

Abstract
A number of in vitro studies have suggested that costimulatory molecules B7-1 and B7-2 and their receptor CD28 can promote clonal deletion, and limited in vivo studies have indicated that CD28 is involved in the clonal deletion of some T cells. However, the significance of B7-mediated clonal deletion in preventing autoimmune diseases has not been studied systematically. Here we report that the perinatal blockade of B7-1 and B7-2 substantially inhibits the clonal deletion of T cells in the thymus and leads to an accumulation of T cells capable of inducing fatal multiorgan inflammation. These results reveal a critical role for costimulatory molecules B7-1 and B7-2 in deleting pathogenic autoreactive T cells in the thymus. The critical role of B7-1 and B7-2 in T cell clonal deletion may explain, at least in part, the paradoxical increase of autoimmune disease in mice deficient for this family of costimulatory molecules, such as cytotoxic T lymphocyte associated molecule 4, CD28, and B7-2. The strong pathogenicity of the self-reactive T cells supports a central hypothesis in immunology, which is that clonal deletion plays an important role in preventing autoimmune diseases.
Keywords: autoimmunity, clonal deletion, B7-1 and B7-2, CD28, CTLA-4
Introduction
Mice with a targeted mutation in one of the B7 receptors, CD28 or cytotoxic T lymphocyte–associated molecule 4 (CTLA-4)* (1, 2), have either a rapid onset of fatal autoimmune lymphoproliferative disease (3, 4), or accelerated autoimmune diabetes in a predisposed nonobese diabetic (NOD) background (5). More recently, autoimmune neuropathy was reported in NOD mice with a targeted mutation of B7-2 (6). The phenotypes of the CTLA-4–deficient mice have been attributed to CTLA-4's negative regulatory function by many (3, 4), although not by all investigators (7, 8). The enhanced autoimmunity in mice with mutations of CD28 or B7-2, the prototypic costimulatory molecules, suggests that the autoimmune phenotype caused by mutations in these genes cannot necessarily be attributed to functions of these molecules in T cell activation, as their blockade in adult mice clearly reduces the incidence and severity of diabetes (9). A largely unexplored issue is whether the blockade of costimulatory molecules during T cell development may increase the number and spectrum of potential autoreactive T cells, and if so, whether this effect contributes, at least in part, to the paradoxical autoimmune phenotypes of these mice.
Since the identification of B7-1 and B7-2 as the prototypic costimulatory molecules in T cell activation, there has been considerable interest in their potential contribution in T cell development. Several lines of evidence indicate that costimulatory molecules can contribute to the negative selection of T cells. First, B7-1 and B7-2 are expressed at significant levels in the thymic dendritic cells (10) and medulla epithelial cells (11, 12). Given the accepted roles for these cells in the clonal deletion of T cells (13, 14), it is reasonable to test whether B7-1 and B7-2 on them contributes to the efficacy of T cell clonal deletion. Second, agonistic anti-CD28 mAb enhanced the death of immature T cells induced by ligation of the TCR–CD3 complex (15). Third, the blockade of B7-1 and B7-2 in thymic organ culture reduced the efficacy of clonal deletion in thymic organ culture (16). Fourth, clonal deletion induced by low doses of antigenic peptide or bacteria superantigen was reduced in CD28−/− mice (17). Fifth, a recent report showed that clonal deletion induced by endogenous viral superantigen was significantly affected by the targeted mutation of CD28−/− (18). Collectively, these studies demonstrated that B7-1 and B7-2 can promote the clonal deletion of autoreactive T cells, although the biological significance of B7-mediated T cell clonal deletion has not been systematically analyzed.
Although T cell development proceeds throughout much of one's life span, the perinatal period is the most critical in establishing the T cell repertoire. Here we tested the effect of a transient blockade of both B7-1 and B7-2 during the perinatal period on the accumulation of pathogenic self-reactive T cells. Using transgenic mice that express both antigen and TCR, we report that perinatal treatment with anti–B7-1 and anti–B7-2 prevents T cell clonal deletion in vivo. In addition, in mice with polyclonal T cell repertoire, we show that clonal deletion by viral superantigen (VSAg) is substantially reduced by perinatal anti-B7 treatment. More importantly, the T cells, rescued by the anti-B7 mAbs, cause severe autoimmune destruction in syngeneic recipients upon adoptive transfer. These results reveal a critical role of B7-1 and B7-2 in tolerizing pathogenic autoreactive T cells and suggest that this function may contribute, at least in part, to the paradoxical increase in autoimmune disease in mice with targeted mutations of costimulatory molecules.
Materials and Methods
Experimental Animals.
Recombinase activating gene 1 (RAG-1)–deficient and wild-type C57BL/6j mice were purchased from The Jackson ImmunoResearch Laboratories and maintained in the University Laboratory Animal Research Facility at Ohio State University (Columbus, OH) under specific pathogen-free conditions. Transgenic mice expressing tumor antigen P1A/TCR specific for tumor antigen P1A have been described elsewhere (19). Mice at day 16 of pregnancy, and those 2-, 3-, or 6-wk-old, were used for the current study. Nu+/nu+ C57BL/6j littermates were purchased from Taconic and used as the recipients of the adoptive transfer at 14-d-old.
Perinatal Blockade of B7-1 and B7-2 Using Specific mAbs.
On days 16 and 19 of pregnancy, the female mice were injected intraperitoneally with 100 μg of mAb per mouse of anti–B7-1 (3A12, hamster IgG) and anti–B7-2 (GL-1, rat IgG2a) mAbs. The newborn mice were injected intraperitoneally with the same mAbs at a reduced dose (25 μl of mAb per mouse) on days 0, 3, 6, and 9. Control mice were injected with an equivalent amount of normal hamster IgG (Rockland Inc.) and rat IgG (Sigma-Aldrich). The spleen cells and thymocytes were either analyzed by flow cytometry or used for adoptive transfer.
Flow Cytometry.
Both cell surface markers and intracellular cytokine production were analyzed by flow cytometry. Cell surface markers CD25, CD44, CD69, and CD62L, and Vβ3, Vβ5, Vβ8, Vβ11, and Vβ12 were analyzed with conjugated mAbs purchased from BD PharMingen. For intracellular cytokine production, spleen cells were stimulated with 40 ng/ml of PMA and 10 μM of ionomycin (both from Sigma-Aldrich) for 6–9 h. 5 μM of monensin (Sigma-Aldrich) was added 1 h after culture. The intracellular IL-2, IL-4, IL-10, or IFN-γ were analyzed using a kit from BD PharMingen according to the manufacturer's instructions.
Adoptive Transfer.
The given numbers of thymocytes from mice that received either anti-B7s or control IgG during the perinatal period were incubated with anti-CD8 mAbs (clone 2.43, rat IgG2b, 5 μg/ml) at 4°C for 30 min. After the removal of unbound mAbs, the antibody-coated thymocytes were removed by anti-Ig–coated magnetic beads as instructed by the manufacturer (Biosource International). The CD4 T cell–enriched thymocytes were injected intraperitoneally into syngeneic RAG-1−/− mice. In other experiments, spleen T cells were purified by the depletion of MHC class II+ and FcR+ cells using mAbs to I-A (M5/114, rat IgG2b) and to Fcγ receptors (CD16/CD32; 2.4G2, rat IgG2b) and adoptively transferred into either RAG-1−/− or nu+/nu+ C57BL/6j mice.
Histological Analysis.
Mouse organs were fixed with 10% buffered formalin and were paraffin embedded. Tissue sections were stained with hematoxylin and eosin (H&E), and examined under a microscope. In some experiments, frozen sections were prepared and stained with 2 μg/ml of antibodies specific for different subsets of leukocytes, Mac-1 (M1/70, rat IgG2b), CD4 (GK1.5, rat IgG2b), CD8, or variable regions of the TCRβ chain (Vβ3 and Vβ11). Pathological score (0–4) is based on the average numbers/size of inflammatory lesions per organ section: 0, no infiltrates; 1, 1 to 2 lesions; 2, 2 to 3 lesions; 3, 4 to 5 lesions; and 4, >6 lesions or inflammation covering more than 50% of the organ section.
Acute Syngeneic GVHD.
Spleens or CD4 T cell–enriched thymocytes harvested from mice perinatally treated with anti-B7 were incubated with a mixture of FITC–anti-Vβ3, -Vβ5, -Vβ11, and -Vβ12 mAbs for 15 min at 4°C. After removing the unbound mAbs, anti-FITC beads were added. The cells bound to the MACS® beads were enriched using the MACS® system™ (Miltenyi Biotec), according to the manufacturer's instructions. After purification, analysis indicated that among enriched spleen cells, 80% of the CD4 T cells and 90% of the CD8 cells had expressed the Vβ of interest. The purified cells were injected intravenously with 106 spleen T cells per mouse into the lethally irradiated mice that had each received 107 bone marrow cells from syngeneic mice. Both Vβ3-, Vβ5-, Vβ11-, Vβ12-expressing (3 × 106 per mouse, enriched) and nonexpressing (3 × 107 per mouse, depleted) thymocytes were used for adoptive transfer. About 50% of the enriched thymocytes expressed the Vβ of interest, whereas the depleted population contained no detectable Vβ3-, Vβ5-, Vβ11-, and Vβ12-expressing cells. The thymocytes were injected intravenously in conjunction with 107 bone marrow cell. 3–4 wk after the adoptive transfer, the recipient mice were killed and analyzed for the TCR representation, pathology, and tissue infiltration by Vβ3- and Vβ11-expressing T cells.
Results
Perinatal Blockade of B7-1 and B7-2 Inhibited T Cell Clonal Deletion of Antigen-specific T Cells In Vivo.
We have recently produced a transgenic mouse line that overexpresses tumor antigen P1A in the thymus and in the spleen, and another transgenic line that expresses H-2Ld–restricted, P1A-specific TCR (P1CTL) (19). We reported that thymic expression of P1A led to the clonal deletion of P1A-specific T cells in the P1CTL+/P1A+ mice (19). To test the effect of B7 blockade in T cell development, we bred P1A transgenic mice with the TCR transgenic mice and injected anti-B7 or control antibodies from E16 to day 10 after birth. The effects of anti-B7 mAbs on the littermates that expressed either P1CTL TCR alone or both TCR and the P1A in the thymus were analyzed by flow cytometry on day 15 after birth. As shown in Fig. 1 a, perinatal treatment with anti-B7 mAbs substantially increased the number of T cells in the double transgenic mice. Within the control IgG-treated group, expression of P1A led to the clonal deletion of P1CTL, as indicated by a reduction in the total number of thymocytes and a specific reduction of the number of CD4+CD8+ thymocytes (Fig. 1 b). Both effects were reversed by perinatal treatment with anti-B7 mAbs. These results demonstrated that anti-B7 mAbs prevented the clonal deletion of the P1A-specific T cells.
Figure 1.

Anti-B7 blockade in the perinatal period (from E16 to day 10 after birth) significantly reduced the efficacy of T cell clonal deletion, analyzed on day 15 after birth. (a) Anti-B7 mAb increased the total number of thymocytes in transgenic mice expressing both the T cell receptor and its specific antigen. Data shown are summarized from three independent experiments. Each point represents one double transgenic mouse. (b) Thymocyte composition in TCR single transgenic (P1CTL+/P1A−) or TCR P1A double transgenic (P1ACTL+/P1A+) mice. Transgenic thymocytes were analyzed by three-color flow cytometry using anti-Vα8, anti-CD4, and anti-CD8 mAbs. The number of thymocytes is given at the bottom of each panel, and the percentage of the Vα8+ cells is provided in the parentheses. Data shown are profiles of gated Vα8+ T cells and are representative of three independent experiments. (c) The subset distribution of gated Vα8− T cells from either anti-B7 or control Ig-treated double transgenic mice. The percentage of the gated population is presented at the bottom of each panel.
Interestingly, the anti-B7 treatments also increased the number of T cells and altered the ratio of CD8+CD4−/CD4+CD8− thymocytes in the P1CTL+/P1A− mice (Fig. 1 b, left). Because P1A is an unmutated tumor antigen with limited expression in normal mice (19), including thymic medulla epithelial cells (20) that can mediate negative selection (11, 13), it is possible that anti-B7 treatment may decrease the clonal deletion induced by endogenous P1A antigen. A partial deletion of CD8+CD4− T cells can lead to a relative increase in the proportion of CD4+CD8− T cells in the thymus, as observed in the control group. It should also be noted that in this (Fig. 1 b, which depicts the TCR+ cells only) and other TCR-transgenic models (21, 22), clonal deletion caused a substantial increase in the percentage of CD4−CD8−TCR+ cells. The specific decrease in the proportion of the CD4−CD8− T cells (Fig. 1 b) by anti-B7 is consistent with the notion that anti-B7 blocked the clonal deletion of P1A-specific T cells. Thus, B7-1 and B7-2 are critical for the clonal deletion of P1A-specific autoreactive T cells in the thymus.
It has been demonstrated that strong peripheral immune response can lead to antigen-independent deletion of thymocytes, particularly the immature CD4+CD8+ thymocytes (23). An interesting issue is whether anti-B7 increases the number of thymocytes by preventing the antigen-dependent clonal deletion or by inhibiting the peripheral T cell activation. To address this issue, we took advantage of the fact that allelic exclusion at TCR Vα locus is incomplete, and we analyzed the composition of Vα8− thymocytes that utilize endogenous Vα genes and are therefore not specific for P1A. As shown in Fig. 1 c, the subset distribution of the Vα8− cells was normal in the double transgenic mice and was not affected by anti-B7 treatment. Thus, the effect of anti-B7 is primarily to inhibit the antigen-dependent T cell clonal deletion in the thymus.
To test whether the autoreactive T cells migrated to the periphery, we analyzed the number and the phenotypes of the transgenic T cells in the spleens on day 15 after birth. As shown in Fig. 2 , although anti-B7 treatment did not significantly alter the number of transgenic T cells in the P1A and P1CTL of double transgenic mice (P = 0.16), it substantially increased the percentage and numbers (Fig. 2) of CD8+Vα8+ T cells (P = 0.02). In P1CTL single transgenic mice, anti-B7 somewhat increased the number of transgenic T cells. The biological significance of this occurrence is not yet clear. Because the T cells constituted a small fraction of the spleen cells, the total number of spleen cells was not significantly affected by anti–B7-1 and anti–B7-2. As shown in Fig. 2, the P1A transgene caused a drastic reduction in the percentage of CD8+Vα8+ T cells in the spleen. This effect was substantially blocked by the anti-B7 treatment. In the mice that received anti-B7 treatment, the accumulated transgenic T cells exhibited phenotypes of activated T cells, as they were larger in size and higher in the level of CD44 (Fig. 2, bottom). These results demonstrated that the transgenic T cells in the spleen of anti–B7-treated mice were stimulated by antigen in vivo. Because this was not observed in anti–B7-treated mice that expressed transgenic TCR, but not the P1A antigen, the high number of CD44high-transgenic T cells was induced primarily by the transgenic P1A antigen. Thus, the T cells rescued by anti-B7 treatment may have responded to the P1A antigen. In contrast, the T cells in the spleens of control IgG-treated mice were smaller in size and were comprised of substantially less CD44high subset, which suggested that these T cells were hyporesponsive to the endogenous P1A antigen.
Figure 2.

Anti-B7 mAb treatment increases the amount of mature CD4−CD8+Vα8+T cells in the spleen. Top panels show the expression of the CD8 coreceptor, whereas the lower panels show the expression of CD44 activation markers. Among the P1A and P1CTL double transgenic mice, anti–B7- and the control Ig-treated groups have a similar number of Vα8+ transgenic T cells. However, the number of Vα8+CD8+ mature T cells is increased by 2.5-fold in the anti–B7-treated group. Data shown are the contour plots of gated Vα8+ T cells and are representative of three independent experiments.
Perinatal Treatment with Anti-B7 Prevents the Effective Clonal Deletion of T Cells Specific for the Viral Superantigens.
The viral superantigens (VSAg) are the most effective inducers of T cell clonal deletion. Although the deletion of T cells for conventional antigens is generally investigated in TCR-transgenic mice due to a low frequency of T cells specific for a given antigen in normal mice, VSAg-reactive T cells can be investigated with wild-type mice. Moreover, the clonal deletion takes place at a late stage in the medulla (24). Here we used wild-type BALB/c mice to investigate the deletion of T cells specific for endogenous viral superantigens. BALB/c mice have integrations of mouse mammary tumor provirus types 6, 8, and 9. As a result, the majority of T cells expressing Vβ3, Vβ5, Vβ11, and Vβ12 have been deleted (25). To test the role for B7-1 and B7-2 in the clonal deletion of T cells reactive to viral superantigens, we treated BALB/c mice during the perinatal period with either control IgG or anti-B7 mAbs. 4 wk after birth, the thymus, spleen, and lymph nodes were harvested, and the clonal deletion of T cells was analyzed using a cocktail of anti-CD4, anti-CD8, and anti-Vβ mAbs. Profiles of the gated single-positive thymocytes from a representative thymus from control IgG- and anti–B7-treated groups are presented in Fig. 3 a, whereas the summary data of mature CD4 or CD8 T cells in the thymus, spleen, and lymph nodes are presented in Fig. 3 b.
Figure 3.

Perinatal B7-blockade rescues VSAg-specific T cells in the thymus and in the periphery. BALB/c mice were treated with either anti-B7 or control IgG during the perinatal period. 4 wk after birth the thymi, spleens, and lymph nodes were harvested and stained with a cocktail of anti-CD4, anti-CD8, and anti-Vβ mAbs. (a) FACS® profiles depict the Vβ subset distribution of gated CD4+CD8− or CD4−CD8+ thymocytes. The numbers given in each panel are the percentage of cells expressing the given Vβs. Data from one representative thymus in each group is presented. (b) Percentages of Vβ expressing CD4+CD8−, CD4−CD8+ thymocytes, spleen cells, and lymph node cells. Data shown are ± SEM from an experiment involving 2 or 3 mice per group that was repeated three times.
Among the CD4+CD8− thymocytes, anti-B7 treatment increased Vβ5+, Vβ11+, and Vβ12+ T cells by three- to fourfold. An almost twofold increase of Vβ3+ CD4 T cells was also found in anti–B7-treated mice. Reflecting the increase in the VSAg-reactive CD4 T cells, a significant reduction in the VSAg-nonreactive Vβ8+CD4 T cells was observed. Among the CD4−CD8+ thymocytes, the deletion of Vβ3+ T cells was not affected by anti-B7 treatment, whereas the effect of anti-B7 on the deletion of Vβ5+ cells was insignificant because the deletion varied widely among individual untreated mice. In contrast, anti-B7 increased the proportion of Vβ11+ CD8 T cells by almost twofold, and that of Vβ12+ cells by threefold. These results clearly demonstrate that B7-1 and B7-2 played a major role in the clonal deletion of a large array of VSAg-specific T cells in the thymus.
The effects of anti-B7 treatment on the number of VSAg-specific T cells in the spleen and lymph node generally recapitulated those in the thymus (Fig. 3 b). Two points are worth noting. First, in contrast to the thymus, the effect of anti-B7 on the Vβ5+ CD8 T cells in the periphery was highly significant. Thus, anti-B7 significantly inhibits clonal deletion of all subsets of VSAg-reactive CD4 cells, and that of all but Vβ3+CD8 T cells. Second, regardless of anti-B7 treatment, the numbers of VSAg-reactive T cells were generally lower in the periphery than in the thymus, although the effect of anti-B7 was more pronounced among the peripheral T cells. Both observations can be due to the fact that the deletion of superantigen-specific T cells is a late event in thymic T cell development (24). Perhaps T cells migrate to the periphery only when the deletion is complete. Alternatively, one can speculate that the clonal deletion of these T cells continues in the periphery. Regardless of which interpretation is correct, our data demonstrated that the deletion of VSAg-specific T cells in the thymus can be prevented by anti-B7, and that the rescued cells can survive and migrate to the periphery.
Pathogenicity of VSAg-specific T Cells Rescued by Perinatal Anti-B7 Treatment.
To directly evaluate the pathogenicity of VSAg-reactive T cells rescued by anti-B7 mAbs, we purified Vβ3, Vβ5, Vβ11, or Vβ12+ T cells from either the spleens or thymi of mice that had received anti–B7-1 and anti–B7-2 mAbs during the perinatal period, and adoptively transferred them (106 spleen T cells per mouse, or 3 × 106 thymocytes enriched for CD4+ Vβ3-, Vβ5-, Vβ11-, or Vβ12-expressing cells) into lethally irradiated syngeneic mice (VSAg+) with syngeneic bone marrow cells (107 per mouse; group A, n = 7). The irradiated recipients were used to ensure the grafting of small numbers of T cells and the presence of B cells, the primary VSAg-expressing cells (26). After depleting the T cells expressing Vβ3, Vβ5, Vβ11, and Vβ12, the remaining Vβ3, Vβ5, Vβ11, Vβ12− thymocytes (3 × 107 per mouse) were also cotransferred (group B, n = 3) with bone marrow cells for control for flow cytometry. A third group of mice (group C, n = 5) that had received bone marrow cells, but not T cells, was also used as a control for pathogenicity of the adoptively transferred T cells. As shown in Fig. 4 a, the mice that received a mixture of Vβ3, Vβ5, Vβ11, or Vβ12+ T cells from anti–B7-treated thymi had a high proportion of T cells expressing these Vβ. The most remarkable subsets were the Vβ11+ and Vβ12+ T cells, which consisted of 38.5% and 12.1% of the CD4 T cells, respectively. These T cells were not newly produced from bone marrow, as the control group that received the Vβ3, Vβ5, Vβ11, Vβ12− T cells and bone marrow had few T cells expressing any of these Vβ chains (Fig. 4 a, left). Thus, the VSAg-specific T cells were not deleted in the mice that expressed the superantigen. Moreover, considering the fact that relatively few T cells (106 per mouse) were injected, and that most of the T cells injected tended to disappear upon adoptive transfer, these T cells must have expanded substantially. Consistent with this, an overwhelming majority of the VSAg-specific T cells exhibited a cell surface phenotype of CD44hi and CD62Llo, which revealed their antigenic experience. The substantial expansion of T cells in the recipient indicated that these cells had not been anergized in anti–B7-treated mice.
Figure 4.
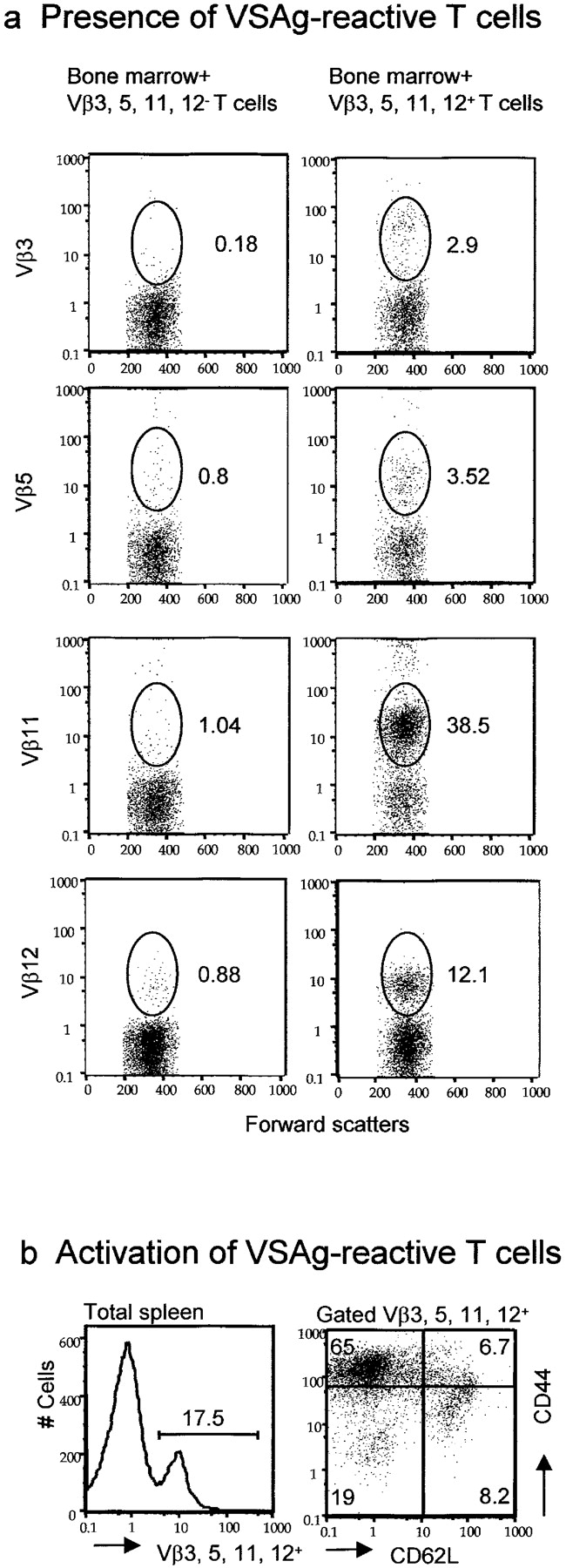
Presence and pathogenicity of VSAg-specific T cells rescued by perinatal treatment with anti-B7 mAbs. Lethally irradiated BALB/c mice (900 rad) were adoptively transferred with 107 per mouse of syngeneic bone marrow cells and 3 × 106 per mouse of thymocytes, enriched for their expressing either Vβ3, Vβ5, Vβ11, or Vβ12. As control, 3 × 107 thymocytes depleted of the Vβ3-, Vβ5-, Vβ11-, and Vβ12-expressing cells were also adoptively transferred together with syngeneic bone marrow cells. 3 wk after adoptive transfer, the recipient mice were killed and the spleen cells were analyzed. (a) Large numbers of VSAg-specific T cells were found in syngeneic VSAg-expressing mice that received bone marrow plus Vβ3, Vβ5, Vβ11, or Vβ12+ cells (right), but not those that received bone marrow plus Vβ3, Vβ5, Vβ11, and Vβ12− cells (left). Data shown were gated CD4+ cells from spleen cells and are representative of the CD4 T cells from all five mice analyzed. The numbers in the panels are the percentage of cells expressing given Vβ among CD4+ T cells. (b) The majority of the VSAg-reactive T cells expressed markers of antigen-experienced T cells. Spleen cells in the chimera mice were stained with FITC-conjugated mAbs specific against Vβ3, Vβ5, Vβ11, or Vβ12. Cychrome–anti-CD44 and PE–anti-CD62L mAbs were added. Data shown in the left panels are histograms for distribution of Vβ3-, Vβ5-, Vβ11-, or Vβ12-expressing cells. The data shown in the right are dot plots depicting the expression of CD44 and CD62L.
On days 7 and 8 of adoptive transfer of Vβ3, Vβ5, Vβ11, Vβ12+ T cells from anti–B7-treated mice, two of the seven recipients developed diarrhea with gastrointestinal hemorrhage. One died of diarrhea 2 d later, and the others survived. At 3–4 wk, the recipients were killed based on their health status and were analyzed for both the presence of the VSAg-specific T cells and for pathological signs of autoimmune destruction. Histological analysis revealed extensive lymphocyte infiltration in multiple organs (Fig. 5 a). All of the six mice that received Vβ3, Vβ5, Vβ11, Vβ12+ T cells and survived had severe inflammation in the lung and intestine. The majority of them (four out of six) also had lymphocyte inflammation in the liver. With the exception of one mouse that had mild inflammation in the lung, no histological lesion was observed in the control mice that had only received bone marrow. To confirm that the VSAg-specific T cells were involved in the inflammation, we also analyzed Vβ3+ and Vβ11+ T cells in the pathological lesions. As shown in Fig. 5 b, massive Vβ3+ and Vβ11+ T cells were found in the inflammatory sites in the intestines, although the number of Vβ3+ cells was less than that of Vβ11+ cells. Thus, the VSAg-specific T cells rescued by perinatal treatment with anti-B7 mAbs are pathogenic to syngeneic hosts. These data directly demonstrated the strong pathogenicity of VSAg-reactive T cells that escape central deletion as a result of B7 blockade, although it is possible that the VSAg are the target antigens recognized by the Vβ3, Vβ5, Vβ11, Vβ12+ T cells.
Figure 5.
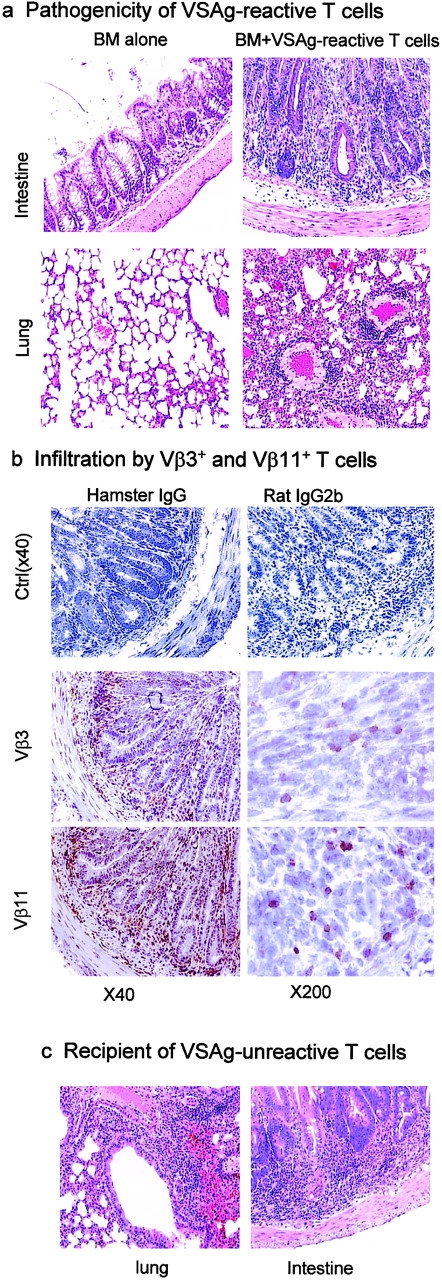
T cells from mice with perinatal B7 blockade caused acute GVHD in syngeneic recipients. Pathogenicity of VSAg-reactive (a and b) and -nonreactive (c) T cells. (a) H&E staining of intestines (top) and lungs (bottom) from mice 4 wk after adoptive transfer of either 107 per mouse of bone marrow cells alone (left) or bone marrow plus 3 × 106 per mouse of VSAg-reactive T cells isolated from the thymi of mice perinatally treated with anti–B7-1 and anti–B7-2 mAbs (right). (b) Infiltration of Vβ3+ and Vβ11+ T cells in the intestine. Intestine sections, of mice that had received both bone marrow cells and VSAg-reactive T cells as described in panel a were stained with anti–Vβ3 (hamster) or anti–Vβ11 (rat) mAbs. Two sections of 40× and 200× are shown for each staining. Isotype controls (40×) for the two mAbs are shown in the top panels. (c) Mice that received thymocytes depleted of Vβ3, Vβ5, Vβ11, and Vβ12+ T cells developed severe inflammation of the lung (left), intestines (right), and liver (unpublished data). The details are as described in panel a, except that after depleting the VSAg-reactive T cells, the remaining CD4-enriched thymocytes (3 × 107 per mouse) and 107 syngeneic bone marrow cells were transferred into lethally irradiated syngeneic VSAg+ recipients.
Interestingly, Vβ3, Vβ5, Vβ11, Vβ12− thymocytes were also pathogenic in the same setting. Two of the three recipients developed diarrhea on days 7 and 8, and one of the two also developed gastrointestinal hemorrhage and died on day 9. Two mice that survived were examined by necropsy. Both mice had severe inflammation in the lung, intestine (Fig. 5 c), and liver (unpublished data). These results suggest that the VSAg-specific T cells are only a subset of the autoreactive T cells rescued by perinatal anti-B7 treatment.
Perinatal Blockade of B7-1 and B7-2 Induces Autoreactive T Cells That Are Lethal in Young Immune-deficient Recipients.
To substantiate the role for B7 in promoting the clonal deletion of autopathogenic T cells, aside from those that are reactive to VSAg, we treated the VSAg-negative C57BL/6 mice from day 16 of embryonic development in the mother to day 10 after birth with either control normal (rat and hamster) IgG or a mixture of anti–B7-1 and anti–B7-2 mAbs. This treatment did not substantially alter the representation of the major subset of thymocytes (unpublished data). At 2–3-wk-old, the percentage of CD4+CD25+ T cells in the thymus of mice treated with anti-B7 mAbs was not significantly different from that of mice treated with control IgG (unpublished data). Although anti-B7 treatment increased the total number of the thymocytes by 30–100%, the number of dendritic cells in the spleen and thymus was not reduced by the anti-B7 treatment (unpublished data). When the mice reached 3-wk-old, and the circulating anti-B7 mAb remained high (unpublished data), male mouse thymocytes were enriched for CD4 T cells and injected intraperitoneally into RAG-1−/− mice that lacked T and B lymphocytes. As shown in Fig. 6 a, top, thymocytes from anti–B7-1– and anti–B7-2–treated mice rapidly killed the syngeneic recipients. The mice that had received a high dose of thymocytes from anti–B7-treated mice all died within 1 wk of adoptive transfer, whereas those that had received the same amount of thymocytes from the control IgG-treated group grew into adult age with no apparent growth retardation. A 10-fold reduction in the number of thymocytes transferred decreased mortality and delayed disease onset. Necropsies revealed that the recipients of thymocytes or T cells from anti–B7-1– and anti–B7-2–treated mice died of severe mononuclear cell inflammation in multiple organs, such as the lung, pancreas, liver, heart, and intestine (Fig. 6 b and unpublished data). For example, in the RAG-1−/− recipients of thymocytes from anti–B7-treated mice, infiltration of lymphocytes and macrophages, but not neutrophils, caused severe destruction of the majority of lung parenchyma. Multiple foci of mononuclear cell infiltration and interstitial edema in the myocardium of the heart were also observed (Fig. 6 b, upper left). In contrast, no infiltration of lymphocytes was observed in any of the organs of the recipients of thymocytes from the control IgG-treated mice, even when analyzed 2 mo after adoptive transfer (Fig. 6 b, bottom). These results demonstrate that the perinatal blockade of B7-1 and B7-2 leads to an accumulation of lethal autoreactive T cells in the thymus.
Figure 6.
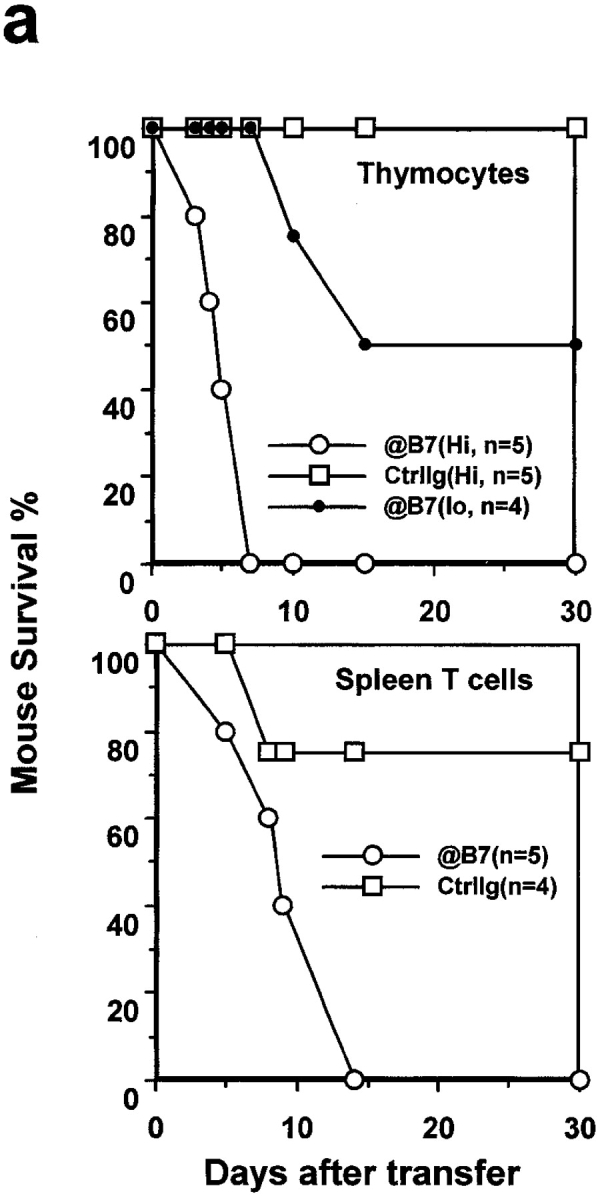
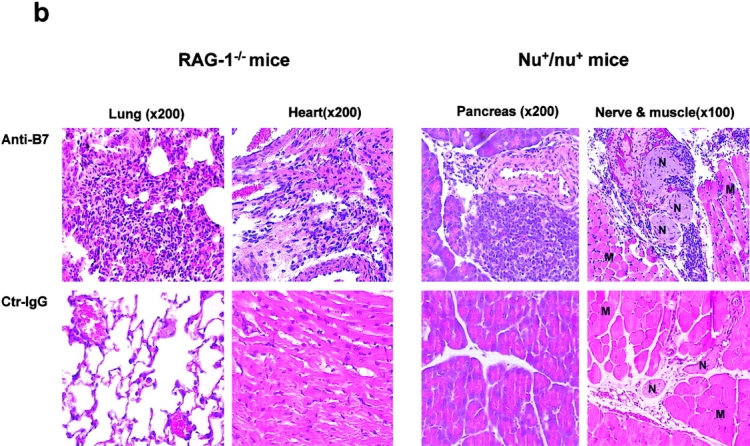
Lethal autoimmune T cells induced by a perinatal blockade of costimulatory molecules B7-1 and B7-2. C57BL/6j mice were treated with a mixture of anti–B7-1 and anti–B7-2, or rat and hamster IgG as control, from day 16 of pregnancy to day 10 after birth. 3 wk after birth, CD4-enriched thymocytes were prepared from anti–B7-treated and control IgG-treated male mice, respectively, and injected in high (hi, 2 × 108 per mouse) or low (lo, 2 × 107 per mouse) doses into 14-d-old syngeneic RAG-1−/− mice. Purified spleen T cells (2 × 107 per mouse) were isolated from both groups of mice and were adoptively transferred intraperitoneally into 2-wk-old, syngeneic nu+/nu+ mice. The recipients were monitored every day, and dead or moribund mice were subject to necropsies. (a) Rapid onset of fatal attack by either CD4-enriched thymocytes (top, n = 5) or spleen T cells (bottom, n = 5 for anti–B7-treated group; n = 4 for control IgG-treated group). (b) Histology of lung and heart sections from a RAG-1−/− recipient (upper left), and a pancreas, a muscle (M) and nerve (N) sections of a nu+/nu+ recipient (upper right). The organs from recipients of T cells from control Ig-treated mice were harvested 2 mo after adoptive transfer (bottom). The data shown are representative of two independent experiments.
To test if the autoreactive thymocytes can exit to the periphery, spleen T cells isolated from the same anti–B7-treated mice were adoptively transferred into syngeneic nu+/nu+ mice. The spleen T cells killed all recipient mice within 2 wk. Massive inflammation of the lung, liver, intestine, muscle, and pancreas was observed in necropsies (Fig. 6 b and unpublished data). For example, all of the mice examined had pancreatitis characterized by interstitial lymphoid infiltration, fibroblast proliferation, the formation of lymphoid aggregates, and the destruction of some pancreatic lobules. Some mice had mononuclear cell infiltration in the muscles and the nerves. In contrast, with the exception of one accidental death, the mice that had received spleen T cells from the control IgG-treated mice were healthy, and grew well into adulthood (Fig. 6 a, bottom). Mice that had received spleen T cells from control IgG-treated mice were histologically normal in all organs examined. Additional experiments indicated that a total of 100 × 106 thymocytes were as efficient as 20 × 106 spleen T cells in inducing lethal autoimmune disease in young nu+/nu+ mice (unpublished data).
Multiple Organ Inflammation and T Cell Activation in Adult Recipients of T Cells from Anti–B7-treated Mice.
When T cells from anti–B7-treated spleens were transferred into the adult RAG-1−/− mice, the inflammations were more chronic and the recipients lived for more than 3 mo after adoptive transfer. During this period, we observed significant growth retardation among the recipients of spleen T cells from anti–B7-treated mice (unpublished data). 3 mo after adoptive transfer, some recipients of the anti–B7-treated T cells became moribund, whereas those that received T cells from the control IgG-treated mice remained healthy. Histological analysis revealed that the recipients of spleen T cells from anti–B7-treated mice developed severe chronic inflammation in multiple organs, including the lung, pancreas, intestine (Fig. 7 a), and liver (unpublished data). Immunohistochemical analysis revealed that the lung infiltrates consisted mainly of CD4 T cells and macrophages, although significant numbers of CD8 T cells were observed (Fig. 7 b). In contrast, no significant inflammation was observed in the recipients of T cells from control IgG-treated mice. The recipients of anti–B7-treated T cells also had significantly higher numbers of T cells in the spleens, lymph nodes, and thymi, and most of these T cells exhibited markers of antigen-experienced cells (Fig. 7 c). Antigen-experienced T cells produce cytokine more rapidly than naive T cells when triggered by either antigen or agonists of the T cell receptors (27). To determine the extent of T cell activation, spleen T cells were analyzed for the production of a panel of cytokines such as IL-2, IL-4, IL-10, and IFN-γ after stimulation with PMA and ionomycin. As shown in Fig. 7 d, in the mice that had received anti–B7-treated T cells, the number of IFN-γ–producing cells was significantly elevated. This elevation was observed in both CD4 and CD8 compartments. Surprisingly, a large number of CD4−CD8− spleen cells expressed IFN-γ. The increase of this population corresponded to a significant increase of the TCRβ+CD4−CD8− T cells in the spleen. The accumulation of antigen-experienced CD4−CD8− T cells recapitulates an important feature of autoimmune mice (28). Interestingly, the number of cells expressing IL-2, IL-4, and IL-10 were low and comparable between the two groups of mice (unpublished data). A selective increase of IFN-γ–expressing cells indicated that anti–B7-treated spleen cells were polarized toward the Th1 subset in the RAG-1−/− recipients.
Figure 7.
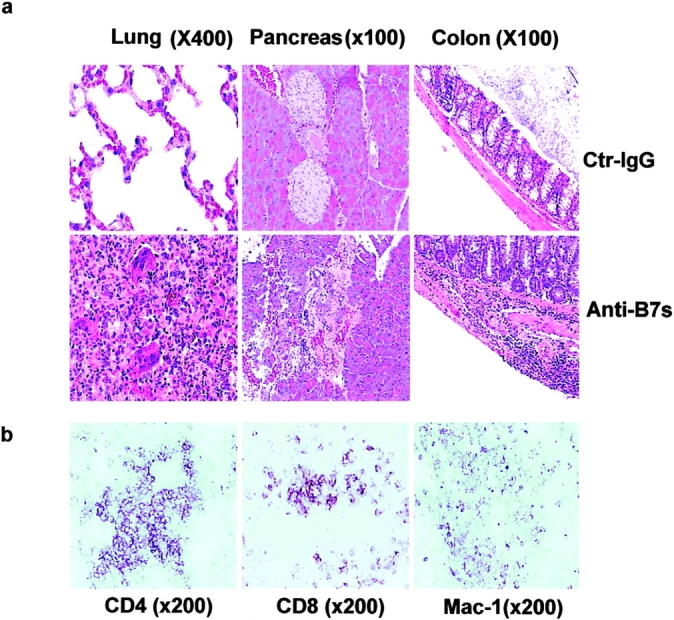


Perinatal blockades of B7-1– and B7-2–induced T cells that cause chronic autoimmune inflammation in adult RAG-1−/− recipients. Spleen T cells were isolated from 3-wk-old female C57BL/6j mice that had received either anti-B7 mAbs or control IgG and injected intraperitoneally (2 × 107 per mouse) into 6-wk-old, syngeneic female RAG-1−/− mice (n = 5 per group). The recipient mice were killed 3 mo after adoptive transfer. (a) H&E staining of lung, liver, and colon sections. In the anti–B7-treated group, multinucleated giant cell granulomas, characteristic of chronic inflammation, were present in the lung. Pancreatic acini were infiltrated by mononuclear cells, and neolymphoid follicles were observed in some mice. The villi of the small intestine were densely infiltrated by lymphocytes (unpublished data), and in some areas inflammatory cells were present in the submucosa and muscularis externa of the colon. The sections presented are from a representative mouse from each group. All mice that received T cells from anti–B7-treated mice developed severe inflammation in the lung, liver, pancreas, and intestine, whereas the recipients of control IgG-treated T cells were histologically normal, with occasional foci or inflammation in the lung. (b) Immunohistochemical analysis of lung sections using anti-CD4, anti-CD8 and anti–Mac-1 antibodies. The pictures were taken from the same anatomic location of three consecutive sections. (c) Numbers and phenotypes of T cells in the spleens and lymph nodes of RAG-1−/− mice that were adoptively transferred with splenic T cells from anti–B7-treated mice. Splenocytes and lymph node cells were isolated, counted, stained with FITC-conjugated anti-TCRαβ, PE-conjugated anti-CD4, and Cychrome–conjugated anti-CD8 (left and middle), or FITC-conjugated anti-TCRαβ, PE-conjugated anti-CD62L, and Cychrome–conjugated anti-CD8, and were analyzed with flow cytometry. The number of CD4−CD8− T cells were obtained by subtracting the CD4+CD8− and CD8+CD4− T cells from the TCR+ T cells. (d) Intracellular cytokine expression in the T cells. Data shown are expressions of IFN-γ from gated CD4, CD8, and CD4−CD8− lymphocytes. No alteration in the number of IL-2–, IL-4–, and IL-10–producing cells was observed (unpublished data). Data shown were representative of two to three independent experiments.
To test if the mice that received anti-B7 mAbs during the perinatal period developed autoimmune diseases, the mice were left untreated after the perinatal period and killed for histological examination 1, 2, and 5 mo after antibody treatment. In comparison to control IgG-treated mice, the anti–B7-treated mice had increases in both the number and severity of pathological lesions in multiple organs as early as 1 mo after the antibody treatments (unpublished data). 2 mo after treatment, the inflammation exacerbated substantially. The pathological and immunological effects of perinatal treatment with anti-B7 mAbs are presented in Fig. 8 . As shown in Fig. 8 a, both male and female mice that received anti-B7 during the perinatal period had severe mononuclear cell infiltration in the lung. The majority of anti–B7-treated female mice had inflammation in the kidney, whereas the control female mice had little kidney inflammation. No inflammation in the kidney was observed in male mice regardless of the treatment during the perinatal period. Corresponding to increased pathological lesions, lymphoproliferation was observed in anti–B7-treated mice. Thus, in comparison with control IgG-treated mice, the T cell numbers in the lymph nodes increased by three- to fivefold (unpublished data). The number of activated T cells, as indicated by CD69hi and CD62Llo phenotypes, also increased significantly (Fig. 8 b). The percentage of IFN-γ–producing cells increased by threefold in anti–B7-treated mice (Fig. 8 b). By month 5, although the numbers of IFN-γ–producing cells remained higher, the pathological lesions were largely resolved in anti–B7-treated mice (unpublished data), perhaps due to immune regulation by the endogenous T cells.
Figure 8.


Autoimmune inflammatory response in mice that received anti-B7 mAbs during the perinatal period. 2 mo after the antibody treatment, the mice were killed and the internal organs were fixed with 10% formalin and examined by H&E staining. (a) Scores of histological lesions in either control (○) or anti–B7-treated mice (•). The scores of lung and kidney lesions were presented. The data shown are summarized from three independent experiments. (b) Phenotypic and functional characterization of the spleen T cells. Spleen cells were analyzed for the cell surface expression of CD69 and CD62L and intracellular expression of IFN-γ. The subsets of T cells were marked by a combination of FITC-conjugated anti-CD4 and Cychrome–conjugated anti-CD8 mAbs. Data from gated CD4 T cells are presented, although similar results were obtained among the gated CD8 T cells. No alteration in the number of IL-2–, IL-4–, and IL-10–producing cells was observed. Data shown are representative of three independent experiments.
Discussion
The perinatal period is the most important for the formation of T cell repertoire. However, the contribution of interactions other than that of TCR–MHC antigen has not been systematically evaluated for this period. Here, we have addressed the biological significance of B7-1– and B7-2–mediated clonal deletion of autoreactive T cells during the perinatal period. Our results reveal that these costimulatory molecules make a substantial contribution for T cell clonal deletion and that the blockade of these interactions results in an accumulation of highly pathogenic autoreactive T cells.
Using a transgenic model, we have shown that perinatal treatment with anti-B7 mAbs prevents the clonal deletion of antigen-specific T cells. Moreover, even in conventional mice with a polyclonal TCR repertoire, anti-B7 prevented the deletion of T cells specific for VSAg. The results demonstrate that costimulatory molecules B7-1 and B7-2 play an important role in T cell clonal deletion. The effect of anti-B7 blockade, in terms of both the spectrums (all four Vβs among CD4 T cells, and three of the four Vβs among CD8 T cells) and the quantity (three- to sixfold increase of VSAg-reactive T cells in the periphery in most cases) of the effect, argues for the critical role of B7-mediated costimulation in the clonal deletion of a significant portion of, although certainly not all, self-reactive T cells, especially in cases in which the antigens are limiting or the affinity of antigen–TCR interaction is low. Our results significantly extend the previous studies on possible contribution of CD28 (15–18, 29–32) and CTLA4 (33) to T cell clonal deletion. As we have reported, the greater contribution of B7-1 and B7-2 than of CD28, as reported by Li and Page (18), raised the possibility that non-CD28 receptor for B7-1 and B7-2 may be involved in clonal deletion.
The contribution of B7 to the clonal deletion of VSAg-reactive T cells prompted us to test whether these T cells are pathogenic in a syngeneic host. We enriched the VSAg-reactive T cells rescued by perinatal treatment of anti–B7-1 and anti–B7-2 blockade and transferred them into irradiated hosts. Our analysis indicated that these VSAg-reactive T cells expanded substantially, and induced severe inflammation in multiple organs. These results directly demonstrate that the VSAg-reactive T cells that escape clonal deletion as a result of B7 blockade are highly pathogenic.
However, the autopathogenic T cells are not limited to VSAg-reactive cells, as anti-B7 also rescues autoreactive T cells in C57BL/6j mice that are pathogenic in the syngeneic VSAg− host (C57BL/6). In three distinct settings, we have shown that perinatal treatment with both anti–B7-1 and anti–B7-2 mAbs induces pathogenic self-reactive T cells in both the thymus and spleen. Upon adoptive transfer, both thymocytes and spleen T cells killed the syngeneic young recipients within 1 to 2 wk. They died of severe multiple organ inflammation. Although the adult recipients survived the acute autoimmune attack, severe chronic autoimmune diseases developed within 3 mo of adoptive transfer. Thus, the adult recipients were less susceptible to the autoreactive T cells. The extreme susceptibility of the young (2–3-wk-old) recipients may explain, at least in part, the lethal lymphoproliferative disease in young scurfy (34) and CTLA4−/− mice (3, 4). The relative contribution of the defective T cell development, as described here, and the potential effect of B7 blockade on the regulatory T cells, as others have postulated (5), to the exacerbated autoimmune disease in mice with targeted mutation of B7-2 (6), CD28 (5), and CTLA-4 (3–5), remains to be resolved. This study described several models that can be used to analyze this important question.
Perhaps the most direct setting in which to evaluate the pathogenicity is the autoimmune phenotype of the mice that received anti-B7 during the perinatal period. Although we observed severe and spontaneous inflammations in multiple organs of these mice, the autoimmune diseases in the adoptive transfer models were more severe than in mice that were directly treated with anti-B7 mAbs during the perinatal period. This difference can be explained, at least in part, by the role of B7 molecules in the activation (35), survival (36), and effector function (37, 38) of self-reactive T cells. In the adoptive transfer model, the injected anti-B7 mAbs were washed away before T cell transfer. However, in the mice that had received antibody treatment, the injected anti–B7-2 mAbs were present at high levels for at least 3 wk after the treatment, whereas anti–B7-1 mAbs were detected for at least 10 wk after the injections (unpublished data). The residual anti-B7 mAbs can inhibit the autoimmune response, as has been reported in many models (39). Moreover, autoreactive T cells may die as a result of TCR engagement in the absence of costimulation (36). Similarly, because the activation and/or effector function of self-reactive T cells requires B7-1 and B7-2 on the antigen-presenting cells in the periphery (40, 41), one would not necessarily expect gross autoimmune disease in mice with targeted mutations of both B7-1 and B7-2 (42, 43), although NOD mice with a mutation of B7-2 do develop spontaneous peripheral polyneuropathy (6). In addition, homeostatic proliferation in the lymphopenic mice may have exacerbated the autoimmune disease. However, because T cells from control IgG-treated mice were not pathogenic in the same model, the homeostatic proliferation was insufficient to cause the strong autoimmunity, as we have observed here.
Because autoreactive T cells accumulate in the thymus as a result of anti-B7 blockade, our results would suggest a B7-dependent central tolerance mechanism that is normally responsible for the elimination of lethal autoreactive T cells. A caveat of this interpretation is that the autoreactive T cells in the thymus of anti–B7-treated mice may have migrated from the periphery. To rule out this possibility, we tested if the autoreactive T cells in the spleen can migrate to the thymus. Our results demonstrated that when T cells from the spleens of anti–B7-treated mice were labeled with fluorescent dye and injected into the control Ig-treated mice, they readily homed to the spleens and lymph nodes, but not to the thymi (unpublished data). Therefore, it is very unlikely that a sufficient number of autoreactive spleen cells can migrate into the thymus at 3–4-wk-old. Given the high efficiency of the thymocytes in inducing inflammation in syngeneic mice, the most likely interpretation is that autoreactive T cells were rescued by anti-B7 in the thymus.
Taken together, our results demonstrate that costimulation by B7-1 and B7-2 plays a critical role in the intrathymic deletion of a wide spectrum of autoreactive T cells, and the cells rescued by B7 blockade are extremely responsive to antigen in vivo. More strikingly, the blockade of B7 leads to an accumulation of highly pathogenic autoreactive T cells. The strong effect of the B7 blockade on T cell clonal deletion suggests that the accumulation of pathologic autoreactive T cells results from a blockade of T cell clonal deletion. The fact that the VSAg-specific T cells rescued by anti-B7 are highly pathogenic in syngeneic mice provides strong support for this hypothesis. Although the frequency of autoreactive T cells that escape clonal deletion as a result of perinatal B7 blockade cannot be accurately determined at this stage, we believe it is likely to be high based on the percentage of VSAg-reactive cells, and on the strong pathogenicity of the thymocytes and spleen T cells from anti–B7-treated mice. The significant increase of pathogenic autoreactive T cells and the proposed role for costimulatory molecules in the activation of regulatory T cells (5) may both contribute to the paradoxical increase of autoimmunity in mice deficient for B7-2 (6), CD28 (5), or CTLA-4 (3, 4).
T cell self-tolerance is induced and maintained by both central and peripheral tolerances. Abundant evidence has established the importance of both processes in eliminating self-reactive T cells (21, 22, 44–46). Although genetic evidence has established the importance of peripheral tolerance in preventing autoimmune attack (47, 48), we are not aware of direct proof for the thesis that defective central tolerance can cause autoimmune disease. Recent studies have indicated that a population(s) of cells in the thymus, termed peripheral antigen-expressing cells, can express antigens previously assumed to be in specific peripheral tissues (20, 49). Expression of these antigens in the thymus correlates to the induction of immune tolerance (50, 51), and susceptibility to autoimmune diseases (50, 52). Our results demonstrate that the perinatal blockade of B7-1 and B7-2 reduced the efficacy of clonal deletion and led to an accumulation of lethal autoreactive T cells in the thymus and spleen. The severity and the high penetration of the disease argue for a strong selection pressure during evolution for an efficient mechanism for central T cell tolerance. It is likely that central tolerance removes the bulk of autoreactive T cells, in which B7 plays a critical role, whereas peripheral mechanisms act as a fail-safe to prevent chronic autoimmune disease, such as those observed in patients or animals with spontaneous mutations that inactivate peripheral tolerance (47, 48).
Acknowledgments
We thank Drs. K. Tung, R. Rudensky, and S. Rath for critical reading of the manuscript, Dr. P. Du for some animal typing, and J. Kiel for editorial assistance.
This work is supported by grants from the National Institutes of Health (AI32981, CA58033, CA69091, and CA82355).
Footnotes
Abbreviations used in this paper: CTLA-4, cytotoxic T lymphocyte–associated molecule 4; H&E, hematoxylin and eosin; NOD, nonobese diabetic; P1CTL, H-2Ld–restricted, P1A-specific TCR; RAG-1, recombinase activating gene 1; VSAg, viral superantigen.
References
- 1.Linsley, P.S., W. Brady, M. Urnes, L.S. Grosmaire, N.K. Damle, and J.A. Ledbetter. 1991. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 174:561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Linsley, P.S., E.A. Clark, and J.A. Ledbetter. 1990. T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB-1. Proc. Natl. Acad. Sci. USA. 87:5031–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tivol, E.A., F. Borriello, A.N. Schweitzer, W.P. Lynch, J.A. Bluestone, and A.H. Sharpe. 1995. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 3:541–547. [DOI] [PubMed] [Google Scholar]
- 4.Waterhouse, P., J.M. Penninger, E. Timms, A. Wakeham, A. Shahinian, K.P. Lee, C.B. Thompson, H. Griesser, and T.W. Mak. 1995. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 270:985–988. [DOI] [PubMed] [Google Scholar]
- 5.Salomon, B., D.J. Lenschow, L. Rhee, N. Ashourian, B. Singh, A. Sharpe, and J.A. Bluestone. 2000. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 12:431–440. [DOI] [PubMed] [Google Scholar]
- 6.Salomon, B., L. Rhee, H. Bour-Jordan, H. Hsin, A. Montag, B. Soliven, J. Arcella, A.M. Girvin, S.D. Miller, and J.A. Bluestone. 2001. Development of spontaneous autoimmune peripheral polyneuropathy in B7-2–deficient NOD mice. J. Exp. Med. 194:677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu, Y. 1997. Is CTLA-4 a negative regulator for T-cell activation? Immunol. Today. 18:569–572. [DOI] [PubMed] [Google Scholar]
- 8.Bachmann, M.F., A. Gallimore, E. Jones, B. Ecabert, H. Acha-Orbea, and M. Kopf. 2001. Normal pathogen-specific immune responses mounted by CTLA-4-deficient T cells: a paradigm reconsidered. Eur. J. Immunol. 31:450–458. [DOI] [PubMed] [Google Scholar]
- 9.Lenschow, D.J., S.C. Ho, H. Sattar, L. Rhee, G. Gray, N. Nabavi, K.C. Herold, and J.A. Bluestone. 1995. Differential effects of anti–B7-1 and anti–B7-2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J. Exp. Med. 181:1145–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sacedon, R., A. Vicente, A. Varas, E. Jimenez, J.J. Munoz, and A.G. Zapata. 1999. Glucocorticoid-mediated regulation of thymic dendritic cell function. Int. Immunol. 11:1217–1224. [DOI] [PubMed] [Google Scholar]
- 11.Degermann, S., C.D. Surh, L.H. Glimcher, J. Sprent, and D. Lo. 1994. B7 expression on thymic medullary epithelium correlates with epithelium-mediated deletion of V beta 5+ thymocytes. J. Immunol. 152:3254–3263. [PubMed] [Google Scholar]
- 12.Nelson, A.J., S. Hosier, W. Brady, P.S. Linsley, and A.G. Farr. 1993. Medullary thymic epithelium expresses a ligand for CTLA4 in situ and in vitro. J. Immunol. 151:2453–2461. [PubMed] [Google Scholar]
- 13.Webb, S.R., and J. Sprent. 1990. Tolerogenicity of thymic epithelium. Eur. J. Immunol. 20:2525–2528. [DOI] [PubMed] [Google Scholar]
- 14.Matzinger, P., and S. Guerder. 1989. Does T-cell tolerance require a dedicated antigen-presenting cell? Nature. 338:74–76. [DOI] [PubMed] [Google Scholar]
- 15.Punt, J.A., B.A. Osborne, Y. Takahama, S.O. Sharrow, and A. Singer. 1994. Negative selection of CD4+CD8+ thymocytes by T cell receptor-induced apoptosis requires a costimulatory signal that can be provided by CD28. J. Exp. Med. 179:709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Page, D.M. 1999. Cutting edge: thymic selection and autoreactivity are regulated by multiple coreceptors involved in T cell activation. J. Immunol. 163:3577–3581. [PubMed] [Google Scholar]
- 17.Kishimoto, H., Z. Cai, A. Brunmark, M.R. Jackson, P.A. Peterson, and J. Sprent. 1996. Differing roles for B7 and intercellular adhesion molecule-1 in negative selection of thymocytes. J. Exp. Med. 184:531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li, R., and D.M. Page. 2001. Requirement for a complex array of costimulators in the negative selection of autoreactive thymocytes in vivo. J. Immunol. 166:6050–6056. [DOI] [PubMed] [Google Scholar]
- 19.Sarma, S., Y. Guo, Y. Guilloux, C. Lee, X.-F. Bai, and Y. Liu. 1999. Cytotoxic T lymphocytes to an unmutated tumor antigen P1A: normal development but restrained effector function. J. Exp. Med. 189:811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Derbinski, J., A. Schulte, B. Kyewski, and L. Klein. 2001. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat. Immunol. 2:1032–1039. [DOI] [PubMed] [Google Scholar]
- 21.Sha, W.C., C.A. Nelson, R.D. Newberry, D.M. Kranz, J.H. Russell, and D.Y. Loh. 1988. Positive and negative selection of an antigen receptor on T cells in transgenic mice. Nature. 336:73–76. [DOI] [PubMed] [Google Scholar]
- 22.Kisielow, P., H. Bluthmann, U.D. Staerz, M. Steinmetz, and H. von Boehmer. 1988. Tolerance in T-cell-receptor transgenic mice involves deletion of nonmature CD4+8+ thymocytes. Nature. 333:742–746. [DOI] [PubMed] [Google Scholar]
- 23.Martin, S., and M.J. Bevan. 1997. Antigen-specific and nonspecific deletion of immature cortical thymocytes caused by antigen injection. Eur. J. Immunol. 27:2726–2736. [DOI] [PubMed] [Google Scholar]
- 24.Surh, C.D., and J. Sprent. 1994. T-cell apoptosis detected in situ during positive and negative selection in the thymus. Nature. 372:100–103. [DOI] [PubMed] [Google Scholar]
- 25.Abe, R., M. Foo-Phillips, and R.J. Hodes. 1991. Genetic analysis of the Mls system. Formal Mls typing of the commonly used inbred strains. Immunogenetics. 33:62–73. [DOI] [PubMed] [Google Scholar]
- 26.Jarvis, C.D., R.N. Germain, G.L. Hager, M. Damschroder, and L.A. Matis. 1994. Tissue-specific expression of messenger RNAs encoding endogenous viral superantigens. J. Immunol. 152:1032–1038. [PubMed] [Google Scholar]
- 27.Cho, B.K., C. Wang, S. Sugawa, H.N. Eisen, and J. Chen. 1999. Functional differences between memory and naive CD8 T cells. Proc. Natl. Acad. Sci. USA. 96:2976–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen, P.L., and R.A. Eisenberg. 1991. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu. Rev. Immunol. 9:243–269. [DOI] [PubMed] [Google Scholar]
- 29.Amsen, D., C. Revilla Calvo, B.A. Osborne, and A.M. Kruisbeek. 1999. Costimulatory signals are required for induction of transcription factor Nur77 during negative selection of CD4(+)CD8(+) thymocytes. Proc. Natl. Acad. Sci. USA. 96:622–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amsen, D., and A.M. Kruisbeek. 1996. CD28-B7 interactions function to co-stimulate clonal deletion of double-positive thymocytes. Int. Immunol. 8:1927–1936. [DOI] [PubMed] [Google Scholar]
- 31.Noel, P.J., M.L. Alegre, S.L. Reiner, and C.B. Thompson. 1998. Impaired negative selection in CD28-deficient mice. Cell. Immunol. 187:131–138. [DOI] [PubMed] [Google Scholar]
- 32.Kishimoto, H., and J. Sprent. 1999. Several different cell surface molecules control negative selection of medullary thymocytes. J. Exp. Med. 190:65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cilio, C.M., M.R. Daws, A. Malashicheva, C.L. Sentman, and D. Holmberg. 1998. Cytotoxic T lymphocyte antigen 4 is induced in the thymus upon in vivo activation and its blockade prevents anti–CD3-mediated depletion of thymocytes. J. Exp. Med. 188:1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Godfrey, V.L., J.E. Wilkinson, E.M. Rinchik, and L.B. Russell. 1991. Fatal lymphoreticular disease in the scurfy (sf) mouse requires T cells that mature in a sf thymic environment: potential model for thymic education. Proc. Natl. Acad. Sci. USA. 88:5528–5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oliveira-dos-Santos, A.J., A. Ho, Y. Tada, J.J. Lafaille, S. Tonegawa, T.W. Mak, and J.M. Penninger. 1999. CD28 costimulation is crucial for the development of spontaneous autoimmune encephalomyelitis. J. Immunol. 162:4490–4495. [PubMed] [Google Scholar]
- 36.Boise, L.H., A.J. Minn, P.J. Noel, C.H. June, M.A. Accavitti, T. Lindsten, and C.B. Thompson. 1995. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 3:87–98. [DOI] [PubMed] [Google Scholar]
- 37.Chang, T.T., C. Jabs, R.A. Sobel, V.K. Kuchroo, and A.H. Sharpe. 1999. Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. J. Exp. Med. 190:733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allison, J., L.A. Stephens, T.W. Kay, C. Kurts, W.R. Heath, J.F. Miller, and M.F. Krummel. 1998. The threshold for autoimmune T cell killing is influenced by B7-1. Eur. J. Immunol. 28:949–960. [DOI] [PubMed] [Google Scholar]
- 39.Aruffo, A., and D. Hollenbaugh. 2001. Therapeutic intervention with inhibitors of co-stimulatory pathways in autoimmune disease. Curr. Opin. Immunol. 13:683–686. [DOI] [PubMed] [Google Scholar]
- 40.Mandelbrot, D.A., A.J. McAdam, and A.H. Sharpe. 1999. B7-1 or B7-2 is required to produce the lymphoproliferative phenotype in mice lacking cytotoxic T lymphocyte–associated antigen 4 (CTLA-4). J. Exp. Med. 189:435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tivol, E.A., S.D. Boyd, S. McKeon, F. Borriello, P. Nickerson, T.B. Strom, and A.H. Sharpe. 1997. CTLA4Ig prevents lymphoproliferation and fatal multiorgan tissue destruction in CTLA-4-deficient mice. J. Immunol. 158:5091–5094. [PubMed] [Google Scholar]
- 42.Borriello, F., M.P. Sethna, S.D. Boyd, A.N. Schweitzer, E.A. Tivol, D. Jacoby, T.B. Strom, E.M. Simpson, G.J. Freeman, and A.H. Sharpe. 1997. B7-1 and B7-2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity. 6:303–313. [DOI] [PubMed] [Google Scholar]
- 43.Yu, X., S. Fournier, J.P. Allison, A.H. Sharpe, and R.J. Hodes. 2000. The role of B7 costimulation in CD4/CD8 T cell homeostasis. J. Immunol. 164:3543–3553. [DOI] [PubMed] [Google Scholar]
- 44.Kappler, J.W., N. Roehm, and P. Marrack. 1987. T cell tolerance by clonal elimination in the thymus. Cell. 49:273–280. [DOI] [PubMed] [Google Scholar]
- 45.Schwartz, R.H. 1989. Acquisition of immunologic self-tolerance. Cell. 57:1073–1081. [DOI] [PubMed] [Google Scholar]
- 46.Miller, J.F., and G. Morahan. 1992. Peripheral T cell tolerance. Annu. Rev. Immunol. 10:51–69. [DOI] [PubMed] [Google Scholar]
- 47.Watanabe-Fukunaga, R., C.I. Brannan, N.G. Copeland, N.A. Jenkins, and S. Nagata. 1992. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 356:314–317. [DOI] [PubMed] [Google Scholar]
- 48.Lenardo, M., K.M. Chan, F. Hornung, H. McFarland, R. Siegel, J. Wang, and L. Zheng. 1999. Mature T lymphocyte apoptosis--immune regulation in a dynamic and unpredictable antigenic environment. Annu. Rev. Immunol. 17:221–253. [DOI] [PubMed] [Google Scholar]
- 49.Hanahan, D. 1998. Peripheral-antigen-expressing cells in thymic medulla: factors in self-tolerance and autoimmunity. Curr. Opin. Immunol. 10:656–662. [DOI] [PubMed] [Google Scholar]
- 50.Smith, K.M., D.C. Olson, R. Hirose, and D. Hanahan. 1997. Pancreatic gene expression in rare cells of thymic medulla: evidence for functional contribution to T cell tolerance. Int. Immunol. 9:1355–1365. [DOI] [PubMed] [Google Scholar]
- 51.Klein, L., T. Klein, U. Ruther, and B. Kyewski. 1998. CD4 T cell tolerance to human C–reactive protein, an inducible serum protein, is mediated by medullary thymic epithelium. J. Exp. Med. 188:5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klein, L., M. Klugmann, K.A. Nave, V.K. Tuohy, and B. Kyewski. 2000. Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat. Med. 6:56–61. [DOI] [PubMed] [Google Scholar]