Cμ (containing Cμ1-4, and membrane and secreted exons) was replaced by the corresponding region from pSV5-Cμm, from which the secreted exon and polyadenylation site had been deleted (18). The EcoRI-XhoI fragment of the resulting plasmid, pICEM-Cμm, was then inserted into pBKS-II. The VDJ-enhancer unit was then excised as an EcoRI fragment and inserted at the EcoRI site of calf intestinal phosphatase (CIP)-treated pBKS-Cμm; correct orientation was confirmed by restriction digest. The resulting VH186.2–mIgM construct (termed mIgM hereon) encodes membrane-bound, but not secreted, IgMa.
The mIgM construct was tested by transfection into the CH1 cell line, which expresses IgMb using λ light chain. Although FACS® analysis confirmed the presence of NP-binding IgMa on transfectants, no IgMa was detectable by ELISA in culture supernatant. Transgenic (Tgic) mice resulting from microinjection of the purified DNA construct were initially identified by Southern blot analysis.
Mice.
Mice bearing the mIgM transgene (Tg) were initially backcrossed with MRL/lpr mice four times, fixing homozygosity for lpr. The resultant Tg-positive mice were then backcrossed to the JHD/lpr strain to fix homozygosity of the JHD mutation, a neo insertion in the JH locus (fixation occurred after backcross one). This eliminated the expression of endogenous Ig heavy chains. All subsequent Tg-positive mice were generated by backcrossing to the JHD/lpr strain. Through this breeding strategy, all the Tgic animals used in this study had >98.9% MRL genes. The resulting mIgM.MRL/MpJ-Faslpr mice (termed mIgM mice hereon) have functional B cells that do not secrete appreciable Ig (see below). All animals used in this study were obtained from our colony at the Yale University School of Medicine (New Haven, CT) and were housed under specific pathogen-free conditions. All Tgic mice and controls analyzed in this study were aged 24 wk or more.
A PCR detecting the rearranged VH186.2 V(D)J segment of the mIgM Tg was used to identify the Tg-positive mice. The oligonucleotides used for this PCR were VH186.2 5′ (5′-TGCTCTTCTTGGCAGCAAC-3′ [5′ primer]) and VH186.2 3′ (5′-TGAGGAGACTGTGAGAGTG-3′ [3′ primer]). Amplification conditions were 94°C for 2 min and 35 cycles of 30 s each at 94°C, 54°C, and 72°C, followed by a 7-min incubation at 72°C. Homozygosity for lpr was detected by PCR as previously described (13).
Reagents and Antibodies.
The following mAbs were used as FACS® reagents in this study: CD19 (1D3-biotin; PharMingen); B220 (RA3-6B2-FITC); CD4 (H129.19-CyChrome; PharMingen); CD44 (Pgp-1-FITC); CD62L (Mel-14–biotin); and anti– mouse Fcγ receptor (2.4G2). Streptavidin-conjugated PE (Molecular Probes) was added as a secondary step for the biotinylated reagents. Pgp-1, Mel-14, and RA3-6B2 were purified from hybridoma supernatants on protein G columns (Amersham Pharmacia Biotech) after ammonium sulfate precipitation, and were conjugated as described (19). The antibodies were verified by comparison with commercially available antibodies with the same specificities.
The following mAbs were used as standard controls for the ELISAs: T183 (IgM; Sigma Chemical Co.), 4G7 (IgG1; reference 19), Hy1.2 (IgG2a; reference 20), Pl9-10 (IgG2b), Pl9-11 (IgG3), Pl9-10 (anti-double stranded [ds]DNA; reference 21), 400tμ23 (RF; reference 19), and LG4-1 (anti-chromatin; reference 22). 23.3 culture supernatant was used as the source of anti-NP IgG2a for the rheumatoid factor ELISAs (19). Polyclonal anti–mouse κ conjugated to FITC (Southern Biotechnology Associates, Inc.) was used in the immunofluorescence assays to detect glomerular Ig deposition.
Cell Preparation and Flow Cytometry.
FACS® analysis was conducted as previously described (16).
ELISA.
For anti-Ig ELISA, Falcon 96-well, flat-bottomed plates (Becton Dickinson) were coated overnight at 4°C with the primary antibody (antiisotype). Plates were then blocked with 1% BSA in PBS for 1 h at room temperature. Serum dilutions along with the standard were allowed to incubate for 2 h. The secondary antibody (antiisotype conjugated to alkaline phosphatase; Southern Biotechnology Associates, Inc.) was then incubated in the plate for 1 h at room temperature.
For autoantibody ELISA, the assays were conducted in the same manner as indicated above. However, the following were used to coat the plates with autoantigen: poly-l-lysine and purified calf thymus DNA (anti-dsDNA), NP/anti-NP IgG2a complexes (RF), and purified calf thymus chromatin (antichromatin). The secondary antibody was goat polyclonal anti–mouse κ conjugated to biotin, followed by streptavidin-alkaline phosphatase (Southern Biotechnology Associates, Inc.). The colorimetric data from the pNPP substrate was quantified using Bio Kinetics Reader EL 340 (Bio-Tek Instruments). DeltaSoftIII software (BioMetallics, Inc.) was used to analyze this data.
Tissue Preparation.
Kidneys were bisected and fixed in 10% buffered formalin and embedded in paraffin, and sections were stained with hematoxylin and eosin. For immunofluorescent evaluation, kidney halves were lightly fixed in a 0.7% paraformaldehyde-lysine-periodate solution overnight at 4°C. Then, each sample was incubated in a 30% sucrose phosphate buffer at room temperature for >2 h and frozen in OCT compound (Tissue-Tek). 7-μm sections were cut using a Cryocut 1800 cryostat (Reichert-Jung) and stained overnight with an anti–mouse κ conjugated with FITC (Southern Biotechnology Associates, Inc.). Fluorescence was visualized using a Nikon Optiphot microscope (Nikon, Japan) at 200× or 400× and was photographed with an exposure time of 5–6 s on Elite ASA 400 film (Eastman Kodak Co.).
Renal Scoring.
The severity of nephritis and vasculitis was graded based on a semiquantitative scale using the parameters described in Table I of reference 23. In brief, a 0–4+ scale was used for each compartment (glomerular, interstitial, and vascular) with the pathology graded according to specified criteria as absent, mild, moderate, or severe. For comparative purposes, all of the tissue sections were scored by one observer (M.P.M.), who was blinded to their origin, in three sessions for the unmanipulated mice. A proportion of samples was reread a second time, with generally excellent concordance (<1 point average difference). In rare cases of disagreement, the sections were evaluated a third and final time and the average of the scores was used.
Table I.
Lymphoid Organ Weight and Total Cell Numbers of mIgM, MRL/lpr, and JHD Mice
| Genotype | Spleen | Lymph node | ||||||
|---|---|---|---|---|---|---|---|---|
| Weight | Cell No. | Weight | Cell No. | |||||
| mg | ×106 | mg | ×106 | |||||
| mIgM | 366 ± 107* | 181 ± 43* | 11 ± 5 | 4 ± 3 | ||||
| MRL/lpr | 696 ± 358* | 224 ± 162* | 175 ± 150* | 59 ± 67* | ||||
| JHD | 148 ± 80 | 82 ± 72 | 32 ± 37 | 12 ± 17 | ||||
Splenic and inguinal lymph node organ weights and total cell numbers were obtained from mIgM, MRL/lpr (B cell–intact), and JHD (B cell– deficient) mice. Averages and one standard deviation were calculated from samples consisting of 6–8-mo-old animals. Sample sizes for spleen and lymph nodes are as follows: mIgM weight and cell number (n = 6), MRL/lpr weight and cell number (n = 25), JHD weight (n = 9), and JHD cell number (n = 8).
P < 0.05 for comparisons to B cell–deficient JHD mice.
Statistics.
Mortality curves were plotted using the Kaplan-Meier method and examined for significance using the Mantel-Cox logrank test. All other statistical tests used the nonparametric Mann-Whitney U statistic. The statistical analyses were conducted using StatView 4.5 (Abacus Software) for the Macintosh. P < 0.05 was considered significant.
Results
B Cells Are Restored in Tgic mIgM Mice.
Tgic mice with the mIgM construct, which lacked the secreted exons, were created as described in Materials and Methods. A diagram of the Tg is shown in Fig. 1. Mice bearing the mIgM Tg were backcrossed at least seven times to the B cell–deficient JHD strain, placing the Tgs onto the autoimmune MRL/lpr background. This also established homozygosity of the JHD mutation, which prevented the development of B cells expressing endogenous Ig. As shown in Fig. 2, the mIgM Tg restored B cell maturation. The percentages of B cells in the Tg mice were comparable with those in wild-type animals (average spleen percentage: B cell–intact − 15 ± 7%, mIgM – 16 ± 8).
Figure 1.

Tg schematic. The mIgM heavy chain Tg is depicted. The DNA directing secretion (μs) and the transcription termination site (pAs) have been deleted. VH186.2 is the rearranged heavy chain V(D)J segment. E indicates the heavy chain intronic enhancer. Much of the switch region has been deleted; a small region of residual switch region is indicated.
Figure 2.

The Tg restores splenic B cells in JHD mice. Spleen cells from JHD (A), mIgM (B), and MRL/lpr (C) mice were analyzed via flow cytometry. Live (propidium iodide–negative) cells were analyzed for CD19 (PE) and B220 (FITC) expression. B cells are B220+/CD19+ (upper right quadrant). Percentages of B cells among live splenocytes are shown. In MRL/lpr mice, B220+/CD19− cells are T cells.
mIgM Mice Have Greater Splenomegaly than JHD Mice.
JHD mice at 4–6 mo of age have reduced lymphoid organ weights and cell numbers compared with B cell–intact animals (13, 16). Table I lists the cell numbers and organ weights of the mIgM Tgic animals along with age-matched B cell–intact and B cell–deficient MRL/lpr mice. Splenic weight and cell number were greater in mIgM mice than in B cell–deficient animals (weight: 2.5-fold increase, P < 0.02; cell number: 2.2-fold increase, P < 0.002). Since B cells generally comprise only 15–20% of splenocytes in older MRL/lpr mice, this 2.2-fold difference in cell number cannot be accounted for simply by B cells per se. Most of the increased cell number is attributable, instead, to T cells, consistent with our previous reports on the effect of B cells on T cell activation and expansion. Although significantly greater than JHD/lpr mice, splenomegaly in the mIgM Tgics was decreased when compared with control MRL/lpr mice. Similarly, in the lymph nodes restoration of B cells in the Tgic mice had no effect on weight and cell number. The phenotype of partial restoration of T cell accumulation mediated by the Tgic mice most likely reflects the restricted repertoire enforced by a single VH in the Tg. A partial or reduced disease phenotype has been found in several other conventional Ig or TCR Tgic strains associated with the partial repertoire restriction imposed by allelic exclusion by the Tgs (24, 25).
Spontaneous T Cell Activation in mIgM Mice Occurs in the Absence of Circulating Ig.
The majority of T cells in MRL/ lpr mice have an activated/memory phenotype (16, 26). However, in B cell–deficient MRL/lpr animals, the percentage of naive cells is increased, whereas the percentage of memory T cells is decreased (16). Furthermore, B cells are required for the accumulation of most memory T cells since there are 5–10 times as many memory T cells in B cell–intact mice compared with B cell–deficient mice. In principle, B cells could be exerting this effect on memory T cell development either directly (e.g., via presentation of [auto]antigens) or indirectly (e.g., via autoantibody-mediated inflammation causing the release of autoantigens).
To distinguish between these possibilities and to determine the mechanism by which B cells promote spontaneous T cell activation, we analyzed CD4+ T cells from the spleens of mIgM mice for expression of CD44 and CD62L (Fig. 3). The T cell activation profiles of mIgM animals resembled B cell–intact mice rather than B cell–deficient mice. When compared with JHD animals, the percentages of memory cells (CD44high, CD62Llow) were significantly greater (1.6-fold increase, P < 0.0005) in mIgM mice (Fig. 3 D). Furthermore, percentages of naive cells (CD44low, CD62Lhigh) were reduced by 90% (P < 0.002). The number of memory CD4+ T cells in the mIgM mice was greater (5.5-fold increase, P < 0.003) than that of B cell–deficient animals and was similar to the B cell–intact, control MRL/ lpr mice (Fig. 3 E). As in the spleen, the percentage of naive cells in the lymph nodes was decreased and the percentage of memory cells was increased in mIgM Tgic mice compared with B cell–deficient animals (data not shown). However, there was no statistical difference in memory CD4+ cell number since lymph nodes were not generally enlarged in the mIgM mice. For CD8+ T cells, a similar pattern of naive cell reduction and memory cell augmentation was observed (data not shown). Since the accumulation of memory and activated T cells proceeded efficiently in the absence of secreted antibody (see below) in the mIgM mice, we conclude that B cells directly promote T cell activation and accumulation, rather than indirectly via antibody-mediated tissue damage.
Figure 3.

Spontaneous splenic CD4+ T cell activation is restored by the mIgM Tg. Three-color FACS® analysis was conducted on splenocytes. Representative FACS® contour plots are shown for JHD (A), MRL/lpr (B), and mIgM (C) mice. CD4high gated cells were analyzed for CD44 (FITC) and CD62L (PE) expression. Percentages of total CD4+ T cells (D) were obtained for the naive (CD44lowCD62Lhigh), activated (CD44highCD62Lhigh), and memory (CD44highCD62Llow) subsets as depicted in parts A–C. Cell numbers (E) were calculated by multiplying total cell percentages of each activation subset by the total spleen cell number for each mouse. Columns show the averages of percentages (D) or cell numbers (E) of a cohort of mice. Error bars represent one standard deviation. Sample sizes for cell percentages are: JHD (n = 17), MRL/lpr (n = 26), mIgM (n = 6) mice. Sample sizes for cell numbers are: JHD (n = 9), MRL/lpr (n = 25), mIgM (n = 5). All mice were 24–32 wk of age. Asterisks indicate significant differences of P < 0.05 with the B cell–deficient group, as determined by the two-tailed Mann-Whitney U test.
mIgM Mice Develop Renal Lesions in the Absence of Circulating Ig.
MRL/lpr mice characteristically develop spontaneous nephritis (Fig. 4, E and F), whereas B cell–deficient JHD mice do not (Fig. 4, G and H) (13). mIgM mice, on the other hand, have significant cellular infiltrates in the renal interstitium and around the vessels, despite the inhibition of secreted Ig (Fig. 4, A–D). These infiltrates were predominantly composed of T cells (data not shown).
Figure 4.
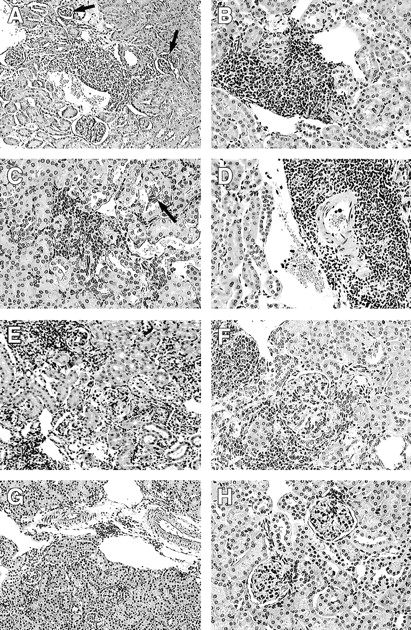
Lupus nephritis occurs in the absence of circulating Ig. Kidneys were fixed in formalin, and sections were stained with hematoxylin and eosin: (A) 23 wk mIgM, (B) 30 wk mIgM, (C and D) 42 wk mIgM, (E) 31 wk MRL/lpr, (F) 30 wk MRL/lpr, and (G and H) 45 wk JHD. Cellular interstitial and perivascular infiltrates are observed in mIgM (A–D) and MRL/lpr (E and F), but not in JHD mice, even up to 45 wk of age (G and H). Black arrows point to glomerular lesions in the mIgM mice (A and C).
Renal disease in the glomeruli, interstitium, and vessels was scored blindly to assess severity (Fig. 5, A–C). IN and vasculitis scores of the mIgM mice were greater than those of the JHD animals (interstitium, P < 0.03; vessels, P < 0.03) and comparable to those of the MRL/lpr mice. There were no statistically significant differences in glomerular scores between the JHD and mIgM strains, both of which were lower than MRL/lpr mice (P < 0.02). Nevertheless, focal lesions that resembled glomerular atrophy were observed in some glomeruli of the mIgM mice (Fig. 4, A and C), whereas these lesions were never observed in the JHD strain (Fig. 4, G and H). This picture is different than the typical proliferative GN picture seen in mild to moderate MRL/lpr disease and may reflect a previously unappreciated antibody-independent mode of glomerular disease perhaps related to adjacent interstitial or vascular disease.
Figure 5.

Kidney disease scores and mortality in MRL/lpr mice. Kidney samples were examined via light microscopy and scored blindly for disease on a semiquantitative scale from 0 (normal) to 4 (maximum). Scores for the glomeruli (A), interstitium (B), and vessels (C) are shown above. Each point represents an individual animal. Black horizontal bars indicate the median score. All animals were 24–30 wk of age. Comparisons to JHD mice were conducted, and statistical significance is indicated in each graph. Sample sizes for the mortality graph (D) are: MRL/lpr (n = 203), JHD (n = 267), and mIgM (n = 71). The JHD mice had greater survival than either MRL/lpr (P < 0.0001) or mIgM (P < 0.0001) mice. Survival curves were generated as described in Materials and Methods using the Kaplan-Meier method and examined for statistical significance using the Mantel-Cox logrank tests.
mIgM Mice Have Greater Mortality than JHD Mice.
In our colony, 50% mortality for B cell–intact MRL/lpr mice occurred at 32 wk (n = 203) (Fig. 5 D). The JHD strain, with markedly reduced nephritis, vasculitis, and T cell activation, had a significantly greater life span (n = 267, P < 0.0001). The mortality of mIgM mice (50% at 56 weeks, n = 71) was accelerated when compared with the JHD mice (P < 0.0001), demonstrating that the restoration of B cells in the absence of circulating, soluble autoantibody has a direct and relevant effect on disease expression. However, in keeping with the fact that restoration of B cells with a restricted repertoire leads to milder nephritis and somewhat less T cell activation than in wild-type MRL/lpr animals, mIgM mice also had prolonged survival compared with the control MRL/lpr strain (P < 0.0007).
mIgM Mice Do Not Have Significant Serum Antibody.
To confirm the absence of secreted antibody in the mIgM strain, serum Ig and autoantibody levels were measured (Fig. 6). Some animals had no detectable Ig as expected (baseline dots in Fig. 6). However, others had trace quantities of some Ig isotypes; these concentrations were 100– 1,000 times lower than MRL/lpr controls. Notably, there were mIgM animals that did not have any detectable serum Ig, yet developed renal disease. Finally, there was no significant total anti-dsDNA, RF, or antichromatin detected in the serum for most of the mIgM mice (Fig. 6 B).
Figure 6.

Serum Ig levels. Sera were obtained from mIgM, MRL/lpr, and JHD mice. Ig levels as determined by ELISA for isotypes IgM, IgG1, and IgG2a are shown on the logarithmic y axis in A. Anti-dsDNA, anti-IgG2a rheumatoid factor, and antichromatin Ig are shown in B. Each point represents one mouse. Horizontal bars indicate the median Ig level.
Ig Deposition Is Not Detected in the Kidneys of mIgM Mice.
Using immunofluorescence, we examined the kidneys of two mIgM mice that had detectable circulating Ig. In both, there were no observable Ig deposits in the glomeruli, interstitium, or vessels, consistent with the low to absent levels of serum Ig and autoantibodies. These mIgM kidneys were indistinguishable in this regard from kidneys of the B cell–deficient, negative controls, whereas MRL/lpr mice showed intense staining, as expected (Fig. 7).
Figure 7.

Lack of renal antibody deposition in mIgM mice. Mice were assayed for Ig deposition, as determined by anti–mouse κ-FITC: (A) MRL/lpr, (B) JHD, and (C) mIgM. Deposition was observed in MRL/lpr mice (A), whereas none was detected in JHD (B) and mIgM (C) mice. All animals were 29–36 wk of age.
Discussion
SLE is a complex disease that may have multiple pathogenic manifestations. In addition to the classic GN, vasculitis, IN, arthritis, and skin disease are often seen. The key finding of this work is that in the absence of circulating Ig and renal antibody deposition, the mIgM strain developed IN, vasculitis, and focal glomerular atrophy. Thus, these studies demonstrate for the first time an antibody-independent mechanism for renal and vascular disease in a murine model of SLE. Disease promoted by B cells in the absence of Ab is biologically relevant in that IN, vasculitis, and, importantly, mortality were all enhanced by B cells alone. To the extent that these results are generalizable to other murine models and to human SLE, the data suggest a wider view of lupus pathogenesis, which is generally thought to be solely antibody mediated (1).
Renal cellular infiltrates in MRL/lpr mice, which are not observed in the JHD strain, contain significant numbers of T cells (Chan, O.T.M., and M.J. Shlomchik, unpublished observation, and references 6, 27). These cells are restored in mIgM mice (Fig. 4 and immunocytochemistry data not shown). It is important to emphasize that interstitial disease and vasculitis are prominent features in diseased kidneys of many SLE patients (7, 9, 12). Indeed, interstitial injury may correlate best with overall outcome. Moreover, cellular infiltration is a prominent feature in other SLE manifestations, such as some skin lesions and sialoadenitis (10, 28). It is worth noting that cellular infiltration in the MRL/lpr model is not due to the Faslpr mutation since Fas-intact MRL/MpJ-+Fas-lpr/+Fas-lpr mice also develop cellular infiltrates (references 3 and 5, and Chan, O.T.M., unpublished data).
Although overall statistical analysis of blind readings in a system that emphasized generalized disease did not show a difference in glomerular disease, this masks the fact that focal glomerular lesions were seen in mIgM mice (Fig. 4). Focal glomerular lesions have never been noted in JHD mice (13). The nature of these lesions has not yet been defined; they could be due to adjacent vascular damage, periglomerular infiltrates, and/or local release of toxic cytokines (8, 29). However, it seems that in the absence of marked glomerular disease, an antibody-mediated component is missing from the mIgM mice, as might have been predicted. The relationship of IN and GN has been controversial and the subject of speculation in the literature (30); but, until now, linkage could not be addressed in any experimental way in either humans or murine models. The present data show that IN and vasculitis can proceed without severe GN, but that some elements of GN might be exacerbated or even caused by surrounding cellular infiltrates as suggested by focal glomerular necrosis observed. In fact, several recent studies of experimental GN models that were thought to be solely antibody mediated have now demonstrated an important role for T cells (31–34). From all these studies, one could propose that there is synergy and cooperation among B and T cells to mediate a variety of pathogenic outcomes and that T cells do play a direct role in mediating disease that was previously thought to be only antibody dependent.
The mIgM Tgic mice accumulated memory T cells in their spleens at levels similar to that of wild-type animals, in contrast to B cell–deficient mice. Thus, T cell activation is antibody-independent, but exactly how B cells promote T cell activation and cellular infiltration into tissues such as kidney and vessels is not completely clear. However, it is likely that B cells primarily act as autoantigen-presenting cells for the amplification of autoreactive T cells. In this scenario, autoreactive T cells may initially be activated by other APCs, such as dendritic cells; however, as B cell autoimmunity progresses and expanded clones of self-reactive B cells accumulate (35, 36), these cells become increasingly important APCs for T cells (37–41). B cells are known to be extraordinarily efficient APCs for antigens that can bind to their surface Ig (42–44). Indeed, B cells that accumulate in MRL/lpr mice chiefly have specificity for self-IgG (RFs) or DNA/histone (36). Such B cells bind particles (either immune complexes or nucleosomes) that are likely to contain multiple proteins, which could stimulate self-reactive T cells. Thus, these B cells are well suited to obtain T cell help. As certain autoreactive B cells expand and dominate the B cell repertoire, their relative importance in promoting disease via T cell activation (possibly including breaking peripheral T cell tolerance) (45, 46) and autoantibody secretion would escalate. This might represent an element of a positive feedback circuit that leads to fulminant disease.
Mamula, Janeway, and colleagues first suggested that B cells might be important autoantigen-presenting cells and might promote the breakdown of peripheral T cell tolerance (45–47). Using a cross-immunization scheme in normal mice, they were able to elicit autoreactive T cells in a fashion that was likely to be B cell dependent. They also suggested that this might be a mechanism for epitope spreading, a common phenomenon in both lupus and organ-specific autoimmunity. These studies were conducted in normal mice via immunization with heterologous proteins in Freund's adjuvant. Our work now lends support to the idea that these mechanisms are actually operating in spontaneously autoimmune animals.
Although we favor a role for B cells as APCs, we cannot formally rule out that B cells are also acting as producers of cytokines that promote T cell activation or pathology. B cells are known to produce cytokines under certain circumstances (48–51). However, since B cells are not considered major producers of cytokines, it is difficult to envision B cells as supplying rate-limiting quantities of cytokines, particularly in a scenario in which there is massive T cell activation. In addition, immunohistochemical analyses revealed few B cells in the proximity of the renal infiltrates of MRL/lpr mice (data not shown), making it very unlikely that B cells are the source of cytokines responsible for direct parenchymal injury.
Although IN and vasculitis were marked in mIgM mice compared with their B cell–deficient MRL/lpr counterparts, the median renal disease scores of the mIgM mice were not as high as those of the control MRL/lpr mice. Similarly, mIgM mice had substantially accelerated mortality compared with B cell–deficient MRL/lpr mice; but these Tgic mice had delayed mortality when compared with the wild-type. The lack of circulating autoantibody could certainly contribute to milder disease. Particularly for IN and vasculitis, it is likely that milder disease in mIgM mice is also due to a reduced Ig repertoire resulting from the fixed heavy chain Tg. It is possible that this restriction limited the generation of autoreactive B cells in the Tgic animals, thereby inhibiting the full potential of the B cell compartment to recognize self-antigens, activate them, and present these autoantigens to T cells. We predict that disease would have been more severe had a full Ig repertoire been expressed, but thus far the technology to produce such a mouse is not readily at hand.
The importance in SLE of cell-mediated autoimmune pathogenesis revealed by our studies is reminiscent of present views of organ-specific autoimmune diseases such as diabetes. It is intriguing that B cell deletion also inhibited diabetes in the nonobese diabetic (NOD) mouse (52–54). This result was interpreted as due to lack of APC function, a point that was not actually proven but could potentially be demonstrated directly using our Tgs on the NOD background. In any case, the similarities suggest that organ-specific and systemic autoimmunity may not need to be distinguished as much on the basis of pathogenic mechanism, but rather in extent of autoimmune targeting. Our studies further support the rationale for directly targeting B cells in the therapy of SLE (13, 16) and, by extension of this logic, to other autoimmune diseases.
Acknowledgments
We thank Dr. Joseph Craft and Dr. Mark Mamula for critically reading this manuscript, and Brian Kinlan for his technical help.
This work was supported by National Institutes of Health (NIH) grant R01-AR44077. A.M. Haberman was supported by a grant from the Donaghue Foundation. O.T.M. Chan and L.G. Hannum were supported by NIH training grant AI07019.
Abbreviations used in this paper
- dsDNA
double stranded DNA
- GN
glomerulonephritis
- IN
interstitial nephritis
- mIgM
membrane IgM
- NOD
nonobese diabetic
- NP
(4-hydroxy-3-nitro-phenyl) acetyl
- Tg
transgene
- Tgic
transgenic
References
- 1.Kotzin B. Systemic lupus erythematosus. Cell. 1996;85:303–306. doi: 10.1016/s0092-8674(00)81108-3. [DOI] [PubMed] [Google Scholar]
- 2.Hewicker M, Trautwein G. Glomerular lesions in MRL mice. A light and immunofluorescence microscopic study. J Vet Med. 1986;33:727–739. doi: 10.1111/j.1439-0450.1986.tb00093.x. [DOI] [PubMed] [Google Scholar]
- 3.Hewicker M, Trautwein G. Sequential study of vasculitis in MRL mice. Lab Anim. 1987;21:335–341. doi: 10.1258/002367787781363408. [DOI] [PubMed] [Google Scholar]
- 4.Jabs DA, Prendergast RA. Reactive lymphocytes in lacrimal gland and vasculitic renal lesions of autoimmune MRL/lpr mice express L3T4. J Exp Med. 1987;166:1198–1203. doi: 10.1084/jem.166.4.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexander EL, Moyer C, Travlos GS, Roths JB, Murphy ED. Two histopathologic types of inflammatory vascular disease in MRL/Mp autoimmune mice. Arthritis Rheum. 1985;28:1146–1155. doi: 10.1002/art.1780281011. [DOI] [PubMed] [Google Scholar]
- 6.Moyer CF, Strandberg JD, Reinisch CL. Systemic mononuclear-cell vasculitis in MRL/Mp-lpr/lpr mice. A histologic and immunocytochemical analysis. Am J Pathol. 1987;127:229–242. [PMC free article] [PubMed] [Google Scholar]
- 7.Alexopoulos E, Seron D, Hartley RB, Cameron JS. Lupus nephritis: correlation of interstitial cells with glomerular function. Kidney Int. 1990;37:100–109. doi: 10.1038/ki.1990.14. [DOI] [PubMed] [Google Scholar]
- 8.D'Amico, G. 1998. Tubulo-interstitial damage in glomerular diseases: its role in the progression of the renal damage. Nephrol. Dial. Transplant. 13(Suppl 1):80–85. [DOI] [PubMed]
- 9.O'Dell JR, Hays RC, Guggenheim SJ, Steigerwald JC. Tubulointerstitial renal disease in systemic lupus erythematosus. Arch Intern Med. 1985;145:1996–1999. [PubMed] [Google Scholar]
- 10.Sontheimer, R.D., and T.T. Provost. 1996. Systemic lupus erythematosus. In Cutaneous Manifestations of the Rheumatic Diseases. R.D. Sontheimer and T.T. Provost, editors. Williams and Wilkins, Baltimore, MD. 1–73.
- 11.Abe S, Amagasaki Y, Iyori S, Konishi K, Kato E, Sakaguchi H. Significance of tubulointerstitial lesions in biopsy specimens of glomerulonephritis patients. Am J Nephrol. 1989;9:30–37. doi: 10.1159/000167931. [DOI] [PubMed] [Google Scholar]
- 12.Nath KA. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am J Kidney Dis. 1992;20:1–17. doi: 10.1016/s0272-6386(12)80312-x. [DOI] [PubMed] [Google Scholar]
- 13.Shlomchik MJ, Madaio MP, Ni D, Trounstine M, Huszar D. The role of B cells in lpr/lpr-induced autoimmunity. J Exp Med. 1994;180:1295–1306. doi: 10.1084/jem.180.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen J, Trounstine M, Alt FW, Young F, Kurahara C, Loring JF, Huszar D. Immunoglobulin gene rearrangement in B cell deficient mice generated by targeted deletion of the JH locus. Int Immunol. 1993;5:647–656. doi: 10.1093/intimm/5.6.647. [DOI] [PubMed] [Google Scholar]
- 15.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 16.Chan O, Shlomchik MJ. A new role for B cells in systemic autoimmunity: B cells promote spontaneous T cell activation in MRL-lpr/lprmice. J Immunol. 1998;160:51–59. [PubMed] [Google Scholar]
- 17.Bruggemann M, Muller HJ, Burger C, Rajewsky K. Idiotypic selection of an antibody mutant with changed hapten binding specificity, resulting from a point mutation in position 50 of the heavy chain. EMBO (Eur Mol Biol Organ) J. 1986;5:1561–1566. doi: 10.1002/j.1460-2075.1986.tb04397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peterson ML, Perry RP. Regulated production of μs and μm RNA requires linkage of the poly (A) addition sites and is dependent on the length of the μm-μs intron. Proc Natl Acad Sci USA. 1986;83:8883–8887. doi: 10.1073/pnas.83.23.8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shlomchik MJ, Zharhary D, Camper S, Saunders T, Weigert M. A rheumatoid factor transgenic mouse model of autoantibody regulation. Int Immunol. 1993;5:1329–1341. doi: 10.1093/intimm/5.10.1329. [DOI] [PubMed] [Google Scholar]
- 20.Nose M, Okuda T, Gidlund M, Ramstedt U, Okada N, Okada H, Heyman B, Kyogoku M, Wigzell H. Mutant monoclonal antibodies with select alteration in complement activation ability. Impact on immune complex functions in vivo. J Immunol. 1988;141:2367–2373. [PubMed] [Google Scholar]
- 21.Losman MJ, Fasy TM, Novick KE, Monestier M. Relationships among antinuclear antibodies from autoimmune MRL mice reacting with histone H2A-H2B dimers and DNA. Int Immunol. 1993;5:513–523. doi: 10.1093/intimm/5.5.513. [DOI] [PubMed] [Google Scholar]
- 22.Losman JA, Fasy TM, Novick KE, Massa M, Monestier M. Nucleosome-specific antibody from an autoimmune MRL/Mp-lpr/lpr mouse. Arthritis Rheum. 1993;36:552–556. doi: 10.1002/art.1780360417. [DOI] [PubMed] [Google Scholar]
- 23.Chan O, Madaio MP, Shlomchik MJ. The roles of B cells in murine lupus. Ann NY Acad Sci. 1997;815:75–87. doi: 10.1111/j.1749-6632.1997.tb52046.x. [DOI] [PubMed] [Google Scholar]
- 24.Perkins DL, Listman JA, Marshak-Rothstein A, Kozlow W, Kelly VR, Finn PW, Rimm IJ. Restriction of the TCR repertoire inhibits the development of memory T cells and prevents autoimmunity in lprmice. J Immunol. 1996;156:4961–4968. [PubMed] [Google Scholar]
- 25.Rubio CF, Kench J, Russell DM, Yawger R, Nemazee D. Analysis of central B cell tolerance in autoimmune-prone MRL/lprmice bearing autoantibody transgenes. J Immunol. 1996;157:65–71. [PubMed] [Google Scholar]
- 26.Giese T, Davidson WF. Evidence for early onset, polyclonal activation of T cell subsets in mice homozygous for lpr. J Immunol. 1992;149:3097–3106. [PubMed] [Google Scholar]
- 27.Tarkowski A, Jonsson R, Sanchez R, Klareskog L, Koopman WJ. Features of renal vasculitis in autoimmune MRL lpr/lpr mice: phenotypes and functional properties of infiltrating cells. Clin Exp Immunol. 1988;72:91–97. [PMC free article] [PubMed] [Google Scholar]
- 28.Murata H, Kita Y, Sakamoto A, Matsumoto I, Matsumura R, Sugiyama T, Sueishi M, Takabayashi K, Iwamoto I, Saitoh Y, et al. Limited TCR repertoire of infiltrating T cells in the kidneys of Sjogren's syndrome patients with interstitial nephritis. J Immunol. 1995;155:4084–4089. [PubMed] [Google Scholar]
- 29.Brown Z, Robson RL, Westwick J. Regulation and expression of chemokines: potential role in glomerulonephritis. J Leukocyte Biol. 1996;59:75–80. doi: 10.1002/jlb.59.1.75. [DOI] [PubMed] [Google Scholar]
- 30.Yee J, Kuncio GS, Neilson EG. Tubulointerstitial nephritis following glomerulonephritis. Semin Nephrol. 1991;11:361–366. [PubMed] [Google Scholar]
- 31.Li S, Holdsworth SR, Tipping PG. Antibody independent crescentic glomerulonephritis in μ chain deficient mice. Kidney Int. 1997;51:672–678. doi: 10.1038/ki.1997.97. [DOI] [PubMed] [Google Scholar]
- 32.Diaz Gallo, C., A.M. Jevnikar, D.C. Brennan, S. Florquin, A. Pacheco-Silva, and V.R. Kelley. Autoreactive kidney-infiltrating T-cell clones in murine lupus nephritis. Kidney Int. 1992;42:851–859. doi: 10.1038/ki.1992.360. [DOI] [PubMed] [Google Scholar]
- 33.Penny MJ, Boyd RA, Hall BM. Role of T cells in the mediation of Heymann nephritis. II. Identification of Th1 and cytotoxic cells in glomeruli. Kidney Int. 1997;51:1059–1068. doi: 10.1038/ki.1997.148. [DOI] [PubMed] [Google Scholar]
- 34.Huang XR, Tipping PG, Apostolopoulos C, Oettinger C, D'Souza M, Milton G, Holdsworth SR. Mechanisms of T cell-induced glomerular injury in anti-glomerular basement membrane (GBM) glomerulonephritis in rats. Clin Exp Immunol. 1997;157:134–142. doi: 10.1046/j.1365-2249.1997.4091307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shlomchik MJ, Marshak-Rothstein A, Wolfowicz CB, Rothstein TL, Weigert MG. The role of clonal selection and somatic mutation in autoimmunity. Nature. 1987;328:805. doi: 10.1038/328805a0. [DOI] [PubMed] [Google Scholar]
- 36.Shan H, Shlomchik MJ, Marshak-Rothstein A, Pisetsky DS, Litwin S, Weigert MG. The mechanism of autoantibody production in an autoimmune MRL/ lpr mouse. J Immunol. 1994;153:5104–5120. [PubMed] [Google Scholar]
- 37.Kurt-Jones EA, Liano D, Hayglass KA, Benacerraf B, Sy M-S, Abbas AK. The role of antigen-presenting B cells in T cell priming in vivo: studies of B cell- deficient mice. J Immunol. 1988;140:3773–3778. [PubMed] [Google Scholar]
- 38.Ron Y, Sprent J. T cell priming in vivo: a major role for B cells in presenting antigen to T cells in lymph nodes. J Immunol. 1987;138:2848–2856. [PubMed] [Google Scholar]
- 39.Janeway CA, Jr, Ron J, Katz ME. The B cell is the initiating antigen-presenting cell in peripheral lymph nodes. J Immunol. 1987;138:1051–1055. [PubMed] [Google Scholar]
- 40.Chesnut RW, Grey HM. Studies on the capacity of B cells to serve as antigen-presenting cells. J Immunol. 1981;126:1075–1079. [PubMed] [Google Scholar]
- 41.Hayglass KT, Naides SJ, Scott CF, Benacerraf B, Sy M-S. T cell development in B cell-deficient mice. IV. The role of B cells as antigen-presenting cells in vivo. J Immunol. 1986;136:823–829. [PubMed] [Google Scholar]
- 42.Lanzavecchia A. Antigen-specific interactions between T and B cells. Nature. 1985;314:537. doi: 10.1038/314537a0. [DOI] [PubMed] [Google Scholar]
- 43.Roosnek E, Lanzavecchia A. Efficient and selective presentation of antigen–antibody complexes by rheumatoid factor B cells. J Exp Med. 1991;173:487–489. doi: 10.1084/jem.173.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Constant S, Schweitzer N, West J, Ranney P, Bottomly K. B lymphocytes can be competent antigen-presenting cells for priming CD4+T cells to protein antigens in vivo. J Immunol. 1995;155:3734–3741. [PubMed] [Google Scholar]
- 45.Mamula MJ, Lin R-H, Janeway CA, Jr, Hardin JA. Breaking T cell tolerance with foreign and self co-immunogens: a study of autoimmune B and T cell epitopes of cytochrome c. J Immunol. 1992;149:789–795. [PubMed] [Google Scholar]
- 46.Lin R-H, Mamula MJ, Hardin JA, Janeway CA., Jr Induction of autoreactive B cells allows priming of autoreactive T cells. J Exp Med. 1991;173:1433–1439. doi: 10.1084/jem.173.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mamula MJ, Fatenejad S, Craft J. B cells process and present lupus autoantigens that initiate autoimmune T cell responses. J Immunol. 1994;152:1453–1461. [PubMed] [Google Scholar]
- 48.Spencer NFL, Daynes RA. IL-12 directly stimulates expression of IL-10 by CD5+ B cells and IL-6 by both CD5+ and CD5−B cells: possible involvement in age-associated cytokine dysregulation. Int Immunol. 1997;9:745–754. doi: 10.1093/intimm/9.5.745. [DOI] [PubMed] [Google Scholar]
- 49.Ware CF, Crowe PD, Grayson MH, Androlewicz MJ, Browning JL. Expression of surface lymphotoxin and tumor necrosis factor on activated T, B, and natural killer cells. J Immunol. 1992;149:3881–3888. [PubMed] [Google Scholar]
- 50.Kouskoff V, Famiglietti S, Lacaud G, Lang P, Rider JE, Kay BK, Cambier JC, Nemazee D. Antigens varying in affinity for the B cell receptor induce differential B lymphocyte responses. J Exp Med. 1998;188:1453–1464. doi: 10.1084/jem.188.8.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fu YX, Huang G, Wang Y, Chaplin DD. B lymphocytes induce the formation of follicular dendritic cell clusters in a lymphotoxin alpha-dependent fashion. J Exp Med. 1998;187:1009–1018. doi: 10.1084/jem.187.7.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Noorchashm H, Noorchashm N, Kern J, Rostami SY, Barker CF, Naji A. B-cells are required for the initiation of insulitis and sialitis in nonobese diabetic mice. Diabetes. 1997;46:941–946. doi: 10.2337/diab.46.6.941. [DOI] [PubMed] [Google Scholar]
- 53.Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, Fleming SA, Leiter EH, Shultz LD. B lymphocytes are essential for the initiation of T cell–mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Ig μ null mice. J Exp Med. 1996;184:2049–2053. doi: 10.1084/jem.184.5.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Falcone M, Lee J, Patstone G, Yeung B, Sarvetnick N. B lymphocytes are crucial antigen-presenting cells in the pathogenic autoimmune response to GAD65 antigen in nonobese diabetic mice. J Immunol. 1998;161:1163–1168. [PubMed] [Google Scholar]