

Abstract
Interaction between inducible costimulator (ICOS) and its ligand is implicated in the induction of cell-mediated and humoral immune responses. However, the molecular details of this interaction are unknown. We report here a mutagenesis analysis of residues in ICOS that are critical for ligand binding. A three-dimensional model of the extracellular immunoglobulin-like domain of ICOS was used to map the residues conserved within the CD28 family. This analysis identified a surface patch containing the characteristic “PPP” sequence and is conserved in human and mouse ICOS. Mutations in this region of human ICOS reduce or abolish ligand binding. Our results suggest that the ligand binding site in ICOS maps to a region overlapping yet distinct from the CD80/CD86 binding sites in CD28 and cytotoxic T lymphocyte antigen (CTLA)-4. Thus, the analysis suggests that differences in ligand binding specificity between these related costimulatory molecules have evolved by utilization of overlapping regions with different patterns of conserved and nonconserved residues. Two site-specific mutants generated in the course of our studies bound ICOS ligand with higher avidity than wild-type ICOS. An S76E mutant protein of ICOS blocked T cell costimulatory function of ICOS ligand and inhibited T cell response to allogeneic antigens superior to wild-type ICOS. Our studies thus identified critical residues involving in ICOS receptor–ligand interaction and provide new modulators for immune responses.
Keywords: ICOS, CD28, molecular model, mutagenesis, avidity
Introduction
Receptor–ligand interactions of costimulatory molecules are required for an optimal activation of T cells stimulated by MHC–peptide complexes expressed on APC. The interactions between CD28/cytotoxic T lymphocyte antigen (CTLA)*-4 on T cells with their ligands B7–1 and/or B7–2 have been intensively studied. CD28 triggering increases antigen-specific proliferation of CD4+ T cells, enhances production of cytokines, stimulates differentiation of effector function of CD8+ T cells (1, 2), and promotes T cell survival (3). Signaling through CTLA-4, however, is thought to deliver a negative signal that inhibits T cell proliferation, IL-2 production, and cell cycle progression (4, 5).
Recent studies indicate that other members of the CD28 family and their B7-like ligands also participate in the regulation of cellular and humoral immune response. One of these new members is inducible costimulator (ICOS) (H4), a CD28-like receptor and member of the Ig superfamily (IgSF) (6, 7). Similar to CTLA-4, ICOS is induced during T cell activation, whereas CD28 is expressed constitutively on resting T cells. In addition to activated T cells, mature thymocytes and thymus NK T cells also express ICOS. In HIV-infected patients, ICOS is highly expressed in phases characterized by high levels of viremia, and its expression can be induced in vitro by the HIV envelope glycoprotein gp120 (8). In independent studies, several laboratories including ours have identified murine and human ligands of ICOS (9–15). Engagement of ICOS by either specific antibodies or its natural ligand B7-H2 (B7h, B7RP-1, GL50, LICOS) costimulates T cell proliferation and increases production of cytokines (6, 9). While costimulation of T cells through ICOS induces both Th1 and Th2 cytokines, the effect of ICOS appears to preferentially stimulate the functions of Th2 effector cells (16, 17). Blocking this interaction with either ICOSIg fusion protein or specific mAb significantly or partially inhibits dendritic cell–mediated allogeneic responses and antigen-specific T cell proliferation to tetanus toxin (18), experimental allergic encephalomyelitis, lung mucosal inflammation, and allograft rejection (19–21). Mice with a genetic knockout of ICOS exhibit profound deficiencies in T cell–dependent B cell response, impaired germinal center formation, and decreased Th2 cytokine secretion (22–24). These results implicate ICOS and its ligand in the regulation of cell-mediated and humoral immune responses.
ICOS is a type I glycoprotein with 24 and 17% amino acid identity to CD28 and CTLA-4, respectively. Murine and human ICOS share 69% amino acid identity. The overall length and the relative position of the transmembrane segment are similar to those of CD28 and CTLA-4. Moreover, cysteine residues that are critical for the formation of the intra- and intermolecular disulfide bonds are conserved in ICOS and CD28/CTLA-4. ICOS contains a single extracellular V-like Ig domain similar to CD28 and CTLA-4 (6, 11). The V-like domains of CD28 and CTLA-4 contain a strictly conserved MYPPPY sequence motif that maps to the CDR3-analogous loop and is critical for recognition of their ligands B7–1 and B7–2 (25). However, this hexapeptide motif is not conserved in ICOS.
With the aid of a three-dimensional model, we have analyzed regions of ICOS that are involved in the interaction with B7-H2. By homologue replacement and site-specific mutagenesis, we have identified residues that are important or essential for the interaction between ICOS and B7-H2. Furthermore, two ICOS mutant proteins were identified having increased avidity and improved blocking capacity compared with wild-type ICOS.
Materials and Methods
Cell Culture.
Stably transfected B7-H2+ Chinese hamster ovary (CHO) cells were maintained in CHO-SF II medium (GIBCO BRL) supplemented with 10% heat-inactivated FBS (HyClone). COS cells were grown in DMEM (GIBCO BRL) supplemented with 10% FBS, 25 mM Hepes, 2 mM l-glutamine, 1 mM sodium pyruvate, 1% MEM nonessential amino acids, and 100 U/ml penicillin G, and 100 μg/ml streptomycin sulfate.
Ig-fusion Proteins.
The fusion proteins with extracellular domain of human B7-H2 linking to either mouse IgG2a or human IgG1 Fc portion (B7-H2mIg/B7-H2Ig) were produced in stably transfected CHO cells and purified by protein G affinity column as described previously (9). ICOSIg was prepared by transiently transfecting COS cells with plasmid cDNA created by fusing the cDNA of the extracellular domain of human ICOS in frame to the CH2-CH3 portion of human IgG1. The transfected COS cells were cultured in serum-free DMEM media and concentrated supernatants were used as sources for Ig fusion proteins. The Ig proteins were also purified with protein G column as indicated.
Residue Mapping.
The solution structure of the monomeric extracellular IgV-like domain of CTLA-4 (26) and molecular models of human CD28 (27) and human ICOS (28) were used to study residue conservation in human and mouse ICOS relative to CTLA-4 and CD28. The molecular models of CD28 and ICOS were reported previously (27, 28) and applied herein for residue mapping. Computer graphical analysis was performed using InsightII (MSI). Selected residues were subjected to a two-step mutagenesis analysis. In the first (homologue replacement) step, protein segments predicted to contain residues important for ligand binding were replaced with corresponding regions of human CD28. In the second step, selected residues in these regions were targeted by PCR-based site–specific mutagenesis.
ICOSIg Mutants.
Homologue replacement mutant plasmids were designed to introduce CD28 cDNA sequences into ICOSIg while, at the same time, deleting the equivalent region from ICOS. All mutants of ICOSIg (see Table I) were constructed by two-step PCR where ICOSIg cDNA was used as the template. Overlapping oligonucleotide primers were synthesized encoding the desired mutations and two flanking 5′ and 3′ primers were designed to contain EcoRI and BglII restriction sites, respectively. Appropriate regions of cDNA were initially amplified using the corresponding overlapping and flanking primers. Then, using the flanking 5′ and 3′ primers, fragments whose sequences overlapped were fused together and amplified. PCR products were digested with EcoRI and BglII and ligated into EcoRI/Bgl II–digested pHIg vectors (9). To verify that the desired mutations but no other mutations were introduced, each mutant was sequenced using an ABI Prism 310 Genetic Analyzer. Plasmids were transfected into COS cells and serum-free supernatants were harvested and used as sources for Ig fusion proteins or purified by protein G column.
Table I.
Ligand-binding Sites of ICOS and High Avidity Mutants with Improved Function
| Substitutionsa
|
Percentage of B7-H2d binding activity | ||
|---|---|---|---|
| Mutant | Nucleic acidsb | Amino acidsc | |
| ICOSIg | − | − | 100 |
| RP49-52 | ICOS145∼156/CD28145∼156 | ICOS49∼52/CD2849∼52 | 9 |
| RP64-78 | ICOS190∼234/CD28199∼243 | ICOS64∼78/CD2867∼81 | <0.1 |
| RP114-119 | ICOS340∼357/CD28349∼366 | ICOS114∼119/CD28117∼122 | <0.1 |
| Q49S | CAG/TCG | Q49/S | 33 |
| Q50S | CAA/TCA | Q50/S | 6 |
| F51S | TTT/TCT | F51/S | <0.1 |
| H52S | AAA/AGC | K52/S | 200 |
| D64S | GAT/TCT | D64/S | 6 |
| K67S | AAG/AGC | K67/S | <0.1 |
| K69S | AAA/AGC | K69/S | 3 |
| S76E | TCC/GAA | S76/E | 300 |
| F114S | TTT/TCT | F114/S | <0.1 |
| D115S | GAT/TCT | D115/S | <0.1 |
| P117S | CCT/TCT | P117/S | <0.1 |
| F119S | TTT/TCT | F119/S | <0.1 |
The regions, nucleotides, or amino acids in front of the slash were replaced with the corresponding regions, nucleotides, or amino acids after the slash.
Nucleotides are numbered from the A of the initiation codon.
Amino acids are numbered from the initiation methionine.
Specific Binding activities were determined for each of the indicated fusion proteins. The concentration of a fusion protein required to give an arbitrary A405 the same as that found for ICISIg was determined from the linear region of binding curves and expressed as a percentage of specific binding activity of ICOSIg. Values represent the average of three determinations from each binding curve. Values are representative of three experiments.
ELISA.
To quantitate Ig fusion proteins in culture media or purified proteins, microtiter plates were coated with 2 μg/ml goat anti–human IgG (Sigma-Aldrich) overnight at 4°C. Wells were blocked for 1 h with blocking buffer (10% FBS in PBS), then washed with PBS containing 0.05% Tween 20 (PBS-Tween). COS cell culture media supernatants were added and incubated for 2 h at room temperature. Known concentrations of ICOSIg were also added to separate wells on each plate for the generation of the standard curve. After washing, HRP-conjugated goat anti–human IgG (TAGO, Inc.) diluted at 1:2,000 was added and incubated for 1 h at room temperature. Wells were then washed and incubated with 3,3′,5,5′ tetramethylbenzidine substrate for 5–15 min before stopping the reaction by the addition of 0.5 M H2SO4. Absorbance was measured at wavelengths of 405 nm on a microtiter plate reader. Concentrations of Ig fusion proteins were determined by comparison with the linear range of a standard curve of ICOSIg. Data from triplicate wells were determined and the standard deviations from the mean were <10%. Experiments were repeated at least three times. The ability of ICOSIg mutants to bind B7-H2 was measured using capture ELISA assay. Recombinant B7-H2mIg fusion proteins were coated on microtiter plates at 5 μg/ml overnight at 4°C. Wells were blocked, washed with PBS-Tween, and COS cell culture media or purified proteins added and incubated for 2 h at room temperature. After washing, HRP-conjugated goat anti–human IgG was added followed by tetramethylbenzidine substrate and absorbance read at 405 nm.
Flow Cytometry.
B7-H2+ CHO cells at 105 were incubated in FACS® buffer (PBS, 3% FBS, 0.02% NaN3) with an equal amount of fusion proteins in COS cell culture media on ice for 45 min. B7-H2Ig fusion protein was used as negative control. The cells were washed and further incubated with FITC-conjugated goat anti–human IgG (BioSource) on ice for 30 min. Fluorescence was analyzed by a FACScaliber™ flow cytometry (Becton Dickinson) with CellQUEST™ software (Becton Dickinson).
Surface Plasmon Resonance Analysis.
All experiments were performed on a BIAcore™ 3000 instrument (Biacore) at 25°C using 0.1 M Hepes, pH 7.4, containing 0.15 M NaCl, 0.005% surfactant P20 as the running buffer. B7-H2Ig fusion protein was covalently coupled by primary amine groups to the carboxymethylated dextran matrix on a CM5 sensor chip (Biacore) via random amine coupling chemistry. The dextran surface was activated with N-ethyl-N-dimethylaminopropyl carbodiimid/N-hydroxysuccinimide, followed by injection of 20 μg/ml B7-H2Ig protein diluted in 10 mM sodium acetate buffer, pH 4.5. The protein was exposed to the activated surface until 3,800 response units (RU) were observed. The excess active groups on the dextran were blocked with 1 M ethanolamine. A control sensor surface was prepared by activating and blocking a separate flow cell without protein immobilization. Wild-type and mutants of ICOSIg were diluted to 500 nM in running buffer, and were injected across the immobilized surfaces at a flow rate of 30 μl per min for 2.66 min. Analysis of the experimental data was performed using the BIAevaluation software v3.2 (BIAcore).
T Cell Proliferation Assays.
PBMCs were isolated from peripheral blood of health donors by centrifugation over Ficoll-Hypaque and allowed to adhere to plastic dishes for 2 h. Enrichment of T cells was performed by passing nonadherent PBMCs of a healthy donor through nylon wool columns (Robbins Scientific, Co.) as described previously (9). For the costimulation assay, flat-bottomed 96-well microplates were first coated with 50 μl of anti-CD3 mAb (model no. 555336; BD PharMingen) at 5 ng/ml at 4°C overnight. After intensive washing with PBS, the plates were further coated with 50 μl of B7-H2Ig (5 μg/ml) at 37°C for 2 h and then the purified T cells (2 × 105 cells per well) were added with S76E mutant protein or ICOSIg at the indicated concentrations to the wells in triplicate. Both S76E and ICOSIg were purified by protein G affinity column as described previously (9). The cells were cultured for 72 h and 3[H]-TdR at 1.0 μCi per well was added during the last 9 h. The incorporation of 3[H]-TdR was counted by MicroBeta Trilux liquid scintillation counter (Wallac). Allogeneic mixed lymphocyte reaction (MLR) were set up by coculturing 105 γ-irradiated adherent cells (3,000 rad 137Cs) with 2 × 105 purified allogeneic T cells in the presence of various concentrations of S76E mutant protein or ICOSIg. Adherent cells were prepared by incubation of purified PBMCs of health donors for 2 h in plastic dishes. These cells were cocultured in 96-well round-bottom microtiter plates for 5 d. T cell proliferation was assessed after the addition of 1 μCi per well 3[H]-TdR for the final 9 h.
Results
Two- and Three-Dimensional Sequence Analysis of ICOS.
An alignment of extracellular Ig-domain sequences of the extended CD28 family is shown in Fig. 1 a. Human and mouse ICOS share a number of conserved residues with CD28 and/or CTLA-4. The region corresponding to the “MYPPPY” motif in CD28 and CTLA-4, which is critical for B7 binding and function of these proteins, is most conserved within this family. However, in mouse and human ICOS, only the PPP sequence is conserved, consistent with the finding that ICOS does not bind CD80 or CD86 (11, 29). However, the alignment identifies other regions of ICOS-specific residue conservation. FACS® analysis shows that both human and mouse ICOSIg bind B7-H2 at a high level, although mouse ICOSIg appears to bind less well than its human counterpart (data not shown). This suggests that the ligand binding sites in human and mouse ICOS are conserved and that similar residues contribute to the binding of B7-H2 ligand. To project specific residue conservation into three dimensions, we have mapped residues conserved in the CD28 family on the solution structure of human CTLA-4 (26) and molecular models of human CD28 (27) and human ICOS (28) that were built previously based on the CTLA-4 structure, as shown in Fig. 1 b. The analysis suggests that both CD28/CTLA-4–specific and ICOS-specific residue conservation is concentrated on the upper part of the A′GFCC′ face of the domain, proximal to the conserved MYPPPY or PPP motifs. Some residue conservation is also observed on the opposite (BED) face of the domain. However, this region is masked by N-linked glycosylation sites in ICOS and CD28 and should thus not be available for ligand binding. By contrast, no glycosylation sites are found within the conserved regions on the A′GFCC′ face of the domain. It was previously shown that the surface batch strictly conserved in CD28 and CTLA-4 is critical for CD80 and CD86 binding (26). However, the region of ICOS-specific residue conservation only partly overlaps with the ligand binding site in CD28 and CTLA-4. In the ICOS model, the spatially continuous region of conserved residues is essentially formed by four discontinuous sequence segments, 49–52, 64–68, 75–78, and 114–119 (Fig. 1 a). Thus, on the basis of residue mapping, these regions in ICOS were considered likely to play an important role in ligand binding, and a mutagenesis analysis was performed to assess this possibility.
Figure 1.

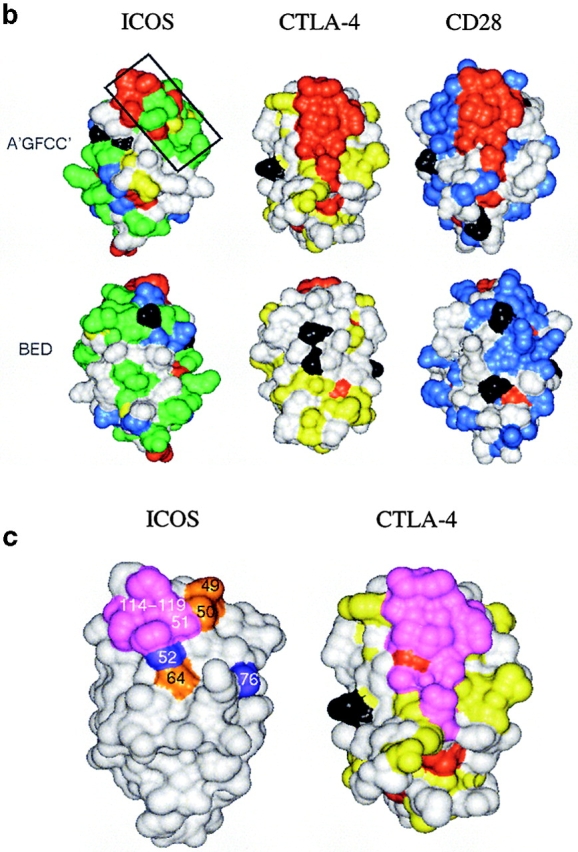
Analysis of residue conservation in the extended CD28 family and mapping of mutagenesis sites. (a) Structure-oriented sequence alignment. Sequences are shown of the extracellular Ig-domains of CD28, CTLA-4, and ICOS from different species. b, bovine; h, human; m, mouse; r, rat. β–strands observed in the solution structure of human CTLA-4 are labeled. Assignments of residues to the A- and C′′-strands are tentative. Residue numbers are given for human ICOS. Ig V-set consensus residues and other hydrophobic core residues are shown in lower case. These residues are important for maintaining structural integrity but are not available for ligand binding. Other residues conserved in CD28, CTLA-4, and ICOS are shown in red. Residues conserved only in CD28 or CD28 and ICOS are shown in blue, residues conserved only CTLA-4 or CTLA-4 and ICOS are shown in yellow, and residues conserved only in ICOS are shown in green. The most conservative residue replacements (e.g., Y/F, R/K, E/Q) are taken into account. Residues conserved in CD28 and CTLA-4 and critical for CD80/CD86 binding are labeled with asterisks. Potential N-linked glycosylation sites are boxed. The positions of ICOS residues subjected to site-specific mutagenesis are labeled with exclamation marks. (b) Three-dimensional analysis of residue conservation. A molecular model of the extracellular domain of human ICOS, the structure of human CTLA-4, and a model of human CD28 are shown in equivalent orientation and with solvent-accessible surface representation. Residues conserved are mapped and color-coded according to a. N-linked glycosylation sites are also mapped and colored black. Two views are shown that focus on the opposite β–sheet surfaces of the Ig domains (top: A′GFCC′ face; bottom: BED face; related by ∼180 degree rotation around the vertical axis). In this orientation, the “PPP” (ICOS) and “MYPPPY” (CD28/CTLA-4) motifs are at the top of the domains and the COOH termini at the bottom. The boxed region in ICOS (top left image) was selected as the likely B7-H2 binding site on the basis of residue conservation. Some ICOS-specific residue conservation is also observed on the BED face of the domain (bottom left). However, this region is masked by a glycosylation site (black), similar to the corresponding regions in CTLA-4 and CD28. (c) Residues important for ligand binding. ICOS and CTLA-4 are shown in the same orientation as in b, and residues are highlighted according to their importance for ligand binding. As identified in previous mutagenesis studies (see reference 26 and references therein), residues on the A′GFCC′ face in CTLA-4 critical for CD80/CD86 binding are colored magenta. All of these residues are strictly conserved in CD28. The positions of ICOS mutants that caused either abolished, reduced, or improved ligand binding are colored in magenta, gold, and blue, respectively. Mutated ICOS residues are numbered.
Mutagenesis Analysis of the Interaction between ICOS and B7-H2.
As a first step, the discontinuous sequence segments contributing to the formation of the conserved surface patch discussed above were replaced in human ICOS with corresponding regions of human CD28 according to Fig. 1 a. Regions 64–68 and 75–78 encompassing the C′-C′′ region were combined so that a total of three segment swap mutants were generated (RP49–52, RP64–78, and RP114–119). These homologue replacement constructs were expressed as Ig fusion proteins. The concentrations of mutant Ig fusion proteins in serum-free COS cell culture media were determined by sandwich ELISA using anti–human Ig mAbs. All three hybrid mutant proteins were expressed and bound by antiserum for ICOS at levels comparable to wild-type ICOS. The ability of each ICOSIg mutant to bind to B7-H2 was determined by sandwich ELISA using B7-H2mIg and anti–human Ig mAb (Fig. 2 a) and by FACS® analysis of the ICOSIg binding to CHO cells transfected to express B7-H2 (data not shown). Similar results were obtained by both methods. As summarized in Table I, mutant RP49–52 bound to B7-H2 but considerably less than wild-type ICOSIg, while the mutants RP64–78 and RP114–119 showed a complete loss of binding. Since all three mutants displayed either significantly reduced (RP49–52) or abolished binding capacity to B7-H2 (RP64–78 and RP114–119), the results provided a first indication that residues in these regions were either directly (i.e., as contact residues) or indirectly (i.e., maintaining local structural integrity) important for binding.
Figure 2.
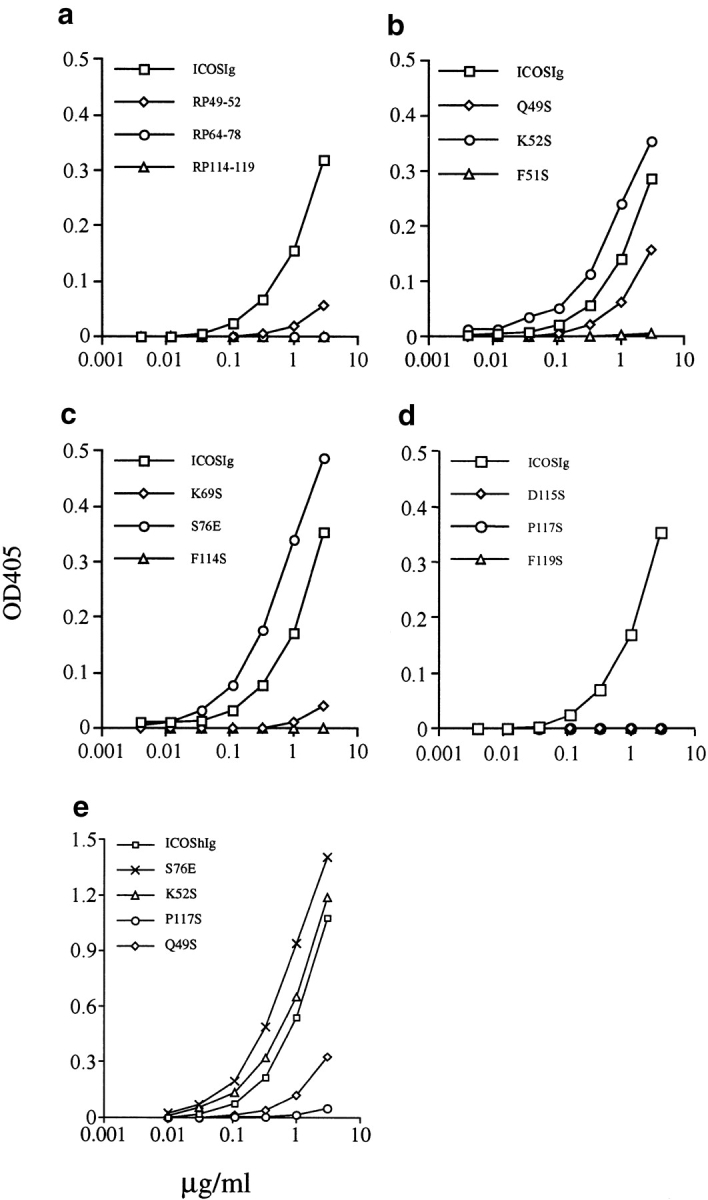
Analysis of the binding ability of mutant ICOS to B7-H2. Binding of wild-type and mutant ICOSIg to plate-bound B7-H2Ig (mIg) fusion protein was measured by ELISA. The concentrated supernatants (a–d) or purified proteins (e) from the cultures of transfected COS cells by indicated ICOS mutants were used. All determinations represent the mean of triplicate samples and differed from the mean by <10%. Shown is a representative of three independent experiments.
In the next step, we attempted to determine whether individual residues in these segments are important for B7-H2 binding. Therefore, four site-specific mutants were generated in each of the three targeted regions. These experiments are summarized in Table I. Conserved residues were selected on the basis of model analysis. Each selected residue mapped to a position on the surface of our molecular model (and was thus predicted to be available for ligand binding). The majority of selected residues were mutated to serine, which is more polar than alanine and therefore more compatible with solvent-exposed positions. The PCR-generated mutant constructs were verified by DNA sequencing, transfected into COS cells, and tested for binding by B7-H2. In the capture ELISA, mutation of Q49S reduced the binding to B7-H2 only slightly. However, mutants Q50S, D64S, and K69S displayed almost complete loss of binding. The most drastic effects were observed with mutants F51S, K67S, F114S, D115S, P117S, and F119S, which completely abrogated B7-H2 binding capacity. By contrast, two other mutants in this region, K52S and S76E, bound B7-H2 two- or threefold better than wild-type ICOSIg in both ELISA and FACS® analysis (Fig. 2 and Table I). Therefore, our results suggest critical roles of several residues conserved in human and mouse ICOS for ligand binding and, in addition, provide clues for further genetic engineering of this receptor to improve its biological activity.
Binding Analysis by BIAcore.
The binding of wild-type and representative mutants of ICOSIg to B7-H2 was analyzed by surface plasmon resonance using a BIAcore 3000 instrument. For comparison, B7-H2Ig protein was immobilized on a sensor chip and equal concentration of ICOSIg and mutant proteins in fluid phase was passed over the chip. The binding response of each analyte (measured in arbitrary RU) was calculated by subtracting the response seen in the control surface from the response seen in the B7-H2Ig–coated surface. Equilibrium binding curves showed results equivalent to the ELISA. In the applied concentration, S76E and wild-type ICOSIg have a significant difference in the association phase while another mutant, P117S, which did not measurably bind by ELISA showed minimal binding to the chips. Binding of S76E reached equilibrium faster than wild-type ICOS while their dissociation rates were similar (Fig. 3 and data not shown). In addition, as shown in Fig. 3, the saturation binding level was higher for the mutant than wild-type, then confirming its higher avidity.
Figure 3.

The surface plasmon resonance analysis of binding of mutant ICOS to B7-H2. A sensorgram overlay for binding of ICOSIg and representative mutants to immobilized B7-H2Ig is shown. Wild-type and mutants of ICOSIg (500 nM) were injected separately for 2.66 min at 30 μl per min through a sensor chip with or without B7-H2Ig immobilized. RU were calculated by subtracting the response seen in the control from the response seen in the B7-H2Ig-coated surface. Shown is a representative of three independent experiments.
Increased Blockade of T Cell Responses by ICOS Mutant Protein S76E.
We also tested ICOSIg mutant S76E to determine whether its improved avidity correlates with an increased ability to block the function of B7-H2. For these experiments, mutant S76E was selected because it had higher apparent avidity than K52S. We showed previously that immobilized B7-H2Ig costimulates T cell proliferation and cytokine production in the presence of suboptimal doses of anti-CD3 mAb (mimicking the TCR signal) (9) and we now have tested the effect of S76E in this system. As shown in Fig. 4 a, wild-type ICOSIg at 1–10 μg/ml significantly blocked the costimulatory effect of immobilized B7-H2Ig on purified human T cells. Inclusion of S76E protein in the culture more strongly inhibited the costimulatory effect of B7-H2Ig, and blocking by S76E was ∼10-fold more effective than by wild-type ICOSIg. Interestingly, the effect of the S76E protein was observed even at 0.1 μg/ml while wild-type ICOSIg at the same concentration did not inhibit T cell proliferation. On the basis of these findings, mutant S76E displays clearly improved B7-H2 blocking ability.
Figure 4.
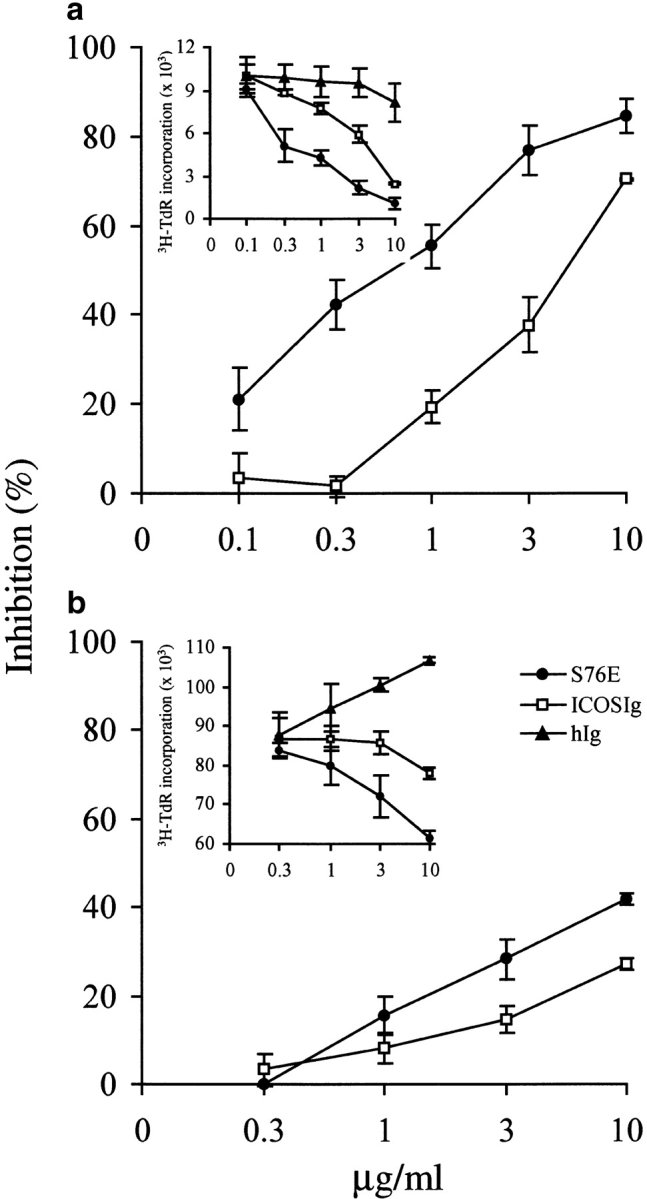
Increased blockage of T cell responses by ICOS S76E mutant protein. (a) S76E has improved blocking effects on T cell costimulatory functions of B7-H2. Human T cells were cultured for 3 d with immobilized anti-CD3 mAb and B7-H2Ig fusion protein in the presence of a soluble form of S76E mutant protein and ICOSIg at the indicated concentrations for 3 d. 3[H]-TdR was added during the last 9 h of culture. (b) S76E more strongly inhibits T cell responses to allogeneic antigens. Human T cells purified by nylon wool were cocultured with irradiated allogeneic adherent cells in the presence of indicated concentrations of S76E mutant protein and ICOSIg for 5 d. 3[H]-TdR was added during the last 9 h of culture. The results were also expressed as percentage of inhibition. Percentage of inhibition (%) = 100 × (average cpm of control Ig-treated group − cpm of sample group)/(average cpm of control Ig-treated group). Shown are the representatives of five independent experiments.
To further explore the blocking effects of S76E mutant protein in more physiological conditions, we also tested the S76E mutant protein in one-way MLR to allogeneic antigens. Inclusion of wild-type ICOSIg inhibited T cell proliferation in MLR by up to 25% at 10 μg/ml compared with the control Ig. However, S76E blocked T cell responses up to 43% (P < 0.01) at the same concentration (Fig. 4 b). Our results thus demonstrate that mutant S76E is superior to wild-type ICOSIg in the inhibition of T cell responses to allogeneic antigens.
Discussion
We have applied a three-dimensional model of the extracellular domain of ICOS to map conserved regions and thus provide a rational basis for the selection of residues for mutagenesis. Our subsequent mutagenesis analysis has identified several residues conserved in mouse and human ICOS, but not CD28 or CTLA-4, that are critical for B7-H2 binding. On the basis of these studies, we have generated a preliminary outline of the ligand-binding site by mapping mutated ICOS residues according to the binding characteristics of their mutants (Fig. 1 c). The underlying rationale has been that mutations of selected residues either affect binding directly or indirectly (i.e., by inducing local structural perturbations). On the basis of the expression and antiserum binding profiles of our mutants, the presence of gross structural perturbations or misfolding as a consequence of specific mutations was highly unlikely (consistent with the well-known stability and sequence tolerance of the Ig-fold). In fact, the finding that mutation of selected residues led to differentially reduced or improved ligand binding suggests that regions conserved in mouse and human ICOS, and targeted in this study, are directly involved in B7-H2 binding. Although the MYPPPY motif is not conserved in ICOS, residues in this region (114–119) are also a major determinant of binding, similar to CD28 and CTLA-4. However, different from CD28 and CTLA-4, the predicted binding site in ICOS extends more in the direction of the C′-C′′ region (Fig. 1 c). Thus, we conclude that the ligand-binding site in ICOS is, in terms of its location and residue composition, overlapping yet distinct from the one in CD28 and CTLA-4. This view is consistent with the arrangement of glycosylation sites in the CD28 family and rationalizes why CD28/CTLA-4 and ICOS bind distinct ligands. Interactions between members of the CD28 and B7 families involve conserved structural motifs, Ig variable-type folds, that have in part evolved to mediate different binding specificities, which is well illustrated by binding characteristics of ICOS. The evolutionary relationship between members of the CD28 family is clearly manifested by conservation of their molecular topology, the finding that corresponding regions of the Ig-fold are employed for ligand binding, and the presence of some residual residue conservation outside protein core positions. These include, for example, the PPP motif in ICOS that corresponds to the MYPPPY motif in CD28 and CTLA-4, and that is also important for binding and function. However, although structurally similar, ICOS has departed from CD28 and CTLA-4 by using in part different and not conserved sets of protein surface residues for ligand binding, thus providing a molecular rationale for the modulation of specificity within this receptor family. It is anticipated that similar mechanisms will determine the specificity of ligands belonging to the expanding B7 family.
Our experiments have identified two ICOS mutant proteins with improved binding to B7-H2 and this has enabled us to compare functional characteristics of such mutant forms with wild-type ICOS. We have found that mutant ICOSIg with improved binding ability also more effectively inhibits T cell costimulatory functions of B7-H2 in vitro, thus providing an example for the potential of engineering specific cell surface receptor–ligand interactions to modulate and improve immunoregulatory functions. We also generated an additional double mutant combining the two single-site mutants with increased avidity, K52S and S76E. It was anticipated that this protein would have further increased avidity. However, although this ICOSIg double mutant was readily expressed and we verified that no other spurious mutations were present in the construct, the protein did, surprisingly, not measurably bind to B7-H2 (data not shown). Since the single-site mutations alone did not compromise structural integrity and mapped to distant positions on the surface of the Ig-fold, the presence of global structural defects or direct interactions between the mutated residues is highly unlikely. Thus, the reason for the loss of binding remains unclear and we can only speculate that simultaneous introduction of both changes is not compatible with the formation of the receptor-ligand interface. However, it is clear from our study that S76E has improved blocking function compared with that of wild-type ICOSIg, representing an ∼10-fold increase of blocking capacity. Thus, in this case, increased avidity of an ICOS mutant protein correlates with improved biological effects. In allogeneic MLR, the effect of wild-type ICOSIg appears to be incomplete, a phenomenon that is consistent with the involvement of other costimulatory molecules. However, the ICOS mutant protein tested here increased blocking to a level of >40%. Our results support the notion that the interaction between ICOS and its ligand represents a major costimulatory interaction during the induction of T cell responses to allogeneic antigens.
While our results suggest the possibility that ICOS mutant proteins with improved ligand binding may manipulate T cell responses more efficiently than wild-type in vivo, this remains to be confirmed in a clinical setting. An alternative approach would be to engineer the mouse homologue of human ICOS and test this possibility in mouse models. As described previously, mouse and human ICOS have conserved ligand binding characteristics, and this should make it possible to develop such alternative models and provide further clues for the generation of mutant forms for effective manipulation of T cell responses in vivo.
Acknowledgments
We thank Patricia L. Caffes in the Biomedical Mass Spectrometry and Functional Proteomics Core in Mayo Clinic for BIAcore analysis, Dallas Flies and Julie S. Lau for their expert technical assistance, and Kathy Jensen for editing the manuscript.
This work was supported by the Mayo Foundation, National Institutes of Health grants CA79915 and CA85721, and by Albany Molecular Research, Inc. (formerly New Chemical Entities, Inc.).
L. Chen and J. Bajorath contributed equally to this work.
Footnotes
Abbreviations used in this paper: CHO, Chinese hamster ovary; CTLA, cytotoxic T lymphocyte antigen; ICOS, inducible costimulator.
References
- 1.Lenschow, D.J., T.L. Walunas, and J.A. Bluestone. 1996. CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 14:233–258. [DOI] [PubMed] [Google Scholar]
- 2.Chambers, C.A., and J.P. Allison. 1997. Co-stimulation in T cell responses. Curr. Opin. Immunol. 9:396–404. [DOI] [PubMed] [Google Scholar]
- 3.Rathmell, J.C., and C.B. Thompson. 1999. The central effectors of cell death in the immune system. Annu. Rev. Immunol. 17:781–828. [DOI] [PubMed] [Google Scholar]
- 4.Krummel, M.F., and J.P. Allison. 1996. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 183:2533–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walunas, T.L., C.Y. Bakker, and J.A. Bluestone. 1996. CTLA-4 ligation blocks CD28-dependent T cell activation. J. Exp. Med. 183:2541–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hutloff, A., A.M. Dittrich, K.C. Beier, B. Eljaschewitsch, B. Kraft, I. Anagnostopoulos, and R.A. Kroczek. 1999. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature. 397:263–266. [DOI] [PubMed] [Google Scholar]
- 7.Buonfiglio, D., M. Bragardo, V. Redoglia, R. Vaschetto, F. Bottarel, S. Bonissoni, T. Bensi, C. Mezzatesta, C.A. Janeway, Jr., and U. Dianzani. 2000. The T cell activation molecule H4 and the CD28-like molecule ICOS are identical. Eur. J. Immunol. 30:3463–3467. [DOI] [PubMed] [Google Scholar]
- 8.Lucia, M.B., F. Bottarel, D. Buonfiglio, T. Bensi, S. Rutella, C. Rumi, L. Ortona, C.A. Janeway, Jr., R. Cauda, and U. Dianzani. 2000. Expression of the novel T cell activation molecule hpH4 in HIV-infected patients: correlation with the disease status. AIDS Res. Hum. Retroviruses. 6:549–557. [DOI] [PubMed] [Google Scholar]
- 9.Wang, S., G. Zhu, A.I. Chapoval, H. Dong, K. Tamada, J. Ni, and L. Chen. 2000. Costimulation of T cells by B7-H2, a B7-like molecule that binds ICOS. Blood. 96:2808–2813. [PubMed] [Google Scholar]
- 10.Swallow, M.M., J.J. Wallin, and W.C. Sha. 1999. B7h, a novel costimulatory homolog of B7.1 and B7.2, is induced by TNFα. Immunity. 11:423–432. [DOI] [PubMed] [Google Scholar]
- 11.Yoshinaga, S.K., J.S. Whoriskey, S.D. Khare, U. Sarmiento, J. Guo, T. Horan, G. Shih, M. Zhang, M.A. Coccia, T. Kohno, et al. 1999. T-cell co-stimulation through B7RP-1 and ICOS. Nature. 402:827–832. [DOI] [PubMed] [Google Scholar]
- 12.Ling, V., P.W. Wu, H.F. Finnerty, K.M. Ban, V. Spaulding, L.A. Fouser, J.P. Leonard, S.E. Hunter, R. Zollner, J.L. Thomas, et al. 2000. Identification of GL50, a novel B7-like protein that functionally binds to ICOS receptor. J. Immunol. 164:1653–1657. [DOI] [PubMed] [Google Scholar]
- 13.Brodie, D., A.V. Collins, A. Laboni, J.A. Fennelly, L.M. Sparks, X.-N. Xu, P.A. van der Merwe, and S.J. Davis. 2000. LICOS, a primordial costimulatory ligand? Curr. Biol. 10:333–336. [DOI] [PubMed] [Google Scholar]
- 14.Yoshinaga, S.K., M. Zhang, J. Pistillo, T. Horan, S.D. Khare, K. Miner, M. Sonnenberg, T. Boone, D. Brankow, T. Dai, et al. 2000. Characterization of a new human B7-related protein: B7RP-1 is the ligand to the co-stimulatory protein ICOS. Int. Immunol. 12:1439–1447. [DOI] [PubMed] [Google Scholar]
- 15.Mages, H.W., A. Hutloff, C. Heuck, K. Buchner, H. Himmelbauer, F. Oliveri, and R.A. Kroczek. 2000. Molecular cloning and characterization of murine ICOS and identification of B7h as ICOS ligand. Eur. J. Immunol. 30:1040–1047. [DOI] [PubMed] [Google Scholar]
- 16.McAdam, A.J., T.T. Chang, A.E. Lumelsky, E.A. Greenfield, V.A. Boussiotis, J.S. Duke-Cohan, T. Chernova, N. Malenkovich, C. Jabs, V.K. Kuchroo, et al. 2000. Mouse inducible costimulatory molecule (ICOS) expression is enhanced by CD28 costimulation and regulates differentiation of CD4+ T cells. J. Immunol. 165:5035–5040. [DOI] [PubMed] [Google Scholar]
- 17.Coyle, A.J., S. Lehar, C. Lloyd, J. Tian, T. Delaney, S. Manning, T. Nguyen, T. Burwell, H. Schneider, J.A. Gonzalo, M. Gosselin, L.R. Owen, C.E. Rudd, and J.C. Gutierrez-Ramos. 2000. The CD28-related molecule ICOS is required for effective T cell-dependent immune responses. Immunity. 13:95–105. [DOI] [PubMed] [Google Scholar]
- 18.Aicher, A., M. Hayden-Ledbetter, W.A. Brady, A. Pezzutto, G. Richter, D. Magaletti, S. Buckwalter, J.A. Ledbetter, and E.A. Clark. 2000. Characterization of human inducible costimulator ligand expression and function. J. Immunol. 164:4689–4696. [DOI] [PubMed] [Google Scholar]
- 19.Rottman, J., T. Smith, J.R. Ronra, K. Ganley, T. Bloom, R. Silva, B. Pierce, J.C. Gutierrez-Ramos, E. Ozkaynak, and A.J. Coyle. 2001. The costimulatory molecule ICOS plays an important role in the immunopathogenesis of EAE. Nat. Immunol. 2:605–611. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalo, J.A., J. Tian, T. Delaney, J. Corcoran, J.B. Rottman, J. Lora, A. Al-Garawi, R. Kroczek, J.C. Gutierrez-Ramos, and A.J. Coyle. 2001. ICOS is critical for T helper cell-mediated lung mucosal inflammatory responses. Nat. Immunol. 2:597–604. [DOI] [PubMed] [Google Scholar]
- 21.Ozkaynak, E., W. Gao, N. Shemmeri, C. Wang, J.C. Gutierrez-Ramos, J. Amaral, S. Qin, J.B. Rottman, A.J. Coyle, and W.W. Hancock. 2001. Importance of ICOS-B7RP-1 costimulation in acute and chronic allograft rejection. Nat. Immunol. 2:591–596. [DOI] [PubMed] [Google Scholar]
- 22.McAdam, A.J., R.J. Greenwald, M.A. Levin, T. Chernova, N. Malenkovich, V. Ling, G.J. Freeman, and A.H. Sharpe. 2001. ICOS is critical for CD40-mediated antibody class switching. Nature. 409:102–105. [DOI] [PubMed] [Google Scholar]
- 23.Dong, C., A.E. Juedes, U.A. Temann, S. Shresta, J.P. Allison, N.H. Ruddle, and R.A. Flavell. 2001. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature. 409:97–101. [DOI] [PubMed] [Google Scholar]
- 24.Tafurl, A., A. Shahinian, F. Bladt, S.K. Yoshinaga, M. Jordana, A. Wakeham, L.M. Boucher, D. Bouchard, V.S.F. Chan, et al. 2001. ICOS is essential for effective T-helper-cell responses. Nature. 409:105–109. [DOI] [PubMed] [Google Scholar]
- 25.Peach, R.J., J. Bajorath, W. Brady, G. Leytze, J. Greene, J. Naemura, and P.S. Linsley. 1994. Complementarity determining region1 (CDR1)- and CDR2-analogous regions in CTLA-4 and CD28 determine the binding to B7-1. J. Exp. Med. 180:2049–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Metzler, W.J., J. Bajorath, W. Fenderson, S.Y. Shaw, K.L. Constantine, J. Naemura, G. Leytze, R.J. Peach, T.B. Lavoie, L. Mueller, and P.S. Linsley. 1997. Solution structure of human CD152 and delineation of a CD80/CD86 binding site conserved in CD28. Nat. Struct. Biol. 4:527–531. [DOI] [PubMed] [Google Scholar]
- 27.Bajorath, J., W.J. Metzler, and P.S Linsley. 1997. Molecular modeling of CD28 and three-dimensional analysis of residue conservation in the CD28/CD152 family. J. Mol. Graph. Model. 15:135–139. [DOI] [PubMed] [Google Scholar]
- 28.Bajorath, J.A. 1999. Molecular model of inducible costimulator protein and three-dimensional analysis of its relation to the CD28 family of T cell-specific costimulatory receptors. J. Mol. Model. 5:169–176. [Google Scholar]
- 29.Beier, K.C., A. Hutloff, A.M. Dittrich, C. Heuck, A. Rauch, K. Büchner, B. Ludewig, H.D. Ochs, H.W. Mages, and R.A. Kroczek. 2000. Induction, binding specificity and function of human ICOS. Eur. J. Immunol. 30:3707–3717. [DOI] [PubMed] [Google Scholar]