

Abstract
Mycobacterium tuberculosis (MTB) inhibits phagosomal maturation to promote its survival inside macrophages. Control of MTB infection requires CD4 T cell responses and major histocompatibility complex (MHC) class II (MHC-II) processing of MTB antigens (Ags). To investigate phagosomal processing of MTB Ags, phagosomes containing heat-killed (HK) or live MTB were purified from interferon-γ (IFN-γ)–activated macrophages by differential centrifugation and Percoll density gradient subcellular fractionation. Flow organellometry and Western blot analysis showed that MTB phagosomes acquired lysosome-associated membrane protein-1 (LAMP-1), MHC-II, and H2-DM. T hybridoma cells were used to detect MTB Ag 85B(241–256)–I-Ab complexes in isolated phagosomes and other subcellular fractions. These complexes appeared initially (within 20 min) in phagosomes and subsequently (>20 min) on the plasma membrane, but never within late endocytic compartments. Macrophages processed HK MTB more rapidly and efficiently than live MTB; phagosomes containing live MTB expressed fewer Ag 85B(241–256)–I-Ab complexes than phagosomes containing HK MTB. This is the first study of bacterial Ag processing to directly show that peptide–MHC-II complexes are formed within phagosomes and not after export of bacterial Ags from phagosomes to endocytic Ag processing compartments. Live MTB can alter phagosome maturation and decrease MHC-II Ag processing, providing a mechanism for MTB to evade immune surveillance and enhance its survival within the host.
Keywords: Mycobacterium tuberculosis, phagosome, MHC, antigen processing, antigen presentation
Introduction
Mycobacterium tuberculosis (MTB)* is an intracellular pathogen that survives inside macrophage phagosomal compartments. CD4 T cell responses are critical to the control of MTB infection in both animals and humans (1–4) and require processing of mycobacterial Ags to generate peptide–MHC-II complexes. Although the CD4 T cell–dominated immune response controls MTB infection in the majority of otherwise healthy individuals, it does not completely eradicate infection. A small number of bacilli survive inside host macrophages, evading immune responses. The ability of MTB to inhibit MHC expression and Ag presentation may contribute to evasion of immune surveillance (5–12). We have demonstrated that MTB 19-kD lipoprotein inhibits MHC-II expression and Ag processing via a mechanism that is dependent on Toll-like receptor 2 (12). However, this mechanism requires hours to days to establish inhibition and cannot alter Ag processing during initial stages of macrophage infection. Other mechanisms, e.g., the ability of MTB to inhibit phagosome maturation, may apply early after phagocytosis to enhance survival of MTB and inhibit Ag processing.
MTB survives in phagosomes that it modifies to promote its survival. After phagocytosis of other bacteria, phagosomes generally fuse with endosomes and eventually lysosomes to form phagolysosomes, a process known as phagosome maturation. These interactions deliver host molecules such as proteases and proton-ATPase to phagosomes, creating an acidic, catabolic phagosomal environment. In contrast, MTB and some other slow-growing mycobacteria (e.g., Mycobacterium bovis and Mycobacterium avium) inhibit phagosomal maturation, decreasing phagosomal acidification, fusion of phagosomes with lysosomes, and phagosomal acquisition of lysosomal markers and characteristics (13–19). Thus, phagosomes containing live mycobacteria stain intensely for markers of early endosomes or immature phagosomes (e.g., transferrin receptor and rab 5), but only weakly for markers of late endosomes, lysosomes, and mature phagolysosomes (e.g., CD63, lysosome-associated membrane protein [LAMP-1], LAMP-2, and cathepsin D) (15, 20, 21). Loss of maturation is also associated with phagosome retention of tryptophan aspartate–containing coat (TACO), a coat protein of unknown function (22).
CD4 T cell responses to MTB require processing of MTB Ags for presentation by class II MHC (MHC-II) molecules. Newly synthesized MHC-II molecules bind invariant chain in the endoplasmic reticulum and target to endocytic compartments that function in Ag processing, e.g., the MHC class II compartment (MIIC) and class II vesicle (CIIV) (23–29). Despite their specific names and properties, these compartments correspond to conventional endocytic compartments (30). They contain molecules involved in Ag processing, e.g., MHC-II, invariant chain, H2-DM (in mice) or HLA-DM (in humans), and proteases that degrade internalized Ags to produce antigenic peptides. After its delivery to endocytic compartments, invariant chain is degraded, leaving only the class II–associated invariant chain peptide (CLIP) bound to MHC-II (31). H2-DM then catalyzes the replacement of CLIP with antigenic peptide, and the resulting peptide–MHC-II complexes are transported to the cell surface for presentation to CD4 T cells.
While endocytic processing of soluble Ags has been studied extensively, less is known about processing of particulate Ags and the specific roles of phagosomes in this process (phagosomes are defined here to include phagolysosomes). We have recently studied the role of latex-bead phagosomes in Ag processing. Latex bead phagosomes contain MHC-II, invariant chain, and H2-DM, degrade phagosome-associated Ag, and directly mediate the formation of peptide–MHC-II complexes (32–34). In addition, latex-bead phagosomes acquired both newly synthesized and recycling MHC-II molecules, but primarily used newly synthesized molecules for phagocytic Ag processing (35). However, little is known about the antigen processing functions of phagosomes that contain pathogenic bacteria. MTB and M. avium phagosomes have been shown to contain MHC-II molecules (15, 36), but their role in Ag processing remains undefined.
To investigate the role of MTB phagosomes in Ag processing, we used an I-Ab–restricted T hybridoma cell line, BB7, specific for an epitope (241–256) from the 30-kD MTB Ag 85B protein (12). Ag 85B is conserved across mycobacterial species and belongs to the Ag 85 family of proteins that function in cell wall mycolic acid synthesis; it is associated with the bacterial cell wall and is also released into the surrounding medium (37–39). Ag 85B is a major target of human T cell response to MTB and a leading vaccine candidate (38, 40). Presentation of Ag 85B to BB7 T cells required phagocytosis of MTB and H2-DM–dependent intracellular processing. Flow organellometry and Western blotting demonstrated MHC-II and H2-DM in isolated MTB phagosomes derived from IFN-γ activated macrophages. Subcellular fractionation coupled with a T cell assay demonstrated that Ag 85B(241–256)–I-Ab complexes were initially found only in MTB phagosomes and later appeared on the plasma membrane. Phagosomes containing live MTB were found to contain fewer Ag 85B(241–256)–I-Ab complexes than phagosomes containing heat-killed (HK) MTB, indicating that live MTB inhibited phagocytic Ag processing. This is the first direct demonstration that peptide–MHC-II complexes are formed within bacterial phagosomes and export of bacterial Ags from phagosomes to endocytic Ag processing compartments is not required. Moreover, MHC-II Ag processing function is altered by live MTB, providing a mechanism for this intracellular pathogen to evade immune surveillance and survive within the host.
Materials and Methods
Cells and Media
B6D2F1/J and C57BL/6 mice were obtained from The Jackson Laboratory. H2-DM−/− mice (L. Van Kaer, Vanderbilt University, Nashville, TN) were bred under specific pathogen-free conditions. Incubations in the absence of MTB were performed at 37°C in 5% CO2 in standard medium composed of DMEM (Life Technologies) supplemented with 10% decomplemented fetal calf serum (Hyclone), 5 × 10−5 M 2-ME, l-arginine HCl (116 mg/l), l-asparagine (36 mg/l), NaHCO3 (2 g/liter), sodium pyruvate (1 mM), 10 mM HEPES buffer, and antibiotics. For incubations with MTB, standard medium was modified by use of non-decomplemented fetal calf serum, addition of 1% non-decomplemented normal mouse serum (Sigma-Aldrich) and absence of antibiotics. Bone marrow macrophage precursors were harvested from femurs of B6D2F1/J mice and cultured for 7 d in 6-well plates in standard medium supplemented with 20% LADMAC (41) cell-conditioned medium. Macrophages were then stimulated for 48 h with 50 U/ml recombinant IFN-γ (Genzyme). The resulting confluent cultures contained ∼1.5 × 106 cells/well. The T cell hybridoma BB7 (12) recognizes MTB Ag 85B(241–256) bound to I-Ab. The T cell hybridoma DOBW (42) recognizes OVA (323–339) bound to I-Ad (or to a lesser degree I-Ab).
Antibodies
Hybridoma supernatants were prepared from the following cell lines: 1D4B (Developmental Studies Hybridoma Bank, Johns Hopkins University, Baltimore, MD, and University of Iowa, Iowa City, IA), producing a rat IgG2a specific for murine LAMP-1; 34–5-3S (American Type Culture Collection), producing a murine IgG2a which recognizes both I-Ad and I-Ab; 10.2.16 (American Type Culture Collection), producing a murine IgG2a which recognizes I-Ak; Y-3P (American Type Culture Collection), producing a murine IgG2a which recognizes I-Ab. Some Abs were purified by protein A-affinity chromatography. Purified Y-3P was iodinated by the chloramine T method.
Bacteria
MTB H37Ra (American Type Culture Collection) was grown to OD of 0.2 to 0.3 in Middlebrook 7H9 broth (Difco) supplemented with 1% glycerol, 0.05% Tween (Sigma-Aldrich; to prevent clumping) and 10% Middlebrook oleic albumin dextrose catalase (OADC) enrichment (Difco). Bacteria were harvested and used the same day (for all experiments with live MTB) or frozen at −70°C as described (3). Bacterial viability was determined by counting total MTB in Petroff-Hauser chambers and calculating live MTB titers from CFU determinations on Middlebrook 7H11 plates. Additionally, bacterial suspensions were stained with fluorochromic substrates differentiating between live and dead bacteria (BacLight; Molecular Probes). Both analyses revealed MTB viability of ∼90%. HK MTB were prepared by incubating MTB at 80°C for 30 min (killing was confirmed by absence of CFUs). Prior to macrophage infection, all MTB preparations were pelleted, washed, declumped by two passages through an 18-guage needle and three passages through a 22-gauge needle, and centrifuged at 150 g for 5 min to remove clumps. To label HK and live MTB with fluorescein, 109 bacteria were pelleted, resuspended in 1 ml PBS (pH 9.1) and combined with 25 μl of 20 mg/ml FLUOS (Boehringer) in DMSO for 5 min at room temperature. Labeled MTB were washed twice in DMEM and declumped before use. This procedure produced no significant change in viability of MTB. Both HK and live MTB were incubated in medium containing 10% non-heat treated FCS and 1% non-heat treated normal mouse serum with no antibiotics for 20 min at 37°C before incubation with cells.
Ag Processing and Presentation Assays
Macrophages were replated in 96-well flat bottom plates at 2 × 105 cells/well before stimulation with IFN-γ for 2 d. HK or live MTB was added in a final volume of 100 μl. Bacteria were pelleted onto cells by centrifugation at 900 g for 10 min at 37°C. Cells were incubated at 37°C for 10 min (providing a total pulse period of 20 min) and washed in ice-cold DMEM to remove extracellular bacteria. Prewarmed medium was added, and cells were incubated at 37°C for up to 100 min. Macrophages were fixed with 1% paraformaldehyde and washed. T hybridoma cells (105) were added to each well (200 μl total volume) and incubated for 24 h. Supernatants (100 μl) were harvested and assessed for IL-2 using a CTLL-2 proliferation assay, monitored by addition of Alamar blue (Alamar Biosciences) as an indicator dye, and measured as the difference between absorbance at 550 nm and 595 nm after 24 h. Blanks for spectrophotometry were provided by wells containing medium alone (added at initiation of CTLL-2 assay) and Alamar blue (added at the same time as for the other wells). All analyses were performed in triplicate.
Percoll Density Gradient Fractionation for T Cell Assay and Biochemical Analysis
Three 6-well plates containing confluent macrophage cultures were used for each fractionation, and plates were processed as above to achieve a 20 min pulse with MTB (multiplicity of infection [MOI] = 40) and various chase incubations. To identify MIIC endocytic vesicles, macrophages were incubated with soluble OVA (Sigma-Aldrich) at 3 mg/ml for 1 h before the addition of bacteria. Cells were washed, detached by scraping, washed, resuspended in homogenization buffer (0.25 M sucrose and 10 mM HEPES, pH 7.2), and homogenized in a Dounce homogenizer (Kontes Co.) to obtain 80 to 85% lysis (32, 33). Intact cells and nuclei were removed by 3 consecutive spins at 200 g for 5 min at 4°C, and the supernatant (containing phagosomes) was collected. In some experiments, phagosomes were pelleted from the supernatant at 500 g at 4°C for 15 min and resuspended in 1 ml homogenization buffer. 1 ml of sample (homogenate or resuspended phagosomes) was layered on 9 ml of 27% or 40% Percoll in homogenization buffer and centrifuged in a Ti50 fixed angle rotor (Beckman Instruments) at 4°C for 60 min at 36,000 g. The gradients were manually fractionated from the top into 30 fractions of 333 μl, which were divided into replicate aliquots of 10, 30, or 50 μl and frozen at −80°C.
Plasma membrane was marked before homogenization by incubation of macrophages for 60 min at 4°C with 125I-labeled Y-3P (33). β-hexosaminidase activity was measured by combining 50 μl fraction, 150 μl assay buffer (0.1M MES, 0.2% Triton X-100, pH 6.5) and 50 μl p-nitrophenyl-acetyl-b-D-glucosaminide (Sigma-Aldrich; 1.36 mg/ml in water) for 90 min at 37°C (33). Reaction samples (100 μl) were combined with 100 μl stop buffer (0.5 M glycine, pH 10), and OD was determined at 405 nm. To identify fluorescein-labeled bacteria, fractions (50 μl) were transferred to 96-well clear-bottom black plates (Costar) and analyzed on a Spectra Fluor Plus fluorimeter (Tecan). For T cell analysis of Percoll gradient fractions, standard medium and BB7 or DOBW cells (105/well) were added to a final volume of 200 μl in 96-well plates (FCS was added to maintain 10% FCS). After 24 h, T cell responses were assessed as above. Wells containing only equivalent amounts of Percoll, BB7 cells, and medium were used to generate control supernatants and blanks for the CTLL assay.
Preparation of Phagosomes for Flow Organellometry
Two 6-well plates containing confluent macrophage cultures were used for each condition. Fluorescein-labeled bacteria were added (MOI = 40) to achieve 20 min pulse and various chase incubations. Cells were washed, detached by scraping, washed, and resuspended in homogenization buffer with protease inhibitors (1.0 mM phenylmethylsulfonyl fluoride, 1 μg/ml pepstatin, and 20 μg/ml leupeptin). Cells were homogenized, intact cells and nuclei were removed by centrifugation, and phagosomes were pelleted as above. Phagosomes were resuspended and isolated on 27% Percoll density gradients. Fractions containing phagosomes were identified visually and combined with an equal volume of 2% paraformaldehyde for 10 min. The suspension was mixed with an equal volume of 0.4 M lysine in PBS, and phagosomes were washed twice by pelleting and resuspension in 0.5 ml PBS (32). Phagosomes (80–100 μl/well) were stained in 96-well round bottom plates using a buffer containing saponin to allow access to lumenal epitopes (32, 33), pelleted by centrifugation at 1,900 g and analyzed using a COULTER EPICS Elite ESP instrument (Beckman Coulter).
Results
Characterization of MTB Processing and Presentation
Processing of MTB was studied in bone marrow macrophages using an MTB-specific T hybridoma, BB7, which recognizes MTB Ag 85B(241–256)–I-Ab complexes (12). Macrophages processed both soluble Ag 85B (data not shown) and whole MTB (Fig. 1) for presentation to BB7. To confirm that uptake of MTB and intracellular processing were required, macrophages were incubated with or without 10 μg/ml cytochalasin D (to inhibit phagocytic uptake) or 100 μM chloroquine (to inhibit acidification of vacuolar compartments) beginning 15 min before the addition of bacteria. Cytochalasin D and chloroquine both blocked processing of HK MTB (Fig. 1 A), demonstrating that phagocytic uptake and intracellular processing of bacteria were required for production of Ag 85B(241–256)–I-Ab complexes. Processing also required H2-DM, as Ag 85B(241–256)–I-Ab complexes were not generated in macrophages derived from H2-DM−/− mice (Fig. 1 B). Thus, formation of Ag 85B(241–256)–I-Ab complexes required phagocytosis and H2-DM–dependent intracellular processing of MTB bacilli.
Figure 1.


Requirements for processing of MTB bacilli for presentation of MTB Ag 85B. Macrophages were pulsed with HK MTB for 20 min, chased for 10 min at 37°C, and fixed. BB7 T hybridoma cells were used to detect Ag 85B(241–256)–I-Ab complexes. Supernatants were assessed for IL-2 content using a CTLL-2 proliferation assay that was monitored with Alamar blue, an indicator dye. (A) Effects of cytochalasin D (10 μg/ml) and chloroquine (100 μM) on Ag processing. (B) Comparison of processing of HK MTB by B6D2 and H2-DM−/− macrophages. Data points are means of triplicate samples ± SD.
Preparative Isolation of Phagosomes for Flow Analysis by Percoll Density Gradient Centrifugation
Studies of phagosomal Ag processing require analysis of phagosomes with exclusion of other organelles. We have used flow organellometry to analytically separate and characterize latex bead phagosomes without their prior purification (32, 33). However, early studies with MTB phagosomes revealed that contamination of phagosomes with other membranes occurred unless phagosomes were purified on Percoll density gradients before immunolabeling and flow organellometry. Macrophages were initially incubated with soluble OVA to label MIIC vesicles with OVA(323–339)–I-Ad complexes and then incubated with fluorescein-labeled HK MTB for 20 min, followed by a 30-min chase. The plasma membrane was then marked by incubation with 125I-Y-3P (anti–I-Ab) at 4°C, cells were homogenized, and phagosomes and other organelles were separated on 27% Percoll density gradients. Phagosomes consistently appeared in fractions 27–29 in the high-density region of 27% Percoll gradients (Fig. 2 A), whereas plasma membrane radioactivity was found in fractions 8 to 12 in the low-density region (Fig. 2 B). Absence of radioactivity in phagosomal fractions showed that phagosomes were not contaminated by plasma membrane or intact cells (phagosomal fractions contained less than 0.015% of the plasma membrane label). β-hexosaminidase, a lysosomal enzyme, was primarily found in high-density phagolysosomal/lysosomal fractions (Fig. 2 C, fractions 23 to 29). DOBW T hybridoma cells detected OVA(323–339)–I-Ad complexes in MIIC endocytic vesicles in fractions 23 to 28 and on the plasma membrane in fractions 8 to 12 (Fig. 2 D). Thus, the distributions of phagosomes and MIIC vesicles partially overlapped in fractions 27 and 28 on 27% Percoll gradients. Similar results were obtained with phagosomes containing live MTB (data not shown).
Figure 2.
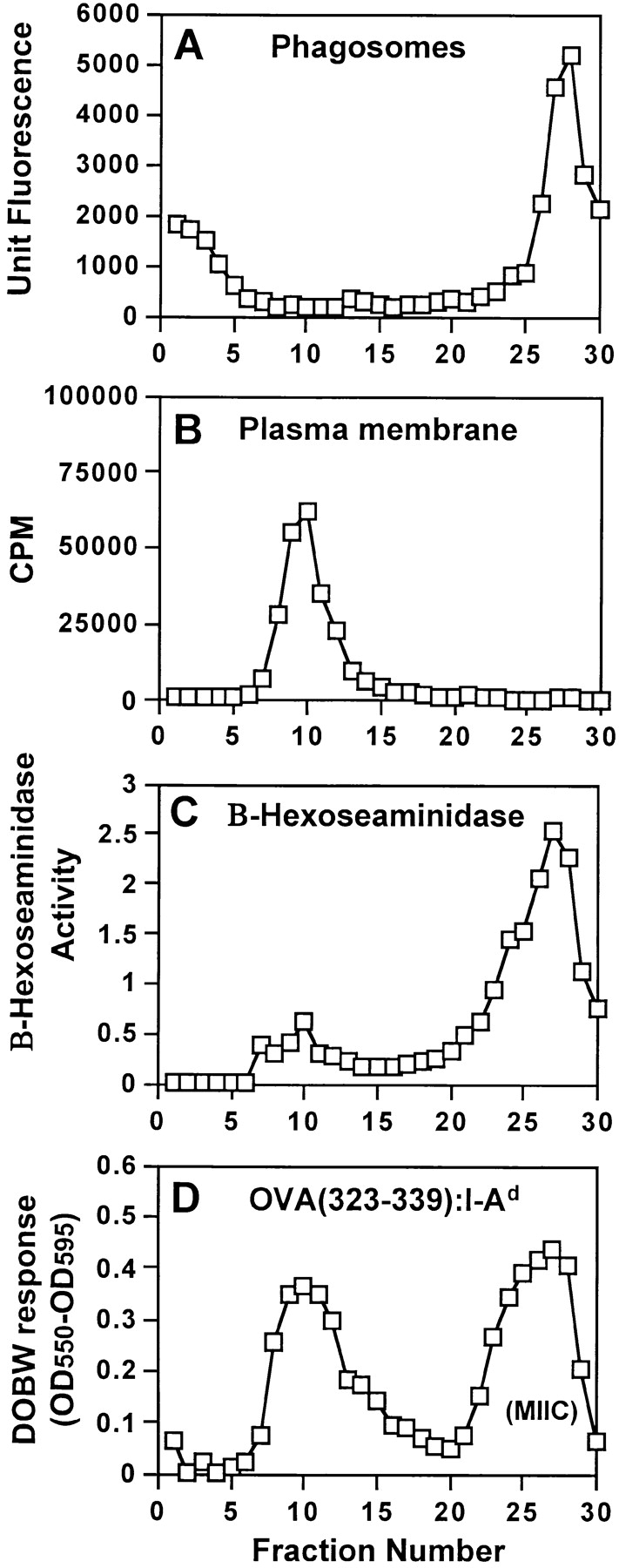
Characterization of MTB phagosomes isolated on 27% Percoll density gradients. Macrophages were incubated with soluble OVA (3 mg/ml) for 1 h, incubated with fluorescein-labeled HK MTB and OVA for 20 min, washed, chased for 30 min at 37°C in the presence of soluble OVA, homogenized, and fractionated on 27% Percoll gradients. (A) Distribution of HK MTB phagosomes detected by fluorimetry. (B) Distribution of plasma membrane radioactivity when macrophage plasma membranes were labeled with 125I-Y-3P (anti–I-Ab) at 4°C before fractionation. (C) B-Hexosaminidase activity (a marker of lysosomal enzyme distribution). (D) Distribution of OVA(323–339)–I-Ad complexes assessed by DOBW T hybridoma assay.
Flow organellometry of MTB phagosomes required further purification of phagosomes before fixation to reduce their contamination with MIIC. Density gradients with a higher percentage of Percoll (e.g., 40%) provided better separation of phagosomes and MIIC vesicles for other types of studies (below), but the higher percentage of Percoll interfered with some procedures for flow organellometry (e.g., fixation and pelleting of phagosomes). Consequently, differential centrifugation (500 g for 15 min at 4°C) was used to pellet phagosomes and separate them from MIIC before isolation of phagosomes on 27% Percoll gradients (MIIC was detected with DOBW T hybridoma cells as in Fig. 2 D). The combination of differential centrifugation with fractionation on 27% Percoll density gradients substantially decreased MIIC contamination of phagosomes (data not shown), and the lack of contamination of phagosomes with other MHC-II–expressing membranes (including MIIC) was confirmed in flow organellometry studies (below).
Assessment of LAMP-1 and MHC-II Expression in Isolated Phagosomes Containing Live or HK MTB by Flow Organellometry
For flow organellometry, gating on optical scatter properties alone was not sufficient to define MTB phagosomes, unlike latex bead phagosomes analyzed in previous studies (32, 33). Therefore, fluorescein-labeled bacteria were used to prepare phagosomes that could be identified during flow organellometry by gating on both scatter properties and fluorescein signal. Macrophages were pulsed with fluorescein-labeled live or HK MTB for 20 min, washed, chased for 10 or 100 min and homogenized. Phagosomes were pelleted, purified on 27% Percoll gradients, fixed, permeabilized with saponin to allow access to lumenal epitopes, and immunolabeled as described previously (32, 33).
Acquisition of LAMP-1 was used as a marker for phagosomal maturation (Fig. 3). After a 10-min chase incubation, HK MTB phagosomes showed positive staining for LAMP-1 with mean fluorescence value (MFV) of 344 (Fig. 3 B). A chase incubation of 100 min increased MFV for LAMP-1 by 125% to 773 (Fig. 3 F). In contrast to the results with HK MTB, phagosomes from macrophages exposed to live MTB showed less acquisition of LAMP-1. Between 10 and 100 min of chase incubation, MFV for LAMP-1 staining of live MTB phagosomes increased from 348 to 453 (by only 30%). Furthermore, much of this increase was due to acquisition of low to moderate levels of LAMP-1 by phagosomes with previously negative or low LAMP-1 expression, as opposed to further acquisition of LAMP-1 to high levels; the MFV of LAMP-1 positive events (gate H2) increased by only 6% (from 454 to 483; Fig. 3, J and N). Thus, phagosomes containing both live and HK MTB acquired at least low levels LAMP-1, but live MTB phagosomes did not acquire the high level of LAMP-1 staining seen with HK MTB phagosomes after longer chase incubations (e.g., 100 min). These data reflect other observations that phagosome acquisition of LAMP-1 is decreased but not eliminated by live MTB (but not HK MTB) (15), consistent with inhibition of phagosome maturation (13) (see Discussion).
Figure 3.


Arrest of phagosomal maturation in MTB phagosomes containing live but not HK MTB. Macrophages were incubated with fluorescein-labeled live or HK MTB for a 20-min pulse with a 10- or 100-min chase incubation. Phagosomes were pelleted, purified on 27% Percoll gradients, and prepared for flow organellometry with staining for LAMP-1 and MHC-II (I-Ab/d). Events were gated for fluorescence and scatter parameters consistent with phagosomes. Staining with isotype-matched negative control Abs was used to define the H1 gate. The H2 gate represents positive events. The H3 gate represents all events. MFV values represent the mean for the H3 gate.
Phagosomes were also evaluated for expression of MHC-II (Fig. 3). After a 10-min chase, HK MTB phagosomes showed positive staining with 34–5-3S (MFV = 16; Fig. 3 D) relative to the isotype-matched negative control Ab (MFV = 3.9). When the chase time was increased to 100 min, no consistent increase in MHC-II was observed (despite the small increase in Fig. 3; evaluation of MFV over three experiments showed no consistent change in MFV between 10 and 100 min of chase incubation). Furthermore, phagosomes containing live MTB also contained MHC-II molecules at levels that remained unchanged between 10 and 100 min of chase (PFig. 3, L and P; MFV of 20.4 for both). Thus, the mean level of MHC-II expression remained relatively constant within this time frame, although individual phagosomes varied widely in MHC-II content.
To confirm that phagosomal staining for MHC-II molecules reflected true phagosomal composition, as opposed to contamination of phagosomes with MHC-II from other membranes during homogenization and subcellular fractionation, the purity of phagosomes was analyzed in cross contamination experiments (Fig. 4). B6D2 macrophages (expressing I-Ab and I-Ad) were exposed to fluorescein-labeled HK MTB and then mixed with naive CBA/J macrophages (expressing I-Ak; not exposed to HK MTB) before homogenization. Immunolabeling of B6D2 phagosomes for I-Ak molecules was used to assess contamination of phagosomes by other membranes expressing MHC-II. B6D2 phagosomes prepared after mixing B6D2 and CBA/J macrophages showed essentially negative staining for I-Ak (MFV = 7.3, 7% in positive gate; Fig. 4 I), similar to negative control B6D2 phagosomes (from B6D2 cells not exposed to CBA/J cells; MFV = 6.0, 9% of events in positive gate; Fig. 4 C). Thus, MFV and percent of events positive for I-Ak indicate that B6D2 phagosomes had essentially no detectable contamination by CBA/J membranes present in the same homogenate. We conclude that our protocol for subcellular fractionation and flow organellometry produced phagosomes with negligible contamination from other MHC-II–expressing membranes, and MHC-II molecules detected by flow organellometry reflected true phagosomal MHC-II content.
Figure 4.

MTB phagosomes have negligible contamination with other MHC-II expressing membranes. B6D2 and CBA/J macrophages were pulsed with fluorescein-labeled HK MTB for 20 min, washed, and chased for 30 min at 37°C. Half of the MTB-infected B6D2 macrophages were mixed with an equal number of naive CBA/J macrophages before homogenization. Homogenates were prepared from MTB-exposed B6D2 macrophages, MTB-exposed CBA/J macrophages, and MTB-exposed B6D2 macrophages mixed with naive CBA/J macrophages. Phagosomes were pelleted, purified on separate 27% Percoll gradients, fixed, and stained for I-Ad/b or I-Ak. Flow organellometry was performed as in Fig. 3.
Western Blot Analysis of Phagosomes for H2-DM and Invariant Chain
Phagosomal levels of H2-DM and invariant chain were determined by Western blot analysis, as the H2-DM–specific antiserum was not effective for flow organellometry. Phagosomes were prepared by differential centrifugation and isolation on 40% Percoll density gradients, which provided better separation of MIIC and phagosomes (below) than the 27% Percoll gradients used to prepare phagosomes for flow organellometry (40% Percoll gradients interfered with the latter procedure). After a 20-min pulse and 10-min chase, phagosomes containing live or HK MTB expressed H2-DM but none of the detectable forms of invariant chain (data not shown; the antibody used, In-1, detects p41 and p33 forms of invariant chain and a degradation intermediate, p10, but not CLIP). The absence of invariant chain from MTB phagosomes suggests that there is little delivery of invariant chain–MHC-II complexes to MTB phagosomes or that invariant chain is rapidly degraded after delivery to MTB phagosomes (it also indicates absence of significant contamination of phagosomes by endoplasmic reticulum, which contains most invariant chain).
Analysis of Subcellular Fractions for Ag 85B(241–256)–I-Ab Complexes Using a T Cell Assay
Although MTB phagosomes were found to contain MHC-II molecules, it was still unclear whether peptide–MHC-II complexes were actually formed within phagosomes or rather within endocytic vesicles (e.g., MIIC) that received phagosome-derived Ag fragments. To determine the site where MTB Ag 85B(241–256)–I-Ab complexes were formed during MTB processing, we used a technique previously developed to detect peptide–MHC-II complexes in latex-bead phagosomes and other organelles (24, 33, 43). This approach uses subcellular fractionation to isolate organelle membranes, which are then disrupted by freezing and thawing to expose the lumenal Ag presenting domain of MHC-II molecules and then probed for specific peptide–MHC-II complexes using T cells. Macrophages were pulsed for 20 min with HK MTB, chased for either 0 or 220 min and fractionated on 27% Percoll gradients. Fractions were incubated with BB7 T hybridoma cells, and detection of Ag 85B(241–256)–I-Ab complexes was monitored by IL-2 secretion (Fig. 5). When macrophages were pulsed with HK MTB for 20 min with no chase incubation, Ag 85B(241–256)–I-Ab complexes were detected primarily in phagosomal fractions with little expression on the plasma membrane (Fig. 5 A), although complexes appeared on the plasma membrane soon thereafter and could be detected at low levels by 20 min in some experiments. With a 20 min pulse and 10–100 min chase incubation, Ag 85B(241–256)–I-Ab complexes were detected in both phagosomal and plasma membrane fractions (Fig. 5 C, and data not shown). When the chase incubation was increased to 220 min, Ag 85B(241–256)–I-Ab complexes were detected primarily in plasma membrane fractions (Fig. 5 B). When 0.5 μM Ag 85B(241–256) peptide was added to the fractions during the T cell assay, BB7 cells responded to all fractions containing MHC-II, providing a positive control and confirming the ability of BB7 cells to respond to all such fractions when Ag 85B(241–256)–I-Ab complexes were present (data not shown). We conclude that Ag 85B(241–256)–I-Ab complexes are formed directly in MTB phagosomes before their expression on the plasma membrane.
Figure 5.

Ag 85B(241–256)–I-Ab complexes are initially found in phagosomes and later appear on the plasma membrane (PM). (A and B) Macrophages were pulsed with HK MTB (MOI = 40) for 20 min, washed, chased for various periods, and fractionated on 27% Percoll density gradients. (C) Macrophages were incubated with soluble OVA for 1 h and then pulsed with HK MTB and OVA for 20 min, washed, chased for 10 min at 37°C in the presence of OVA, and fractionated on 40% Percoll density gradients. Aliquots (50 μl) of each fraction were frozen, thawed, and analyzed for Ag 85B(241–256)–I-Ab and OVA(323–339)–I-Ad complexes using BB7 and DOBW T hybridoma cells, respectively. Diagrams at the top summarize the positions of different compartments in the Percoll gradients.
To confirm that Ag 85B(241–256)–I-Ab complexes were being formed in phagosomes and not MIIC, macrophages were fractionated on 40% Percoll gradients to achieve better separation of phagosomes and MIIC (Fig. 5 C). Prior to fractionation, macrophages were incubated with soluble OVA for 1 h, pulsed with HK MTB for 20 min, chased for 10 min, and fractionated on 40% Percoll gradients. BB7 T hybridoma cells detected Ag 85B(241–256)–I-Ab complexes in phagosomal fractions (25 to 27) and plasma membrane fractions (7 to 10) (Fig. 5 C). DOBW cells detected OVA(323–339)–I-Ad complexes in MIIC fractions (17 to 23) and plasma membrane fractions (6 to 10). As fractions containing MIIC and phagosomes were well separated on 40% Percoll gradients, Ag 85B(241–256)–I-Ab complexes were clearly formed in phagosomes and not late endocytic compartments.
Comparison of Phagosomal Processing of Live versus HK MTB
As previous studies have shown that phagosomal maturation is inhibited to a greater degree by live MTB than HK MTB, we investigated the processing of live versus HK MTB by IFN-γ activated macrophages for presentation to T cells. Macrophages were incubated with live or HK MTB for 20 min, washed, chased for various periods, fixed, and incubated with BB7 T hybridoma cells to assess the presentation of Ag 85B(241–256)–I-Ab complexes. Processing of HK MTB was initiated rapidly, and Ag 85B(241–256)–I-Ab complexes were expressed at low levels on the cell surface by 20 min with high MOI (Fig. 6). At early time points, BB7 cells responded better to macrophages infected with HK than live MTB. However, at later time points (100 min chase), BB7 cells responded similarly to macrophages incubated with either live or HK MTB. We conclude that IFN-γ activated macrophages processed both live and HK MTB, but processing of HK MTB was more rapid and efficient than processing of live MTB. Furthermore, these studies may understate the inhibition of processing that live MTB may achieve in less activated macrophages, since activation of macrophages with IFN-γ may enhance maturation of all phagosomes and decrease differences between live and HK MTB phagosomes, as has been shown in other systems with IFN-γ alone or in combination with other agents (36, 44–47).
Figure 6.
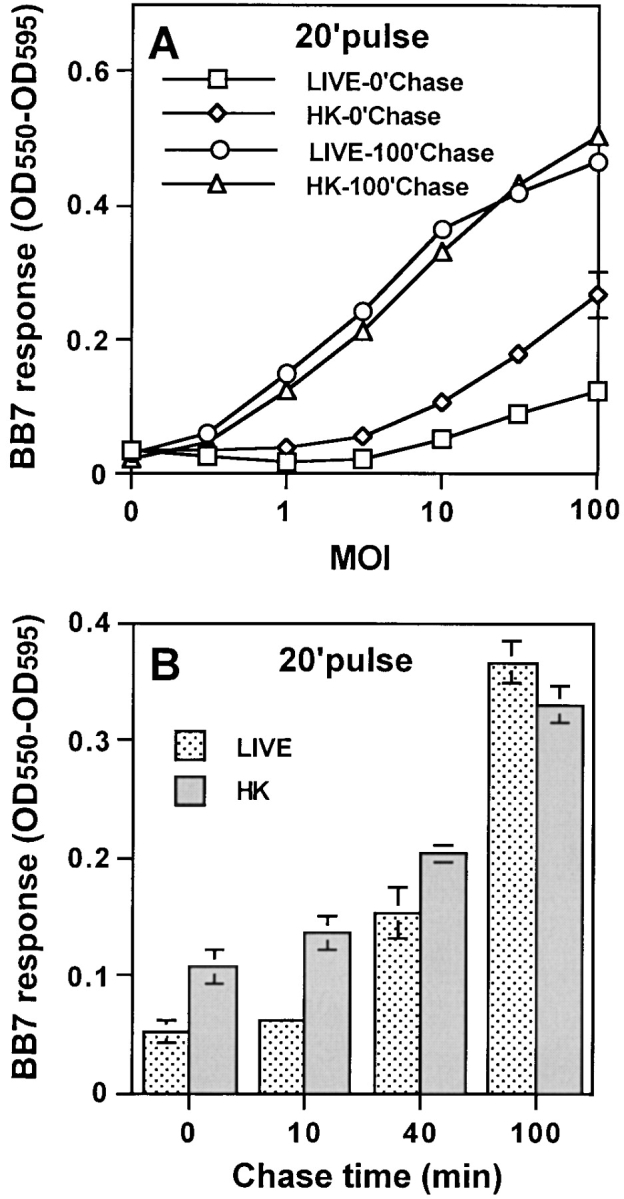
HK MTB is processed more rapidly than live MTB. Macrophages were pulsed with HK or live MTB for 20 min, chased for varying periods at 37°C, and fixed. BB7 T hybridoma cells were used to detect presentation of Ag 85B(241–256)–I-Ab complexes (see Fig. 1). (A) Presentation at varying MOI after 0- or 100-min chase. (B) Presentation after varying chase intervals with MOI = 5. A and B are from a single experiment. Data points are means of triplicate samples ± S.D.
We next determined if differences in processing of live versus HK MTB by macrophages correlated with the efficiency of phagosomal processing as reflected by the levels of Ag 85B(241–256)–I-Ab complexes expressed in the two types of phagosomes. Macrophages were pulsed with live or HK MTB for 20 min at MOI of 13 or 40 bacteria per cell. Infected macrophages were fractionated on 27% Percoll gradients, and Ag 85B(241–256)–I-Ab complexes were detected in fractions with BB7 T hybridoma cells. At MOI of 13, BB7 cells responded only to fractions containing HK MTB phagosomes but not to fractions containing live MTB phagosomes (Fig. 7 A). When the MOI was increased to 40, BB7 cells responded to fractions containing both live and HK MTB phagosomes (Fig. 7 B). Even at this early time point, BB7 cells detected Ag 85B(241–256)–I-Ab complexes in plasma membrane fractions from macrophages pulsed at MOI of 40 with HK MTB, but detectable presentation of complexes in plasma membrane was not achieved with live MTB (Fig. 7 B). The mean uptake of bacteria per macrophage in different experiments was 4–5 at MOI of 13 and 13–15 at MOI of 40 (data not shown), and uptake of live versus HK MTB by macrophages was similar (data not shown). Therefore, when the number of phagosomes in fractions 26 to 28 was low (MOI of 13), phagosomes containing HK MTB expressed sufficient Ag 85B(241–256)–I-Ab complexes to drive detectable BB7 responses, but fractions containing live MTB phagosomes contained too few Ag 85B(241–256)–I-Ab complexes to generate a BB7 response with this number of phagosomes. With the greater number of phagosomes produced at MOI of 40, the total number of complexes in the phagosomal fractions sufficed to generate a BB7 response to both live and HK MTB phagosomes. These observations suggest that phagosomes containing HK MTB generate Ag 85B(241–256)–I-Ab complexes more rapidly and efficiently than phagosomes containing live MTB.
Figure 7.
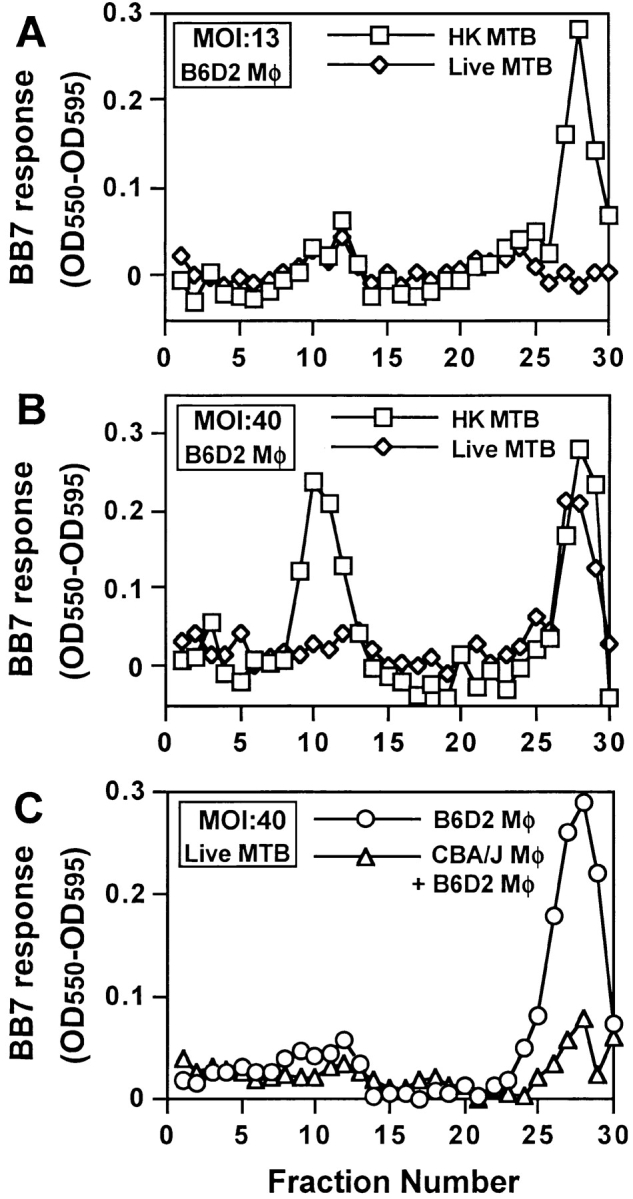
Ag 85B(241–256)–I-Ab complexes are produced in early phagosomes at higher levels with HK MTB than with live MTB. (A and B) Macrophages were pulsed with HK or live MTB for 20 min at a MOI of either 40 or 13 bacteria per cell and fractionated on separate 27% Percoll density gradients. Fractions were processed as in Fig. 5 and analyzed for expression of Ag 85B(241–256)–I-Ab complexes using BB7 T hybridoma cells. (C) B6D2 and CBA/J macrophages were pulsed with live MTB for 20 min at a MOI of 40, washed and chased for 20 min. The MTB-infected CBA/J macrophages were mixed with an equal number of naive B6D2 macrophages before homogenization. The two samples were fractionated on separate 27% Percoll density gradients, and fractions were analyzed for expression of Ag 85B(241–256)–I-Ab complexes using BB7 T hybridoma cells.
Cross contamination control experiments were performed to confirm that Ag 85B(241–256)–I-Ab complexes were formed within intracellular live MTB phagosomes and not after homogenization as an artifact of protease release, extracellular Ag proteolysis and peptide binding to MHC-II. CBA/J macrophages (expressing I-Ak) were pulsed with live MTB (MOI = 40) for 20 min, washed, and chased for an additional 20 min. These cells were mixed with an equal number of naive B6D2 macrophages (expressing I-Ab but not pulsed with live MTB), homogenized, and fractionated on a 27% Percoll gradient to allow for potential formation of Ag 85B(241–256)–I-Ab complexes as an artifact after homogenization. Ag 85B(241–256)–I-Ab complexes were detected in fractions with BB7 T hybridoma cells (Fig. 7 C). These results were compared with BB7 responses to phagosomes from B6D2 macrophages that were similarly pulsed with live MTB. BB7 cells responded strongly to MTB phagosomes from B6D2 macrophages but showed minimal response to phagosomal fractions prepared after mixing infected CBA/J and uninfected B6D2 macrophages (despite the doubled total cell titer and corresponding increase in released proteases in this control protocol). When 0.5 μM Ag 85B(241–256) peptide was added to both sets of fractions during the T cell assay, BB7 cells responded equally well to both phagosomal fractions, confirming the presence of equivalent amounts of I-Ab in these fractions (data not shown). We conclude that Ag 85B(241–256)–I-Ab complexes are indeed generated within phagosomes containing live MTB and not as an experimental artifact arising after homogenization of macrophages.
Discussion
MTB uses multiple mechanisms to evade host defenses, including inhibition of MHC-II expression and Ag processing and presentation (5–12), as well as the secretion of inhibitory cytokines, e.g., IL-10, IL-6, and TGF-β. Some inhibitory mechanisms may not be active during early stages of macrophage infection but may contribute to maintenance of chronic infection. For example, MTB 19 kD lipoprotein produces a Toll-like receptor 2–dependent inhibition of MHC-II Ag processing starting ∼1 d after macrophage infection (12). Other mechanisms, including the ability of MTB to inhibit phagosomal maturation, may be critical for survival of MTB during earlier stages of macrophage infection. As indicated by our current studies, the early-stage mechanisms may also include inhibition of phagosomal Ag processing.
The role of MTB phagosomes in MHC-II processing of MTB Ag was evaluated by subcellular fractionation coupled with either flow organellometry or MTB-specific T cell assays with isolated organelles. Plasma membrane and phagosome fractions were well separated on 27% Percoll gradients (Fig. 2), and no significant plasma membrane contamination of the phagosomal fractions was observed (phagosomal fractions contained <0.015% of plasma membrane marker). As ∼60% of total MHC-II is on the plasma membrane (48), contamination of phagosomes with plasma membrane would have interfered with evaluation of phagosomal MHC-II levels. The distribution of phagosomes and MIIC did overlap on 27% Percoll gradients (Fig. 2). As MIIC contains a significant percentage of intracellular MHC-II molecules and is a site where MHC-II molecules bind peptides, additional steps were taken to separate phagosomes from MIIC. For some experiments, phagosomes were purified on 40% Percoll gradients, which produced good separation of phagosomes and MIIC. As 40% Percoll complicated some procedures, e.g., fixation and pelleting of phagosomes for flow organellometry, other experiments used differential centrifugation to separate phagosomes from smaller membrane structures, e.g., MIIC, followed by fractionation on 27% Percoll gradients. Both approaches provided extremely pure preparations of MTB phagosomes for our analyses.
These studies provide the first use of flow organellometry to analyze bacterial phagosomes (containing MTB, a significant human pathogen). This technique could potentially be adapted for analysis of phagosomes containing other types of bacteria. The use of fluorescein-labeled bacteria allowed identification of bacterial phagosomes by gating on fluorescence and optical parameters (reflecting size). Immunolabeling provided sensitive detection of phagosomal membrane proteins. Relatively few phagosomes are required for this type of analysis, allowing the production of multiple phagosome preparations and their rapid analysis for a variety of membrane proteins. In addition, subpopulations of phagosomes expressing different levels of membrane proteins can be identified.
Phagosomes containing both HK and live MTB were analyzed for LAMP-1 as a marker for phagosome maturation. Other reports have shown that phagosomes containing dead mycobacteria fuse with lysosomes and acquire lysosomal makers (e.g., LAMP-1) in a process termed phagosome maturation. In contrast, phagosomes containing live mycobacteria fuse with early endosomes but resist fusion with lysosomes, i.e., exhibit inhibited phagosome maturation (13–21) (although they do acquire some LAMP-1). Activation of macrophages (as done in our studies to provide for MHC-II expression and Ag presentation) may decrease the ability of live mycobacteria to inhibit phagosome maturation (36, 44–47). In our studies, phagosomes containing HK or live MTB acquired similar low to moderate levels of LAMP-1 at early time points (e.g., 20 min pulse, 10 min chase). As LAMP-1 is expressed on the cell surface and in endosomes as well as lysosomes (although it is most highly expressed in lysosomes), live as well as HK MTB phagosomes could have acquired LAMP-1 from plasma membrane or endosomes without lysosomal fusion. Alternatively, phagosomes could have acquired some LAMP-1 by fusion with lysosomes. However, with longer chase incubations (e.g., 20 min pulse, 100 min chase) phagosomes containing HK MTB acquired high levels of LAMP-1 (consistent with phagosome-lysosome fusion), while live MTB phagosomes lagged in acquisition of LAMP-1. This lag in acquisition of LAMP-1 suggests that live MTB may decrease phagosome maturation even in activated macrophages, although we have not examined other measures of phagosome maturation. In summary, activation of macrophages promotes phagosome maturation and may decrease the degree to which phagosome maturation is inhibited by live MTB, but our data suggest that phagosome maturation is still partially inhibited or delayed even in activated macrophages (this inhibition may not be evident at much later time points [>2 h], as examined in other studies).
Within the time frame of active Ag processing, both live MTB phagosomes and HK MTB phagosomes acquired MHC-II molecules that could potentially bind peptides derived from MTB Ags. There was little change in MHC-II expression by phagosomes from 10 to 110 min of chase (Fig. 3), suggesting that any delivery of MHC-II molecules that occurred was essentially balanced by export of MHC-II from phagosomes (consistent with observations with latex bead phagosomes) (33). Latex bead phagosomes acquire both newly synthesized MHC-II molecules (primarily from intracellular compartments) and preexisting MHC-II molecules (primarily from the plasma membrane), but utilize mostly newly synthesized MHC-II for phagocytic MHC-II Ag processing (35). Ullrich et al. recently showed that M. avium–containing phagosomes contain surface-derived, peptide-loaded MHC-II molecules (36). In our current analysis the source of MHC-II molecules that are present and used in MTB processing is still uncertain and may vary between phagosomes containing live versus HK MTB.
To assess phagosomal Ag processing, macrophages were incubated with HK or live MTB for various periods and fractionated on Percoll density gradients. Ag 85B(241–256)–I-Ab complexes were consistently detected in phagosomal fractions at 20 min (no chase) and in plasma membrane fractions at later time points. Ag 85B(241–256)–I-Ab complexes were never detected in other fractions containing late endocytic compartments, e.g., MIIC, which were found to contain OVA(323–339)–I-Ad complexes after macrophage processing of soluble OVA. Thus, in the time points examined in these studies, MTB peptide–MHC-II complexes were formed directly within phagosomes that contained MTB and not in MIIC after export of MTB Ags from phagosomes.
As phagosomal maturation can be inhibited by live mycobacteria, phagosomal processing of live MTB was also examined. Freshly grown, declumped MTB was used in all experiments to ensure maximum viability of live MTB preparations. Both live and HK MTB were processed and presented to BB7 T cells (Figs. 6 and 7), but processing of MTB Ag in phagosomes formed with live MTB was slower and less efficient than in phagosomes formed with HK MTB. It may seem surprising that live MTB was processed at all, but the use of highly activated macrophages in these studies may have enhanced processing of live MTB and minimized differences between live and HK MTB in phagosomal maturation and Ag processing. Previous studies showed that treatment of macrophages with IFN-γ alone, or with IFN-γ and LPS, enhanced acidification of phagosomes containing live mycobacteria (44–46). Activation of macrophages with IFN-γ substantially increased levels of mature cathepsin D in live mycobacterial phagosomes, rendering the phagosomes more lysosome like (47). Ullrich et al. recently showed that treatment of macrophages with IFN-γ promoted acquisition of MHC-II and H2-DM by live mycobacterial phagosomes (36). Studies comparing phagosomes isolated from macrophages of different states of activation may further clarify the effects of live and HK MTB on phagosomal maturation and MHC-II processing of MTB Ags.
In conclusion, our observations demonstrate clearly that MTB phagosomes are fully competent Ag processing organelles that can mediate the formation of peptide–MHC-II complexes. Live MTB, however, can inhibit both phagosome maturation and MHC-II Ag processing. Additional studies are necessary to understand the mechanisms of this inhibition and the role of macrophage activation in potentially decreasing this inhibition. The ability of MTB to inhibit phagosome maturation and Ag processing may contribute significantly to survival of MTB in host cells, providing a major determinant for the virulence of MTB and its ability to evade host defense mechanisms.
Acknowledgments
We thank David Canaday and Martha Torres for advice, Marilyn Convery for technical assistance, and Mike Sramkoski for help with flow cytometry. Antibodies were generously provided by John Monaco and Andrea Sant.
This work was supported by National Institutes of Health grants AI35726 and AI34343 to C.V. Harding and HL55967 and AI27243 to W.H. Boom.
Footnotes
Abbreviations used in this paper: CLIP, class II–associated invariant chain peptide; HK, heat-killed; LAMP, lysosome-associated membrane protein; MFV, mean fluorescent value; MIIC, MHC class II compartment; MOI, multiplicity of infection; MTB, Mycobacterium tuberculosis.
References
- 1.Barnes, P.F., S.D. Mistry, C.L. Cooper, C. Pirmez, T.H. Rea, and R.L. Modlin. 1989. Compartmentalization of a CD4+ T lymphocyte subpopulation in tuberculous pleuritis. J. Immunol. 142:1114–1119. [PubMed] [Google Scholar]
- 2.Barnes, P.F., A.B. Bloch, P.T. Davidson, and D.E. Snider, Jr. 1991. Tuberculosis in patients with human immunodeficiency virus infection. N. Engl. J. Med. 324:1644–1650. [DOI] [PubMed] [Google Scholar]
- 3.Havlir, D.V., R.S. Wallis, W.H. Boom, T.M. Daniel, K. Chervenak, and J.J. Ellner. 1991. Human immune responses to Mycobacterium tuberculosis antigens. Infect. Immun. 59:665–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsukaguchi, K., K.N. Balaji, and W.H. Boom. 1995. CD4+ alpha-beta T cell and gamma delta T cell reponses to Mycobacterium tuberculosis: similarities and differences in antigen recognition, cytotoxic effector function, and cytokine production. J. Immunol. 154:1786–1796. [PubMed] [Google Scholar]
- 5.Pancholi, P., A. Mirza, N. Bhardwaj, and R.M. Steinman. 1993. Sequestration from immune CD4+ T cells of mycobacteria growing in human macrophages. Science. 260:984–986. [DOI] [PubMed] [Google Scholar]
- 6.Gercken, J., J. Pryjma, M. Ernst, and H.D. Flad. 1994. Defective antigen presentation by Mycobacterium tuberculosis infected monocytes. Infect. Immun. 62:3472–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mazzaccaro, R.J., M. Gedde, E.R. Jensen, H.M. van Santen, H.L. Ploegh, K.L. Rock, and B.R. Bloom. 1996. Major histocompatibility class I presentation of soluble antigen facilitated by Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA. 93:11786–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hmama, Z., R. Gabathuler, W.A. Jefferies, G. de Jong, and N.E. Reiner. 1998. Attenuation of HLA-DR expression by mononuclear phagocytes infected with Mycobacterium tuberculosis is related to intracellular sequestration of immature class II heterodimers. J. Immunol. 161:4882–4893. [PubMed] [Google Scholar]
- 9.Hussain, S., B.S. Zwilling, and W.P. Lafuse. 1999. Mycobacterium avium infection of mouse macrophages inhibits IFN-gamma janus kinase-STAT signaling and gene induction by down-regulation of the IFN-gamma receptor. J. Immunol. 163:2041–2048. [PubMed] [Google Scholar]
- 10.Wojciechowski, W., J. DeSanctis, E. Skamene, and D. Radzioch. 1999. Attenuation of MHC class II expression in macrophages infected with Mycobacterium bovis bacillus Calmette-Guerin involves class II transactivator and depends on the Nramp1 gene. J. Immunol. 163:2688–2696. [PubMed] [Google Scholar]
- 11.Noss, E.H., C.V. Harding, and W.H. Boom. 2000. Mycobacterium tuberculosis inhibits MHC class II antigen processing in murine bone marrow macrophages. Cell. Immunol. 201:63–74. [DOI] [PubMed] [Google Scholar]
- 12.Noss, E.H., R.K. Pai, T.J. Sellati, J.D. Radolf, J. Belisle, D.T. Golenbock, W.H. Boom, and C.V. Harding. 2001. Toll-like receptor 2-dependent inhibition of macrophage class II MHC expression and antigen processing by 19 kD lipoprotein of Mycobacterium tuberculosis. J. Immunol. 167:910–918. [DOI] [PubMed] [Google Scholar]
- 13.Armstrong, J.A., and P.D. Hart. 1971. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J. Exp. Med. 134:713–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barker, L.P., K.M. George, S. Falkow, and P.L.C. Small. 1997. Differential trafficking of live and dead Mycobacterium marinum organisms in macrophages. Infect. Immun. 65:1497–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clemens, D.L., and M.A. Horwitz. 1995. Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. J. Exp. Med. 181:257–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Chastellier, C., T. Lang, and L. Thilo. 1995. Phagocytic processing of the macrophage endoparasite, Mycobacterium avium, in comparison to phagosomes which contain Bacillus subtilis or latex beads. Eur. J. Cell Biol. 68:167–182. [PubMed] [Google Scholar]
- 17.Oh, Y.K., and R.M. Straubinger. 1996. Intracellular fate of Mycobacterium avium: use of dual-label spectrofluorometry to investigate the influence of bacterial viability and opsonization on phagosomal pH and phagosome-lysosome interaction. Infect. Immun. 64:319–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sturgill-Koszycki, S., P.H. Schlesinger, P. Chakraborty, P.L. Haddix, H.L. Collins, A.K. Fok, R.D. Allen, S.L. Gluck, J. Heuser, and D.G. Russell. 1994. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science. 263:678–681. [DOI] [PubMed] [Google Scholar]
- 19.Xu, S., A. Cooper, S. Sturgill-Koszycki, T. van Heyningen, D. Chatterjee, I. Orme, P. Allen, and D.G. Russell. 1994. Intracellular trafficking in Mycobacterium tuberculosis and Mycobacterium avium-infected macrophages. J. Immunol. 153:2568–2578. [PubMed] [Google Scholar]
- 20.Clemens, D.L., and M.A. Horwitz. 1996. The Mycobacterium tuberculosis phagosome interacts with early endosomes and is accessible to exogenously administered transferrin. J. Exp. Med. 184:1349–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clemens, D.L., B.Y. Lee, and M.A. Horwitz. 2000. Deviant expression of Rab5 on phagosomes containing the intracellular pathogens Mycobacterium tuberculosis and Legionella pneumophila is associated with altered phagosomal fate. Infect. Immun. 68:2671–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrari, G., H. Langen, M. Naito, and J. Pieters. 1999. A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell. 97:435–447. [DOI] [PubMed] [Google Scholar]
- 23.Peters, P.J., J.J. Neefjes, V. Oorschot, H.L. Ploegh, and H.J. Geuze. 1991. Segregation of MHC class II molecules from MHC class I molecules in the Golgi complex for transport to lysosomal compartments. Nature. 349:669–676. [DOI] [PubMed] [Google Scholar]
- 24.Harding, C.V., and H.J. Geuze. 1993. Immunogenic peptides bind to class II MHC molecules in an early lysosomal compartment. J. Immunol. 151:3988–3998. [PubMed] [Google Scholar]
- 25.Qiu, Y., X. Xu, A. Wandinger-Ness, D.P. Dalke, and S.K. Pierce. 1994. Separation of subcellular compartments containing distinct functional forms of MHC class II. J. Cell Biol. 125:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rudensky, A.Y., M. Maric, S. Eastmen, L. Shoemaker, P.C. DeRoos, and J.S. Blum. 1994. Intracellular assembly and transport of endogenous peptide-MHC class II complexes. Immunity. 1:585–594. [DOI] [PubMed] [Google Scholar]
- 27.Tulp, A., D. Verwoerd, B. Dobberstein, H.L. Ploegh, and J. Pieters. 1994. Isolation and characterization of the intracellular MHC class II compartment. Nature. 369:120–126. [DOI] [PubMed] [Google Scholar]
- 28.West, M.A., J.M. Lucocq, and C. Watts. 1994. Antigen processing and class II MHC peptide-loading compartments in human B-lymphoblastoid cells. Nature. 369:147–151. [DOI] [PubMed] [Google Scholar]
- 29.Amigorena, S., J.R. Drake, P. Webster, and I. Mellman. 1994. Transient accumulation of new class II MHC molecules in a novel endocytic compartment in B lymphocytes. Nature. 369:113–120. [DOI] [PubMed] [Google Scholar]
- 30.Kleijmeer, M.J., S. Morkowski, J.M. Griffith, A.Y. Rudensky, and H.J. Geuze. 1997. Major histocompatibility complex class II compartments in human and mouse B lymphoblasts represent conventional endocytic compartments. J. Cell Biol. 139:639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cresswell, P. 1996. Invariant chain structure and MHC class II function. Cell. 84:505–507. [DOI] [PubMed] [Google Scholar]
- 32.Ramachandra, L., R.M. Sramkoski, D.H. Canaday, W.H. Boom, and C.V. Harding. 1998. Flow analysis of MHC molecules and other membrane proteins in isolated phagosomes. J. Immunol. Methods. 213:53–71. [DOI] [PubMed] [Google Scholar]
- 33.Ramachandra, L., R. Song, and C.V. Harding. 1999. Phagosomes are fully competent antigen processing organelles that mediate the formation of peptide:class II MHC complexes. J. Immunol. 162:3263–3272. [PubMed] [Google Scholar]
- 34.Ramachandra, L., R.S. Chu, D. Askew, E.H. Noss, D.H. Canaday, N.S. Potter, A. Johnsen, A.M. Krieg, J.G. Nedrud, W.H. Boom, and C.V. Harding. 1999. Phagocytic antigen processing and effects of microbial products on antigen processing and T-cell responses. Immunol. Rev. 168:217–239. [DOI] [PubMed] [Google Scholar]
- 35.Ramachandra, L., and C.V. Harding. 2000. Phagosomes acquire nascent and recycling class II MHC molecules but primarily use nascent molecules in phagocytic antigen processing. J. Immunol. 164:5103–5112. [DOI] [PubMed] [Google Scholar]
- 36.Ullrich, H.J., W.L. Beatty, and D.G. Russell. 2000. Interaction of mycobacterium avium-containing phagosomes with the antigen presentation pathway. J. Immunol. 165:6073–6080. [DOI] [PubMed] [Google Scholar]
- 37.Belisle, J.T., V.D. Vissa, T. Sievert, K. Takayama, P.J. Brennan, and G.S. Besra. 1997. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science. 276:1420–1422. [DOI] [PubMed] [Google Scholar]
- 38.Wiker, H.G., and M. Harboe. 1992. The antigen 85 complex: a major secretion product of Mycobacterium tuberculosis. Microbiol. Rev. 56:648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rambukkana, A., P.K. Das, A. Chand, J.G. Baas, D.G. Groothuis, and A.H. Kolk. 1991. Subcellular distribution of monoclonal antibody defined epitopes on immunodominant Mycobacterium tuberculosis proteins in the 30-kDa region: identification and localization of 29/33-kDa doublet proteins on mycobacterial cell wall. Scand. J. Immunol. 33:763–775. [DOI] [PubMed] [Google Scholar]
- 40.Horwitz, M.A. 1997. A new TB vaccine. The Immunologist. 5:15–20. [Google Scholar]
- 41.Sklar, M.D., A. Tereba, B.D. Chen, and W.S. Walker. 1985. Transformation of mouse bone marrow cells by transfection with a human oncogene related to c-myc is associated with the endogenous production of macrophage colony stimulating factor 1. J. Cell. Physiol. 125:403–412. [DOI] [PubMed] [Google Scholar]
- 42.Harding, C.V. 1992. Electroporation of exogenous antigen into the cytosol for antigen processing and class I major histocompatibility complex (MHC) presentation: weak base amines and hypothermia (18°C) inhibit the class I MHC processing pathway. Eur. J. Immunol. 22:1865–1869. [DOI] [PubMed] [Google Scholar]
- 43.Griffin, J.P., R. Chu, and C.V. Harding. 1997. Early endosomes and a late endocytic compartment generate different peptide-class II MHC complexes via distinct processing mechanisms. J. Immunol. 158:1523–1532. [PubMed] [Google Scholar]
- 44.Schaible, U.E., S. Sturgill-Koszycki, P.H. Schlesinger, and D.G. Russell. 1998. Cytokine activation leads to acidification and increases maturation of Mycobacterium avium-containing phagosomes in murine macrophages. J. Immunol. 160:1290–1296. [PubMed] [Google Scholar]
- 45.Via, L.E., R.A. Fratti, M. McFalone, E. Pagan-Ramos, D. Deretic, and V. Deretic. 1998. Effects of cytokines on mycobacterial phagosome maturation. J. Cell Sci. 111:897–905. [DOI] [PubMed] [Google Scholar]
- 46.Appelberg, R., and I.M. Orme. 1993. Effector mechanisms involved in cytokine-mediated bacteriostasis of Mycobacterium avium infections in murine macrophages. Immunology. 80:352–359. [PMC free article] [PubMed] [Google Scholar]
- 47.Ullrich, H.J., W.L. Beatty, and D.G. Russell. 1999. Direct delivery of procathepsin D to phagosomes: implications for phagosome biogenesis and parasitism by Mycobacterium. Eur. J. Cell Biol. 78:739–748. [DOI] [PubMed] [Google Scholar]
- 48.Harding, C.V., and E.R. Unanue. 1989. Antigen processing and intracellular Ia. Possible roles of endocytosis and protein synthesis in Ia function. J. Immunol. 142:12–19. [PubMed] [Google Scholar]