

Abstract
The development of naive CD4+ T cells into a T helper (Th) 2 subset capable of producing interleukin (IL)-4, IL-5, and IL-13 involves a signal transducer and activator of transcription (Stat)6-dependent induction of GATA-3 expression, followed by Stat6-independent GATA-3 autoactivation. The friend of GATA (FOG)-1 protein regulates GATA transcription factor activity in several stages of hematopoietic development including erythrocyte and megakaryocyte differentiation, but whether FOG-1 regulates GATA-3 in T cells is uncertain. We show that FOG-1 can repress GATA-3–dependent activation of the IL-5 promoter in T cells. Also, FOG-1 overexpression during primary activation of naive T cells inhibited Th2 development in CD4+ T cells. FOG-1 fully repressed GATA-3–dependent Th2 development and GATA-3 autoactivation, but not Stat6-dependent induction of GATA-3. FOG-1 overexpression repressed development of Th2 cells from naive T cells, but did not reverse the phenotype of fully committed Th2 cells. Thus, FOG-1 may be one factor capable of regulating the Th2 development.
Keywords: GATA-3, FOG-1, thymocyte, T lymphocyte, cytokine
Introduction
In development, lineage commitment decisions can be initiated by transient gradients of factors that regulate gene expression. The early effects of transient signaling can become stabilized by feedback pathways such as transcriptional autoactivation (1), in which a transcription factor induces its own expression. For example, autoactivation of Pit-1 and GATA-2 occurs in the developing pituitary after their transient induction by FGF-8 and BMP2/4 (1), stabilizing cellular commitment to expression of specific pituitary hormones. Transcriptional autoactivation also occurs for the transcription factor GATA-1 in hematopoietic development (2), MyoD in muscle development (3, 4), and retinoic acid X receptor (5), suggesting it may be a common developmental strategy for lineage commitment. We recently showed that GATA-3 also exerts transcriptional autoactivation in development of the Th type 2 (Th2) subset of CD4+ T cells (6). In this system, transient signaling by IL-4 receptors during activation of naive T cells causes signal transducer and activator of transcription (Stat)*6-dependent elevation in GATA-3 expression followed by a switch to Stat6-independent GATA-3 autoactivation (6, 7).
Transcriptional autoactivation reinforces developmental choices, but creates a problem of regulating a potentially runaway feedback pathway. For Th2 development, this problem arises because naive T cells express a basal level of GATA-3 that potentially could autoactivate in the absence of IL-4 signaling. However, despite basal GATA-3 expression in naive T cells, Stat6-independent autoactivation is found to occur inefficiently, in ∼5–10% of T cells (6). Therefore, a threshold may exist that must be overcome before autoactivation can efficiently occur.
The physical basis for this threshold is unknown. Conceivably, there may be a requirement for IL-4–induced Stat6 activation in establishing GATA-3–dependent transcription of the GATA-3 gene. This does not appear to be the case, however, since autoactivation of endogenous GATA-3 expression can be established by ectopic GATA-3 even in the absence of IL-4 and in Stat6-deficient T cells (6). A threshold for GATA-3 autoactivation could also be established by factors that partially repress GATA-3 activity. Friend of GATA (FOG)-1 was identified as a GATA-1 interacting factor and shown to enhance GATA-1–dependent transcriptional activity (8). FOG-1 contains multiple zinc finger domains, four of which are capable of interacting with the NH2-terminal zinc finger of the GATA family of transcription factors. In particular, zinc fingers 1, 5, 6, and 9 of FOG-1 interact with the N zinc finger domain of GATA-1. Initially, FOG-1 was found to be capable of activating GATA-1–dependent responses. Subsequent studies have also found that both FOG-1 and FOG-2 can exert repressive activities of GATA-dependent responses (9–11). FOG-1 can interact with GATA-3 in yeast two-hybrid analysis (8), but its effects in T cells have not been examined.
In this study, we examined the effect of FOG-1 in regulating the transcriptional activity of GATA-3 in developing CD4+ T cells. We find that FOG-1 exerts an inhibitory rather than an activating role in regulating GATA-3–dependent transcription in CD4+ T cells. Further, our results indicate that FOG-1 can exert this activity primarily at relatively low levels of GATA-3 expression and selectively in naive T cells, but not in fully differentiated Th2 cells where GATA-3 levels are much higher. FOG-1 appears to selectively regulate GATA-3 autoactivation, and specifically represses the Stat6-independent, GATA-dependent induction of GATA-3 expression, but does not repress IL-4 driven, Stat6-dependent GATA-3 expression.
Materials and Methods
Reagents
Wild-type and Stat6-deficient DO11.10 α/β− TCR transgenic mice have been described previously (12, 13). Cytokines and antibodies were obtained as described previously (6).
T Cell Activation and Phenotype Differentiation
DO11.10 splenocytes were purified by density gradient (Histopaque-1119; Sigma-Aldrich) and activated by 0.3 μM chicken OVA peptide 323–339 at 3 × 106 cells per milliliter in IDME media. For Th1 development, T cells were activated in the presence of IL-12 (10 U/ml) and anti–IL-4 antibody 11B11 (10 μg/ml) (14). For Th2 development, cells were activated in the presence of anti–IL-12 (TOSH) (3 μg/ml; reference 15) and IL-4 (100 U/ml). For passage, T cells were harvested 7 d after the previous activation, washed, and restimulated with 0.3 μM OVA at 1.25 × 105 cells per milliliter and irradiated Balb/c splenocytes (2,000 rads, 2.5 × 106 cells per milliliter).
Constructs and Retroviral Infection
The green fluorescent protein (GFP)-RV vector has been described previously (16). We modified GFP-RV to create GFPR1-RV, allowing EcoR1 cloning of cDNAs into the multiple cloning sites (MCS) as follows. GFP-RV was used as template in a PCR with the following oligonucleotide primers: MCS1: 5′ GGG AGA TCT AAA CTC GAG AAA GAA TTC TAA CGT TAC TGG CCG AAG; and GFP-AS: 5′ GAA TTC GGA TCC TTA CTT GTA CAG CTC GTC C. The PCR product contains the internal ribosomal entry site/GFP cassette from GFP-RV, which now lacks the original 3′ EcoR1 site and making the MCS EcoR1 site unique. This product was digested with BglII and BamHI and ligated with the 5.1-Kb backbone of BglII/BamHI-digested GFP-RV to produce GFPR1-RV. A 3.3-Kb EcoR1 fragment containing full-length FOG-1 cDNA was digested from pMT2-FOG-1 (8) and ligated into the EcoR1 site of GFPR1-RV. Integrity of the full-length FOG-1 cDNA was confirmed by sequencing. A full length murine GATA-3 cDNA was generated using R/m GATA-3 (J.D. Engel, Northwestern University, Evanston, IL; reference 17) as template in a PCR reaction with the following oligonucleotides as primers: GATA-3–5′: GAATTCGTCGACGCTCTGCCTCTCTAACCCAT; and GATA-3–3′: GAATTCGTCGACGGACATGGAGGTGACTGCGGA.
This product was digested with SalI and ligated into XhoI-digested hCD4-RV (18) to generate GATA-3-hCD4-RV. Retrovirus was produced and transfections performed as described previously (13). For coinfection experiments, viral supernatants of FOG-1-RV and GATA-3-hCD4-RV were added together on day 2 Th1 cells and infected T cells were sorted on day 7 for dual expression of GFP and hCD4 using monoclonal anti–human CD4 directly conjugated with R-PE (Caltag).
Northern and Western Blot Analysis
Total RNA was isolated by Rneasy kit (QIAGEN). RNA (5 μg per lane) was electrophoresed and transferred to Zeta probe membrane (Bio-Rad Laboratories). A FOG-1 cDNA probe was generated using the following oligonucleotide primers: FOG1-S: 5′ CTG TCG GCC TTC ACC ACC AA; and FOG-1-AS: 5′ GTG CCT TGT CAG CGG GAA CC. The HPRT, GAPDH, and GATA-3 probes have been described previously (13, 19). Blots were probed with murine monoclonal anti–GATA-3 antibody HG3–31 (Santa Cruz Biotechnology, Inc.), a murine polyclonal anti–FOG-1 serum (8), and polyclonal anti–murine ZAP-70 serum (20).
IL-5 Reporter Analysis
To generate a high copy number IL-5 reporter construct, we obtained the wild-type IL-5 reporter originally generated in the vector pXP1 (21) and moved it into the vector pBS-LUC (22) as follows. The pXP1 IL-5 reporter construct was digested with HindIII and BamHI, and the liberated 1.7 Kb IL-5 promoter fragment ligated into HindIII/BamHI-digested pBS-LUC, generating IL-5-LUC. We used the Renilla luciferase vectors pRL-TK or pRL-CMV (Promega) to normalize Firefly luciferase transfections.
To express GATA-3 in transient transfections, the GATA-3 PCR product generated above using R/m GATA-3 and primers GATA-3–5′ and GATA-3–3′ was digested with SalI and ligated to into XhoI-digested vector pcDNA3.1 (Invitrogen) to generate the plasmid GATA-3-pcDNA (8). 10 × 106 EL-4 cells on ice were electroporated in 1 ml at 960 μF and 320V (22) with combinations of GATA-3-pcDNA and PMT2-FOG-1 as described in the Figure legends. After 12 h, cells were either left untreated or activated by 25 ng/ml PMA and 1 mM Bt2cAMP for 6 h as indicated in the Figure legends and luciferase activity measured as described previously (22).
Analysis of FOG-1–deficient T Cells by RAG2 Blastocyst Complementation
The CJ-7 ES cell line was previously used to generate FOG-1−/+ single targeted and FOG-1−/− doubly targeted ES clones FOG3.31 and FOG3.3.11.10 (23). 10 ES cells were injected into recombination activating gene (RAG)-2–deficient blastocysts as described previously (24). Two chimeras each were analyzed for FOG-1−/+ single targeted and FOG-1−/− doubly targeted ES clones. Analysis of thymocyte subsets (see Fig. 7 A) was performed as described previously (25). To analyze IL-4 production, intracellular staining was performed as described previously (6).
Figure 7.


FOG-1–deficient thymocytes exhibit developmental arrest and inefficient production of mature CD4+ T cells. (A) Thymuses were harvested from 4-wk-old FOG-1−/+ or FOG-1−/− RAG-2 chimeras or age-matched RAG-2−/− mice and analyzed by FACS®. Total thymocyte numbers are shown above the FACS® profiles. Thymocytes were stained for CD4, CD8, CD44, and CD25 expression as described previously (reference 25). Numbers shown are the percentage of live gated cells in each quadrant. (B) Peripheral CD4+ CD62L+ (L-selectin) T cells from the spleens and lymph node of FOG-1−/+ or FOG-1−/− chimeras or unimmunized Balb/c mice were purified by cell sorting using FITC-conjugated rat anti–mouse CD4 and phycoerythrin-conjugated rat anti-CD62L. T cells were activated using plate bound anti-CD3 (5 μg/ml) and anti-CD28 (1 μg/ml) essentially as described previously (reference 43). The indicated conditions cytokines conditions were imposed: Th1, IL-12 (3 μg/ml), and anti–IL-4 (11B11, 10 μg/ml); Th2, IL-4 (100 U/ml), anti–IL-12 (TOSH, 3 μg/ml; reference 15); Neutralized (Neu), anti–IL-12, anti–IL-4, and anti–IFN-γ (H22 10 μg/ml); or without any cytokine or antibody addition (Drift). Cells were allowed to proliferate and expand for 7 d and were harvested, restimulated, and analyzed for IL-4 production by intracellular staining as described previously (6). Numbers in the upper right are the percentage of live gated cells in this quadrant. Similar results were obtained in two experiments
Results
FOG-1 Is Expressed in both Th1 and Th2 by CD4+ T Cells
We first considered FOG-1, FOG-2, BCl-6, and CIITA expression in T cells during the induction of Th1 and Th2 subsets (Fig. 1 A). The mRNAs for BCl-6, FOG-2, and CIITA were essentially undetectable in T cells under either Th1 or Th2 conditions after 2 or 4 d of activation (data not shown). Although CIITA expression in Th1 cells has been reported by one group, it was detected only by RT-PCR, and not by Northern or RNase protection (26). In contrast, FOG-1 mRNA was detectable by Northern blot analysis (Fig. 1 A). Expression was relatively low in naive T cells, induced somewhat by stimulation with anti-CD3 and anti-CD28 antibodies. The induction of both Th1 and Th2 development requires T cell activation, so that the increase in FOG-1 expression seen upon activation may indicate a potential regulatory role for FOG-1 in phenotype development. Expression was essentially the same between Th1- and Th2-inducing conditions and was maintained at relatively similar levels from naive T cells to day 7, not showing significant elevation in expression after Th differentiation (Fig. 1 A). By contrast, GATA-3 expression was barely detectable by Northern blot analysis in naive T cells, but became strongly and selectively expressed during Th2 development. Thus, in early T cells, mRNA for FOG-1 is in relative excess to GATA-3, whereas in differentiated Th2 cells GATA-3 is in relative excess to FOG-1.
Figure 1.


FOG-1 is not selectively expressed in Th1 or Th2 cells. (A) T cells from DO11.10 TCR-transgenic mice were stimulated with 0.3 μM OVA peptide in the presence (+) of either IL-4 (100 U/ml) or IL-12 (10 U/ml) as indicated or with that anti-cytokine antibodies (−) specific for IL-4 (11B11, 10 μg/ml; reference 14) or IL-12 (TOSH 3 μg/ml) (reference 15). Cells were harvested on the indicated day after activation and total RNA prepared. Northern blot analysis for the indicated transcripts was performed as described previously (reference 13). For lanes 1 and 2, naive T cells were purified by cell sorting as described in the Materials and Methods and left untreated or activated with anti-CD3 and anti-CD28 for 20 h and RNA prepared for Northern blot analysis for FOG-1 and GATA-3 expression. (B) T cells activated in the indicated primary as described in A were harvested on day 7, and activated for 12 h with plate-bound anti-CD3 (5 μg/ml) and either left untreated (media) or activated with anti-CD3 (5 μg/ml), and Western blot analysis sequentially performed for FOG-1, GATA-3, and ZAP-70.
We confirmed this result by Western blot analysis, checking whether FOG-1 became differentially expressed in differentiated Th1 or Th2 cells at the level of protein expression (Fig. 1 B). FOG-1 was expressed at essentially the same level in either resting or restimulated cells and in Th1 and Th2 cells (Fig. 1 B, top). As a control, we found that GATA-3 protein is highly expressed in resting and activated Th2 cells, but essentially undetectable in Th1 cells.
FOG-1 Represses GATA-3–dependent Transcriptional Activity
FOG-1 activates GATA-1 transcription at the NF-E2 promoter (8), but represses GATA-1 activity at the M1α and the EKLF promoters (27) in transfection assays and during erythropoiesis in injected Xenopus embryos (10). These results suggest that the activity of FOG-1 may differ in distinct promoter contexts. While FOG-1 can interact with GATA-3 in a yeast two-hybrid system (8), no functional studies of FOG-1 interactions with GATA-3 have been reported.
To test if FOG-1 activates or represses GATA-3–dependent activity in T cells, we used the GATA-3–dependent reporter system based on the IL-5 promoter (28). First, we established the linear range for GATA-3 in which increasing GATA-3 expression caused an increase in PMA/Bt2cAMP-induced reporter activity (Fig. 2 A), consistent with the dose–dependent effects of GATA-3 on the IL-5 promoter (28). Using a linear range of GATA-3 cotransfection, we next asked if FOG-1 could activate or repress GATA-3–dependent IL-5 promoter activity (Fig. 2 B). FOG-1 expression alone had no effect on activating IL-5 reporter activity (Fig. 2 B). As expected, GATA-3 expression increased PMA/Bt2cAMP-inducible reporter activity (Fig. 2 B). Coexpression of FOG-1 with GATA-3 almost completely inhibited GATA-3–induced reporter activity (Fig. 2 B). Inhibition by FOG-1 of GATA-3–dependent reporter activity was dose–dependent (Fig. 2 C), was maximal at 15 μg of FOG-1 plasmid, and saturated at ∼80% inhibition. In summary, FOG-1 represses GATA-3–dependent IL-5 promoter activity.
Figure 2.



FOG-1 inhibits GATA-3–dependent IL-5 promoter activation. (A) 107 EL-4 cells were electroporated with IL-5-Luc (20 μg), pRL-TK (5 μg) and the indicated micrograms of GATA-3-pcDNA (GATA-3) and pcDNA3.1 (Invitrogen). After 16 h, cells were left untreated (open bars) or treated with PMA/Bt2cAMP (closed bars) for 6 h and luciferase activity determined. Values shown are the relative Firefly luciferase activity after normalization by Renilla luciferase activity of pRL-TK. The results were repeated twice. (B) 107 EL-4 cells were transfected with the IL-5-Luc and pRL-TK as above, with additions (+) of GATA-3-pcDNA (6 μg) and pMT2-FOG-1 (20 μg) as indicated. 6 μg of pcDNA3.1 and 20 μg pMT2 were added in replacement (−) to equalize total DNA between samples. After 16 h, cells were left untreated (white bars) or treated with PMA/Bt2cAMP (black bars) for 6 h and luciferase activity determined and analyzed as in A. The experiment was repeated five times with similar results. (C) EL-4 cells were transfected with IL-5-Luc (20 μg), pRL-CMV (5 μg), and the indicated micrograms of GATA-3-pcDNA and pMT2-FOG-1. To equalize DNA between samples, equal amounts of pcDNA3.1, or pMT2 were added in replacement as in B above. Cells were stimulated and analyzed as in A. The data are presented as fold-induction over the unstimulated IL-5 reporter activity in the condition without GATA-3 and FOG addition (lane 1). The experiment was repeated four times with similar results.
FOG-1 Overexpression in Naive T Cells Represses Th2 Development
We wished to determine if FOG-1 also inhibited GATA-3–dependent transcriptional activity in nontransformed T cells. For this we used retroviral gene transfer to express FOG-1 in antigen-activated DO11.10 T cells (Fig. 3 A). First, we asked if expressing FOG-1 early during development would alter acquisition of a Th2-cytokine pattern induced by IL-4 (Fig. 3 B). In T cells activated in the presence of IL-4, retroviral overexpression of FOG-1 partially inhibited IL-5 and IL-4 expression compared with T cells infected with the empty control retrovirus. FOG-1 inhibited IL-4 production by ∼70%, and inhibited IL-5 by ∼50% (Fig. 3 B). In five additional independent experiments, overexpression of FOG-1 consistently inhibited IL-4 expression by between 50 to 80%. While inhibition of IL-4 production was not complete, the effects of FOG-1 were selective for Th2 cells, since IFN-γ production was not affected (data not shown, and Fig. 5).
Figure 3.

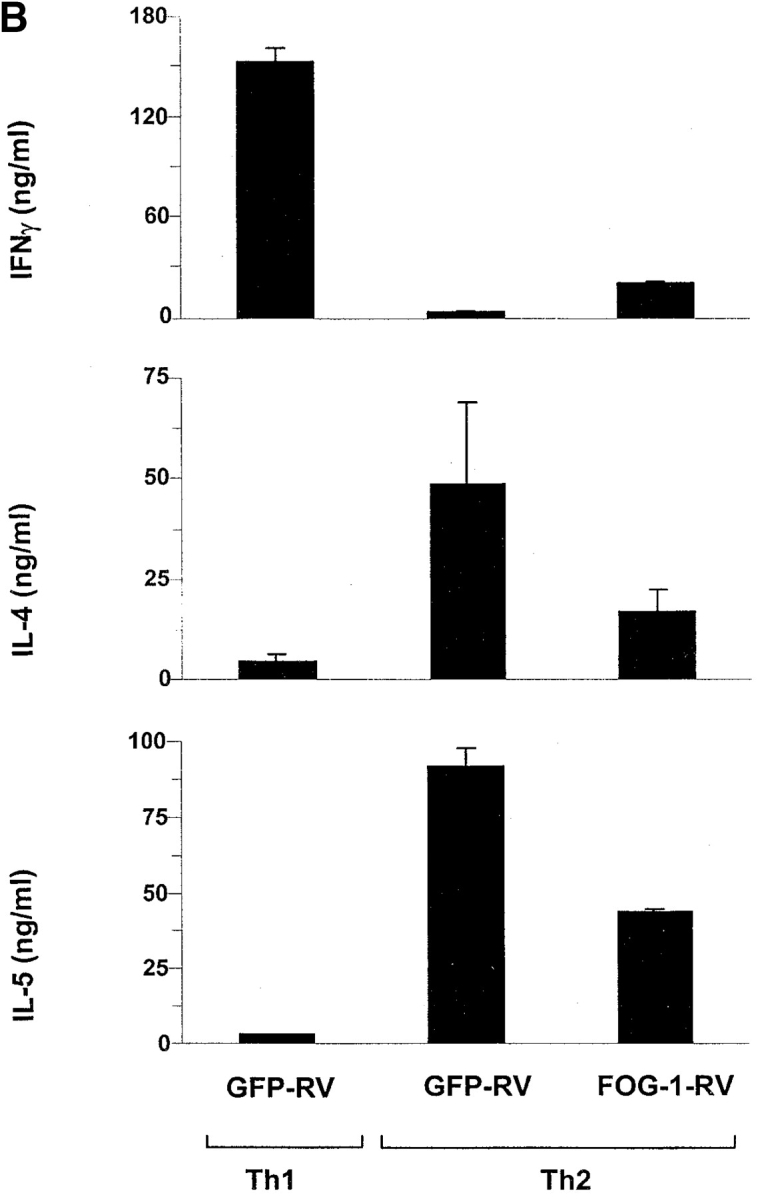

FOG-1 overexpression in naive T cells represses Th2 development. (A) Retroviral constructs. (B) T cells from unimmunized DO11.10 TCR transgenic mice were activated using 0.3 μM OVA peptide and conditions inducing either Th1 (10 U/ml IL-12 and 10 μg/ml anti–IL-4) or Th2 (100 U/ml IL-4 and 3 μg/ml anti–IL-12) development and infected by GFP-RV or FOG-1-RV virus after 36 h. CD4+ GFP+ T cells were purified by cell sorting on day 7 and restimulated with OVA, and expanded for 7 d. T cells were restimulated (1.25 × 105 cells per milliliter) with 0.3 μM OVA peptide, and cytokines measured by ELISA as described previously. Similar results were obtained in six independent experiments. (C) Cells in B were expanded for 1 wk and activated with PMA (50 ng/ml) and ionomycin (1 μM) overnight. Whole cell lysates (5 × 106 cells per lane) were analyzed by Western blot analysis for expression of FOG-1 and ZAP-70.
Figure 5.

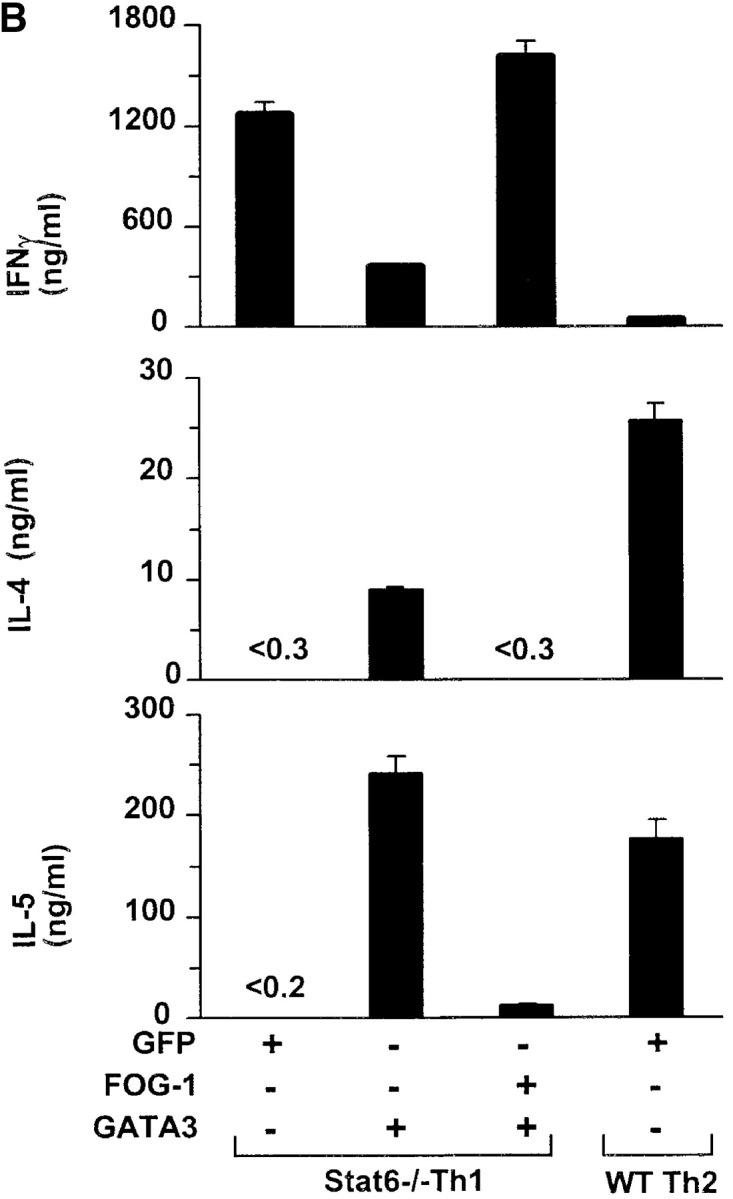
FOG-1 represses GATA-3–induced Th2 in Stat6-deficient T cells. T cells from unimmunized DO11.10 TCR transgenic mice were activated using 0.3 μM OVA peptide and conditions inducing either Th1 (10 U/ml IL-12 and 10 μg/ml anti–IL-4) or Th2 (100 U/ml IL-4 and 3 μg/ml anti–IL-12) development as indicated. After 36 h, cells were infected (+) with GFP-RV, FOG-1-RV, or GATA-3-RV as indicated. Retrovirally infected T cells were purified for the appropriate markers on day 7 by cell sorting, and expanded once with OVA and APCs for an additional 7 d. T cells were purified a second time by cell sorting, immediately stimulated with OVA/APC as described in Fig. 3, and cytokine production measured by ELISA. Similar results were obtained in six independent experiments. (B). Stat6-deficient (Stat6−/−) or wild-type (WT) DO11.10 T cells from unimmunized mice were activated using 0.3 μM OVA peptide and conditions inducing either Th1 (10 U/ml IL-12 and 10 μg/ml anti–IL-4) or Th2 (100 U/ml IL-4 and 3 μg/ml anti–IL-12) development as indicated. After 36 h, T cells were infected (+) with GFP-RV, FOG-1-RV, or GATA-3-RV as indicated. Cells were purified for the appropriate retroviral markers on day 7 by cell sorting and expanded once. Then (on day 14), T cells were purified a second time by cell sorting and activated as above, and cytokines measured by ELISA.
As a control, we verified by Western blot analysis that the retroviral expression of FOG-1 significantly increased FOG-1 protein content in these cells (Fig. 3 C). This experiment also verified that the level of endogenous FOG-1 is similar between Th1 and Th2 cells as found above (Fig. 3 C, compare lanes 1 and 2). Thus, overexpression of FOG-1 repressed IL-4-induced Th2 development, consistent with it acting as a repressor of GATA (ROG)-3–dependent activity in the transient transfection system.
We next asked if FOG-1 could repress Th2 cytokines in fully differentiated cells. T cells that had undergone 1 wk of previous differentiation were infected with FOG-1–expressing and control retroviruses (Fig. 4). At this time point, overexpression of FOG-1 did not significantly repress Th2 cytokine production (Fig. 4 A), even though FOG-1 protein levels were significantly elevated as before (Fig. 4 B). This result suggests that FOG-1 can act as a repressor primarily during an early step in Th2 development.
Figure 4.
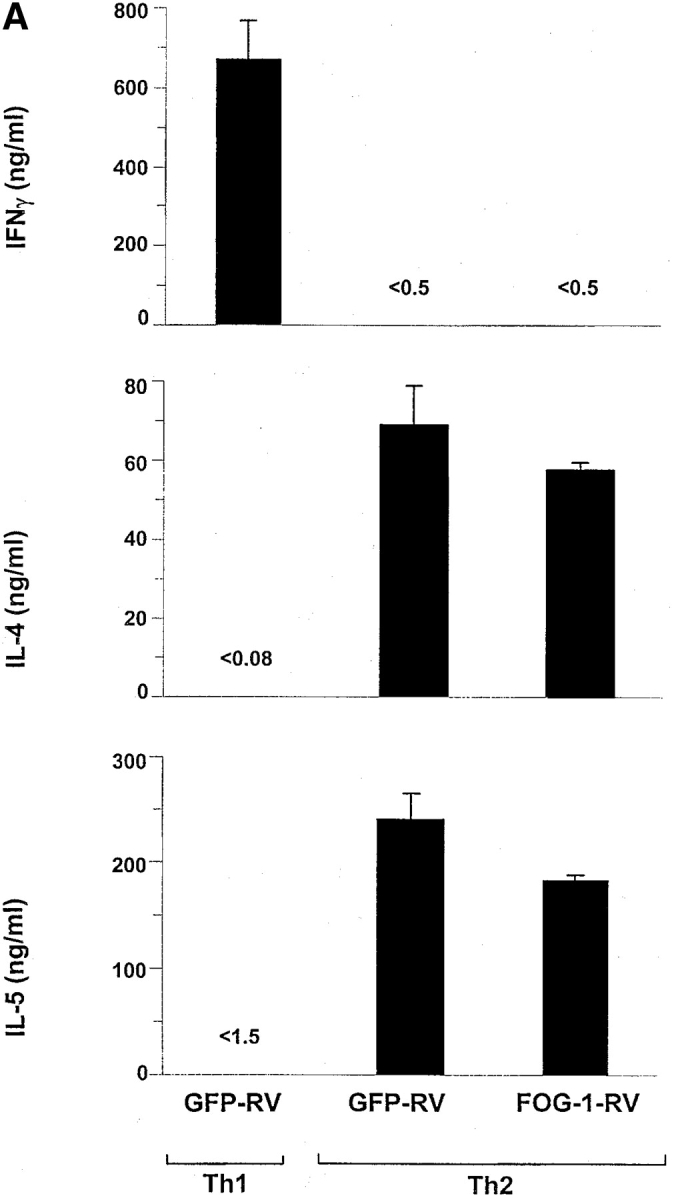

FOG-1 overexpression in differentiated Th2 cells does not repress cytokine production. T cells from unimmunized DO11.10 TCR transgenic mice were activated using 0.3 μM OVA peptide and conditions inducing either Th1 (10 U/ml IL-12 and 10 μg/ml anti–IL-4) or Th2 (100 U/ml IL-4 and 3 μg/ml anti–IL-12) development and allowed to develop for 7 d. T cells were then restimulated with OVA/APCs and infected with GFP-RV or FOG-1-RV virus after 36 h. CD4+ GFP+ T cells were purified by cell sorting after another 6 d. Cells were then immediately activated (1.25 × 105 cells per milliliter with 0.3 μM OVA and irradiated APCs) and cytokine production measured by ELISA. (B) Cells in A were expanded for 1 wk and activated with PMA (50 ng/ml) and ionomycin (1 μM) overnight. Whole cell lysates (5 × 106 cells per lane) were analyzed by Western blot analysis for expression of FOG-1 and ZAP-70. Similar results were obtained in three additional independent experiments.
FOG-1 Inhibits GATA-3–induced Th2 Development
FOG-1 could repress Th2 development either by inhibiting Stat6-dependent processes or a GATA-3–dependent process. To distinguish these possibilities, we performed a series of coinfection experiments in which FOG-1 and GATA-3 were independently expressed by different retroviral vectors. Since retroviral GATA-3 expression can induce Th2 development independently of Stat6 and in Stat6-deficient cells (6, 13), coexpressing FOG-1 with GATA-3 allows a direct test of its role in the GATA-3–dependent Th2 development.
First, we performed coinfection studies in wild-type T cells that contain Stat6, activated in Th1-inducing conditions, where Stat6 activation may be prevented using a neutralizing anti–IL-4 antibody. T cells activated in Th1-inducing conditions were infected with combinations of FOG-1 and GATA-3 retroviruses on day 2 and infected cells purified by two-color cell sorting on day 7. Under Th1 conditions, the GATA-3 retrovirus induced IL-4 and IL-5 production and inhibited IFN-γ production (Fig. 5 A) consistent with previous findings (6, 13, 29). In contrast, the FOG-1 retrovirus did not induce Th2 development or inhibit IFN-γ production (Fig. 5 A). However, when FOG-1 and GATA-3 both expressed in a dual infection, the FOG-1 significantly inhibited the extent of GATA-3–induced Th2 development (Fig. 5 A).
In the above experiment, IL-4 induced by the retroviral GATA-3 may not be completely neutralized by antibody, allowing for potential Stat6-dependent induction of GATA-3 in T cells. To eliminate this possibility, we repeated these coinfection experiments using Stat6-deficient DO11.10 T cells also activated under Th1-inducing conditions (Fig. 5 B). In this setting, retroviral GATA-3 expression strongly induced Th2 development, with high IL-4 and IL-5 production (Fig. 5 B). However, when FOG-1 and GATA-3 were now coexpressed, FOG-1 almost completely inhibited GATA-3–induced Th2 development (Fig. 5 B), with IL-4 production nearly undetectable, and with >10-fold reduced IL-5. Additionally, FOG-1 coexpression with GATA-3 increased IFN-γ production fourfold in Stat6-deficient T cells. These results suggest that FOG-1 can repress GATA-3–induced Th2 development.
FOG-1 Inhibits GATA-3 Autoactivation, but not Stat6–dependent GATA-3 Induction
Since retroviral GATA-3 can induce expression of the endogenous GATA-3 gene (6), we wanted to ask whether FOG-1 could repress GATA-3 autoactivation. Northern blot analysis can distinguish between the retrovirally derived and the endogenous cellular transcripts for both GATA-3 and FOG-1. On Northern blots, endogenous GATA-3 transcripts migrate at ∼3.8 Kb and retroviral GATA-3 transcripts migrate at 4.6 Kb. Endogenous FOG-1 migrates at 3.4 Kb and retroviral FOG-1 at 6.5 Kb.
We first examined wild-type T cells that express Stat6, in which FOG-1 only incompletely blocked GATA-3–induced Th2 development (Fig. 6 A). T cells were activated in Th1-inducing conditions, infected on day 2 with FOG-1 or GATA-3 retroviruses, purified by two-color cell sorting on day 7, expanded, and analyzed by Northern and Western blot analysis (Fig. 6 A). Retroviral GATA-3 infection induced the expression of endogenous GATA-3 transcripts (Fig. 6 A, lane 3), consistent with previous findings (6). Retroviral FOG-1 did not induce endogenous GATA-3 transcription (Fig. 6 A, lane 2). Coinfection of FOG-1 with GATA-3 reduced the relative level of induction of endogenous GATA-3 both as measured by Northern blot analysis (Fig. 6 A, lane 4) and also as measured by Western blot analysis (Fig. 6 B, compare lane 3 and 4).
Figure 6.



FOG-1 represses Stat6-independent GATA-3 autoactivation. (A and B) Cells analyzed for cytokine expression in Fig. 5 A were expanded for 7 d, activated with PMA and ionomycin for 12 h, and total RNA prepared. Northern blot analysis (A) for expression of the retroviral and endogenous FOG-1 and GATA-3 transcripts, and GAPDH was performed. Western blot analysis (B) was performed for GATA-3 and ZAP-70. (C) Cells analyzed for cytokine expression in Fig. 5 B were expanded for 7 d, activated with PMA and ionomycin for 12 h, and total RNA prepared. Northern blot analysis for retroviral and endogenous GATA-3 expression, and GAPDH expression was performed.
To directly test whether FOG-1 can repress IL-4–dependent and Stat6-dependent induction of GATA-3, we also examined GATA-3 protein expression of FOG-1–infected cells activated in the presence of IL-4 (Fig. 6 B, lane 6). Importantly, in these Th2-inducing conditions, retroviral FOG-1 expression did not reduce GATA-3 levels. Thus, in T cells expressing Stat6, FOG-1 partially represses GATA-3 autoactivation induced by retroviral GATA-3, but this repression may be incomplete since some IL-4 produced by some T cells (Fig. 3 B) could potentially induce endogenous GATA-3 through the Stat6-dependent pathway. However, in this condition, the transcriptional effect of GATA-3 that is expressed can still be inhibited by FOG-1, since we observed 50–80% decrease in IL-4 production (Fig. 3 B).
To completely eliminate Stat6-dependent induction of GATA-3 and examine FOG-1 repression of GATA-3 autoactivation in isolation, we repeated the experiment now using Stat6-deficient T cells (Fig. 6 C). As expected, T cells activated in Th1 conditions and infected by empty retrovirus showed no expression of endogenous GATA-3 (Fig. 6 C, lane 1). Retroviral GATA-3 again strongly induced the expression of endogenous GATA-3 transcripts (Fig. 6 C, lane 2). However, now in Stat6-deficient T cells, the coexpression of FOG-1 with GATA-3 caused a very significant reduction in the level of endogenous GATA-3 transcription induced by retroviral GATA-3 (Fig. 6 C, compare lanes 2 and 3). This result suggests that FOG-1 can repress the transcriptional activity of the GATA-3 protein in driving the expression of the endogenous GATA-3 locus.
Analysis of FOG-1–deficient T Cells Using RAG-2 Blastocyst Complementation
Embryonic lethality prevents direct analysis of mature FOG-1–deficient T cells (23). Therefore, we attempted to generate normal mature T cells by producing chimeras using complementation of RAG-2−/− blastocysts with ES cells targeted at both FOG-1 alleles (23) (Fig. 7). In such chimeras, mature T and B cells are exclusively derived from FOG-1–targeted ES cells. For controls, we generated chimeras with an ES cell targeted on only one FOG-1 allele (23).
Overall thymocyte numbers in FOG-1−/− chimeras were greatly reduced relative to FOG-1−/+ chimeras (Fig. 7 A). FOG-1−/− chimeras were nearly devoid of CD4+CD8+ (DP) thymocytes (2%) (Fig. 7 A, top) compared with control FOG-1−/+ chimeras which showed the normal majority of DP thymocytes (82%). In FOG-1−/− chimeras, CD4−CD8− (DN) thymocytes were the majority, similar to unreconstituted RAG-2−/− mice. Analysis of DN thymocytes showed a block in the transition from CD44−/CD25+ to CD44−/CD25− in FOG-1−/− thymocytes, indistinguishable to RAG-2−/− thymocytes, whereas this transition occurred normally in FOG-1−/+ thymocytes (Fig. 7 A, bottom). Endogenous RAG-2−/− DN thymocytes are present in all these chimeras, and without additional analysis we cannot precisely conclude at which step before the DP stage the FOG-1−/− thymocytes development arrests. Some single positive CD4+ or CD8+ thymocytes T cells are present in FOG-1−/− thymuses despite the lack of DP thymocytes, distinguishing them from totally unreconstituted RAG-2−/− thymuses, although it is unclear how these thymocytes developed in the absence of a DP population. Finally, mature B cells were identified at similar numbers in the spleens of FOG-1−/− chimeras and FOG-1−/+ chimeras (data not shown). These results could either indicate a requirement for FOG-1 in DP thymocyte development, or reflect poor T cell reconstitution by this particular FOG-1−/− ES clone.
Despite poor thymocyte development in FOG-1−/− chimeras, we attempted to identify potential effects on Th1/Th2 development by purifying the peripheral CD4+ T cells from FOG-1−/− and FOG-1−/+ chimeras. Peripheral CD4+ T cell numbers in FOG-1−/− chimeras were significantly lower than in FOG-1-/+ chimeras, preventing isolation of cells sufficient for direct biochemical (Northern and Western) blot analysis, but sufficient for in vitro activation (Fig. 7 B). Activation of these T cells by antibodies, rather than by antigen, was necessary since these thymocytes are not on a TCR-transgenic background, but on the 129/Sv genetic background of CJ-7 ES cell (23). Thus, we included activation of Balb/c T cells as a control for this difference in genetic background and activation method.
Naive FOG-1−/−, FOG-1−/+, or Balb/c T cells were purified by cell sorting and activated with plate bound anti-CD3/CD28 antibodies under various initial conditions (Fig. 7 B). In Th1 conditions, each population failed to acquire IL-4 production (Fig. 7 B, Th1). Similarly low levels of IL-4 production were seen when all cytokines (e.g., IL-4, IL-12, and IFN-γ) were neutralized (Fig. 7 B, Neu). In Th2 conditions, Balb/c T cells acquired a strong Th2 phenotype, with 49% cells positive for IL-4 production. However, both the FOG-1−/− and FOG-1−/+ T cells became only weakly positive for IL-4 production, with 6.5 and 11% of cells producing IL-4 under these same Th2 conditions (Fig. 7 B). FOG-1−/− T cells were less positive for IFN-γ intracellular staining under Th1 conditions (24%) compared with FOG-1−/+ T cells (40%; data not shown). These results could indicate a general problem with cytokine production in FOG-1–deficient T cells, related either to abnormal reconstitution or to the absence of FOG-1.
More importantly, differences in genetic background prevent strong conclusions from these RAG-2 chimera studies. Even under Th2-inducing conditions, 129/sv-derived T cells showed much less efficient IL-4 production than Balb/c T cells, independently of FOG-1 deficiency. This effect could be due to differences in genetic background (30–35). Further, in unmanipulated conditions (Fig. 7 B, Drift), Balb/c T cells became 39% positive, whereas FOG-1−/− or FOG-1−/+ T cells became essentially negative, at 2% and <1% positive for IL-4 production, demonstrating the sensitivity of default Th2 development to variations in genetic background. While, we intended to test if FOG-1 deficiency caused constitutive GATA-3 autoactivation, we can only conclude that FOG-1–deficient T cells do not acquire a default Th2 phenotype under these in vitro conditions, and were unable to directly analyze GATA-3 expression. Thus, we must temper our conclusions due to possible artifacts induced by poor thymocyte development in these chimeras and to differences in genetic background between 129/sv and Balb/c T cells for default Th2 development.
Discussion
Th2 development can be divided into two phases (7); an initial phase in which a transient excess of IL-4 induces GATA-3 expression in a Stat6-dependent manner (13) followed by a phase in which GATA-3 promotes its own transcription (i.e., autoactivation) in a Stat-6–independent manner, stabilizing GATA-3 expression and Th2 development (6). Since naive activated T cells express detectable levels of GATA-3 even without IL-4 treatment (6), we wondered whether T cells contain factors capable of controlling GATA-3–dependent transcription and thereby generating a threshold for GATA autoactivation. In this study we considered factors known to interact with GATA family members. FOG-1 and FOG-2 could potentially repress GATA-3–dependent transcription. However FOG-2 was not detectable in T cells. FOG-1 is detectable in both naive and differentiated CD4+ T cells. FOG-1 was expressed slightly higher in activated naive T cells than resting naive T cells, but was similar between Th1 and Th2 conditions. Thus, we wondered whether FOG-1 could act to inhibit GATA-3–dependent transcriptional activity in T cells. Our findings suggest that FOG-1 can repress GATA-3–dependent promoter activity, and repress the GATA-3–dependent induction of Th2 development, but does not block Stat6-dependent processes, such as IL-4–induced transcription of GATA-3.
We have not examined all possible factors that could regulate GATA-3 autoactivation. The zinc finger protein ROG has been reported to exert an inhibitory effect in T cells that may be related to its ability to interact with GATA proteins (36). ROG was identified initially as a GATA-3–interacting protein, and when overexpressed in T cell clones exerted repression of both Th1 and Th2 cytokines, with effects shown for IFN-γ, IL-4, and IL-5 (36). ROG-1 is induced within 24 h of activation under both Th1 and Th2 conditions, so conceivably could also participate in regulating GATA-3–dependent autoactivation (36). However, so far the effects of introducing ROG by overexpression have been examined in fully differentiated T cell clones, and not in early developing primary T cells. Thus, the effect that ROG could potentially have on GATA-3 expression has not been examined.
Other factors could also help regulate GATA-3–dependent transcriptional activity, including but not limited to CIITA (26), BCl-6 (37), and T-bet (38), since each of these factors opposes Th2 cytokine expression in one way or another. At present, these factors are not thought to directly regulate GATA-3 transactivation, but rather to exert independent effects on downstream targets. For example, GATA-3 levels were reported to be independent of CIITA expression (26). However, it will be interesting to determine their potential for directly regulating GATA-3 autoactivation as well.
FOG-1 contains repetitive zinc finger motifs, with zinc fingers 1, 5, 6, and 9 capable of interacting with GATA-1 N finger (39). FOG-1 may inhibit GATA-dependent transcriptional activity in part by recruiting CtBP2, a local transcriptional repressor (40). A short protein motif, PIDLS, located between zinc fingers 6 and 7 of FOG-1 mediates interaction with CtBP2 (40). Not all repression may involve CtBP2, however, since the NH2 terminus of FOG-2 exerts transcriptional inhibition independently of its ability to bind CtBP2 (41).
We propose that FOG-1 may be one factor that can regulate GATA-3–dependent transcriptional activity in T cells. This effect could occur either by recruiting CtBP2 (or other corepressors) into the GATA-3 transcriptional complex, or by direct repression of GATA-3 transcriptional activity. In either model, the level of inhibition caused by FOG-1 depends on relative amounts of FOG-1 and GATA-3. When GATA-3 is low, as in naive T cells, fixed levels of FOG-1 might bind a larger fraction of GATA-3, and more effectively repress GATA-3–dependent transcription. Our data showed that FOG-1 only repressed GATA-3–induced processes, including GATA-3 autoactivation and Stat6-independent Th2 development, but not the IL-4–induced, Stat6-driven GATA-3 expression. In this way, FOG-1 might regulate GATA-3 activity in naive T cells, but not block the Stat6-dependent IL-4–driven induction of GATA-3. Importantly, FOG-1 expression is not strongly induced in Th2 development, so that higher levels of GATA-3 in Th2 cells results in a smaller FOG-1–bound fraction, and less repression. This is consistent with our finding that overexpression of FOG-1 in differentiated Th2 cells did not inhibit IL-4 or IL-5 production (Fig. 4 A). Since germline deletion of FOG-1 (23) and GATA-3 (42, 43) are both lethal, testing these models for T cells in vivo is difficult. We attempted to analyze FOG-1–deficient T cells for altered Th2 development using RAG-2 blastocyst complementation, but we are unable to draw firm conclusions from these experiments. More definitive tests of FOG-1's role in regulating Th2 development will likely require a inducible deletion of FOG-1.
Acknowledgments
We thank Sheila H. Ranganath for helpful comments and discussions, and Dominic Fenoglio for help with cell sorting.
S.G. Katz is supported by a predoctoral fellowship of the Howard Hughes Medical Institute. This work was supported by grants from the National Institutes of Health. K.M. Murphy and S.H. Orkin are Investigators of the Howard Hughes Medical Institute.
Footnotes
Abbreviations used in this paper: FOG, friend of GATA; GFP, green fluorescent protein; RAG, recombination activating gene; ROG, repressor of GATA; Stat, signal transducer and activator of transcription.
References
- 1.Dasen, J.S., S.M. O'Connell, S.E. Flynn, M. Treier, A.S. Gleiberman, D.P. Szeto, F. Hooshmand, A.K. Aggarwal, and M.G. Rosenfeld. 1999. Reciprocal interactions of Pit1 and GATA2 mediate signaling gradient-induced determination of pituitary cell types. Cell. 97:587–598. [DOI] [PubMed] [Google Scholar]
- 2.Tsai, S.F., E. Strauss, and S.H. Orkin. 1991. Functional analysis and in vivo footprinting implicate the erythroid transcription factor GATA-1 as a positive regulator of its own promoter. Genes Dev. 5:919–931. [DOI] [PubMed] [Google Scholar]
- 3.Steinbach, O.C., A. Ulshofer, A. Authaler, and R.A. Rupp. 1998. Temporal restriction of MyoD induction and autocatalysis during Xenopus mesoderm formation. Devel. Biol. 202:280–292. [DOI] [PubMed] [Google Scholar]
- 4.Aurade, F., C. Pinset, P. Chafey, F. Gros, and D. Montarras. 1994. Myf5, MyoD, myogenin and MRF4 myogenic derivatives of the embryonic mesenchymal cell line C3H10T1/2 exhibit the same adult muscle phenotype. Differentiation. 55:185–192. [DOI] [PubMed] [Google Scholar]
- 5.Barger, P.M., and D.P. Kelly. 1997. Identification of a retinoid/chicken ovalbumin upstream promoter transcription factor response element in the human retinoid X receptor γ2 gene promoter. J. Biol. Chem. 272:2722–2728. [DOI] [PubMed] [Google Scholar]
- 6.Ouyang, W., M. Lohning, Z. Gao, M. Assenmacher, S. Ranganath, A. Radbruch, and K.M. Murphy. 2000. Stat6-independent GATA-3 autoactivation directs IL-4-independent Th2 development and commitment. Immunity. 12:27–37. [DOI] [PubMed] [Google Scholar]
- 7.Murphy, K.M., W. Ouyang, S. Ranganath, and T.L. Murphy. 1999. Bi-stable transcriptional circuitry and GATA-3 auto-activation in Th2 commitment. Cold Spring Harbor Symp. 64:585–588. [DOI] [PubMed] [Google Scholar]
- 8.Tsang, A.P., J.E. Visvader, C.A. Turner, Y. Fujiwara, C. Yu, M.J. Weiss, M. Crossley, and S.H. Orkin. 1997. FOG, a multitype zinc finger protein, acts as a cofactor for transcription factor GATA-1 in erythroid and megakaryocytic differentiation. Cell. 90:109–119. [DOI] [PubMed] [Google Scholar]
- 9.Tevosian, S.G., A.E. Deconinck, A.B. Cantor, H.I. Rieff, Y. Fujiwara, G. Corfas, and S.H. Orkin. 1999. FOG-2: a novel GATA-family cofactor related to multitype zinc-finger proteins Friend of GATA-1 and U-shaped. Proc. Natl. Acad. Sci. 96:950–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deconinck, A.E., P.E. Mead, S.G. Tevosian, J.D. Crispino, S.G. Katz, L.I. Zon, and S.H. Orkin. 2000. FOG acts as a repressor of red blood cell development in Xenopus. Development. 127:2031–2040. [DOI] [PubMed] [Google Scholar]
- 11.Svensson, E.C., R.L. Tufts, C.E. Polk, and J.M. Leiden. 1999. Molecular cloning of FOG-2: a modulator of transcription factor GATA-4 in cardiomyocytes. Proc. Natl. Acad. Sci. 96:956–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaplan, M.H., U. Schindler, S.T. Smiley, and M.J. Grusby. 1996. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 4:313–319. [DOI] [PubMed] [Google Scholar]
- 13.Ouyang, W., S.H. Ranganath, K. Weindel, D. Bhattacharya, T.L. Murphy, W.C. Sha, and K.M. Murphy. 1998. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. 9:745–755. [DOI] [PubMed] [Google Scholar]
- 14.Ohara, J., and W.E. Paul. 1985. Production of a monoclonal antibody to and molecular characterization of B-cell stimulatory factor-1. Nature. 315:333–336. [DOI] [PubMed] [Google Scholar]
- 15.Tripp, C.S., M.K. Gately, J. Hakimi, P. Ling, and E.R. Unanue. 1994. Neutralization of IL-12 decreases resistance to Listeria in SCID and C.B-17 mice. Reversal by IFN-γ. J. Immunol. 152:1883–1887. [PubMed] [Google Scholar]
- 16.Ranganath, S., W. Ouyang, D. Bhattarcharya, W.C. Sha, A. Grupe, G. Peltz, and K.M. Murphy. 1998. GATA-3-dependent enhancer activity in IL-4 gene regulation. J. Immunol. 161:3822–3826. [PubMed] [Google Scholar]
- 17.Ko, L.J., M. Yamamoto, M.W. Leonard, K.M. George, P. Ting, and J.D. Engel. 1991. Murine and human T-lymphocyte GATA-3 factors mediate transcription through a cis-regulatory element within the human T-cell receptor δ gene enhancer. Mol. Cell. Biol. 11:2778–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farrar, J.D., J.D. Smith, T.L. Murphy, S. Leung, G.R. Stark, and K.M. Murphy. 2000. Selective loss of type I interferon-induced STAT4 activation caused by a minisatellite insertion in mouse STAT2. Nat. 1:65–69. [DOI] [PubMed] [Google Scholar]
- 19.Szabo, S.J., A.S. Dighe, U. Gubler, and K.M. Murphy. 1997. Regulation of the interleukin (IL)-12R β 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J. Exp. Med. 185:817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan, A.C., M. Iwashima, C.W. Turck, and A. Weiss. 1992. ZAP-70: a 70 kd protein-tyrosine kinase that associates with the TCR ζ chain. Cell. 71:649–662. [DOI] [PubMed] [Google Scholar]
- 21.Zhang, D.H., L. Cohn, P. Ray, K. Bottomly, and A. Ray. 1997. Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J. Biol. Chem. 272:21597–21603. [DOI] [PubMed] [Google Scholar]
- 22.Szabo, S.J., J.S. Gold, T.L. Murphy, and K.M. Murphy. 1993. Identification of cis-acting regulatory elements controlling interleukin-4 gene expression in T cells: roles for NF-Y and NF-ATc. Mol. Cell. Biol. 13:4793–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsang, A.P., Y. Fujiwara, D.B. Hom, and S.H. Orkin. 1998. Failure of megakaryopoiesis and arrested erythropoiesis in mice lacking the GATA-1 transcriptional cofactor FOG. Genes Dev. 12:1176–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen, J., R. Lansford, V. Stewart, F. Young, and F.W. Alt. 1993. RAG-2-deficient blastocyst complementation: an assay of gene function in lymphocyte development. Proc. Natl. Acad. Sci. 90:4528–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng, A.M., I. Negishi, S.J. Anderson, A.C. Chan, J. Bolen, D.Y. Loh, and T. Pawson. 1997. The Syk and ZAP-70 SH2-containing tyrosine kinases are implicated in pre-T cell receptor signaling. Proc. Natl. Acad. Sci. 94:9797–9801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gourley, T., S. Roys, N.W. Lukacs, S.L. Kunkel, R.A. Flavell, and C.H. Chang. 1999. A novel role for the major histocompatibility complex class II transactivator CIITA in the repression of IL-4 production. Immunity. 10:377–386. [DOI] [PubMed] [Google Scholar]
- 27.Fox, A.H., C. Liew, M. Holmes, K. Kowalski, J. Mackay, and M. Crossley. 1999. Transcriptional cofactors of the FOG family interact with GATA proteins by means of multiple zinc fingers. EMBO J. 18:2812–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang, D.H., L. Yang, and A. Ray. 1998. Differential responsiveness of the IL-5 and IL-4 genes to transcription factor GATA-3. J. Immunol. 161:3817–3821. [PubMed] [Google Scholar]
- 29.Lee, H.J., N. Takemoto, H. Kurata, Y. Kamogawa, S. Miyatake, A. O'Garra, and N. Arai. 2000. GATA-3 induces T helper cell type 2 (Th2) cytokine expression and chromatin remodeling in committed Th1 cells. J. Exp. Med. 192:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsieh, C.S., S.E. Macatonia, A. O'Garra, and K.M. Murphy. 1995. T cell genetic background determines default T helper phenotype development in vitro. J. Exp. Med. 181:713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorham, J.D., M.L. Guler, and K.M. Murphy. 1997. Genetic control of interleukin 12 responsiveness: implications for disease pathogenesis. J. Mol. Med. 75:502–511. [DOI] [PubMed] [Google Scholar]
- 32.Gorham, J.D., M.L. Guler, R.G. Steen, A.J. Mackey, M.J. Daly, K. Frederick, W.F. Dietrich, and K.M. Murphy. 1996. Genetic mapping of a murine locus controlling development of T helper 1/T helper 2 type responses. Proc. Natl. Acad. Sci. 93:12467–12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guler, M.L., J.D. Gorham, W.F. Dietrich, T.L. Murphy, R.G. Steen, C.A. Parvin, D. Fenoglio, A. Grupe, G. Peltz, and K.M. Murphy. 1999. Tpm1, a locus controlling IL-12 responsiveness, acts by a cell-autonomous mechanism. J. Immunol. 162:1339–1347. [PubMed] [Google Scholar]
- 34.Guler, M.L., J.D. Gorham, W.F. Dietrich, R.G. Steen, C. Parvin, D. Fenoglio, A. Grupe, G. Peltz, and K.M. Murphy. 1999. Loci influencing development of Th responses. Identification from in vitro analysis. Microbes. Infect. 1:79–88. [DOI] [PubMed] [Google Scholar]
- 35.Guler, M.L., J.D. Gorham, C.S. Hsieh, A.J. Mackey, R.G. Steen, W.F. Dietrich, and K.M. Murphy. 1996. Genetic susceptibility to Leishmania: IL-12 responsiveness in TH1 cell development. Science. 271:984–987. [DOI] [PubMed] [Google Scholar]
- 36.Miaw, S.C., A. Choi, E. Yu, H. Kishikawa, and I.C. Ho. 2000. ROG, repressor of GATA, regulates the expression of cytokine genes. Immunity. 12:323–333. [DOI] [PubMed] [Google Scholar]
- 37.Dent, A.L., A.L. Shaffer, X. Yu, D. Allman, and L.M. Staudt. 1997. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 276:589–592. [DOI] [PubMed] [Google Scholar]
- 38.Szabo, S.J., S.T. Kim, G.L. Costa, X.K. Zhang, C.G. Fathman, and L.H. Glimcher. 2000. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 100:655–669. [DOI] [PubMed] [Google Scholar]
- 39.Tsai, S.F., D.I. Martin, L.I. Zon, A.D. D'Andrea, G.G. Wong, and S.H. Orkin. 1989. Cloning of cDNA for the major DNA-binding protein of the erythroid lineage through expression in mammalian cells. Nature. 339:446–451. [DOI] [PubMed] [Google Scholar]
- 40.Turner, J., and M. Crossley. 1998. Cloning and characterization of mCtBP2, a co-repressor that associates with basic Kruppel-like factor and other mammalian transcriptional regulators. EMBO J. 17:5129–5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Svensson, E.C., G.S. Huggins, F.B. Dardik, C.E. Polk, and J.M. Leiden. 2000. A functionally conserved N-terminal domain of the friend of GATA-2 (FOG-2) protein represses GATA4-dependent transcription. J. Biol. Chem. 275:20762–20769. [DOI] [PubMed] [Google Scholar]
- 42.Pandolfi, P.P., M.E. Roth, A. Karis, M.W. Leonard, E. Dzierzak, F.G. Grosveld, J.D. Engel, and M.H. Lindenbaum. 1995. Targeted disruption of the GATA3 gene causes severe abnormalities in the nervous system and in fetal liver haematopoiesis. Nat. Gen. 11:40–44. [DOI] [PubMed] [Google Scholar]
- 43.Carter, L.L., and K.M. Murphy. 1999. Lineage-specific requirement for signal transducer and activator of transcription (Stat)4 in interferon γ production from CD4+ versus CD8+ T cells. J. Exp. Med. 189:1355–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]