

Abstract
Autophagy is a degradative pathway by which cells sequester nonessential, bulk cytosol into double-membrane vesicles (autophagosomes) and deliver them to the vacuole for recycling. Using this strategy, eukaryotic cells survive periods of nutritional starvation. Under nutrient-rich conditions, autophagy machinery is required for the delivery of a resident vacuolar hydrolase, aminopeptidase I, by the cytoplasm to vacuole targeting (Cvt) pathway. In both pathways, the vesicle formation process requires the function of the starvation-induced Aut7 protein, which is recruited from the cytosol to the forming Cvt vesicles and autophagosomes. The membrane binding of Aut7p represents an early step in vesicle formation. In this study, we identify several requirements for Aut7p membrane association. After synthesis in the cytosol, Aut7p is proteolytically cleaved in an Aut2p-dependent manner. While this novel processing event is essential for Aut7p membrane binding, Aut7p must undergo additional physical interactions with Aut1p and the autophagy (Apg) conjugation complex before recruitment to the membrane. Lack of these interactions results in a cytosolic distribution of Aut7p rather than localization to forming Cvt vesicles and autophagosomes. This study assigns a functional role for the Apg conjugation system as a mediator of Aut7p membrane recruitment. Further, we demonstrate that Aut1p, which physically interacts with components of the Apg conjugation complex and Aut7p, constitutes an additional factor required for Aut7p membrane recruitment. These findings define a series of steps that results in the modification of Aut7p and its subsequent binding to the sequestering transport vesicles of the autophagy and cytoplasm to vacuole targeting pathways.
Keywords: autophagy, lysosome, protein targeting, vacuole, yeast
Introduction
Stress conditions elicit an array of specialized responses in eukaryotic cells. To survive nutritional deprivation, cells must quickly mobilize the transport of nonessential cytoplasmic materials to the principal degradatory organelle, the vacuole/lysosome, for subsequent digestion and reuse in essential biosynthetic processes (reviewed in Kim and Klionsky 2000). Accordingly, the level of intracellular degradation increases dramatically when cells encounter starvation conditions with >85% of the degradative capacity of the cell mediated by vacuolar proteolysis (Teichert et al. 1989). Sustained starvation conditions result in turnover of nearly half of all cellular proteins in the vacuole.
The starvation-induced autophagy pathway provides the molecular machinery to sequester bulk cytoplasm destined for vacuolar degradation (Klionsky and Ohsumi 1999). The delivery mechanism entails enwrapping cytoplasmic material inside double-membrane vesicles called autophagosomes (see Fig. 9 B; Takeshige et al. 1992). Fusion of the outer vesicle membrane with the vacuole results in the release of the inner single-membrane autophagic body into the vacuolar lumen. The subsequent breakdown of the autophagic body by resident vacuolar hydrolases results in the digestion and recycling of cytoplasmic cargo. Genetic analysis of the autophagy (Apg) mutants (apg, aut) (Tsukada and Ohsumi 1993; Thumm et al. 1994) in Saccharomyces cerevisiae indicates an overlap with the cytoplasm to vacuole targeting (Cvt) pathway that is used to deliver the resident hydrolase aminopeptidase I (API) (Klionsky et al. 1992; Harding et al. 1995). Consistent with the genetic overlap between the two pathways (Harding et al. 1996; Scott et al. 1996), the Cvt pathway shares common mechanistic features with autophagy, including the formation of double-membrane transport vesicles (Cvt vesicles) and the breakdown of the single-membrane vesicles (Cvt bodies) in the vacuolar lumen (Baba et al. 1997; Scott et al. 1997). Therefore, precursor API (prAPI) uses the Cvt pathway during nutrient-rich conditions and the autophagy pathway during starvation conditions for import into the vacuole.
Figure 9.
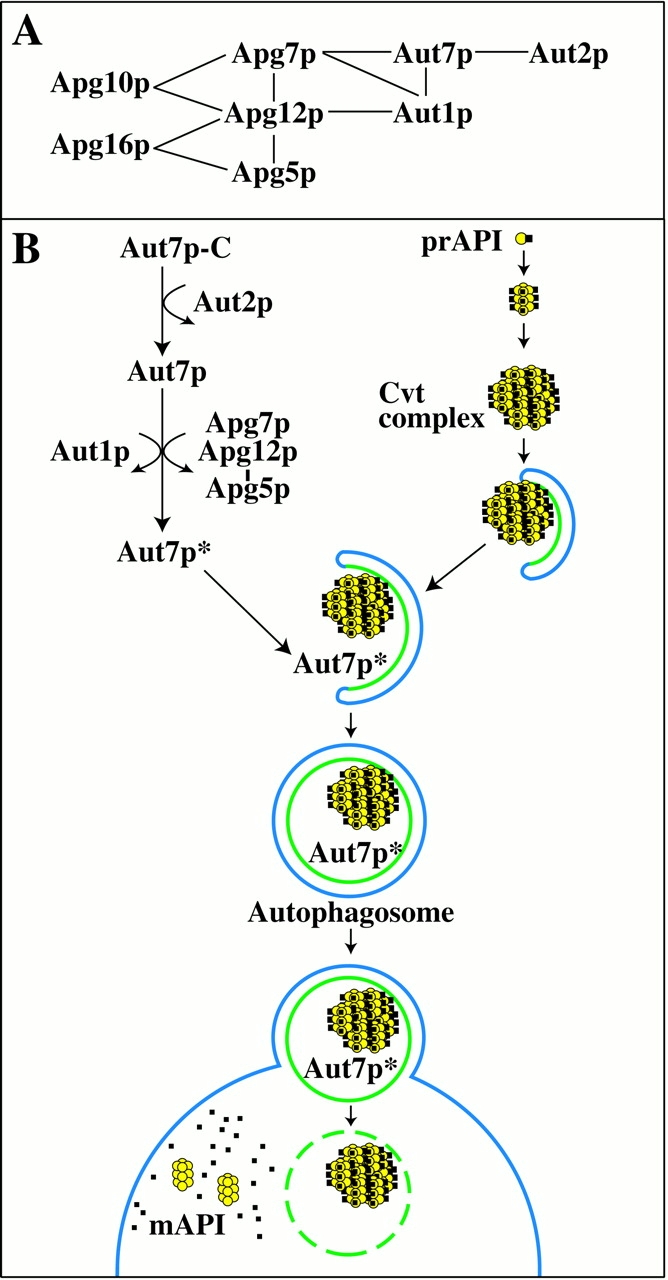
(A) Molecular interactions between autophagy components. Apg5p, 7p, 10p, 12p, and 16p constitute the Apg conjugation system. This covalent protein-modification system is essential for the Cvt and autophagy pathways. Interactions between Apg conjugation components and Aut7p, Aut1p, and Aut2p have also been recently demonstrated. Details are discussed in the text. (B) A model of Aut7p membrane binding in the context of prAPI transport. In summary, we have defined three discrete events that lead to the membrane binding of Aut7p. First, Aut7p is synthesized in the cytosol and subsequently cleaved in an Aut2p-dependent manner. Once cleaved, Aut1p and the Apg conjugation system further interact with Aut7p to facilitate its binding to the membrane. These steps required for Aut7p membrane binding are presented in the context of prAPI import by the autophagy pathway. Details are discussed in the text.
Analysis of the autophagy and cvt mutants indicates that many of the characterized components are required at an early stage(s) in vesicle formation. Mutants defective in this part of the pathway all possess a phenotype in which prAPI binds to a pelletable membrane but remains accessible to exogenous protease treatment, indicating that a completed vesicle has not yet formed (Kim et al. 1999; Kirisako et al. 1999; George et al. 2000; Huang et al. 2000; Noda et al. 2000). Among the requirements for this stage of vesicle formation and/or completion is a novel Apg conjugation system comprised of Apg5p, Apg7p, Apg10p, Apg12p, and Apg16p (Mizushima et al. 1998, Mizushima et al. 1999; Kim et al. 1999; Shintani et al. 1999; Tanida et al. 1999; George et al. 2000). Apg7p shares homology with the E1 ubiquitin activating enzyme Uba1p (Kim et al. 1999; Tanida et al. 1999). Through ATP hydrolysis, Apg7p forms a thioester bond to Apg12p. The activated Apg12p is then transferred to Apg10p (Shintani et al. 1999), a protein conjugating enzyme, and ultimately forms a covalent isopeptide linkage to Apg5p. Apg16p is required to form a multimeric complex with the Apg12p-Apg5p conjugate. Although mutants in the Apg conjugation system are defective in Cvt/autophagic vesicle formation, the specific function of this covalent modification system remains to be determined.
Autophagosomes are substantially larger than Cvt vesicles that form under vegetative conditions (Baba et al. 1997). To accommodate the significant increase in size, a corresponding increase in membrane material, as well as structural components, are likely required to be recruited to the site of autophagosome formation. Recent studies have identified Aut7p as the first component to be localized to Cvt vesicles and autophagosomes (Kirisako et al. 1999; Huang et al. 2000). AUT7 is allelic to the APG8/CVT5 gene (Harding et al. 1996; Scott et al. 1996), and the aut7 mutant is defective at the step of vesicle formation. Interestingly, Aut7p expression increases significantly under conditions that promote autophagy, suggesting that it may represent a structural component in vesicle formation.
The recruitment of soluble vesicle coat factors to target membranes is a critical step in vesicle formation (Wieland and Harter 1999). In the classical paradigm of COPI vesicle formation, vesicle assembly requires the initial recruitment of a small GTPase (ARF1•GTP) to the target membrane, a reaction catalyzed by a nucleotide exchange factor. This is followed by the recruitment of the preassembled coatomer complex on the membrane through direct interactions with the membrane-bound ARF1•GTP. In addition, the cytoplasmic domains of the p24 family of integral membrane proteins interact with coatomer subunits. This trimeric complex of ARF, coatomer, and p24 are thought to provide the molecular basis for membrane deformation and vesicle budding. It is not known whether analogous components function in the formation of the sequestering vesicle during autophagy or the Cvt pathway.
In this study, we examined the factors required for the recruitment of the soluble Aut7 protein to the membrane, an early step leading to Cvt and autophagic vesicle formation. After synthesis in the cytosol, Aut7p is proteolytically processed in an Aut2p-dependent manner. While this novel processing event is essential for Aut7p membrane binding, Aut7p must undergo additional physical interactions with Aut1p and the Apg conjugation complex. In this study, we assign a functional role for Aut1p and the Apg conjugation system as mediators of Aut7p membrane recruitment.
Materials and Methods
Yeast Strains
The yeast strains used in this study are listed in Table .
Table 1.
Yeast Strains Used in this Study
| Strain | Genotype | |
|---|---|---|
| JKY007 | SEY6210 apg9Δ::HIS3 | Noda et al. 2000 |
| MGY101 | SEY6210 apg5Δ::LEU2 | George et al. 2000 |
| NNY20 | ura3 trp1 leu2 apg1Δ::LEU2 | Matsuura et al. 1997 |
| SEY6210 | MATα his3-Δ200 leu2-3,112 lys2-801 trp1-Δ901 ura3-52 suc2-Δ9 GAL | Robinson et al. 1988 |
| SSY101 | MGY101 × YMT A apg5Δ::LEU2 pep4Δ::HIS3 | George et al. 2000 |
| TVY1 | SEY6210 pep4Δ::LEU2 | Gerhardt et al. 1998 |
| VDY101 | SEY6210 apg7Δ::LEU2 | Kim et al. 1999 |
| WPHY2HA | SEY6210 aut2::AUT2HA | This study |
| WPHYD1 | SEY6210 aut1Δ::HIS5 | This study |
| WPHYD2 | SEY6210 aut2Δ::LEU2 | This study |
| WPHYD7 | SEY6210 aut7Δ::LEU2 | This study |
| YMT A | MATα his3-11,15 leu2-3,112 ura3 pep4::HIS3 | Thumm et al. 1994 |
| YNM101 | MATα ura3 leu2 trp1 his3 apg12Δ::HIS3 | George et al. 2000 |
Antibodies
To prepare antiserum to Aut1p, synthetic peptides corresponding to amino acids 138–156 and 245–263 of Aut1p were synthesized with a Cys residue at the amino terminus and conjugated individually to keyhole limpet hemocyanin (Multiple Peptide Systems). Standard procedures were used to generate antiserum in a New Zealand white rabbit (Harlow and Lane 1999). Antisera against Aut7p and API were prepared as described previously (Klionsky et al. 1992; Huang et al. 2000, respectively). Antiserum against phosphoglycerate kinase (PGK) was provided by Dr. Jeremy Thorner (University of California at Berkeley, Berkeley, CA) (Baum et al. 1978).
Materials and Growth Media
The lipophilic dye FM 4-64 was from Molecular Probes. Ficoll-400 was from Amersham Pharmacia Biotech. Immobilon-P (polyvinylidene fluoride) was from Millipore. Complete™ EDTA-free protease inhibitor cocktail was from Roche Molecular Biochemicals. Oligonucleotides were synthesized by Operon. Oxalyticase was from Enzogenetics. YNB was from DifCo. Copper-free YNB was from Bio101. All restriction enzymes and VentR DNA polymerase were from New England Biolabs, Inc. The pGFP(416)SK and pGFP(426)SK vectors for expressing green fluorescent protein (GFP) fusions were a gift from Dr. Tom Vida (University of Texas Health Sciences Center, Houston, TX). The copper-inducible promoter-based vectors, pCu416, pCu414, and pCu426, were gifts from Dr. Dennis J. Thiele (University of Michigan, Ann Arbor, MI; Labbé and Thiele 1999). The ME3 vector, which contains a triple, tandem repeat of the hemagglutinin (HA) epitope sequences followed by a termination cassette, and then the Schizosaccharomyces pombe HIS5 auxotrophic marker sequences, was a gift from Dr. Neta Dean (State University of New York, Stony Brook, NY). All other reagents were from Sigma-Aldrich.
Yeast strains were grown in synthetic minimal medium (SMD; 0.67% yeast nitrogen base, 2% glucose and auxotrophic amino acids and vitamins as needed). Synthetic minimal medium containing 2% glucose without ammonium sulfate or amino acids (SD-N) was used for nitrogen starvation experiments.
Disruption of the AUT1, AUT2, and AUT7 Chromosomal Loci
The chromosomal AUT1 locus was deleted by a PCR-based, one-step procedure (Noda et al. 2000). The S. pombe HIS5 gene was amplified from the ME3 vector using the following oligonucleotides containing vector sequences outside of the HIS5 marker flanked by AUT1 sequences that encode regions at the beginning and end of the AUT1 open reading frame (ORF): 5′-GTAGTTGGAGAGAATATCTTACCCCCATAACGCACAAATCC-CCCGGGCTGCAGGAATTC-3′ and 5′-GGTCTACCCGTAACGAA-TCATCAATATCGTCTTGTAAATCGTCGACGGTATCGATA-AGC-3′. The PCR product was transformed into the SEY6210 wild type strain and grown on minimal plates lacking histidine. Transformants were then assayed for precursor API accumulation. The correct disruption strain was confirmed by PCR. The PCR-based strategy was also employed to replace the AUT2 and AUT7 chromosomal loci with the LEU2 auxotrophic marker. The 5′ and 3′ oligonucleotides for the AUT2 deletion construct were 5′-GGCGTTTTTGAAGGCTCGTCCGAAGAACCGGCTGCTCTTGGAGAACTTCTAGTATATCC-3′ and 5′-CGATGAAGACGGCCTACCGCCGGCAATGCCTACTGACTGATCGACT-ACGTCGTAAGGCCG-3′, respectively. The 5′ and 3′ oligonucleotides for the AUT7 deletion construct were 5′-GTCTACATTTAAGTCTGAATATCCATTTGAAAAAAGGAAGGCGGAGAACTTCTAG-TATATCC-3′ and 5′-CCTGCCAAATGTATTTTCTCCTGAGTAAGTGACATACAAAAACCATCGACTACGTCGTAAGGCCG-3′, respectively.
Construction of Plasmids
To construct the pAUT1(426) plasmid, the AUT1 ORF and flanking sequences were PCR-amplified from a wild-type yeast genomic DNA preparation. The 5′ and 3′ oligonucleotides used for the amplification were 5′-CAGTGTGCCCATTTTTTACTCGAGTGTCGCCGAATTCGTTGGTAGAGAACC-3′ and 5′-GAAAGTGGTTTTAAGGAAGAAGCTTGTTCCTGCATATTTATAGTTAATTCC-3′, respectively. The 5′ oligonucleotide contains a XhoI site and the 3′ oligonucleotide contains a HindIII site. The AUT1 PCR product was subcloned into pRS426 using the XhoI and HindIII sites. The resulting pAUT1(426) clone contains the AUT1 ORF with 175 bp of upstream and 98 bp of downstream sequences.
The AUT2 clone was also PCR-amplified from a wild-type yeast genomic DNA preparation. The 5′ and 3′ oligonucleotides used for the amplification were 5′- GCTACCACGTGCATGCCTACGTAAGCTTCCTTGATAATGACTATGCC-3′ and 5′-GCGATGACTAGGCATTTTGGACTTCGACTCGAGTTTATACCACCGTTGTCTTAATCTTC-3′, respectively. The 5′ oligonucleotide contains a HindIII site and the 3′ oligonucleotide contains a XhoI site. The AUT2 PCR product was subcloned into pRS416 using the HindIII and XhoI sites. The resulting pAUT2(416) clone contains the AUT2 ORF with 269 bp of upstream and 227 bp of downstream sequences.
To make the AUT2GFP fusion construct, the AUT2 ORF was PCR-amplified using the pAUT2(416) construct as the template. The 5′ and 3′ oligonucleotides used for the amplification were 5′-GCTTCCTTGATAATGAGCTCGCCTTTTTGCC-3′ and 5′-GAATTCCTTACACTAGACTAGTTCATCAATAGG-3′, respectively. The 5′ oligonucleotide contains a SacI site and the 3′ oligonucleotide contains a SpeI site that starts 7 bp upstream of the stop codon. The PCR product was subcloned into the pGFP(416)SK and pGFP(426)SK vectors using the SacI and SpeI sites. The resulting pAUT2GFP(416) and pAUT2GFP(426) constructs contain 253 bp of upstream AUT2 sequences followed by the AUT2 ORF, and then the inframe GFP ORF and termination sequences.
A PCR-based approach was used to create a strain expressing an Aut2HAp fusion protein from the AUT2 chromosomal locus. The 5′ oligonucleotide encoded 40 bp of the COOH-terminal AUT2 ORF followed by the start of the triple HA sequences; the 3′ oligonucleotide contained 40 bp of 3′ untranslated region of AUT2 followed by ME3 vector sequences adjacent to the HIS5 marker: 5′-GGAAACGGTAGGTATTCACAGTCCTATTGATGAAAAATGCTACCCATACGATGTTCCT-3′ and 5′-GGATGATCAAACGAATAAGAACAAAAGAACTGGATACCTCGTCGACGGTATCGATAAG-3′. The ME3 vector was used as the PCR template. The resulting PCR product consisted of 40 bp of the COOH-terminal AUT2 ORF, followed by the triple HA epitope sequences, the termination cassette, the HIS5 auxotrophic marker, and, finally, 40 bp of the AUT2 3′ untranslated region. The PCR product was transformed into the SEY6210 wild-type strain. Chromosomal integration of the PCR product by homologous recombination resulted in an inframe fusion of the triple-HA epitope sequence to the COOH-terminal coding sequence of the AUT2 ORF. Recombinants were selected on minimal plates lacking histidine. A strain expressing the Aut2HAp fusion protein was confirmed by PCR and by anti–HA immunoblots.
The construct for the fusion protein of Aut7p followed by GFP at its COOH terminus [pAUT7GFP(416)] was made by PCR amplifying the AUT7 ORF and upstream sequences using the pAUT7(424) plasmid (Huang et al. 2000) as a template. The 5′ and 3′ oligonucleotides used for the PCR amplification were 5′-GAATTCTAACTGTCGCGGCCGCTAATGTTGTATAATACCCGTG-3′ and 5′-CATTTCTTTTCATATAAAAGAGGATCCGCCAAATGTATTTTCTCC-3′, respectively. The 5′ oligonucleotide contains a NotI site and the 3′ oligonucleotide contains an inframe BamHI site that changes the penultimate arginine codon and the stop codon to glycine and serine codons, respectively. The 0.6-kb PCR product was subcloned into the pCGFP(416) and pCGFP(426) GFP vectors, which have been previously described (Kim et al. 1999). The resulting constructs, pAUT7GFP(416) and pAUT7GFP(426), contain 209 bp of AUT7 upstream sequence, followed by the GFP ORF and actin termination sequences.
To make NH2-terminal GFP fusion proteins under the control of a regulable promoter, the CUP1 promoter cassette from the pCu(416) vector was subcloned into pGFP(416)SK by a SacI–SpeI digest, thus creating the pCuGFP(416) vector. The CUP1-promoter driven construct for the fusion of GFP to the NH2 terminus of Aut7p was then made by PCR amplifying the AUT7 ORF using pAUT7(416) as the template. The 5′ and 3′ oligonucleotides used for the PCR amplification were 5′-CTAGAGACCCGGGGTCTACATTTAAGTCTGAATATCC-3′ and 5′-CGCTTCATTTCTTTTCATACTCGAGACTACCTGCCAAATG-3′, respectively. The 5′ oligonucleotide contains an XmaI site that is in the same reading frame as the end of the GFP ORF of pCuGFP(416) and the 3′ oligonucleotide contains an XhoI site. The PCR product was subcloned into pCuGFP(416) by an XmaI–XhoI digest, resulting in the pCuGFPAUT7(416) plasmid.
Subcellular Fractionation
For subcellular fractionation experiments, strains were grown to midlog phase in SMD medium and spheroplasted as described earlier (Kim et al. 1999) or shifted to SD-N medium for 2 h before the spheroplasting procedure. Spheroplasts were subjected to differential osmotic lysis in TEA lysis buffer (10 mM triethanolamine, pH 7.4, 200 mM sorbitol, 1 mM EDTA) containing the Complete™ EDTA-free protease inhibitor cocktail, 1 mM pepstatin, and 1 mM PMSF. After a preclearing step at 100 g, 4°C, 5 min, the lysate was subjected to low-speed centrifugation at 13,000 rpm in an Eppendorf microcentrifuge, 15 min, 4°C. The low-speed supernatant fraction (S13) was further resolved into high speed supernatant and pellet fractions by centrifugation at 100,000 g, 30 min, 4°C in a TL100 centrifuge (Beckman Coulter). Samples from each step of the differential centrifugation procedure were precipitated with 10% TCA and washed twice with acetone before SDS-PAGE/immunoblot analyses.
Membrane Flotation and Protease Sensitivity Analyses
The procedures for the membrane flotation and protease sensitivity were performed following a previously described protocol (Huang et al. 2000; Noda et al. 2000). In brief, the indicated strains were grown to midlog phase and converted to spheroplasts. The spheroplasts were lysed in PS200 lysis buffer (20 mM Pipes, pH 6.8, 200 mM sorbitol, and 5 mM MgCl2) at a concentration of 20 OD600/ml. A portion was removed for the total fraction (T). The remaining lysates were separated into supernatant (S) and pellet (P) fractions by centrifugation at 5,000 rpm, 5 min, 4°C. For the protease-sensitivity assay, the pellet was resuspended in PS200 lysis buffer in the presence or absence of proteinase K (50 μg/ml) and 0.2% Triton X-100. The resuspended pellets were incubated on ice for 20 min and TCA precipitated. For the membrane flotation analysis, the pellets were resuspended in 100 μl of 15% Ficoll-400 (wt/vol) in PS200 lysis buffer with protease inhibitor cocktail, with or without 0.2% Triton X-100. Overlays with 1 ml of 13% Ficoll-400 and 200 μl of 2% Ficoll-400, both in PS200 lysis buffer, were added, and these step gradients were subjected to centrifugation at 13,000 rpm for 10 min at 25°C. The top 400 μl were collected as the “float” (F) fraction and TCA precipitated. All samples were washed in acetone and subjected to SDS-PAGE/immunoblot analysis.
Confocal and Conventional Fluorescence Microscopy
For confocal microscopy analysis of yeast strains expressing the GFPAut7p fusion protein under the control of the CUP1 copper-inducible promoter, cells were grown to midlog phase in SMD medium lacking copper and uracil. GFPAut7p fusion protein expression was induced with 10 or 50 μM CuSO4 for 2–3 h, followed by FM 4-64 labeling at a final concentration of 10 μg/ml for 20 min. The FM 4-64–labeled cells were incubated in YPD for 90 min to allow uptake of the dye, and then viewed directly or shifted to SD-N medium for 3 h to induce autophagy. Cells were examined on a Leica DM IRB confocal microscope. Eight scans of each optical section were averaged for the final images, except in Fig. 2B and Fig. C (below), where single scans of each image were taken.
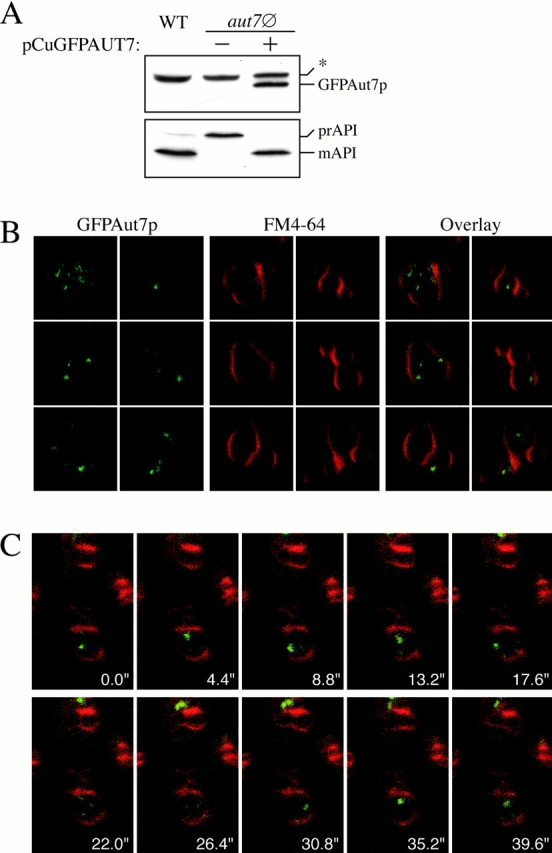
Figure 2.
A GFP fusion to the NH2 terminus of Aut7p (GFPAut7p) serves as a functional marker for the Cvt and autophagy pathways. (A) The aut7Δ (WPHYD7) strain was transformed with a plasmid encoding GFP fused to the NH2 terminus of Aut7p and under the control of the CUP1 copper-inducible promoter (pCuGFPAUT7). Cells were grown in SMD medium lacking copper until midlog phase, and then induced with 50 μM CuSO4 for 3 h. Protein extracts were prepared as described in Materials and Methods and examined by immunoblots with anti–Aut7p (top) and anti–API (bottom) antiserum. *Background band. GFPAut7p is expressed as a full-length fusion protein and complements the prAPI defect in aut7Δ cells. (B) GFPAut7p labels Cvt bodies inside the vacuole. The pep4Δ (TVY1) strain was transformed with pCuGFPAUT7 and grown to midlog phase in SMD medium lacking copper. GFPAut7p expression was induced with 10 μM CuSO4 for 3 h. After labeling the vacuoles with FM 4-64, the cells were examined on a Leica DM IRB confocal microscope. In nutrient-rich conditions, GFPAut7p accumulates inside Cvt bodies in pep4Δ cells. (C) Time-series examination of Cvt bodies. GFPAut7p-expressing pep4Δ (TVY1) cells were treated as in B. A single optical plane of cells was followed over the time course indicated. (D) GFPAut7p labels autophagic bodies inside the vacuole. GFPAut7p expression was induced with 50 μM CuSO4 for 3 h in pep4Δ (TVY1) cells. After labeling the vacuoles with FM 4-64, the cells were shifted to SD-N for 3 h. In starvation conditions, GFPAut7p accumulates inside autophagic bodies in pep4Δ cells.

Results
Aut7p Membrane Binding Requires an Aut2p-dependent Proteolytic Cleavage Event
Aut7p is the first component to be identified on Cvt vesicles and autophagosomes and, as such, serves as a valuable vesicle marker for the pathway (Lang et al. 1998; Kirisako et al. 1999; Huang et al. 2000). To study the role of Aut7p in the vesicle formation process, we constructed a GFP fusion protein to the COOH terminus of Aut7p. Based on previous studies (Kirisako et al. 1999; Huang et al. 2000), the Aut7pGFP fusion was predicted to be localized to both Cvt vesicles and autophagosomes destined for degradation in the vacuole. A wild-type strain was transformed with the pAUT7GFP fusion construct and examined by confocal microscopy (Fig. 1 A). The lipophilic styryl dye FM 4-64 was used to label vacuoles. Contrary to previous findings with native Aut7p, the Aut7pGFP fusion protein was not localized to Cvt vesicles, but instead appeared to be distributed uniformly in the cytosol (Fig. 1 A). Shifting the cultures to medium lacking nitrogen (SD-N) to induce autophagy and Aut7 recruitment to autophagosomes did not occur as the fluorescent signal from Aut7pGFP remained diffusely localized in the cytosol (data not shown). Nevertheless, the Aut7pGFP fusion plasmid complemented the prAPI accumulation phenotype in the aut7 null strain, suggesting that the fusion protein was functional in some capacity to rescue the transport defect of the aut7 mutation (Fig. 1 B).
Figure 1.
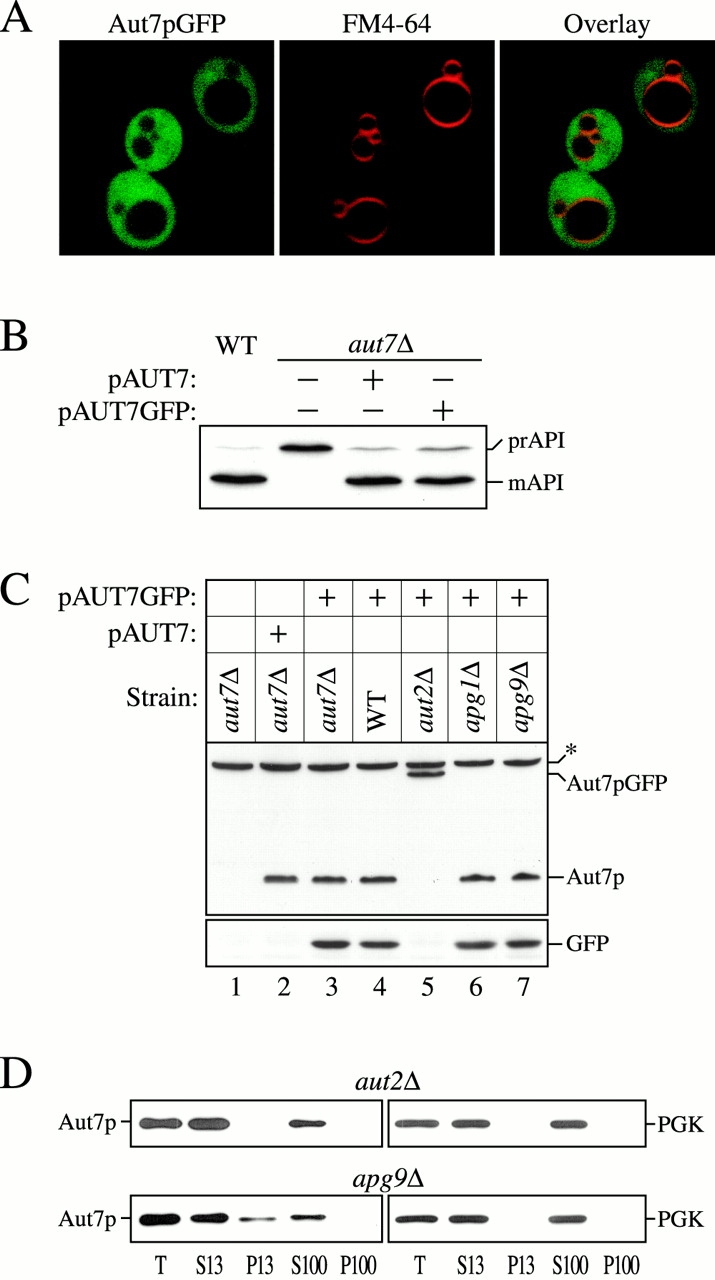
Aut2p-dependent proteolytic processing of Aut7p is required for its membrane binding. (A) Localization of Aut7pGFP. Wild-type (WT; SEY6210) cells transformed with a multicopy plasmid encoding GFP fused to the COOH terminus of Aut7p (Aut7pGFP) were grown in SMD to midlog phase and labeled with FM 4-64 to identify vacuoles. The labeled cells were viewed directly with a Leica DM IRB confocal microscope (see Materials and Methods). The cells show a diffuse cytosolic GFP pattern due to the Aut7p COOH-terminal cleavage event, which liberates GFP into the cytosol (see below). (B) WT (SEY6210) and aut7Δ (WPHYD7) strains transformed with either the centromeric AUT7 plasmid (pAUT7) or AUT7GFP plasmid were grown to midlog phase in SMD. Protein extracts were prepared and analyzed by immunoblots using antiserum to API. The positions of the precursor and mature forms of API are indicated. Both the pAUT7 and pAUT7GFP plasmids rescue the prAPI transport defect in aut7Δ. (C) WT (SEY6210), aut7Δ (WPHYD7), aut2Δ (WPHYD2), apg1Δ (NNY20), and apg9Δ (JKY007) strains transformed with either the centromeric pAUT7 or pAUT7GFP plasmids were grown to midlog phase in SMD. Protein extracts were prepared and analyzed by immunoblots using antiserum to Aut7p (top) or GFP (bottom). *Background band. The COOH-terminal processing of Aut7pGFP is dependent on Aut2p. (D) Aut7p subcellular fractionation pattern. Cells from aut2Δ and the apg9Δ control strain were grown to midlog phase in SMD, and then shifted to SD-N autophagy induction conditions for 2 h before converting them to spheroplasts. The spheroplasts were lysed in TEA osmotic lysis buffer (see Materials and Methods). After a preclearing centrifugation step at 100 g for 5 min to remove unlysed spheroplasts, the total lysate (T) was separated into low-speed supernatant (S13) and pellet (P13) fractions. The S13 fraction was further separated into 100,000 g supernatant (S100) and pellet (P100) fractions. The fractionated samples were subjected to immunoblot analysis using antiserum to Aut7p and the cytosolic marker protein PGK. The proteolytic cleavage of Aut7p is required for its subsequent association with a P13 membrane fraction.
Concomitant with the confocal microscopy analysis of the Aut7pGFP fusion protein, we wanted to confirm that the fusion protein was expressed properly (Fig. 1 C). Yeast strains were transformed with the pAUT7GFP plasmid and grown under vegetative conditions to midlog phase in SMD medium. Protein extracts were prepared from each strain and subjected to immunoblot analysis with anti–Aut7p and –GFP antibodies. In both wild-type and aut7Δ cells transformed with the pAUT7GFP plasmid, a proteolytic processing event cleaves the Aut7pGFP fusion into separate Aut7p and GFP proteins (Fig. 1 C, 3 and 4). The Aut7p moiety resulting from cleavage appeared identical in electrophoretic mobility to the Aut7p expressed from the pAUT7 plasmid (Fig. 1 C, compare 2 with 3), suggesting that the cleavage occurred near the COOH terminus of the protein. Therefore, proteolytic cleavage of the Aut7pGFP fusion releases the GFP domain into the cytosol, consistent with the confocal microscopy observations in Fig. 1 A, while the Aut7p portion of the fusion protein functions to complement the prAPI transport defect of the aut7Δ strain.
We next wanted to identify components that could mediate the proteolytic processing of Aut7p. One potential candidate was Aut2p, which has been demonstrated to interact with Aut7p by affinity chromatography (Lang et al. 1998). Immunoblot analysis of the Aut7pGFP fusion protein in the aut2 null genetic background indicated that the fusion protein remained intact, while in other apg mutants, such as apg1Δ and apg9Δ, Aut7p processing still occurred. These findings indicate that Aut2p mediates the COOH-terminal proteolytic cleavage of Aut7p. Because these strains were grown in rich medium, high expression of the endogenous Aut7p was not induced. As a result, the only detectable Aut7p in our experimental conditions was derived from the pAUT7 and pAUT7GFP plasmids. Taken together, the Aut2p-mediated cleavage of the Aut7pGFP fusion protein agrees with the recent study by Kirisako et al. 2000 demonstrating that Aut2p/Apg4p functions as a novel cysteine endopeptidase that proteolytically removes the COOH-terminal arginine residue of Aut7p.
Aut7p is a hydrophilic, soluble protein that is recruited to forming autophagosomes. To determine whether the COOH-terminal processing of Aut7p was required for its membrane association, subcellular fractionations were performed. Aut7p in the aut2Δ strain exists in the uncleaved form, while in other apg mutants, such as apg9Δ, Aut7p exists in the proteolytically modified state (Fig. 1 C). Therefore, cells from aut2Δ and apg9Δ strains were grown to midlog phase under vegetative conditions, shifted to nitrogen starvation conditions for 2 h to induce autophagy and Aut7p expression, and then converted to spheroplasts. The spheroplasts were then subjected to differential osmotic lysis conditions that allow lysis of the plasma membrane while retaining the integrity of subcellular compartments. The lysed spheroplasts were then centrifuged to separate the lysates into low-speed supernatant and pellet fractions (S13 and P13, respectively). The S13 fraction was then further separated into high-speed supernatant and pellet fractions (S100 and P100, respectively). In the apg9Δ strain, where Aut7p has undergone the proteolytic cleavage event, a significant population of Aut7p appeared in the low-speed pellet fraction (Fig. 1 D). In contrast, the uncleaved Aut7p in the aut2Δ strain was distributed entirely in the soluble supernatant fractions. Taken together, these results indicate that the Aut2p-mediated COOH-terminal processing of Aut7p is required for Aut7p membrane association, in agreement with recent findings by Kirisako et al. 2000.
A GFP Fusion to the NH2 Terminus of Aut7p is an In Vivo Marker for both the Cvt Pathway and Autophagy
Cleavage of GFP from the COOH terminus of Aut7p provided a useful diagnostic tool for examining the novel proteolytic processing reaction that modifies the Aut7 protein. However, the Aut7pGFP construct could not be used to follow localization of Aut7p. We next explored whether a GFP fusion to the NH2 terminus of Aut7p would provide an intact, functional, fusion protein that could be used to monitor the Cvt and autophagy pathways in vivo. The NH2-terminal GFPAut7p fusion was placed under the control of a copper-inducible promoter (pCuGFPAUT7) and transformed into aut7Δ cells. Expression of the GFP Aut7p fusion protein was induced by the addition of 50 μM CuSO4 for 3 h before immunoblot analysis. The expressed GFPAut7p fusion protein was maintained in its full-length form and complemented the prAPI accumulation phenotype in the aut7Δ strain, indicating that it was an intact, functional fusion protein (Fig. 2 A).
Previous studies have demonstrated that Aut7p is localized to both Cvt vesicles and autophagosomes that are targeted to the vacuole (Kirisako et al. 1999, Kirisako et al. 2000; Huang et al. 2000). Further, these studies demonstrated the requirement for Aut7p in the formation/completion of transport vesicles in both the Cvt pathway and autophagy. The fusion of these double-membrane transport vesicles with the vacuole releases the single-membrane Cvt or autophagic body into the vacuole lumen (Takeshige et al. 1992; Scott et al. 1997). The subsequent breakdown of these autophagic bodies is dependent on resident hydrolases, as strains deficient in vacuolar proteases such as proteinase A (pep4Δ) accumulate these Aut7p-containing, single-membrane vesicles inside the vacuole. If the GFPAut7p fusion protein follows the itinerary of Cvt vesicles and autophagosomes, then it would also accumulate inside Cvt and autophagic bodies in a pep4Δ strain under nutrient-rich and starvation conditions, respectively. To test this hypothesis, pep4Δ cells transformed with the pCuGFPAUT7 plasmid were grown in nutrient rich conditions to detect Cvt bodies or in nitrogen starvation conditions (SD-N) to observe autophagic bodies inside the vacuole (Fig. 2, B–D).
To examine Cvt bodies, fusion protein expression was induced with a relatively low concentration of CuSO4 (10 μM) for 3 h to reflect the lower level of Aut7p expression under nutrient-rich conditions. After labeling the vacuoles with FM 4-64, the cells were examined by confocal microscopy. Under these nutrient-rich conditions, the GFPAut7p fusion protein was localized to multiple Cvt bodies inside the FM 4-64–labeled vacuoles (Fig. 2 B). To demonstrate the dynamic nature of these accumulated Cvt bodies inside the vacuole, we performed a time-series analysis of a single focal plane of pep4Δ cells expressing GFPAut7p under nutrient-rich conditions (Fig. 2 C). The sequential video frames, taken every 4.4 s, show GFPAut7p-labeled Cvt bodies inside FM 4-64–labeled vacuoles moving in and out of the focal plane of the optical section. These findings demonstrate that GFPAut7p, like the endogenous Aut7 protein, is recruited to Cvt vesicles that are transported to the vacuole via the Cvt pathway.
To view autophagic bodies, GFPAut7p expression was induced with 50 μM of CuSO4 for 3 h and shifted to SD-N for 3 h to induce autophagy. Under these conditions, the FM 4-64–labeled vacuoles were full of GFPAut7p-labeled autophagic bodies (Fig. 2 D). Taken together, these observations confirm that the GFPAut7p fusion protein provides a useful in vivo marker for both the Cvt pathway and autophagy. Consistent with previous studies, the autophagic bodies were much larger than the Cvt bodies, and allowed for easier detection by confocal microscopy (compare Fig. 2B with D). Furthermore, because autophagy conditions induce the expression of Aut7p, thus facilitating its immunodetection, all subsequent Aut7p experiments were performed under autophagy conditions.
The Aut2p-mediated proteolytic processing of Aut7p is a requisite step in its subsequent binding to the P13 membrane fraction (Fig. 1 D). We next characterized the nature of the P13 membrane fraction to which Aut7p binds by examining the GFPAut7p marker by confocal microscopy. The apg1Δ and aut2Δ strains were transformed with the pCuGFPAUT7 plasmid and grown to midlog phase in SMD. GFPAut7p expression was induced with 50 μM CuSO4 for 3 h. After labeling vacuoles with FM 4-64, the cells were shifted to SD-N medium to induce autophagy. In the apg1Δ strain, where Aut7p processing occurs (Fig. 1 C), GFPAut7p localized to prominent punctate structures and was also seen in a diffuse cytosolic population (Fig. 3). These punctate structures were always detected in a perivacuolar region and generally numbered from one to several structures per cell. With a few notable exceptions (discussed below), a population of GFPAut7p displaying a punctate pattern was found in all of the apg mutants examined (data not shown). In contrast, GFPAut7p in the aut2Δ strain, where Aut7p processing did not occur, remained entirely diffuse in the cytosol (Fig. 3). This result supports the fractionation data (Fig. 1 D), as well as the findings of Kirisako et al. 2000, and show that the Aut2p-mediated processing of Aut7p is a requisite step for its subsequent membrane association.
Figure 3.

Aut2p is required for the GFPAut7p association to punctate, perivacuolar structures. The apg1Δ (NNY20) and aut2Δ (WPHYD2) strains were transformed with the pCuGFPAUT7 plasmid. The cells were grown to midlog phase in SMD medium. GFPAut7p expression was then induced with 50 μM CuSO4 for 3 h. Cells were then labeled with FM 4-64, shifted to SD-N medium for 3 h, and examined as described in Fig. 2. In apg1Δ cells, GFPAut7p is recruited to punctate structures, whereas in the Aut7p processing–defective aut2Δ cells, GFPAut7p appears uniformly distributed in the cytoplasm.
The physical interaction between Aut2p and Aut7p suggested that the localization of Aut2p might display the same membrane-binding pattern observed for Aut7p. To examine the subcellular localization of Aut2p, a fusion construct of AUT2 followed by a triple HA epitope tag was integrated at the AUT2 locus. The Aut2HAp integration was functional, as prAPI maturation in the aut2::AUT2HA strain appeared similar to the wild-type strain (Fig. 4 A). To determine the subcellular fractionation pattern of Aut2HAp, cells expressing Aut2HAp were grown to midlog stage in SMD, and then shifted to SD-N medium for 2 h before spheroplasting. The spheroplasts were then lysed in differential osmotic lysis buffer and separated into S13 and P13 fractions. The S13 fraction was further resolved into S100 and P100 fractions. Immunoblot analysis revealed Aut2HAp to be entirely in the supernatant fractions, along with the PGK cytosolic marker (Fig. 4 B). The cytosolic fractionation pattern of Aut2HAp was also observed in cells grown under vegetative conditions (data not shown). The Aut2HAp cytosolic fractionation pattern was also supported by an Aut2pGFP fusion protein, which complemented the prAPI defect in the aut2Δ strain (Fig. 4 A), and exhibited a diffuse cytosolic localization pattern (data not shown). Further, unlike Aut7p, Aut2p expression was not induced in starvation conditions, suggesting that it may act in a catalytic manner (data not shown). Taken together, the findings above indicate that the soluble autophagy component, Aut2p, acts to mediate the cleavage of Aut7p in the cytosol. After proteolytic processing, Aut7p binds to a punctate, perivacuolar, membrane structure that can be fractionated into a low-speed pellet by centrifugation.
Figure 4.
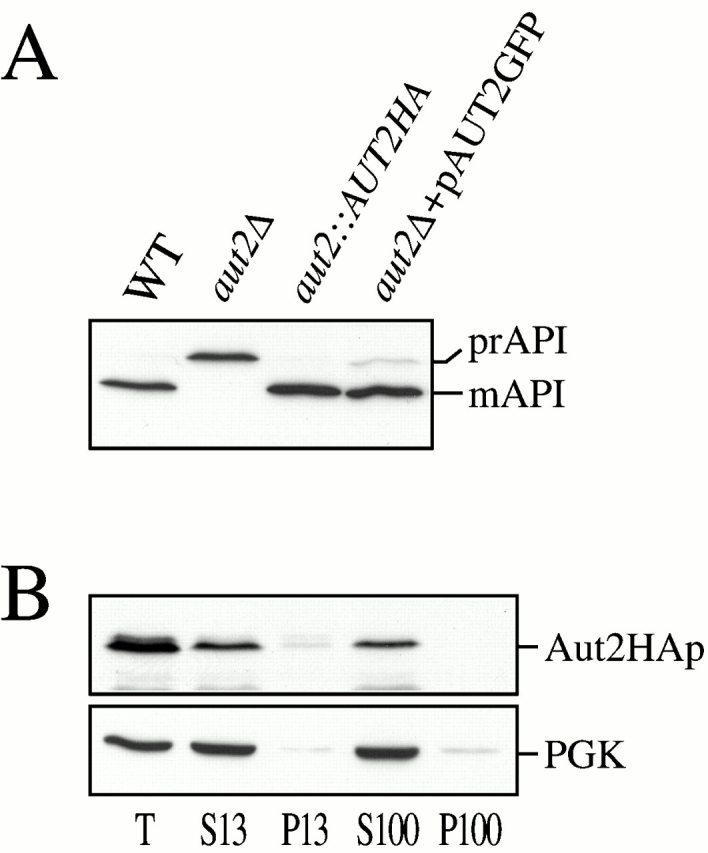
Characterization of Aut2p. (A) API immunoblots of WT (SEY6210), aut2Δ (WPHYD2), WT with a chromosomal integration of an AUT2HA fusion at the AUT2 locus (WPHY2HA, aut2::AUT2HA), and aut2Δ transformed with a centromeric plasmid encoding GFP fused to the COOH terminus of Aut2p (pAUT2GFP). Protein extracts were prepared as described in Materials and Methods. Both Aut2HAp and Aut2pGFP are functional fusion proteins as prAPI maturation appears normal. (B) Aut2HAp subcellular fractionation pattern. Cells expressing the Aut2HAp from the AUT2 chromosomal locus were grown in SMD to midlog phase, and then shifted to SD-N for 2 h before spheroplasting. The spheroplasts were lysed in TEA osmotic lysis buffer as described in Materials and Methods. The precleared total lysate (T) was separated into low-speed supernatant and pellet fractions (S13 and P13). The S13 fraction was further resolved into high-speed supernatant and pellet fractions (S100 and P100) as described in Fig. 1 D and Materials and Methods. Immunoblot analysis was performed with antisera to HA and the cytosolic marker protein, PGK. Aut2HAp is a soluble protein and localizes to the supernatant fractions.
Aut1p Function Is Required for Aut7p Recruitment to the Membrane
Comprehensive two-hybrid analyses of the yeast genome have recently elucidated a number of novel interactions among autophagy components (see Fig. 9 A; Ito et al. 2000; Uetz et al. 2000). These included a strong interaction between Aut7p and Aut1p (Ito et al. 2000). A previous phenotypic analysis of the aut1Δ strain demonstrated both defects in prAPI transport as well as starvation-induced transport of bulk cytoplasm to the vacuole (Schlumpberger et al. 1997). We next investigated the significance of the Aut1p–Aut7p two-hybrid interaction by examining the role of Aut1p on Aut7p recruitment to the membrane. The aut1Δ strain was grown to midlog phase and shifted to SD-N medium to induce autophagy and Aut7p expression. Subcellular fractionations and centrifugations separated the lysate into S13, P13, S100, and P100 fractions. Immunoblot analysis with antiserum to Aut7p indicated that Aut7p remained entirely in a cytosolic fraction (Fig. 5 A). These observations were also supported by examination of the GFPAut7p fusion protein in the aut1Δ background (Fig. 5 B). The aut1Δ strain transformed with pCuGFPAUT7 was induced with 50 μM CuSO4 at midlog phase for 3 h, labeled with FM 4-64 to mark the vacuoles, and shifted to SD-N medium for 3 h before examination by confocal microscopy. The GFPAut7p fusion protein was distributed diffusely in the cytoplasm (Fig. 5 B).
Figure 5.

Characterization of Aut1p and its role in Aut7p membrane binding. (A) Aut7p subcellular fractionation pattern in aut1Δ. Cells from the aut1Δ (WPHYD1) strain were grown to midlog phase in SMD and shifted to SD-N medium for 2 h before spheroplasting. The spheroplasts were then lysed osmotically as described in Materials and Methods. The precleared total lysate (T) was separated into low-speed supernatant and pellet fractions (S13 and P13); the S13 fraction was further separated into high-speed supernatant and pellet fractions (S100 and P100). Immunoblot analysis was performed using antiserum to Aut7p and the cytosolic marker PGK. Deletion of AUT1 prevents Aut7p binding to the P13 membrane fraction. (B) Localization of GFPAut7p in aut1Δ. The aut1Δ (WPHYD1) strain was transformed with the pCuGFPAUT7 plasmid. Cells were grown to midlog phase in SMD medium, induced with 50 μM CuSO4 for 3 h, labeled with FM 4-64, shifted to SD-N medium for 3 h, and examined as described in Materials and Methods. GFPAut7p appears uniformly distributed in the cytoplasm of the aut1Δ cells. For comparison, see GFPAut7p localization in apg1Δ (Fig. 3). (C) Aut1p subcellular fractionation pattern. WT (SEY6210) cells were grown to midlog phase in SMD and shifted to SD-N medium for 2 h before spheroplasting. The subcellular fractionation procedure used to examine Aut1p was identical to the method described in A. Immunoblot analysis was performed using antisera to Aut1p and the cytosolic marker PGK. Aut1p is a soluble protein and appears in the supernatant fractions.
To better understand the role of Aut1p in Aut7p function, we also examined the subcellular location of Aut1p (Fig. 5 C). Wild-type cells were grown to midlog phase, and then shifted to SD-N medium for 2 h before converting them into spheroplasts. Differential osmotic lysis and centrifugation steps separated the lysate into S13, P13, S100, and P100 fractions. Immunoblot analysis with antiserum to Aut1p indicated that Aut1p was localized to the supernatant fractions in both starvation conditions (Fig. 5 C) and vegetative growth conditions (data not shown). We also attempted to follow localization of Aut1p by fusing it to GFP. However, neither NH2- nor COOH-terminal GFP fusions to Aut1p complemented the prAPI defect in the aut1Δ strain, and therefore were not further analyzed (data not shown). These findings demonstrate that Aut1p is predominantly a cytosolic autophagy component whose function is required for Aut7p membrane localization.
Aut1p, 2p, and 7p Are Involved in Autophagosome Vesicle Formation
Thus far, we have characterized Aut1p and Aut2p in terms of their distinct roles in Aut7p membrane association. We next examined the function of these components in the context of known steps in the transport of prAPI via the Cvt/autophagy pathways. Upon synthesis in the cytosol, prAPI oligomerizes (Kim et al. 1997) and subsequently forms a large pelletable Cvt complex (Baba et al. 1997; Scott et al. 1997). Biochemical and morphological studies indicate that the Cvt complex associates with a membrane source that ultimately forms Cvt vesicles under vegetative growth conditions, and autophagosomes in starvation conditions.
To determine the step of the pathway in which Aut1p and Aut2p are required, we analyzed the state of prAPI in the null strains by protease sensitivity assays and membrane flotation gradients. The aut1Δ and aut2Δ strains were grown to midlog phase and converted into spheroplasts. After osmotic lysis of the spheroplasts, the lysates were separated into supernatant and pellet fractions by a 5,000 rpm centrifugation procedure. Under these conditions, prAPI appeared entirely in the pellet fraction (Fig. 6). The prAPI-bound pellet fraction was subjected to exogenous protease treatment in the presence or absence of detergent. The protease sensitivity assays indicated that prAPI in the aut1Δ and aut2Δ strains was accessible to exogenous proteases even in the absence of detergent treatment (Fig. 6), suggesting that prAPI in these strains was not protected by a completed vesicle.
Figure 6.

Protease-sensitivity and membrane-flotation analyses. The aut1Δ, aut2Δ, aut7Δ, and ypt7Δ strains were grown to midlog phase and converted into spheroplasts. The spheroplasts were lysed in PS200 osmotic lysis buffer. The total lysate (T) was resolved into supernatant (S) and pellet (P) fractions by centrifugation at 5,000 rpm. For the protease sensitivity assay, the pellet fractions were subjected to protease treatment in the absence or presence of 0.2% Triton X-100, as described in Materials and Methods. For the membrane flotation assay, the pellet fraction was subjected to centrifugation in a Ficoll step gradient with or without detergent, as described in Materials and Methods. Membrane-associated proteins were collected in the float (F) fraction. All samples were subjected to immunoblot analysis with antisera to API and PGK. The aut1Δ, aut2Δ, and aut7Δ mutants all accumulate precursor API at a protease-sensitive, membrane-associated stage.
In the membrane flotation gradient analysis, membrane-associated proteins migrate to the top of a step gradient and are collected as the “float” (F) fraction (see Materials and Methods). The prAPI-containing pellets in the aut1Δ and aut2Δ strains were subjected to flotation step gradients. The collected float fractions from both the aut1Δ and aut2Δ strains contained prAPI. However, treating the pellets with detergent prevented prAPI from floating through the gradient, suggesting that the buoyant population of prAPI was membrane associated (Fig. 6). The aut7Δ and ypt7Δ strains were included in these analyses as controls. Ypt7p is a rab guanosine triphosphatase required for vesicle fusion with the vacuole and thus the ypt7Δ strain accumulates prAPI in a protease-protected, membrane-enclosed state (Kim et al. 1999). By contrast, while prAPI in the aut7Δ strain is membrane associated (Huang et al. 2000), it remains accessible to exogenous protease treatment (Kirisako et al. 1999; Huang et al. 2000). Taken together, these findings indicate that Aut1p, Aut2p, and Aut7p are all required at the step(s) of vesicle formation and/or completion.
The Apg Conjugation System Functions to Recruit Aut7p to the Membrane
In addition to the interactions between Aut7p and Aut2p (Lang et al. 1998), and Aut7p and Aut1p (Ito et al. 2000), the recent two-hybrid analysis of the yeast genome revealed extensive associations between components of the Apg conjugation machinery and Aut7p and Aut1p (see Fig. 9 A; Uetz et al. 2000). Specifically, associations between Aut7p and Apg7p, Aut1p and Apg7p, and Aut1p and Apg12p were identified. While the basic details of the Apg conjugation process have been determined (Mizushima et al. 1998, Mizushima et al. 1999; Shintani et al. 1999), the exact function of such a protein-modification system in the autophagy pathway remains to be elucidated. Therefore, we examined whether the Apg conjugation system might function to mediate the recruitment of autophagy components, such as Aut7p, to the membrane. The null strains of three principal conjugation components, Apg5p, Apg7p, and Apg12p, were transformed with the pCuGFPAUT7 plasmid expressing the GFPAut7p fusion protein. At midlog growth stage, GFPAut7p expression was induced with 50 μM CuSO4 for 3 h. After labeling vacuoles with FM 4-64, the cells were shifted to SD-N medium for 3 h to induce autophagy, and then analyzed by confocal microscopy. In the apg9Δ control strain, a population of GFPAut7p was concentrated to punctate structures in the perivacuolar region (Fig. 7 A), similar to the GFPAut7p pattern observed in the apg1Δ strain (Fig. 3). However, in the apg5Δ, apg7Δ, and apg12Δ conjugation mutants, no punctate structures were detected, as GFPAut7p appeared diffusely localized in the cytosol (Fig. 7 A).
Figure 7.
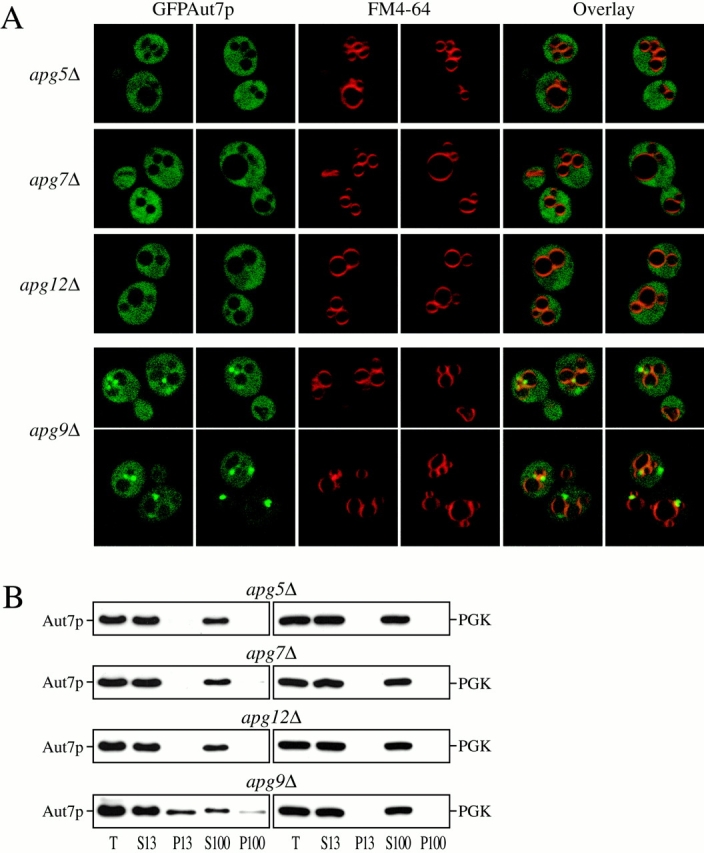
The Apg conjugation system is required for Aut7p membrane binding. (A) The Apg conjugation mutants apg5Δ (MGY101), apg7Δ (VDY101), apg9Δ (JKY007), and apg12Δ (YNM101) strains were transformed with the pCuGFPAUT7 plasmid. GFPAut7p expression was induced at midlog stage with 50 μM CuSO4 for 3 h. Cells were then labeled with FM 4-64, shifted to SD-N medium for 3 h, and examined by confocal microscopy as described in Materials and Methods. In apg9Δ cells, GFPAut7p is recruited to punctate, perivacuolar structures, whereas GFPAut7p appears uniformly distributed in the apg5Δ, apg7Δ, and apg12Δ conjugation mutants. (B) Aut7p subcellular fractionation pattern in the Apg conjugation mutants. Cells from apg5Δ, apg7Δ, apg9Δ, and apg12Δ strains were grown to midlog phase in SMD, and then shifted to SD-N for 2 h to induce autophagy. After spheroplasting and osmotic lysis in TEA osmotic lysis buffer, the precleared total lysate (T) was separated into low-speed supernatant (S13) and pellet (P13) fractions. The S13 fraction was further separated into 100,000 g supernatant (S100) and pellet (P100) fractions. The fractionated samples were subjected to immunoblot analysis using antisera to Aut7p and PGK. The apg5Δ, apg7Δ, and apg12Δ conjugation mutants are defective in Aut7p binding to the P13 pellet fraction. Essentially identical results were seen with the apg10 and apg16Δ mutant strains (data not shown).
Localization of Aut7p in the Apg conjugation strains by subcellular fractionations supports the confocal microscopy observations (Fig. 7 B). The apg5Δ, apg7Δ, and apg12Δ conjugation mutants and the apg9Δ control strain were grown to midlog phase and shifted to SD-N medium for 2 h before the subcellular fractionation procedure, which separated the lysates into S13, P13, S100, and P100 fractions. Immunoblot analysis of these samples revealed that Aut7p from the conjugation mutants appeared entirely in the supernatant fractions, along with the PGK cytosolic marker, while a significant population of Aut7p binds to the P13 fraction from the apg9Δ control strain (Fig. 7 B). We also examined the Aut7p fractionation pattern in the apg10 and apg16Δ conjugation mutants. Apg10p acts at an intermediate step in the Apg12p-Apg5p reaction, while Apg16p mediates the dimerization of the final Apg12p-Apg5p conjugate (Fig. 9 A; Shintani et al. 1999; Mizushima et al. 1999). The distribution of GFPAut7p and the fractionation pattern of Aut7p in the apg10 and apg16Δ strains appeared identical to that of the apg5Δ, apg7Δ, and apg12Δ strains (data not shown). These confocal microscopy and subcellular fractionation findings support the role of the Apg conjugation system in mediating Aut7p binding to a punctate, perivacuolar membrane compartment.
Aut7p Processing in the Conjugation Mutants and Apg Conjugation in the aut1 and aut2 Mutants
Because Aut1p and the Apg conjugation system are both required for Aut7p binding, we examined whether the defect in aut1Δ and the Apg conjugation mutants was at the step of Aut7p COOH-terminal proteolytic processing. While Aut7pGFP remained intact in the aut2Δ background, the fusion protein was cleaved in both the aut1Δ and Apg conjugation mutants (Fig. 8 A). This finding indicates that although Aut7p membrane binding requires both Aut1p and the Apg conjugation system, the Aut7p COOH-terminal processing event occurs independently of their function. Conversely, Aut1p and Aut2p may be required for the Apg conjugation reaction, which involves the formation of a thioester bond between Apg12p and Apg7p before the transfer of Apg12p to Apg5p (Mizushima et al. 1998). The transfer reaction ultimately results in the covalent linkage between Apg12p and Apg5p via an isopeptide bond. Mutations in the Apg conjugation components Apg5p, Apg7p, Apg10p, or Apg12p prevent the formation of the final Apg12p–Apg5p conjugate. To examine the presence of the Apg12p–Apg5p conjugate, aut1Δ, aut2Δ, apg7Δ, and wild-type cells were transformed with a plasmid encoding an HA-tagged Apg5p fusion protein (George et al. 2000). In wild-type cells, as well as in the aut1Δ and aut2Δ cells, immunoblot analysis with antiserum to HA detected Apg5HAp in both the free form and as part of the Apg12p–Apg5p conjugate. This result indicates that Aut1p and Aut2p are not required for the Apg conjugation reactions (Fig. 8 B). As expected, the Apg12p–Apg5p conjugate did not form in the apg7Δ strain.
Figure 8.
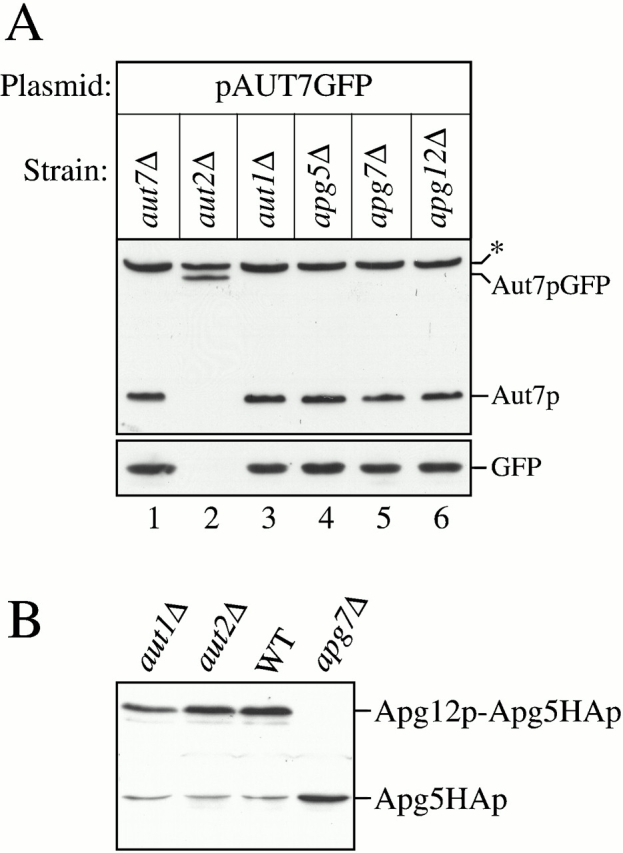
Aut7p COOH-terminal processing and Apg12p–Apg5p conjugate formation analyses. (A) The aut7Δ (WPHYD7), aut2Δ (WPHYD2), aut1Δ (WPHYD1), apg5Δ (MGY101), apg7Δ (VDY101), and apg12Δ (YNM101) strains were transformed with the centromeric pAUT7GFP plasmid. Cells were grown to midlog phase in SMD. Protein extracts were prepared and analyzed by immunoblots using antisera to Aut7p (top) and GFP (bottom). *Background band. Aut1p and the Apg conjugation components (Apg5p, Apg7p, and Apg12p) are not required for Aut7p COOH-terminal processing. (B) The aut1Δ, aut2Δ, wild-type (SEY6210), and apg7Δ strains were transformed with a plasmid encoding an Apg5HAp fusion protein. Protein extracts from cells grown to midlog phase were prepared and analyzed by immunoblots with antiserum to the HA epitope. The Apg12p–Apg5p conjugate can be detected in the aut1Δ, aut2Δ, and wild-type strains, but not in the apg7Δ strain.
Discussion
A key early step in the membrane expansion and autophagic vesicle-formation processes may be defined by the recruitment of starvation-induced components to a membrane compartment involved in the formation of autophagic vesicles. The Aut7 protein is the first characterized transport vesicle component that is upregulated during starvation conditions (Kirisako et al. 1999; Huang et al. 2000). The membrane association of the hydrophilic Aut7 protein is an important requirement for transport vesicle formation and completion. In this study, we have characterized the steps and components required for Aut7p membrane binding. We have identified a function for Aut2p in mediating a proteolytic processing event at the COOH terminus of Aut7p (Fig. 1 C). The Aut7p processing reaction is rapid, occurring with a half time of <1 min by radioactive pulse-chase labeling analysis (data not shown). This modification is a requisite step for the subsequent membrane binding of Aut7p, as the Aut7 protein is found exclusively in a soluble fraction in an aut2Δ strain defective for proteolytic processing (Fig. 1 D). Similarly, a GFPAut7p fusion protein that functions as an in vivo marker for the autophagy pathway displays a diffuse cytosolic staining pattern in the aut2Δ mutant (Fig. 3). In contrast, in most of the autophagy mutants that possess the capacity to process Aut7p (e.g., apg1Δ and apg9Δ; Fig. 3 and Fig. 7 A, respectively), the GFPAut7p fusion protein localizes to prominent, punctate perivacuolar structures in addition to its cytosolic distribution. These punctate structures may represent a previously described intermediate compartment that appears during autophagosome formation (Kirisako et al. 1999).
To further understand the role of Aut2p in the Aut7p cleavage reaction, we examined the Aut2 protein. Both subcellular fractionations (Fig. 4 B) and confocal microscopy of the Aut2pGFP fusion protein (data not shown) indicate that Aut2p is a cytosolic, soluble protein. These findings indicate that the Aut2p-mediated cleavage of Aut7p most likely occurs in the cytosol. In addition, the observation that Aut2p expression does not increase under starvation conditions (data not shown) suggests that Aut2p is not a stoichiometric partner of Aut7p, but, rather, interacts with Aut7p in a catalytic manner to facilitate Aut7p proteolysis. Recently, Kirisako et al. 2000 have demonstrated that Aut2p/Apg4p is a cysteine protease that cleaves the ultimate residue (arginine) from the COOH terminus of Aut7p. Moreover, the Aut7p C-terminal cleavage was determined to be required for its subsequent membrane association. Our subcellular fractionation results (Fig. 1) as well as the localization of the GFPAut7p fusion protein in the aut2Δ background confirm these observations.
The recent two-hybrid analyses of the S. cerevisiae genome have presented an array of interactions between Apg components (Ito et al. 2000; Uetz et al. 2000). These interactions included the association of Aut7p with Aut1p. In addition, Aut7p and Aut1p both interact with components of the Apg conjugation system that covalently links Apg12p to Apg5p through the action of Apg7p (Mizushima et al. 1998). Specifically, Aut7p associates with Apg7p while Aut1p interacts with both Apg7p and Apg12p (Fig. 9 A). Based on these findings, we investigated the role of Aut1p and the Apg conjugation components in Aut7p function by examining Aut7p membrane binding in the aut1Δ strain and in null mutants defective in the conjugation reaction. Both subcellular fractionations of the endogenous Aut7 protein and confocal microscopy analysis of the GFPAut7p fusion protein indicate that Aut1p and the Apg conjugation system are required for Aut7p membrane recruitment. Aut7p appears exclusively in the soluble, supernatant fractions in the aut1Δ and Apg conjugation mutants, while a significant amount of Aut7p binds to a low-speed pellet fraction in the other apg mutant strains (Fig. 5 A and 7 B, respectively). In agreement with the fractionation results, the GFPAut7p fusion protein is diffusely distributed in the cytosol in the aut1Δ and in all of the Apg conjugation mutant strains. These findings establish a role for Aut1p and the Apg conjugation system in mediating Aut7p recruitment to an intermediate, punctate membrane compartment during autophagosome formation. Interestingly, the requirement of the conjugation component Apg16p, which facilitates the multimerization of the already-conjugated Apg12p–Apg5p species, suggests that it is not the conjugation reaction per se that plays a role in membrane binding of Aut7p, but rather some function of the multimeric Apg conjugation complex. Finally, as stated above, two-hybrid studies link Aut7p with Apg7p. It is interesting to note that the fluorescent staining pattern of GFPAut7p is similar to that of Apg7pGFP (Kim et al. 1999). Both proteins display a cytosolic distribution along with punctate perivacuolar structures. It therefore appears likely that both proteins are recruited to the same, as yet unidentified, intermediate compartment during autophagosome formation.
Although Aut1p interacts with Apg7p and Apg12p of the conjugation system, defects in Aut1p do not prevent the final conjugation of Apg12p to Apg5p, indicating that Aut1p is not directly required for the Apg conjugation reactions (Fig. 8 B). Whether Aut1p and the conjugation system act in concert or independently through parallel pathways to recruit Aut7p to the membrane remains to be determined. However, neither Aut1p nor the Apg conjugation components are required for the Aut2p-mediated processing of Aut7p (Fig. 8 A). Taken together, these findings suggest that Aut1p and the conjugation system act at a post-processing step in Aut7p membrane recruitment.
Kirisako et al. 2000 have recently investigated the role of Apg7p, the E1 ubiquitin-activating enzyme homologue of the Apg conjugation system, in the Aut7p membrane-binding step. While the function of Apg7p in forming an ATP hydrolysis-dependent thioester linkage to Apg12p has been well characterized (Mizushima et al. 1998; Tanida et al. 1999), Kirisako et al. 2000 also demonstrated that Apg7p is required for Aut7p membrane association after its Aut2p-mediated processing step. In their study, the initial removal of the COOH-terminal arginine residue of Aut7p by Aut2p resulted in a loosely membrane-bound conformation. Then, by a ubiquitination-like mechanism, Apg7p (and potentially other factors) mediate the conjugation of Aut7p to an as-yet unidentified “X” factor on the membrane, thus converting Aut7p from a loosely membrane-bound state to a tightly membrane-bound one. Furthermore, Kirisako et al. 2000 demonstrated that Aut7p in the tightly membrane-associated form was subsequently liberated from the X factor through an Aut2p-mediated cleavage reaction, thus completing the Aut7p membrane-association cycle. In this study, we demonstrate that the entire collection of Apg conjugation components, including Apg5p, Apg7p, Apg10p, Apg12p, and Apg16p, are all required for Aut7p membrane binding. The exact function of the other Apg conjugation components in mediating Aut7p membrane binding remains to be elucidated. It is possible that the Apg conjugation system involving Apg12p–Apg5p acts indirectly by affecting the membrane compartment to which Aut7p initially binds. Future studies will be aimed at determining the role of these proteins in Aut7p membrane recruitment. However, it appears that Apg7p may be involved in two parallel pathways that are both required for Aut7p membrane binding: one that involves Apg10p in the conjugation of Apg12p–Apg5p and a second pathway that uses a second conjugating enzyme to add the X factor directly to Aut7p.
Other recent studies have offered two views of Aut7p function. An initial study by Lang et al. 1998 proposed that Aut2p and Aut7p functioned as attachment proteins that link autophagosomes to the microtubule cytoskeleton for vectorial delivery to the vacuole. According to this model, defects in Aut2p and Aut7p would result in the cytoplasmic accumulation of prAPI-containing autophagosomes. An alternative view of Aut7p function was subsequently proposed (Kirisako et al. 1999; Huang et al. 2000). These latter studies demonstrated that precursor API in the aut7Δ strain existed in a membrane-bound, protease-accessible state, suggesting that Aut7p was required at a step before vesicle completion. Our results indicate that prAPI in the aut1Δ and aut2Δ strains also exists in a membrane-associated but protease-accessible state, identical to the aut7Δ prAPI phenotype (Fig. 6). Because Aut1p and Aut2p are required for Aut7p function, the common prAPI phenotype exhibited by the aut1, aut2, and aut7 null mutants supports the view that Aut1p, Aut2p, and Aut7p all act at the same step(s) of vesicle formation and/or completion.
The findings from this study can be incorporated into the current model of prAPI import by the Cvt and autophagy pathways (Fig. 9 B). Immediately after synthesis, prAPI oligomerizes into a dodecamer, and then assembles into a higher-order Cvt complex. The Cvt complex then associates with a membrane of unknown origin. Interestingly, while deletions of AUT1, AUT2, and the Apg conjugation apparatus all prevent Aut7p membrane binding, prAPI in all cases retains its ability to bind to a pelletable membrane fraction (Fig. 6). While these autophagy components are not required for prAPI membrane binding, additional factors may play a role in this process. The Aut2p-mediated cleavage of Aut7p generates a binding-competent form of this protein. However, both Aut1p and the Apg conjugation components are essential in facilitating Aut7p membrane binding. Our previous results indicated that the initial binding of Aut7p results in a protease-accessible form of the protein (Huang et al. 2000; Fig. 9 B). Upon vesicle completion, Aut7p travels with prAPI to the vacuole. Subsequent fusion with the vacuole releases the prAPI and Aut7p-containing autophagic bodies into the vacuole lumen. The autophagic bodies are then degraded by resident hydrolases, thus allowing prAPI to be processed to the mature form. In this final step, both Aut7p and bulk cytoplasmic cargo are degraded by vacuolar proteases and recycled for essential biosynthetic processes required to survive starvation conditions.
The Cvt and autophagy pathways share a common set of molecular components. However, the autophagosomes that are formed under starvation conditions are much larger than their Cvt vesicle counterparts formed under vegetative conditions. These observations suggest that formation of the autophagosome requires an expansion of the enwrapping membrane, as well as the increased expression of structural components. Aut7p may represent one member in a family of such structural components localized to these autophagic transport vesicles. Accordingly, the level of Aut7p expression in starvation versus vegetative conditions appears to reflect the 10–20-fold increase in vesicle surface area between autophagosomes and Cvt vesicles (Huang et al. 2000). In support of this hypothesis, a block in the upregulation of Aut7p results in the formation of abnormally small autophagosomes (Abeliovich et al. 2000).
In this study, we have characterized three events that are required for Aut7p recruitment to the membrane, a critical step in the formation of autophagic vesicles. In addition, these data define a function of the Apg conjugation system in recruiting structural components to the forming autophagosome. Further studies will be aimed at determining the specific modification effected by the conjugation machinery, the function of Aut1p in membrane binding of Aut7p, and the source of the membrane compartment to which Aut7p is recruited. These studies will begin to provide detailed mechanistic information about the formation of the sequestering vesicle used in the Cvt and autophagy pathways.
Acknowledgments
The authors thank Chao-Wen Wang for technical assistance, Dr. Neta Dean for her gift of the ME3 plasmid, and Drs. Hagai Abeliovich, Benjamin Lin, Sidney Scott, and Sarah Teter for helpful advice and critical reading of the manuscript.
This work was supported by Public Health Service grant GM53396 from the National Institutes of Health to D.J. Klionsky.
Footnotes
Abbreviations used in this paper: Apg, autophagy; API, aminopeptidase I; Cvt, cytoplasm to vacuole targeting; GFP, green fluorescent protein; HA, hemagglutinin; ORF, open reading frame; PGK, phosphoglycerate kinase; SMD, synthetic minimal medium; TEA, tetraethylammonium.
References
- Abeliovich H., Dunn W.A., Jr., Kim J., Klionsky D.J. Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J. Cell Biol. 2000;151:1025–1033. doi: 10.1083/jcb.151.5.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M., Osumi M., Scott S.V., Klionsky D.J., Ohsumi Y. Two distinct pathways for targeting proteins from the cytoplasm to the vacuole/lysosome. J. Cell Biol. 1997;139:1687–1695. doi: 10.1083/jcb.139.7.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum P., Thorner J., Honig L. Identification of tubulin from the yeast Saccharomyces cerevisiae . Proc. Natl. Acad. Sci. USA. 1978;75:4962–4966. doi: 10.1073/pnas.75.10.4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George M.D., Baba M., Scott S.V., Mizushima N., Garrison B.S., Ohsumi Y., Klionsky D.J. Apg5p functions in the sequestration step in the cytoplasm-to-vacuole targeting and macroautophagy pathways. Mol. Biol. Cell. 2000;11:969–982. doi: 10.1091/mbc.11.3.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt B., Kordas T.J., Thompson C.M., Patel P., Vida T. The vesicle transport protein Vps33p is an ATP-binding protein that localizes to the cytosol in an energy-dependent manner. J. Biol. Chem. 1998;273:15818–15829. doi: 10.1074/jbc.273.25.15818. [DOI] [PubMed] [Google Scholar]
- Harding T.M., Hefner-Gravink A., Thumm M., Klionsky D.J. Genetic and phenotypic overlap between autophagy and the cytoplasm to vacuole protein targeting pathway. J. Biol. Chem. 1996;271:17621–17624. doi: 10.1074/jbc.271.30.17621. [DOI] [PubMed] [Google Scholar]
- Harding T.M., Morano K.A., Scott S.V., Klionsky D.J. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J. Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow E., Lane D. Using AntibodiesA Laboratory Manual 1999. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: pp. 61–99 [Google Scholar]
- Huang W.-P., Scott S.V., Kim J., Klionsky D.J. The itinerary of a vesicle component, Aut7p/Cvt5p, terminates in the yeast vacuole via the autophagy/Cvt pathways. J. Biol. Chem. 2000;275:5845–5851. doi: 10.1074/jbc.275.8.5845. [DOI] [PubMed] [Google Scholar]
- Ito T., Tashiro K., Muta S., Ozawa R., Chiba T., Nishizawa M., Yamamoto K., Kuhara S., Sakaki Y. Toward a protein–protein interaction map of the budding yeasta comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc. Natl. Acad. Sci. USA. 2000;97:1143–1147. doi: 10.1073/pnas.97.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Dalton V.M., Eggerton K.P., Scott S.V., Klionsky D.J. Apg7p/Cvt2p is required for the cytoplasm-to-vacuole targeting, macroautophagy, and peroxisome degradation pathways. Mol. Biol. Cell. 1999;10:1337–1351. doi: 10.1091/mbc.10.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Klionsky D.J. Autophagy, cytoplasm-to-vacuole targeting pathway, and pexophagy in yeast and mammalian cells. Annu. Rev. Biochem. 2000;69:303–342. doi: 10.1146/annurev.biochem.69.1.303. [DOI] [PubMed] [Google Scholar]
- Kim J., Scott S.V., Oda M., Klionsky D.J. Transport of a large oligomeric protein by the cytoplasm to vacuole protein targeting pathway. J. Cell Biol. 1997;137:609–618. doi: 10.1083/jcb.137.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisako T., Baba M., Ishihara N., Miyazawa K., Ohsumi M., Yoshimori T., Noda T., Ohsumi Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell Biol. 1999;147:435–446. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisako T., Ichimura Y., Okada H., Kabeya Y., Mizushima N., Yoshimori T., Ohsumi M., Takao T., Noda T., Ohsumi Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 2000;151:263–276. doi: 10.1083/jcb.151.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J., Cueva R., Yaver D.S. Aminopeptidase I of Saccharomyces cerevisiae is localized to the vacuole independent of the secretory pathway. J. Cell Biol. 1992;119:287–299. doi: 10.1083/jcb.119.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J., Ohsumi Y. Vacuolar import of proteins and organelles from the cytoplasm. Annu. Rev. Cell. Dev. Biol. 1999;15:1–32. doi: 10.1146/annurev.cellbio.15.1.1. [DOI] [PubMed] [Google Scholar]
- Labbé S., Thiele D.J. Copper ion inducible and repressible promoter systems in yeast. Methods Enzymol. 1999;306:145–153. doi: 10.1016/s0076-6879(99)06010-3. [DOI] [PubMed] [Google Scholar]
- Lang T., Schaeffeler E., Bernreuther D., Bredschneider M., Wolf D.H., Thumm M. Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:3597–3607. doi: 10.1093/emboj/17.13.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura A., Tsukada M., Wada Y., Ohsumi Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae . Gene. 1997;192:245–250. doi: 10.1016/s0378-1119(97)00084-x. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Noda T., Ohsumi Y. Apg16p is required for the function of the Apg12p–Apg5p conjugate in the yeast autophagy pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:3888–3896. doi: 10.1093/emboj/18.14.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Noda T., Yoshimori T., Tanaka Y., Ishii T., George M.D., Klionsky D.J., Ohsumi M., Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- Noda T., Kim J., Huang W.-P., Baba M., Tokunaga C., Ohsumi Y., Klionsky D.J. Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J. Cell Biol. 2000;148:465–480. doi: 10.1083/jcb.148.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.S., Klionsky D.J., Banta L.M., Emr S.D. Protein sorting in Saccharomyces cerevisiaeisolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol. Cell. Biol. 1988;8:4936–4948. doi: 10.1128/mcb.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlumpberger M., Schaeffeler E., Straub M., Bredschneider M., Wolf D.H., Thumm M. AUT1, a gene essential for autophagocytosis in the yeast Saccharomyces cerevisiae . J. Bacterial. 1997;179:1068–1076. doi: 10.1128/jb.179.4.1068-1076.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott S.V., Baba M., Ohsumi Y., Klionsky D.J. Aminopeptidase I is targeted to the vacuole by a nonclassical vesicular mechanism. J. Cell Biol. 1997;138:37–44. doi: 10.1083/jcb.138.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott S.V., Hefner-Gravink A., Morano K.A., Noda T., Ohsumi Y., Klionsky D.J. Cytoplasm to vacuole targeting and autophagy employ the same machinery to deliver proteins to the yeast vacuole. Proc. Natl. Acad. Sci. USA. 1996;93:12304–12308. doi: 10.1073/pnas.93.22.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T., Mizushima N., Ogawa Y., Matsuura A., Noda T., Ohsumi Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:5234–5241. doi: 10.1093/emboj/18.19.5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshige K., Baba M., Tsuboi S., Noda T., Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992;119:301–311. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I., Mizushima N., Kiyooka M., Ohsumi M., Ueno T., Ohsumi Y., Kominami E. Apg7p/Cvt2pa novel protein-activating enzyme essential for autophagy. Mol. Biol. Cell. 1999;10:1367–1379. doi: 10.1091/mbc.10.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichert U., Mechler B., Müller H., Wolf D.H. Lysosomal (vacuolar) proteinases of yeast are essential catalysts for protein degradation, differentiation, and cell survival. J. Biol. Chem. 1989;264:16037–16045. [PubMed] [Google Scholar]
- Thumm M., Egner R., Koch M., Schlumpberger M., Straub M., Veenhuis M., Wolf D.H. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae . FEBS Lett. 1994;349:275–280. doi: 10.1016/0014-5793(94)00672-5. [DOI] [PubMed] [Google Scholar]
- Tsukada M., Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae . FEBS Lett. 1993;333:169–174. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- Uetz P., Giot L., Cagney G., Mansfield T.A., Judson R.S., Knight J.R., Lockshon D., Narayan V., Srinivasan M., Pochart P. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae . Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Wieland F., Harter C. Mechanisms of vesicle formationinsights from the COP system. Curr. Opin. Cell Biol. 1999;11:440–446. doi: 10.1016/s0955-0674(99)80063-5. [DOI] [PubMed] [Google Scholar]