

Abstract
During an immune response naive T helper (Th) cells differentiate into two functionally distinct subsets, Th1 and Th2, based on their cytokine secretion profile and immunomodulatory function. c-Jun amino terminal kinase (JNK) regulates Th cell differentiation by activating a transcriptional program required for cytokine production. We have recently identified a TNFR superfamily death domain–containing molecule, death receptor (DR)6, which potently activates JNK. T cells from DR6-deficient mice are substantially impaired in JNK activation. When DR6−/− mice were challenged with protein antigen, their T cells hyperproliferate and display a profound polarization toward a Th2 response whereas Th1 differentiation is not equivalently affected. In addition, DR6−/− T cells showed preference toward Th2 differentiation in vitro. The phenotype seen in the DR6−/− mice is not due to the apoptotic pathway. Therefore, DR6, working through JNK, rather than apoptosis, functions to attenuate the Th2 response. This is the first demonstration of a role in the activation and differentiation of Th cells by DR6 in particular and DRs in general.
Keywords: hyperproliferation, apoptosis, TNFR-superfamily, polarization, Th2
Introduction
When activated by APCs, naive CD4+ Th cells undergo clonal expansion and secrete IL-2 (1). After 4–5 d of proliferation, a significant reprogramming of gene expression occurs that is mediated by the cytokine microenvironment, allowing for the emergence of predominantly Th1 or Th2 cell populations (2–6). Th1 cells are responsible for cell-mediated inflammatory reactions as they secrete cytokines such as IFN-γ and TNF-β (3, 7). In contrast, Th2 cells mediate B cell activation and differentiation by virtue of secreting IL-4, IL-5, IL-9, IL-10, and IL-13, all of which promote humoral immunity (2, 8). An intracellular signaling pathway that regulates Th cell differentiation has recently been found to involve c-Jun amino terminal kinase (JNK)*-1, a MAP kinase that phosphorylates and activates a number of transcription factors including c-Jun, Jun D, Elk-1, ATF, and Sap-1 (9–11). The most direct proof for the involvement of JNK-1 in the balance of Th1 and Th2 immune responses comes from the study of JNK-1 null mice (12). After keyhole limpet haemocyanin (KLH) immunization, JNK-1–deficient mice display an enhanced Th2 type cytokine response, suggesting that JNK-1 normally functions to suppress the production of Th2 type cytokines. Signals from both the TCR–CD3 complex and costimulatory receptors such as CD28 are required for JNK activation (12). Our studies suggest that death receptor (DR)6 fulfills an important costimulatory role for JNK-1 activation in T cells.
Many members of the TNF ligand superfamily and their cognate receptors play a critical role in organogenesis of lymphoid organs and homeostatic regulation of immune effector cells (13). DR6 was identified as a member of the TNFR family based on the presence of characteristic extracellular cysteine-rich domains. These 40 amino acid pseudorepeats typically consist of six highly conserved cysteines. The cytoplasmic domains of TNFR are less well conserved with the exception of death domains (DDs) that are present in a subset of receptors termed DRs, including TNFR1, CD95, EDAR, DR3, DR4, DR5, and DR6 (14–20). The DD is typically composed of ∼60 amino acids forming a globular structure of six conserved α helices with a characteristic “greek key” topology. Upon ligand engagement, one major signaling event for DRs is the recruitment of DD-containing adaptors through homotypic association. In contrast to most DD-containing receptors in the family, even upon overexpression, DR6 does not induce cell death in the apoptosis sensitive indicator cell line, MCF-7, suggesting an alternate function. Consistent with this, DR6 does not convincingly associate with DD-containing adaptor molecules including TRADD, FADD, RAIDD, and RIP that mediate downstream signaling from activated DRs (21). Regardless, upon overexpression, which for TNFR family members mimics ligand activation, DR6 was found to be a potent inducer of JNK activation.
Activation of the JNK pathway and DR6's expression in lymphoid organs such as thymus, spleen and lymphoid cells (21), suggested a possible role for DR6 in immune function. To explore this possibility we generated DR6-deficient mice. When these mice were challenged with protein antigen, their T cells hyperproliferated, and DR6−/− T cells showed preference toward Th2 differentiation in vitro. Although the DR6 is a DD-containing receptor, the DR6 null mutation had no effect on apoptosis; the phenotype seen in the DR6−/− mice was independent of apoptosis. These results support the notion that DR6, working through JNK regulates the differentiation of Th cells.
Materials and Methods
Generation of DR6-deficient Mice
The targeting construct containing 12.8 kb of 5′ homology and 4.0 kb of 3′ homology was linearized and used to electroporate 107 ES cells. After positive/negative selection with G418 and gancyclovir, surviving ES clones were screened for homologous recombination by Southern blot analysis. Genomic DNA was digested with Sac1 and hybridized with a 241-bp probe from a region just upstream to the 1.8-kb homology arm. Targeted ES clones were used to generate chimeric animals by injection into C57BL/6 blastocysts. Male chimeras were bred with C57BL/6 females and tail biopsies of agouti-colored offspring screened to identify germline transmission. Heterozygous mice were interbred to generate DR6-deficient animals. Expression of DR6 gene was evaluated by reverse transcription (RT)-PCR analysis using total RNA isolated from livers and a custom pair of forward (5′GGCATCGAGGAAGGGACAGTG3′) and reverse primers (5′TTGATGTCGAAATGCTTGTGAGC3′).
Taqman™ PCR
Quantitative RT-PCR was performed using Taqman™ assay reagents (Applied Biosystems). Total RNA was extracted using QIAGEN RNeasy columns and digested with DNase 1 to remove contaminating genomic DNA. All Taqman™ reactions were performed in duplicate and normalized to rpl19, a ribosomal housekeeping gene. Primer sequences used for mouse DR6 Taqman™ analysis were: forward primer, 5′CACCACGCAGTTGGAAAC3′, reverse primer, 5′GAATTCTCAAGTTTCACGTTAGGA3′ and probe primer, 5′ACTGGCTCTCCCCATGAGCCC 3′.
Flow Cytometry
Single cell suspension from various tissues was stained with conjugated mAbs (BD PharMingen) for 30 min at 4°C. Fluorescence was analyzed using a FACScan™ instrument and associated CELLQuest™ software (Becton Dickinson).
In Vitro T Cell Assays
For polyclonal stimulation of T cells, 5 × 105 purified CD4+ T cells were cultured in the presence of different concentrations of Con A or anti-CD3 alone or in the presence of anti-CD28 in 96-well plates in triplicate. Proliferation was measured by addition of 1 μCi of [3H]thymidine (ICN Biomedicals) for the last 18 h of a 5-d culture and incorporation of radioactivity assayed by liquid scintillation counting.
In Vivo T Cell Activation
For anti-KLH responses, mice were immunized with 100 μg KLH in saline, in a 1:1 emulsion with CFA (Difco Laboratories) in the hind footpads. After 9 d, popliteal lymph nodes were removed and cell suspensions were prepared. The recovered lymph node cells were cultured (5 × 105 cells per well) with indicated concentrations of KLH and proliferation assessed by uptake of [3H]thymidine as described previously. Assays for cytokine production by T cells were conducted by culturing 5 × 105 draining lymph node cells either from KLH-primed wild-type or DR6-deficient mice in the presence of KLH in two sets of 96-well plates in triplicate at a final volume of 200 μl. From one set of plates, after overnight culture, 150 μl of culture supernatant was removed for determination of IL-2. From the second set of plates, after 96 h of culture, 150 μl of culture supernatant was removed for determination of IL-4 and IFN-γ by ELISA using antibodies from BD PharMingen.
Serum Ig Concentrations
Unimmunized mice were bled at 12 wk of age and serum was analyzed for various Ig isotypes by ELISA using a kit from BD PharMingen.
Generation of Antigen-specific Antibodies
For trinitrophenyl (TNP)-specific antibodies, 5-wk-old wild-type or DR6-deficient mice were immunized intraperitoneally with TNP-KLH in CFA and 10 d later mice were bled and serum was analyzed for TNP-specific antibodies using standard ELISA protocols.
In Vitro Differentiation of T Cells
Naive T cells from the spleen and lymph nodes of wild-type or DR6-deficient littermates were purified using MACS® CD4+ microbeads (Miltenyi Biotec) according to the manufacturer's instructions. Purified T cells (106 cells per milliliter) were cultured in the presence of plastic-immobilized anti-CD3 (5 μg/ml) and anti-CD28 (1 μg/ml) plus IL-12 (3.5 ng/ml; R & D Systems) and 500 ng/ml IL-4 antibody (BD PharMingen) to promote Th1 cell differentiation or IL-4 (103 U/ml; R & D Systems) and 500 ng/ml anti–IFN-γ (BD PharMingen) for Th2 differentiation. After 4 d, cells were exhaustively washed, counted, and restimulated at 106 cells per milliliter with plastic-immobilized anti-CD3 (5 μg/ml). Supernatants were harvested 24 h later and analyzed for the presence of cytokines.
Apoptosis Assays
Thymocytes were resuspended at 5 × 106 cells per milliliter in high glucose DMEM containing 10% FCS (complete medium [CM]) and plated in a final volume of 200 ul in 96-well flat-bottomed tissue culture plates and the following proapoptotic stimuli administered: medium alone; soluble FLAG-tagged FasL and FLAG antibody at 1 ug/ml each; plate-bound anti-CD3 and anti-CD28 (BD PharMingen) both at 20 ug/ml; etoposide at 100 uM; dexamethasone (Sigma-Aldrich) at 2 uM. 24 h later, thymocytes were harvested, stained with annexin V and propidium iodide, and analyzed by flow cytometry.
Measurement of Activation-induced Cell Death
For activation-induced cell death (AICD) in splenocytes, cells from wild-type and DR6-deficient mice were isolated and washed twice in CM. Cells were stimulated by the addition of ConA (5 μg/ml) and 50 U/ml of recombinant mouse IL-2 (100 IU/ml; Genzyme) for 48 h, followed by culture in CM containing murine recombinant IL-2 for 48 h. Cells were then Lympholyte-M purified and washed twice in CM. The activated lymphocytes were treated with either plate-bound α-CD3 (5 μg/ml) or soluble FLAG-tagged FasL and α-FLAG M2 (Sigma-Aldrich) at 1 μg/ml each for 24 h. Treated cells were harvested and percentage of cell death determined using annexin V and PI staining of cells by flow cytometry.
Measurement of JNK Activity in Splenocytes
Purified splenic T cells from wild-type or DR6-deficient mice were stimulated with plate-bound anti-CD3 (5 μg/ml) and anti-CD28 (1 μg/ml) for 48 h. JNK activity was measured with JNK assay kit using GST-Jun as substrate (New England Biolabs, Inc.).
Results
Cloning and Expression of DR6
To investigate the physiological function of DR6, a knockout strategy was devised, the first step of which was cloning a murine cDNA encoding DR6. Murine and human DR6 displayed 88% protein sequence identity (Fig. 1 A). Examination of mouse tissues by Northern blot analysis demonstrated the presence of a 4-kb DR6 transcript in adult lymphoid tissues including PBLs, thymus, and spleen. The transcript could also be detected at varying levels in every adult or fetal tissue analyzed (Fig. 1 B). To further characterize the expression pattern of DR6 in various lymphoid cell populations, we performed real time quantitative RT-PCR by Taqman™ assay using total RNA extracted from various cell populations. Our analysis indicated that DR6 was present on CD4+, CD8+, CD19+, and CD14+ cells, suggesting that DR6 is specifically expressed on T cells, B cells, and monocytes (data not shown). DR6 expression was also determined in Th0 and differentiated Th1 and Th2 cells. DR6 mRNA was found in Th0 cells as well as in Th1 and Th2 cells; however, levels of DR6 transcript were twofold higher in Th2 cells (Fig. 1 C). These results reinforced the notion that DR6 may be important in immune function and potentially regulate Th cell responses.
Figure 1.


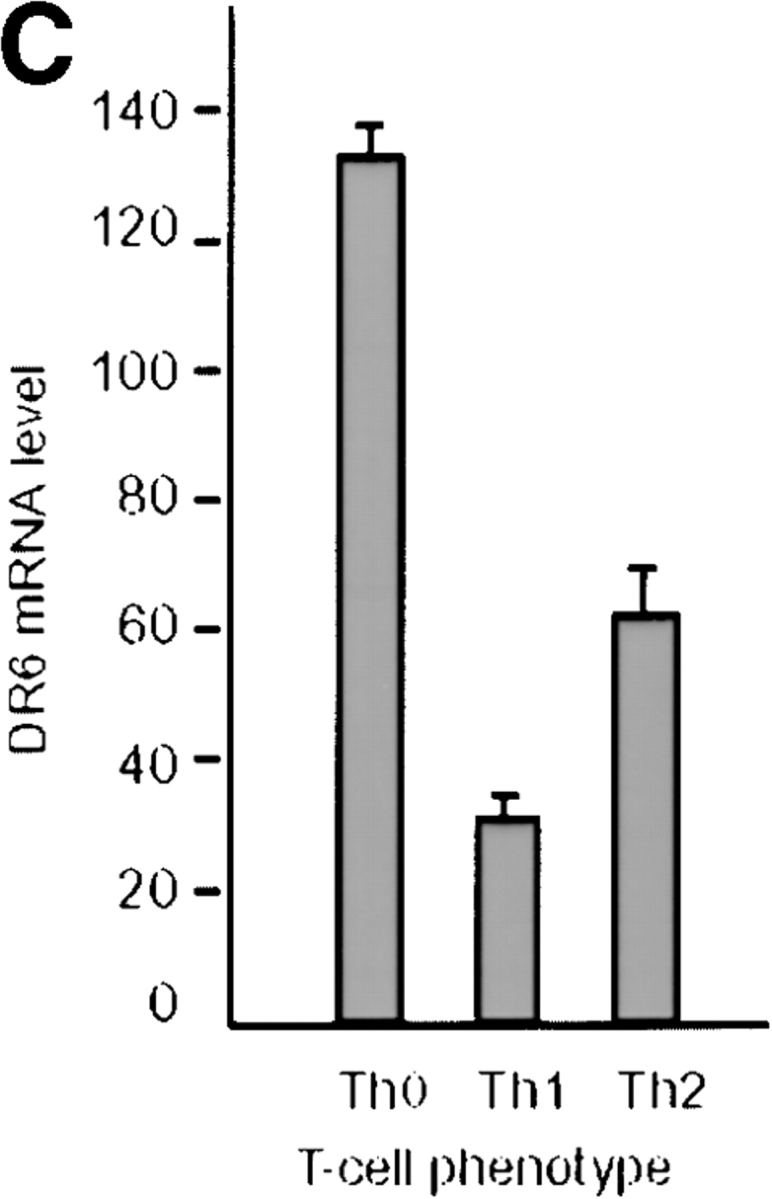
Amino acid sequence and tissue distribution of DR6. (A) Amino acid sequence of mouse and human DR6. (B) Northern blot analysis of human DR6. Human multiple tissue Northern blots (CLONTECH) were probed with a cDNA fragment corresponding to the cytoplasmic domain of DR6. (C) Expression of mouse DR6 in T cells. Expression of DR6 mRNA in undifferentiated T cells (Th0) or T cells differentiated for 3 d into Th1 or Th2 cells. DR6 mRNA levels were determined by Taqman™ real-time quantitative PCR and normalized to rpl19, a ribosomal housekeeping gene.
Generation of DR6-deficient Mice
To create DR6-deficient mice, a gene-targeting vector was constructed such that homologous recombination would delete the entire extracellular and transmembrane domain of the endogenous mouse DR6 gene. The 3.9-kb deletion included exons 2 and 3, resulting in a mutant DR6 allele lacking amino acid residues Q33 to G415 (Fig. 2 A). Three targeted ES cell lines were injected into C57BL/6 blastocysts. Founder mice with germline transmission were interbred to produce homozygous DR6-deficient mice, which were born at the expected Mendelian ratio. Inactivation of the DR6 gene locus was verified by Southern blot analysis (Fig. 2 B) and loss of gene expression was demonstrated by RT-PCR analysis of total liver RNA (Fig. 2 C). DR6-deficient mice were viable, fertile, and detailed histological analysis did not reveal any obvious abnormalities (data not shown).
Figure 2.



Targeting of the DR6 gene by homologous recombination. (A) Structure of the mouse DR6 gene and gene targeting vector. Exons 2 and 3 corresponding to the extracelluar and transmembrane regions of mDR6 were replaced by a neomycin resistance gene under the control of the PGK promoter. B, BamHI; S, SacI; Sp, SpeI. (B) Southern blot analysis. Genomic DNA digested with Sac I was hybridized with a [32P]-labeled probe derived from sequence 5′ to the targeting vector. (C) RT-PCR analysis of total RNA isolated from livers of DR6 null and wild-type mice. PCR products representing DR6 and GAPDH are shown in the top and bottom panel, respectively. ctl, no RNA control.
Lymphocyte Populations in DR6-deficient Mice
Initial profiling of immune cells revealed a slight, though statistically significant (P < 0.05) increase in the percentage of CD3+ T cells in the thymus and PBL compartments of the knockout mice (Fig. 3 A). We further measured thymic weight and compared the total number of thymocytes in DR6−/− mice to control wild-type littermates. There was no significant change in the size of the thymus or in the number of thymocytes (data not shown). Additional analysis showed that both CD4+ and CD8+ T cells were almost elevated twofold in the PBL compartment (Fig. 3 B and C). Flow cytometry analysis of cells stained with mAbs to B220, Gr.1, NK1.1, F4/80, and CD14 did not indicate any gross differences in B cells, neutrophils, NK cells, or monocyte/macrophages in lymphoid organs of DR6-deficient mice when compared with age- and sex-matched controls (data not shown).
Figure 3.

Effect of DR6 on T cell development. Total T cells and CD8+ T subpopulations were determined in thymus (Thy), blood (PBL), spleen (Sp), and LNs from DR6 knockout (black bars) and wild-type (white bars) mice. Organ-specific (A) total CD3+ T cells, (B) CD4+ cells, and (C) CD8+ cells. Lymphoid organs were harvested from mice and single cell suspensions stained with mAbs and analyzed by flow cytometry.
In Vitro T Cell Responses
Given the significant increase in T cell numbers in DR6-deficient mice, this phenotype was further characterized by measuring their proliferative capacity. Purified CD4+ T cells from DR6-deficient and control wild-type littermates were stimulated with Con A, a polyclonal T cell stimulator or with α-CD3 mAb alone or α-CD3 plus α-CD28. Subsequently, proliferative response and IL-2 production were measured. T cells from DR6-deficient mice hyperproliferated and produced increased amounts of IL-2 as compared with wild-type cells regardless of the nature of the proliferative stimulus (Fig. 4). These data indicated that CD4+ T cells lacking DR6 were intrinsically hyperresponsive to mitogenic, TCR, and costimulatory signals consistent with the notion that DR6 normally functions to suppress T cell activation and cytokine production.
Figure 4.

In vitro activation of T cells. For in vitro responses of T cells, purified CD4+ T cells from DR6−/− mice (black squares) or wild-type littermates (black circles) were cultured in the presence of different concentrations of ConA or anti-CD3 alone or in the presence of anti-CD28 and proliferation measured by 3[H]thymidine incorporation. IL-2 levels were determined by ELISA. (A) ConA induced proliferation and (B) IL-2 production, (C) anti-CD3 induced proliferation, and (D) IL-2 production, (E) anti-CD3 plus anti-CD28–induced proliferation, and (F) IL-2 production.
In Vivo T Cell Responses
To determine the influence of DR6 on in vivo T cell responses, wild-type and DR6-deficient mice were immunized with KLH and 9 d later draining LNs (DLNs) were harvested. After in vitro stimulation of DLN cells with KLH, cytokine production and recall proliferative ability of these cells was assessed. T cells from primed DR6-deficient mice had increased proliferative responses with production of higher levels of IL-2 and IFN-γ (Fig. 5 A–C). Additionally, production of IL-4 was markedly enhanced in DLN cells from DR6-deficient mice, levels being almost twofold higher (Fig. 5 D). These results indicate that DR6-deficient mice are hyperresponsive to in vivo antigenic challenge. This is consistent with our in vitro data reinforcing the notion that DR6 functions to suppress T cell responsiveness in vivo. Although T cells from DR6-deficient mice produced somewhat increased levels of various cytokines, the level of IL-4 was significantly elevated, underscoring a role for DR6 in the Th2 response. The in vivo KLH/CFA challenge experiments were designed to examine the influence of antigenic challenge on general T cell responses and not Th differentiation which typically requires immunization with KLH in alum. However, even under these sub-optimal conditions, we did see some preference for Th2 differentiation.
Figure 5.




Antigen-induced in vivo responses in DR6-deficient mice. Cytokine production and proliferative response of lymph node cells from wild-type (white circles) and DR6-deficient (white squares) mice immunized with KLH in CFA. Lymph node cells were collected 9 d after immunization, cultured in the presence of KLH, and analyzed for their capacity to proliferate (A) or produce IL-2 (B), IFN-γ (C), and IL-4 (D). Data is presented as the mean SD values derived from five animals in each group.
In Vivo Humoral Responses
Since Th2 cells regulate humoral immunity and Ig production, particularly IgG1 and IgE antibodies while the Th1 response contributes to the production of IgG2a and, to a lesser extent, IgG3 antibodies, serum Ig levels were determined in DR6-deficient mice. Naive DR6-deficient mice had Ig levels comparable to wild-type littermates (Fig. 6 A). However, upon in vivo challenge with TNP-conjugated KLH, DR6-deficient mice developed markedly increased titers of TNP-specific IgG1 and IgE (Fig. 6 B), whereas levels of IgM and IgG2a were only slightly elevated. The levels of IgG1 were increased by ∼200% in DR6-deficient mice, consistent with preferential Th2 cell differentiation and hyperproliferation. This marked polarization toward a Th2 response was also substantiated by the significant increase in IL-4 production in KLH-primed DR6−/− T cells (Fig. 5 D).
Figure 6.


Effect of DR6 on Ig subclasses. Serum was collected from wild-type (black bars) and DR6-deficient mice (white bars) and subclass concentrations determined by ELISA (A). Serum was obtained from wild-type and DR6-deficient mice immunized with TNP-KLH in CFA and levels of IgM, IgG1, IgE, and IgG2a determined by TNP-specific ELISA (B). Data is presented as the mean SD values derived from five animals in each group.
In Vitro Differentiation of T Cells
Since DR6-deficient mice exhibited an enhanced Th2 response in vivo, we investigated the role of DR6 in mediating Th differentiation in vitro. Purified naive CD4+ T cells from wild-type and DR6-deficient mice were differentiated into either Th1 cells by activating them with α-CD3 plus α-CD28 in the presence of antibodies to IL-12 and IL-4 or into Th2 cells in the presence of IL-4 and antibody to IFN-γ. When differentiated into Th1 cells, DR6-deficient lymphocytes produced levels of IFN-γ equivalent to wild-type controls (Fig. 7). In contrast, DR6-deficient lymphocytes grown in the presence of IL-4 and anti–IFN-γ produced markedly enhanced Th2 cytokines such as IL-4 itself. Cytokine production was also determined by staining the T cells for intracellular IFN-γ and IL-4. A significantly higher number of T cells produced IL-4 (data not shown), indicating that the IL-4 producing T cells pool was expanded in DR6−/− mice. Taken together, our results indicate that DR6 plays a major role in Th cell differentiation both in vivo and in vitro.
Figure 7.

In vitro induction of Th cell differentiation and effect of DR6 on cell death. T cells purified from spleens of wild-type (black bars) or DR6-deficient (white bars) mice were differentiated into Th1 or Th2 cells with anti-CD3 plus anti-CD28. Production of IFN-γ and IL-4 was determined by ELISA. Data represent the mean SD of pools of five mice per group. ND, not detected.
DR6 Does Not Regulate T Cell Differentiation through Apoptosis
Since DR6 possesses an intracellular DD, this raised the possibility that it may regulate T cell activation and differentiation by modulating apoptosis. To address this, AICD of thymocytes and splenic T cells from DR6-deficient mice was measured in response to various apoptotic stimuli. No appreciable difference was observed in susceptibility of splenocytes or thymocytes to AICD or death induced by other proapoptotic stimuli (Fig. 8 A and B). Therefore, it was unlikely that decreased apoptosis accounts for the increased proliferative potential of DR6-deficient T cells. In sum, our data are consistent with an intrinsic, cell autonomous growth inhibitory role for DR6, removal of which, as in the knockout cells leads to a hyperproliferative response.
Figure 8.

Effects of DR6 on thymocyte apoptosis. (A) Apoptosis was induced in thymocytes from wild-type (black bars) and DR6-deficient (white bars) mice with soluble FLAG-tagged FasL and α-FLAG antibody, plate-bound α-CD3 plus α-CD28, etoposide, and dexamethasone. Percentage of surviving cells were analyzed using PI/FITC-annexin V staining of cells by flow cytometry 24 h after treatment. (B) Effects of DR6 on AICD. Splenocytes from wild-type and DR6-deficient mice were activated with Con A for 2 d, followed by culture in medium containing murine recombinant IL-2 for 2 d. The activated lymphocytes depleted of dead cells were treated with either soluble FLAG-tagged FasL and α-FLAG antibody or plate-bound α-CD3 for 24 h. Percentage of surviving cells were analyzed by PI/FITC-annexin V staining by flow cytometry 24 h after treatment. Abrogation of JNK activation in DR6-deficient CD4+ T cells (c) Purified splenic CD4+ T cells from wild-type (WT) or DR6-deficient (KO) mice were stimulated with anti-CD3 and anti-CD28 for 48 h. JNK activity was measured using GST-Jun as substrate. Expression of JNK was confirmed by anti-JNK Western blot analysis.
DR6 Regulates Differentiation of T Cells by Activation of the JNK Pathway
Given the role of the JNK-1 pathway in Th cell differentiation and the polarized Th2 response observed in both JNK-1(12) and DR6 null mice (this study), we asked if JNK activity was correspondingly reduced in DR6-deficient T cells. Purified CD4+ T cells from wild-type or DR6-deficient mice were stimulated with α-CD3 plus α-CD-28 and JNK activity measured using recombinant c-Jun as substrate. In DR6-deficient T cells, JNK activity was significantly reduced (Fig. 8 C), consistent with the notion that attenuation of this MAP kinase contributes to the polarized Th cell differentiation observed in JNK-deficient mice (12).
Discussion
A significant role for effector Th1 and Th2 cells in adaptive immunity has been well recognized in recent years. Although considerable progress has been made in defining the rules for differentiation of naive CD4+ T cells into Th1 and Th2 cells, a complete understanding of the molecules that regulate this process has remained elusive. In this study, we demonstrate the role of a recently identified novel DD-containing TNFR family member, DR6, in controlling the activation and differentiation of Th cells. The differentiation of Th cells is influenced by a number of factors including cytokine environment, antigen dose, and costimulatory signals. The exact mechanism by which such disparate factors influence Th differentiation is unknown except that JNK, a MAPK that phosphorylates c-Jun and enhances AP-1 transcriptional activity, is an integral component of the final common pathway. Activation of JNK in T cells requires engagement of both the T cell receptor and costimulatory receptors such as CD28. Our work suggests that DR6 also provides costimulatory support for activation of the JNK pathway in T cells. The most compelling evidence for this comes from the observation that the enhanced Th2 response seen in JNK null T cells is also found in DR6−/− T cells. Furthermore, the induction of JNK activity upon stimulation with α-CD3 and α-CD28 is substantially attenuated in CD4+ T cells from DR6-deficient mice.
The current model for differentiation of CD4+ T cells into Th1 and Th2 cells suggests that the cytokine microenvironment plays a crucial role (4, 22, 23). The most critical cytokine for Th1 development is IL-12 as mice deficient in IL-12, IL-12 receptor β1, or the associated signaling molecule Stat4 possess markedly reduced Th1 responses (24). Conversely, IL-4, acting via Stat6, is the primary inducer of Th2 development (25), as mice deficient in either IL-4, IL-4 receptor, or the associated downstream signaling adaptor Stat6 do not develop Th2 responses. Besides promoting growth and differentiation of their own subset Th1 and Th2 cytokines also cross-regulate by inhibiting the development of the opposing subset. For example, IL-4 downregulates IL-12 receptor expression on developing Th cells, thereby committing them to a Th2 lineage (26, 27). Similarly, IFN-γ promotes Th1 development by inducing IL-12 and inhibiting Th2 cell growth (26, 28, 29). This study suggests an exception to the current model of Th cell differentiation, as DR6-deficient mice possess an enhanced Th2 response but normal Th1-associated responses. Therefore, it appears that in this particular instance Th2 type cytokines are not counterregulating the Th1 response. The exact mechanism for this is unclear but probably has to do with the nature of the signaling pathways engaged by DR6. Although DR6 is a DD-containing receptor, it appears not to mediate apoptosis but rather activation of the JNK pathway. To a much lesser extent, DR6 also activates the nuclear factor κB pathway, but it is difficult to assess whether this is of physiological significance. The precise signaling pathway used by DR6 to activate the JNK pathway awaits definition. Since known DD signaling adapters such as TRADD and FADD do not associate with DR6, it is likely that a yet to be elucidated intermediary adaptor(s) provide the physical link between DR6 and JNK. Besides identification of components of the DR6 signaling complex, an equally important question is the nature of the ligand for DR6. It is tempting to speculate that activated T cells are the autocrine source of the ligand since the substantial difference in JNK activity seen between wild-type and DR6 knockout CD4+ T cell cells (Fig. 8 C) is observed with a purified CD4+ T cell population that lacks contaminating APCs. Identification of the ligand may have potential therapeutic value as our work suggests that activating the DR6 pathway will suppress the evolution of a Th2 response and may therefore represent a novel strategy to treat Th2-mediated diseases such as chronic asthma.
Acknowledgments
We thank the Genentech microinjection lab, DNA sequencing lab, and the Laboratory of Animal Resources for support.
H. Zhao, M. Yan, and H. Wang contributed equally to this paper.
Footnotes
Abbreviations used in this paper: AICD, activation-induced cell death; CM, complete medium; DD, death domain; DLN, draining LN; DR, death receptor; JNK, c-Jun amino terminal kinase; KLH, keyhole limpet haemocyanin; TNP, trinitrophenyl; RT, reverse transcription.
References
- 1.Jain, J., C. Loh, and A. Rao. 1995. Transcriptional regulation of the IL-2 gene. Curr. Opin. Immunol. 7:333–342. [DOI] [PubMed] [Google Scholar]
- 2.Mosmann, T.R., H. Cherwinski, M.W. Bond, M.A. Giedlin, and R.L. Coffman. 1986. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 136:2348–2357. [PubMed] [Google Scholar]
- 3.Schmitt, E., P. Hoehn, C. Huels, S. Goedert, N. Palm, E. Rude, and T. Germann. 1994. T helper type 1 development of naive CD4+ T cells requires the coordinate action of interleukin-12 and interferon-γ and is inhibited by transforming growth factor-β. Eur. J. Immunol. 24:793–798. [DOI] [PubMed] [Google Scholar]
- 4.O'Garra, A. 1998. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity. 8:275–283. [DOI] [PubMed] [Google Scholar]
- 5.Kamogawa, Y., L.A. Minasi, S.R. Carding, K. Bottomly, and R.A. Flavell. 1993. The relationship of IL-4- and IFN-γ-producing T cells studied by lineage ablation of IL-4-producing cells. Cell. 75:985–995. [DOI] [PubMed] [Google Scholar]
- 6.Constant, S.L., C. Dong, D.D. Yang, M. Wysk, R.J. Davis, and R.A. Flavell. 2000. JNK1 is required for T cell-mediated immunity against Leishmania major infection. J. Immunol. 165:2671–2676. [DOI] [PubMed] [Google Scholar]
- 7.Seder, R.A., and W.E. Paul. 1994. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu. Rev. Immunol. 12:635–673. [DOI] [PubMed] [Google Scholar]
- 8.Le Gros, G., S.Z. Ben-Sasson, R. Seder, F.D. Finkelman, and W.E. Paul. 1990. Generation of interleukin 4 (IL-4)–producing cells in vivo and in vitro: IL-2 and IL-4 are required for in vitro generation of IL-4–producing cells. J. Exp. Med. 172:921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cavigelli, M., F. Dolfi, F.X. Claret, and M. Karin. 1995. Induction of c-fos expression through JNK-mediated TCF/Elk-1 phosphorylation. EMBO J. 14:5957–5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gupta, S., T. Barrett, A.J. Whitmarsh, J. Cavanagh, H.K. Sluss, B. Derijard, and R.J. Davis. 1996. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 11.Ip, Y.T., and R.J. Davis. 1998. Signal transduction by the c-Jun N-terminal kinase (JNK)—from inflammation to development. Curr. Opin. Cell. Biol. 10:205–219. [DOI] [PubMed] [Google Scholar]
- 12.Dong, C., D.D. Yang, M. Wysk, A.J. Whitmarsh, R.J. Davis, and R.A. Flavell. 1998. Defective T cell differentiation in the absence of Jnk1. Science. 282:2092–2095. [DOI] [PubMed] [Google Scholar]
- 13.Locksley, R.M., N. Killeen, and M.J. Lenardo. 2001. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 104:487–501. [DOI] [PubMed] [Google Scholar]
- 14.Tartaglia, L.A., T.M. Ayres, G.H. Wong, and D.V. Goeddel. 1993. A novel domain within the 55 kd TNF receptor signals cell death. Cell. 74:845–853. [DOI] [PubMed] [Google Scholar]
- 15.Chinnaiyan, A.M., K. O'Rourke, G.L. Yu, R.H. Lyons, M. Garg, D.R. Duan, L. Xing, R. Gentz, J. Ni, and V.M. Dixit. 1996. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science. 274:990–992. [DOI] [PubMed] [Google Scholar]
- 16.Monreal, A.W., B.M. Ferguson, D.J. Headon, S.L. Street, P.A. Overbeek, and J. Zonana. 1999. Mutations in the human homologue of mouse δλ cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat. Genet. 22:366–369. [DOI] [PubMed] [Google Scholar]
- 17.Headon, D.J., and P.A. Overbeek. 1999. Involvement of a novel TNF receptor homologue in hair follicle induction. Nat. Genet. 22:370–374. [DOI] [PubMed] [Google Scholar]
- 18.Sheridan, J.P., S.A. Marsters, R.M. Pitti, A. Gurney, M. Skubatch, D. Baldwin, L. Ramakrishnan, C.L. Gray, K. Baker, W.I. Wood, et al. 1997. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 277:818-821. [DOI] [PubMed] [Google Scholar]
- 19.Pan, G., J. Ni, Y.F. Wei, G. Yu, R. Gentz, and V.M. Dixit. 1997. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 277:815–818. [DOI] [PubMed] [Google Scholar]
- 20.Pan, G., K. O' Rourke, A.M. Chinnaiyan, R. Gentz, R. Ebner, J. Ni, and V.M. Dixit. 1997. The receptor for the cytotoxic ligand TRAIL. Science. 276:111–113. [DOI] [PubMed] [Google Scholar]
- 21.Pan, G., J.H. Bauer, V. Haridas, S. Wang, D. Liu, G. Yu, C. Vincenz, B. Aggarwal, J. Ni, and V.M. Dixit. 1998. Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS Lett. 431:351–356. [DOI] [PubMed] [Google Scholar]
- 22.Abbas, A.K., K.M. Murphy, and A. Sher. 1996. Functional diversity of helper T lymphocytes. Nature. 383:787–793. [DOI] [PubMed] [Google Scholar]
- 23.Constant, S.L., and K. Bottomly. 1997. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu. Rev. Immunol. 15:297–322. [DOI] [PubMed] [Google Scholar]
- 24.Gately, M.K., L.M. Renzetti, J. Magram, A.S. Stern, L. Adorini, U. Gubler, and D.H. Presky. 1998. The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annu. Rev. Immunol. 16:495–521. [DOI] [PubMed] [Google Scholar]
- 25.Nelms, K., A.D. Keegan, J. Zamorano, J.J. Ryan, and W.E. Paul. 1999. The IL-4 receptor: signaling mechanisms and biologic functions. Annu. Rev. Immunol. 17:701–738. [DOI] [PubMed] [Google Scholar]
- 26.Szabo, S.J., A.S. Dighe, U. Gubler, and K.M. Murphy. 1997. Regulation of the interleukin (IL)-12R β2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J. Exp. Med. 185:817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ouyang, W., S.H. Ranganath, K. Weindel, D. Bhattacharya, T.L. Murphy, W.C. Sha, and K.M. Murphy. 1998. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. 9:745–755. [DOI] [PubMed] [Google Scholar]
- 28.Ma, X., J.M. Chow, G. Gri, G. Carra, F. Gerosa, S.F. Wolf, R. Dzialo, and G. Trinchieri. 1996. The interleukin 12 p40 gene promoter is primed by interferon γ in monocytic cells. J. Exp. Med. 183:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gajewski, T.F., J. Joyce, and F.W. Fitch. 1989. Antiproliferative effect of IFN-γ in immune regulation. III. Differential selection of TH1 and TH2 murine helper T lymphocyte clones using recombinant IL-2 and recombinant IFN-γ. J. Immunol. 143:15–22. [PubMed] [Google Scholar]