

Abstract
Salmonella typhimurium causes an invasive disease in mice that has similarities to human typhoid. A type III protein secretion system encoded by Salmonella pathogenicity island 2 (SPI2) is essential for virulence in mice, as well as survival and multiplication within macrophages. Reactive nitrogen intermediates (RNI) synthesized by inducible nitric oxide synthase (iNOS) are involved in the control of intracellular pathogens, including S. typhimurium. We studied the effect of Salmonella infection on iNOS activity in macrophages. Immunofluorescence microscopy demonstrated efficient colocalization of iNOS with bacteria deficient in SPI2 but not wild-type Salmonella, and suggests that the SPI2 system interferes with the localization of iNOS and Salmonella. Furthermore, localization of nitrotyrosine residues in the proximity was observed for SPI2 mutant strains but not wild-type Salmonella, indicating that peroxynitrite, a potent antimicrobial compound, is excluded from Salmonella-containing vacuoles by action of SPI2. Altered colocalization of iNOS with intracellular Salmonella required the function of the SPI2-encoded type III secretion system, but not of an individual “Salmonella translocated effector.” Inhibition of iNOS increased intracellular proliferation of SPI2 mutant bacteria and, to a lesser extent, of wild-type Salmonella. The defect in systemic infection of a SPI2 mutant strain was partially restored in iNOS−/− mice. In addition to various strategies to detoxify RNI or repair damage due to RNI, avoidance of colocalization with RNI is important in adaptation of a pathogen to an intracellular life style.
Keywords: Salmonella typhimurium, SPI2, iNOS, peroxynitrite, innate immunity
Introduction
Phagocytes employ an array of enzymatic activities to inactivate and degrade phagocytosed organisms. Among other antimicrobial effector mechanisms, phagocytes possess enzyme systems that are involved in the production of reactive oxygen intermediates (ROI)* and reactive nitrogen intermediates (RNI) with activities against a wide array of pathogens. Infection experiments with knockout mice defective in gp91phox (ROI production), inducible nitric oxide synthase (iNOS; or NOS2, RNI production), and both enzymes indicated that these enzymes contribute to the control of bacterial infections (1). The combined activity of gp91phox and iNOS is required for the generation of peroxynitrite, one of the most potent antimicrobial effectors of macrophages (2). Although RNI- and ROI-generating pathways are usually highly efficient and result in the killing of phagocytosed bacteria, several intracellular pathogens, such as Salmonella enterica, have evolved strategies to circumvent the action of these bactericidal activities of their host cells.
Infections with S. enterica constitute a significant public health problem in most countries. S. enterica causes self-limited, acute intestinal inflammations, manifesting clinically with diarrhea and vomiting. However, infections with certain serovars of S. enterica can lead to systemic diseases with significant lethality such as typhoid fever. Two hallmarks of pathogenesis of Salmonella infections are the invasion of nonphagocytic cells and the survival and replication within host cells, for example macrophages. Many events during pathogenesis of systemic Salmonella infections have been studied in detail in the model system of murine salmonellosis, a typhoid fever-like infection caused by S. enterica serotype Typhimurium (Salmonella typhimurium) in susceptible mice.
Type III secretion systems (TTSS) are a family of highly evolved protein secretion systems in Gram-negative pathogens. TTSS translocate, in a contact-dependent manner, effector proteins into the eukaryotic host cell (3). The function of a TTSS encoded by Salmonella pathogenicity island 1 (SPI1) is important for interactions of S. typhimurium with the epithelial mucosa, such as invasion of enterocytes (4) and the induction of diarrhea (5). During systemic infections macrophages represent an important site for replication of S. typhimurium (6, 7). Mutant strains of S. typhimurium unable to survive and replicate within macrophages are also severely attenuated in virulence in the animal model of infection (8). Survival and replication of S. typhimurium in macrophages requires the function of a large number of proteins. These functions range from biosynthesis of macromolecules that are limited in the phagosomal environment, enzyme systems involved in the repair of damages due to antimicrobial activities of the host cells, to virulence factors that actively modulate host cell functions (9, 10). Salmonella pathogenicity island 2 (SPI2) harbors genes encoding a second TTSS for virulence proteins in S. enterica (for a review, see reference 11). The function of SPI2 genes is absolutely required for the progression of systemic infections by S. typhimurium in the model of murine salmonellosis (12) and the proliferation within infected organs such as liver and spleen (13). Furthermore, the role of SPI2 in intracellular replication and survival has been demonstrated (14, 15, 16). On the cellular level, the function of SPI2 proteins affects vesicle trafficking in host cells (17). SPI2 is also required to prevent the localization of NADPH-oxidase to Salmonella-containing vesicles (SCVs) of the infected host cell, an activity leading to the evasion of intracellular Salmonella from oxidative damage (18, 19).
Infections with Salmonella induce iNOS expression (for a review, see reference 20) and recent studies demonstrated that the function of iNOS is required to control the proliferation of S. typhimurium in infected organs (21) and within infected macrophages (22). As the function of SPI2 is also required for both phenotypes, we asked if SPI2 is involved in the control of iNOS expression or activity, or may control the production of RNI and their effects on intracellular S. typhimurium. To investigate the cellular microbiology of this antimicrobial effector mechanism in more detail, we established an in vitro system to study the effects of S. typhimurium on expression of iNOS and production of RNI. In this study we report that intracellular S. typhimurium employ the TTSS of SPI2 to prevent the localization of iNOS with SCVs. To our knowledge, this is the first observation that a pathogen can alter the cellular localization of iNOS to protect itself from the damaging effects of RNI and the combined effects of RNI and ROI.
Materials and Methods
Bacterial Strains.
Bacterial strains used in this study are listed in Table I. Salmonella enterica serovar Typhimurium (S. typhimurium) NCTC 12023 was used as wild-type strain and all mutant strains were constructed in the S. typhimurium 12023 background. For colocalization experiments S. typhimurium strains were transformed with plasmid pFPV25.1 constitutively expressing green fluorescent protein (GFP; reference 23).
Table I.
Bacterial Strains Used in This Study
| Strain designation | Relevant characteristics |
Remarks/reference |
|---|---|---|
| S. typhimurium | Wild-type | Identical to ATCC 14028 |
| NCTC 12023 | ||
| P8G12 | ssrB::mTn5 | 13 |
| P2D6 | ssaV::mTn5 | 13 |
| P11D10 | sscA::mTn5 | 13 |
| P7G2 | ssaC::mTn5 | 13 |
| HH101 | sseA::aphT | 15 |
| HH102 | sseB::aphT | 15 |
| HH104 | sseC::aphT | 15 |
| P3H6 | sifA::mTn5 | 44 |
| P7B12 | sipC::mTn5 | 45 |
| MvP374 | ΔsifB::aph | This study |
| MvP375 | ΔsspH1::aph | This study |
| MvP376 | ΔsspH2::aph | This study |
| MvP377 | ΔsseJ::aph | This study |
| MvP378 | ΔsseI::aph | This study |
| MvP379 | ΔslrP::aph | This study |
Construction of Mutations in Salmonella-translocated Effector Genes.
The method of one-step inactivation of chromosomal genes (24) was applied. Briefly, primers with 56 to 65 bases length including 20 bases priming sequences for the template pKD4 and 36 to 45 bases homologous to the respective flanking regions of the gene of interest have been constructed. PCR reactions were performed with the Expand High Fidelity PCR system (Roche) using pKD4 as template. PCR products were purified, digested with DpnI, and gel purified. The PCR products were transformed into S. typhimurium cells harboring pKD46 and grown in the presence of arabinose to induce the Red recombinase. Recombinant clones were selected by plating on Luria-Bertani (LB)-agar containing 50 μg/ml kanamycin. Kanamycin-resistant colonies were further checked for chromosomal insertion of the antibiotic resistance cassette by PCR using primers k1 and k2 (24). The proper insertions of the antibiotic resistance cassettes within the various Salmonella-translocated effector (STE) genes were confirmed by DNA sequencing. Finally, the mutant alleles were moved into a fresh strain background by P22 transduction according to standard procedures (25).
Cell Culture.
The murine macrophage cell line RAW267.4 was obtained from American Type Culture Collection, and maintained in DMEM containing 10% heat-inactivated FCS (Invitrogen/Life Technologies) at 37°C under 5% CO2. The cells were washed gently with PBS and removed from the flasks by scraping. The cells were suspended in 24-well plates for experiments at the density of 5 × 104 cells per well. Peritoneal macrophages were harvested from thioglycollate-elicited peritoneal lavages from C57BL/6 mice and plated in DMEM supplemented with 10% FCS. After 2 h at 37°C, the cells were washed twice with PBS and the adherent cells were incubated overnight in DMEM plus 10% FCS. Cells were always cultured in antibiotic-free media.
Survival of S. typhimurium After In Vitro Exposure to Nitrosative Stress.
Overnight cultures of wild-type S. typhimurium and the ssaV mutant strain were diluted to OD600 of 0.2. S-nitrosoglutathione (GSNO; Calbiochem) and sodium nitrite (NaNO2; Sigma-Aldrich) were prepared freshly in distilled water, adjusted to pH 5.0, and added to 1 ml LB broth at various concentrations. SIN-1 (Calbiochem) was dissolved in distilled water and added to 1 ml LB at various concentrations. Subsequently, 100 μl of the diluted culture was added to each tube. The tubes were incubated at 37°C under shaking conditions. After 3 and 9 h of exposure serial dilutions were made and plated onto Mueller-Hinton agar for the enumeration of surviving bacteria.
Infection of Macrophages.
Bacteria were grown overnight in LB broth in glass test tubes with aeration in a “roller drum” (New Brunswick Scientific). The OD at 600 nm of the cultures was adjusted to 0.2 and 1 ml of the bacterial suspension was centrifuged, washed twice with PBS, and resuspended in DMEM. 10 μl of the bacterial suspension was further added to 1 ml of DMEM. 100 μl of the bacterial suspension was added to the wells at a multiplicity of infection (MOI) of 10 and the plates were centrifuged at 1,200 rpm for 5 min to synchronize the infection. The plates were incubated at 37°C, 5% CO2 for 25 min. Subsequently, the plates were washed three times with PBS and incubated with media containing 100 μg/ml gentamicin for 1 h, followed by media containing 10 μg/ml of gentamicin for the rest of the experiment. For infection of peritoneal macrophages, bacteria were opsonized for 30 min at 37°C with 10% normal mouse serum before infection. Heat-killed bacteria were obtained by incubation at 80°C for 10 min. This procedure did not affect the integrity of the LPS.
Assessment of Intracellular Growth of Bacteria.
Intracellular replication was assessed by means of the standard gentamicin assay (26). After completion of the infection period, the cells were incubated with 100 μg/ml gentamicin for 1 h to kill the extracellular bacteria and subsequently with 10 μg/ml gentamicin for the rest of the time period. 2 and 16 h after infection, the supernatant was removed and cells were washed three times with DMEM (without serum) and lysed with 0.1% Triton X-100. Serial dilutions of the lysates were plated onto LB agar to enumerate intracellular bacteria.
For investigation of intracellular replication by microscopy, the cells were grown on glass coverslips and infected with S. typhimurium strains constitutively expressing GFP. The gentamicin protection was performed as described above, and cells were fixed with 3% para-formaldehyde (PFA).
The inhibitors L-NMMA (l-monomethylarginine; Sigma-Aldrich) or DPI (diphenyleneiodonium chloride) were added after 1 h of gentamicin treatment to avoid the possible effect of the inhibitors on the uptake of bacteria.
Infection Studies.
C57BL/6 and iNOS−/− mice were bred and maintained under specific pathogen-free conditions in accordance with national guidelines for animal studies. Groups of 6–7 mice were infected by intraperitoneal injection of an inoculum consisting of a mixture of 500 cfu wild-type S. typhimurium (NalR, KmS) and 500 cfu of SPI2 mutant strain HH104 (NalS, KmR). At various time points after infection, mice were killed by cervical dislocation and livers and spleens were removed aseptically. Organs were homogenized in 1 ml of PBS and the organ burdens of wild-type S. typhimurium and the SPI2 mutant strain were determined by plating serial dilutions on LB-agar containing 50 μg/ml nalidixic acid or 50 μg/ml kanamycin.
Determination of NO Production.
NO was measured as its end product nitrite using the Griess reagent as described previously (27). Culture supernatants (50 μl) were mixed with 100 μl of Griess reagent (1% sulfanilamide, 0.1% N-(1-napthyl)-ethylenediamine dihydrochloride and 2.5% orthophosphoric acid). After 10 min, absorbance at 570 nm was measured in a microplate ELISA reader (Bio-Rad Laboratories). The concentration of nitrite in the culture supernatant was determined with reference to the standard curve using sodium nitrite. All data represent the mean value of triplicates ± SD.
Immunoblotting.
RAW267.4 cells were seeded in 6-well plates (4 × 105 cells/well) and infected. Cells were lysed in SDS-PAGE sample buffer and boiled for 5 min at 100°C. Aliquots containing equal amounts of protein (20 μg) were loaded onto a 6% gradient polyacrylamide gel, run under reducing conditions, and transferred onto nitrocellulose membranes. The membranes were treated with 5% bovine serum albumin for 1 h to block nonspecific binding, rinsed, and incubated with rabbit polyclonal antibodies against mouse iNOS (raised against peptide amino acids (aa) 1125–1144, BioTrend) for 1 h. The blots were further treated with goat anti rabbit IgG-horseradish peroxidase conjugate (AP-Biotech; dilution 1:3,000) for 1 h. The immune complex on the blots was detected with enhanced chemiluminescence substrate (AP-Biotech) and exposed to Eastman Kodak Co. XAR x-ray film.
Immunofluorescent Staining of iNOS and Peroxynitrite.
RAW 267.4 cells were seeded onto glass coverslips and infected as described before. The antibodies were diluted in PBS containing 5% goat serum, 2% BSA, and 0.1% saponin. For immuno-staining of iNOS, cells were incubated with 1:500 dilutions of rabbit antisera raised against aa 1125–1144 (BioTrend) or aa 1137–1144 of mouse iNOS (donated by Dr. C. Bogdan, University of Erlangen, Erlangen, Germany). For detection of peroxynitrite formation, cells were incubated with a 1:500 dilution of a rabbit antiserum raised against nitrotyrosine (Upstate Biotechnology) for 1 h. After washing with PBS, the cells were incubated with Cy3-conjugated goat anti–rabbit IgG antibody (Dianova) for 30 min. After washing, the cells were inspected for localization of iNOS or peroxynitrite using a fluorescence microscope or a confocal laser-scanning microscope (Leica) using filter sets for FITC (510 nm excitation, 530 nm emission) and TRITC (566 nm excitation, 590 nm emission). Images were further processed using the Leica TCS software package.
Results
Induction of Nitrite Production by RAW267.4 Macrophages Requires Live Salmonella but Does Not Require Bacterial Internalization.
The role of RNI during experimental salmonellosis in murine hosts has recently been demonstrated (21, 22). To elucidate the role of RNI in the cellular microbiology of Salmonella, we first investigated how a cultured macrophage-like cell line reacts to infection with S. typhimurium. RAW267.4 macrophages were infected with S. typhimurium wild-type and the NO response as measured by nitrite accumulation was studied. An increase in the MOI resulted in a gradual increase of the nitrite production by the RAW267.4 cells at early time after infection (Fig. 1 A). 8 h after infection, no significant difference in nitrite production was observed after infection at various MOI in the range of 10 to 150. In contrast, macrophages infected with heat-killed bacteria did not exhibit significant nitrite accumulation after 8 h (Fig. 1 B). After 24 h, accumulation of nitrite was detectable but lower than that observed with live Salmonella. This residual nitrite accumulation may be due to LPS, which is a prime inducer of NO response (28). No nitrite accumulation was detected in macrophage cultures separated from the Salmonella inoculum by a membrane, indicating that contact was required for induction of NO synthesis (Fig. 1 C). However, phagocytosis per se was not required for the NO production, as inhibition of phagocytosis by cytochalasin D did not inhibit the induction of nitrite production by Salmonella (Fig. 1 C). Infection with bacteria grown with aeration to stationary phase or bacteria cultured under O2 limitation resulted in similar levels of NO production (Fig. 1 D). Also, opsonization of the inoculum did not affect the induction of NO production (Fig. 1 D). Phagocytosis of latex beads alone did not result in nitrite production by RAW267.4 cells (data not shown), implying that live Salmonella set a specific signal for NO induction early after infection.
Figure 1.

Induction of iNOS by RAW267.4 macrophages requires live Salmonella. (A) RAW267.4 cells were infected with wild-type Salmonella at various MOI and nitrite in the supernatant was measured by the Griess reaction 4 and 8 h after infection. (B) RAW267.4 macrophages were infected with heat-killed Salmonella and the nitrite was measured 8 and 24 h after infection. The data represent the mean ± SD of three independent experiments done in triplicate. (C) RAW267.4 macrophages were infected with wild-type Salmonella at a MOI of 10 under various experimental conditions as indicated, and nitrite production was measured 8 h after infection. After adding the bacteria to the macrophages and centrifugation, infection took place for 0, 10, 25, or 60 min. In further assays, macrophages were infected for 25 min, but were separated from the bacteria by a 0.4 μM filter, or cytochalasin D was added to the macrophages at 1 μg/ml before infection. not inf., not infected. (D) Effect of bacterial culture conditions on nitrite production. Wild-type S. typhimurium was grown for 16 h in LB media without aeration as standing cultures or with aeration in test tubes in a roller drum. If indicated, the bacteria were opsonized by incubation with 10% normal mouse serum for 30 min. Infection was performed at a MOI of 10 and nitrite production was assayed after 16 h.
Wild-type S. typhimurium and Mutant Strains in SPI2 Induce Similar NO Responses in Macrophages.
The requirement of bacterial viability led us to investigate whether gene products required for intracellular proliferation of S. typhimurium are affecting the NO production. Here, we focused on the role of SPI2 genes. Our experiments revealed no significant difference between the NO response induced by infection with wild-type S. typhimurium and various mutants in the SPI2 locus (Fig. 2 A). The SPI2 mutants induced as high levels of NO accumulation as wild-type Salmonella. Furthermore, no difference in the cellular amounts of iNOS protein was observed in macrophages infected with wild-type S. typhimurium or various SPI2 mutants (Fig. 2 C). These observations indicate that SPI2 function is not directly affecting the NO production or the cellular levels of iNOS.
Figure 2.


Nitrite production by RAW267.4 macrophages infected with wild-type Salmonella and SPI2 mutant strains. (A) RAW267.4 macrophages were infected at a MOI of 10 with different SPI2 mutants and the nitrite was measured in the supernatant after 8 h. (B) Infection was performed as before with wild-type S. typhimurium and an isogenic strain deficient in sipC encoded by SPI1. wt, wild-type. (C) Western blot analysis of iNOS expression in noninfected RAW267.4 macrophages, and macrophages infected with wild-type S. typhimurium or strains harboring mutations in SPI2 genes ssaV or ssrA.
The induction of iNOS expression by effector proteins of SPI1, such as SipC, has been reported recently (29). We observed similar levels of NO synthesis after infection with wild-type Salmonella and different strains deficient in SPI1, such as a sipC strain (Fig. 2 B). SPI1-mediated invasion of macrophages by Salmonella has cytotoxic effects on macrophages (for a review, see reference 30). Under the assay conditions applied in our study, bacteria were grown to stationary phase, thus reduced in expression of SPI1, noninvasive and noncytotoxic. Therefore, in our experimental system mutations in the secretion system of SPI1 or effector proteins of SPI1 are unlikely to affect iNOS expression.
Inhibition of iNOS Enhances Intracellular Proliferation of S. typhimurium and Rescues Mutants Deficient in SPI2.
As iNOS has been recently shown to be one of the potent candidate molecules in controlling the Salmonella infection (22), we further looked at the intracellular growth of wild-type S. typhimurium and SPI2 mutant strains. Irrespective of the high nitrite production, wild-type Salmonella proliferated rapidly inside host cells, while SPI2 mutant strains had reduced rates of intracellular proliferation. Determination of intracellular bacteria in RAW267.4 macrophages at 2 and 16 h after infection indicated a 60-fold increase for S. typhimurium wild-type compared with a 5 to 7-fold increase for mutants in SPI2 genes. L-NMMA, a competitive inhibitor of NO synthases, dose dependently inhibited the nitrite production by macrophages infected with wild type S. typhimurium (Table II). Inhibition of iNOS by L-NMMA increased the intracellular proliferation of the SPI2 mutants (Fig. 3) . This observation indicates that iNOS contributes to the intracellular killing of Salmonella. However, the presence of L-NMMA also resulted in increased intracellular proliferation of the wild-type strain, which suggests that a proportion of wild-type Salmonella was also killed by RNI (Fig. 3).
Table II.
Induction of Nitrite Production by RAW267.4 Macrophages
| Nitrite
|
||
|---|---|---|
| L-NMMA | Noninfected | Infected |
| μM | μM | μM |
| 0 | 0 | 8.9 ± 0.9 |
| 5 | 0 | 2.4 ± 0.3 |
| 10 | 0 | 0.4 ± 0.6 |
| 100 | 0 | 0.0 ± 0.0 |
RAW267.4 macrophages were infected with wild-type S. typhimurium at an MOI of 10. Noninternalized bacteria were removed by washing followed by killing with gentamicin. After infection L-NMMA was added as indicated and 8 h after infection, nitrite levels in the culture supernatant were assayed by the Griess reaction.
Figure 3.

Intracellular proliferation of Salmonella in the presence or absence of iNOS inhibitor L-NMMA. RAW267.4 macrophages were infected with wild-type (wt) S. typhimurium and various mutant strains in SPI2 genes at a MOI of 10. Non-internalized bacteria were removed by washing and killed by gentamicin. L-NMMA was added at a concentration of 10 μM if indicated. The number of intracellular bacteria was determined by plating serial dilutions of macrophage lysates on LB-agar for counting of cfu. Intracellular replication was expressed as the ratio of cfu at 2 to 16 h after infection.
To control if the observed effects are the result of higher susceptibility of SPI2 mutant strains to nitrosative damages, in vitro tests were performed (Fig. 4) . Wild-type S. typhimurium and a representative SPI2 mutant strain (ssaV) were killed to the same extend by GSNO, NaNO2, or SIN-1. Therefore, increased intracellular survival of SPI2 mutant strains in the presence of L-NMMA is not the result of higher sensitivity of these strains to nitrosative damage.
Figure 4.

In vitro survival of S. typhimurium under different nitrosative stress conditions. Wild-type (circles) or SPI2 deficient (ssaV mutant, triangles) S. typhimurium were exposed to various concentrations of GSNO (A) or NaNO2 (B) in LB medium (adjusted to pH 5.0) or SIN-1 (C) in LB medium (adjusted to pH 7.0). At 3 h (open symbols) or 9 h (filled symbols) after exposure serial dilutions were plated and cfu were counted. Results are expressed as mean cfu ± SD of two independent experiments in triplicate.
Localization of iNOS in Salmonella-infected Macrophages Is Dependent on SPI2 Function.
As the previous experiments clearly demonstrated a role of RNI in controlling the intracellular proliferation of Salmonella, we set out to localize iNOS in Salmonella-infected macrophages. RAW267.4 macrophages or peritoneal macrophages were infected with wild-type S. typhimurium and various SPI2 mutants and analyzed by confocal laser-scanning microscopy for the localization of iNOS. In case of macrophages infected with wild-type S. typhimurium, iNOS was only rarely colocalized with the bacteria and a diffuse staining for iNOS was observed (Fig. 5) . In contrast, in macrophages infected with a SPI2 mutant strain defective in ssaV, iNOS frequently colocalized with the bacteria (Fig. 5). The iNOS staining was dense around the SPI2-deficient bacteria. Comparable results were obtained with an antiserum raised against aa 1,125–1,144 (Fig. 5) and an antiserum raised against aa 1137–1144 of mouse iNOS (data not shown). Similar patterns of colocalization with iNOS were obtained with various strains harboring mutations in SPI2 genes sseB, ssaV, ssaJ, ssrA, or ssrB. Previous work demonstrated that the virulence defect of a strain harboring a mutation in sseB can be complemented by a plasmid-born copy of sseB (15). Analysis of a sseB strain harboring psseB revealed the same pattern of iNOS localization as observed for wild type Salmonella (data not shown).
Figure 5.
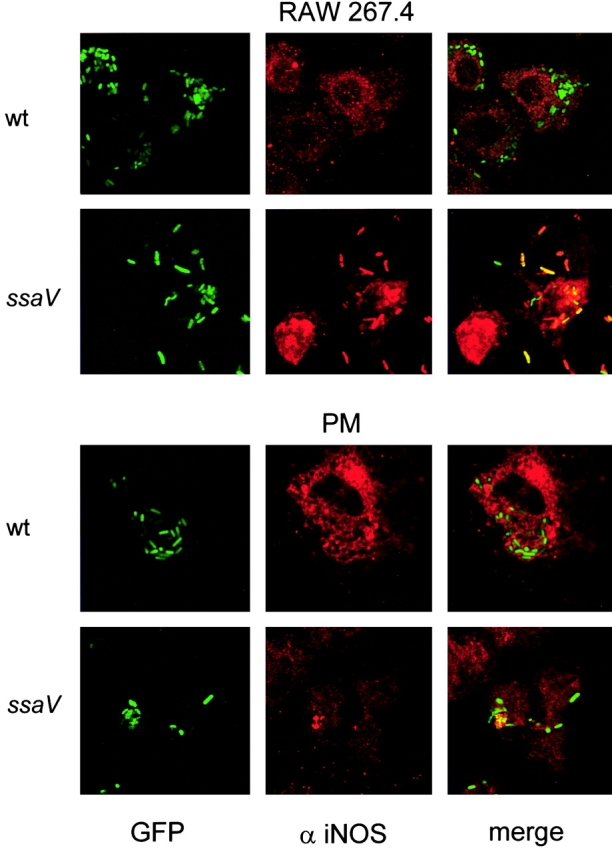
Localization of iNOS in Salmonella-infected macrophages. RAW267.4 macrophages and mouse peritoneal macrophages (PM) were infected with GFP-expressing wild-type (wt) S. typhimurium or SPI2 mutant strain defective in ssaV. Non-internalized bacteria were killed by addition of gentamicin. At 12 h after infection, the cells were fixed and permeabilized for immunostaining. iNOS was detected by incubation with a polyclonal antibody against iNOS and a Cy3-labeled secondary antibody. The localization of iNOS (red) and S. typhimurium (green) was analyzed by fluorescence microscopy (not shown) and confocal laser-scanning microscopy. Colocalization of S. typhimurium and iNOS resulted in orange to red staining of the bacterial cells.
We next investigated whether the altered cellular localization of iNOS is a result of reduced intracellular proliferation of mutant strains in SPI2 or directly related to SPI2 function and analyzed infections with an isogenic strain proficient for SPI2 function but defective in purD. The purD mutant is deficient in purine metabolism and attenuated in virulence (31) and intracellular proliferation (data not shown) to a similar extent as the SPI2-deficient strains. Although the number of intracellular purD bacteria was lower than those of wild-type Salmonella, purD bacteria were not colocalized with iNOS (data not shown).
The proportion of bacterial cells colocalized with iNOS after different time intervals was quantified (Fig. 6 A). At 8 or 16 h after infection, the proportion of wild-type S. typhimurium colocalized with iNOS remained lower than those of SPI2 mutant strains. Similar to a recent report by Webb et al. (32) we did not observe significant colocalization to either strain at earlier time points after infection.
Figure 6.

Colocalization of iNOS with wild-type S. typhimurium and various strains deficient in SPI2 genes or “Salmonella translocated effectors.” (A) RAW267.4 macrophages were infected with wild-type S. typhimurium (wt) and strains deficient in the SPI2-encoded secretion system (ssaV, ssrA, ssaJ). At 2, 8, or 16 h after infection, the percentage of bacteria colocalized with iNOS was scored by examining randomly selected 50 microscopic fields (1,000× magnification) and counted for total green and colocalized yellow or red bacteria. (B) RAW267.4 macrophages were infected with wild-type S. typhimurium, or mutant strains in SPI2 (ssaV) or various STE genes (sspH1, sspH2, sseI, sseJ, sifA, sifB, slrP). 8 h after infection, macrophages were processed for immuno-staining and colocalization was quantified as described in panel A. The data represent two independent experiments.
Our observations indicate that the altered localization of iNOS is an effect of the function of SPI2. SPI2 mutant strains are still able to survive and replicate, to a certain extent, in infected macrophages (15). This result is in line with the observation that a significant percentage of SPI2 mutants (∼40%) was not associated with iNOS. Such bacteria may escape the macrophage defense and be able to replicate intracellularly. On the other hand, a low percentage (∼10%) of wild-type Salmonella colocalized with iNOS, indicating that RNI also control the intracellular proliferation of fully virulent bacteria.
Effect of Mutations in Genes for STEs on iNOS Delivery to Salmonella-containing Phagosomes.
A family for proteins that are translocated by the TTSS of SPI2 is encoded by genes outside of the SPI2 locus (33). The gene products, namely SlrP, SspH1, SspH2, SifA, SifB, SseI, and SseJ are referred to as STEs and have been proposed to interact with host cell functions after translocation into the infected host cell. We set out to analyze if an individual STE protein is required for the prevention of iNOS delivery to SCVs and constructed a set of strains harboring mutations in STE genes. These strains were analyzed for iNOS delivery to SCVs after infection of macrophages. In contrast to strains deficient in the TTSS of SPI2, mutants in STE genes had characteristics similar to wild-type Salmonella, i.e., only 5–10% of the intracellular Salmonella colocalized with iNOS (Fig. 6 B). Colocalization of iNOS to the sifA strain was observed more frequently (∼15%). A role of SifA for maintenance of SCVs was observed (24) and this effector may also, to a certain extend, interfere with iNOS delivery to SCVs.
These observations indicate that a single STE is not sufficient to modify the delivery of iNOS to SCVs. The concerted action of several STE or the function of further, yet unknown effector proteins of the SPI2-encoded TTSS may be required for this phenotype.
Peroxynitrite Formation in Salmonella-infected Macrophages Is Localized with SPI2-deficient Bacteria.
NO per se is not a potent cytotoxic molecule and most potent bactericidal effect of NO appears to be via a reaction with O2 − (2). NO reacts with O2 − to yield peroxynitrite (OONO−). Peroxynitrite generated in the cells cannot be detected directly, but sites of damage due to the reaction of peroxynitrite can be assessed by immunohistochemical staining of nitrotyrosine. In case of macrophages infected with wild-type Salmonella, the nitrotyrosine staining was not, or to a low extent colocalized with the bacteria (Fig. 7) . Similar observations on the nitrotyrosine formation in S. typhimurium–infected macrophages were recently reported (22). In the case of SPI2 mutant strains, nitrotyrosine staining was localized in the vicinity of the bacteria (Fig. 7), indicating events where proteins were targeted by formation of peroxynitrite.
Figure 7.
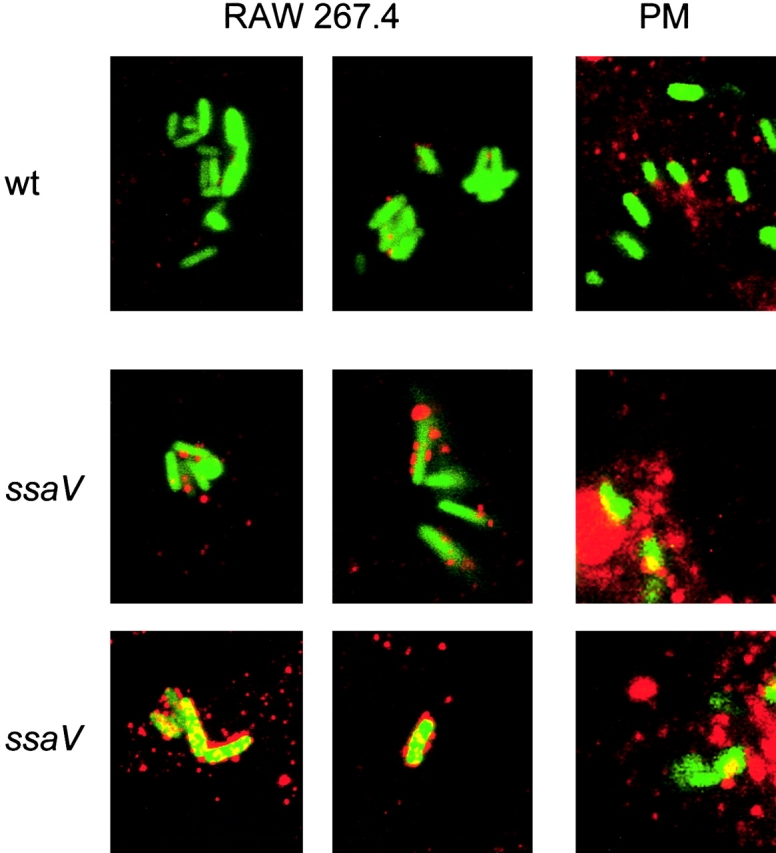
Localization of peroxynitrite in S. typhimurium-infected macrophages. Infection of RAW267.4 macrophages and mouse peritoneal macrophages (PM) was performed for 12 h as described in the legend to Fig. 5. The sites of peroxynitrite formation were detected by immunostaining with an anti-nitrotyrosine antibody and a Cy3-conjugated secondary antibody. Samples were analyzed by confocal laser-scanning microscopy and representative images for the localization of S. typhimurium expressing GFP (green) and peroxynitrite (red) are shown. Various levels of nitrotyrosine formation were observed for the SPI2 mutant strain. wt, wild-type.
Control of Intracellular Proliferation of S. typhimurium in Primary Peritoneal Macrophages.
After infection of primary peritoneal macrophages with S. typhimurium, the nitrite production was higher in primary macrophages (15 ± 1.5 μM nitrite) than in the RAW267.4 cell line (8 ± 0.5 μM nitrite). There was no difference observed in the nitrite production between macrophages infected with wild-type Salmonella or SPI2 mutant strains. Overall, the number of intracellular wild-type Salmonella was lower than those in RAW267.4 macrophages, but again only few events of colocalization with iNOS or nitrotyrosine were observed. On the other hand, in cells infected with SPI2 mutants, colocalization with iNOS or nitrotyrosine was frequently observed, suggesting that in primary macrophages iNOS contributes to the killing of the SPI2 mutants and not of wild-type Salmonella. Inhibition of iNOS by L-NMMA increased the growth of intracellular wild-type Salmonella and SPI2 mutant strains. Furthermore, in infection studies with primary macrophages we used the inhibitor DPI, which inhibits both NADPH oxidase and iNOS. The addition of DPI resulted in a higher recovery of intracellular bacteria than L-NMMA (Fig. 8) . The addition of phenylarsine oxide, which inhibits only NADPH oxidase but not iNOS, resulted in a lower recovery of intracellular bacteria compared with DPI (data not shown). This observation indicates additive effects of ROI and RNI in controlling intracellular proliferation of S. typhimurium in macrophages.
Figure 8.

Intracellular replication in primary macrophages. Peritoneal macrophages were prepared from C57BL/6 mice and infected with S. typhimurium wild-type (wt) or various strains harboring mutations in SPI2 genes. After infection, inhibitors L-NMMA (10 μM) and DPI (10 μM) were added as indicated. The intracellular proliferation was determined as described in the legend of Fig. 3.
Interaction of SPI2 and iNOS during Infection.
The role of iNOS in the control of Salmonella infection was analyzed by comparison of systemic salmonellosis in iNOS+/+ and iNOS−/− mice. Here, we used a mixed infection model in which a mixture of equal amount of wild-type S. typhimurium and a SPI2 mutant strain was applied via the intraperitoneal route. At various time points after infection, the bacterial burden of each strain was determined in infected organs, and the competitive index (CI) for wild-type S. typhimurium versus SPI2 mutants was determined (Table III). The CI is a highly sensitive and reproducible estimate of the virulence of a mutant strain in competition with the wild-type strain (24). In accordance with this previous study, a SPI2 mutant strain was deficient in the colonization of the C57BL/6 (iNOS+/+) mice. At early time points after infection, proliferation of wild-type and SPI2-deficient Salmonella was similar in iNOS+/+ and iNOS−/− mice. However, a dramatic difference was observed at the late phase of infection. At day 5 after infection a CI of < 0.00005 was obtained for iNOS+/+ mice, indicating that the SPI2-deficient strain was out-competed by wild-type S. typhimurium. In contrast, in iNOS−/− mice CI of 1.2 and 0.16 in liver and spleen, respectively, were determined at day 5 after infection. These results demonstrate that iNOS+/+, but not iNOS−/− mice, are able to control the proliferation of SPI2-deficient S. typhimurium, indicating an interaction in vivo of SPI2 and RNI-dependent defense mechanisms of the host.
Table III.
Competitive Indexes (CI) of a SPI2-deficient Mutant versus Wild-type S. typhimurium After Infection of iNOS+/+ and iNOS−/− Mice
| Time after infection (d) |
CI in iNOS+/+
|
CI in iNOS−/−
|
||
|---|---|---|---|---|
| Liver | Spleen | Liver | Spleen | |
| 2 | 0.24 ± 0.056 | 0.075 ± 0.035 | 0.52 ± 0.69 | 0.066 ± 0.091 |
| 3 | 0.0047 ± 0.0026 | 0.00078 ± 0.0001 | 0.0092 ± 0.0016 | 0.0028 ± 0.0005 |
| 5 | <0.000005 | <0.000005 | 1.20 ± 0.49 | 0.16 ± 0.04 |
Discussion
RNI produced by iNOS have important functions in the host defense against intracellular pathogens such as Leishmania spp., Mycobacterium spp., and Salmonella spp. (2, 34). Inhibition of iNOS resulted in increased bacterial burdens in organs and reduced survival of Salmonella-infected mice (35). An increase in the intracellular proliferation of Salmonella was observed in macrophages after inhibition of iNOS (22). Furthermore, it was recently demonstrated that iNOS plays an important role in controlling the growth of Salmonella at the later stages of infection in the model of murine salmonellosis: iNOS−/− mice succumbed to infection with a S. typhimurium strain of intermediate virulence several days after infection, while iNOS+/+ mice survived the infection (21). Inhibition of iNOS also led to increased intracellular replication of Salmonella in macrophages and mutant strains that downregulate the nitrite production also showed increased proliferation in macrophages (36). These evidences suggest a role of iNOS in Salmonella pathogenesis.
Salmonella possesses several systems to detoxify RNI as well as enzymes to repair damages induced by the action of RNI (for a review, see reference 2). Recent observations also indicate that intracellular Salmonella utilize a variety of mechanisms to actively avoid antimicrobial activities of their host cells (17, 18). As RNI are important antimicrobial effector molecules of phagocytes, we analyzed here if S. typhimurium also has mechanisms to prevent synthesis or circumvent the effects of RNI. We are interested to understand the cellular microbiology of SPI2, a virulence factor that is essential for the proliferation of S. typhimurium within infected host tissues and intracellular replication. No difference was observed between wild-type Salmonella and SPI2 mutants in terms of iNOS protein levels or nitrite production, showing that SPI2 is not involved in reduction of RNI production.
The subcellular localization of iNOS was dramatically different in macrophages infected with wild-type Salmonella or SPI2 mutants. We observed that iNOS was closely localized with SPI2 mutant strains, but rarely with wild-type bacteria. As SPI2 mutants show highly reduced intracellular proliferation, the colocalization of iNOS with SPI2 mutants suggests the involvement of nitrogen-derived radicals in the killing of SPI2 mutants. Inhibition of iNOS by the inhibitor L-NMMA resulted in increased proliferation of SPI2-deficient Salmonella in macrophages, indicating a role of RNI in limiting intracellular proliferation within macrophages.
The reaction of RNI with ROI generates peroxynitrite (ONOO−), a reactive molecule with potent antimicrobial activity against S. typhimurium in vitro (2). It was recently reported that peroxynitrite did not colocalize with the wild-type Salmonella and may not be responsible for the killing of intracellular bacteria (22). In fact, we confirm that peroxynitrite rarely colocalizes with wild-type Salmonella. In contrast, nitrotyrosine residues were frequently located in the vicinity of SPI2 mutants, indicating that the peroxynitrite formed can reach the target bacteria and may act as a potent molecule to kill the bacteria. This observation stresses the role of peroxynitrite in eradication of intracellular Salmonella. Production of ROI appears to be a crucial defense mechanism in the early phase of Salmonella infections as mice deficient in the phagocyte oxidase succumb shortly after infection (for a review, see reference 37). During the late phase of infection, the combined activities of ROI and RNI appear to be important for the control of proliferation of Salmonella in infected organs. We assume that peroxynitrite formation is a potent antimicrobial mechanism of macrophages and efficiently inactivates microorganisms that are not adapted to an intracellular lifestyle by virulence factors such as SPI2.
The observations reported here raise important new questions about the mode of delivery of iNOS to SCVs of the host cell and how intracellular Salmonella influence the localization of iNOS. If Salmonella can actively prevent the colocalization of iNOS with SCVs, under normal conditions iNOS may be transported to phagosomes in a vectorial fashion. iNOS of mouse macrophages is reported to reside not only free in the cytosol, but is also present in small vesicles (38). This has led to suggestions that the targeted delivery of such vesicles to the appropriate phagosomes possibly allows the interaction between iNOS and the pathogens, thereby exerting the antimicrobial activity. The association of iNOS with actin has been reported (32), but a recruitment of iNOS to phagosomes containing S. typhimurium was not observed under the specific experimental conditions of the study.
We speculate that transport of iNOS and NADPH-oxidase to phagosomes is tightly controlled to ensure that highly reactive antimicrobial radicals are preferentially produced at the appropriate subcellular location, without causing damage to host cell structures. The first model for the function of SPI2 postulates an inhibition of cellular trafficking, i.e., by inhibition of transport of iNOS-containing vesicles to SCVs. A second explanation of the phenotype described here may be the altered structure of SCVs. Recent observations indicate that intracellular S. typhimurium employ the SPI2 system to interfere with the organization of actin of the host cell and to accumulate actin at SCVs (39). This difference between the phagosomal composition of SCVs containing wild type Salmonella or SPI2 mutants may affect the access of iNOS to the bacteria. The modification of the pathogen-containing vesicle, or coating, induced by the pathogen was also observed in macrophages infected with M. tuberculosis (40).
A large number of STE proteins of the TTSS of SPI2 have been identified (33), and it is likely that the various effectors have different targets and functions in the host cell. However, our study did not indicate a significant role of single STE in protection of intraphagosomal Salmonella against nitrosative damages. STE proteins may have overlapping or partially redundant functions in protection of intracellular Salmonella, or additional, not yet characterized effector proteins of the SPI2 are required for this function.
Various intracellular phenotypes in macrophages have been related to the function of SPI2 of Salmonella: (a) Uchiya et al. reported the inhibition of vesicular trafficking as a function of SPI2 (17). (b) Vazquez-Torres et al. observed that intracellular Salmonella employ the SPI2 system to exclude the NADPH-oxidase from SCVs (18, 19). (c) Finally, we report here that the SPI2 system is involved in altering the localization of iNOS. Further work has to reveal whether the different cellular phenotypes are all consequences of the same function of SPI2, e.g., by a general inhibition of cellular vesicle transport. It is likely that the combined activities of NADPH-oxidase and iNOS at the SCVs are required to generate potent antimicrobial effectors.
Finally, the Salmonella-induced production of RNI might have deleterious effects on the host cells. If high amounts of RNI are produced but proper delivery to phagosomes is inhibited by SPI2, damage to the host cell may occur. Such autotoxicity by incorrectly delivered antimicrobial effector molecules might also contribute to increased intracellular proliferation of Salmonella. It was reported that uric acid, which quenches peroxynitrite formation, improves killing of Salmonella by macrophages (22), suggesting that the peroxynitrite formed cannot reach the target bacteria and becomes cytotoxic to the host cell itself. This effect may enable the spread of Salmonella from infected macrophages and dissemination into other tissues, finally leading to a systemic infection.
Other mechanisms have been described that also contribute to the protection of bacteria against RNI and peroxynitrite. These include superoxide dismutase (41), flavohemoglobin (42), and peroxynitrite reductase (43), that are present in pathogenic bacteria and nonpathogenic relatives. Several of these mechanisms contribute directly to the virulence of S. enterica, while other mechanisms such as superoxide dismutase are duplicated with redundant functions (for a review, see reference 2). In contrast, the SPI2 locus represents an acquisition only present in the S. enterica (13) and is considered as an adaptation to survival and replication in a host organism (10). The large number of bacterial defense mechanisms evolved to counteract antimicrobial activities of RNI and peroxynitrite underline the role of these mechanisms in the innate immunity of the host.
Our observations are summarized in the following model for the interaction of Salmonella with macrophages: macrophages react to the contact with bacteria with expression of iNOS and increased production of RNI. Salmonella that invade macrophages or are taken up by phagocytosis are confronted with the potent antimicrobial activity of RNI produced by iNOS. In contrast to nonpathogenic bacteria, Salmonella employs the TTSS encoded by SPI2 to translocate effector proteins that prevent the colocalization of iNOS and the SCVs, so that RNI cannot react with the phagocytosed bacteria. In addition to bacterial mechanisms that interfere with production of RNI, detoxification of RNI, and repair of damage caused by RNI, the exclusion of RNI- and ROI-producing enzymes appears to be an important virulence mechanism of intracellular pathogens. To our knowledge, S. typhimurium is the first example of a pathogen that actively avoids the delivery of antimicrobial effectors to the pathogen-containing vesicle by means of a TTSS. It will now be of interest to analyze if other intracellular pathogens use similar strategies to evade antimicrobial activities of their host cells.
Acknowledgments
We would like to thank Dr. M. Röllinghoff for generous support of our work and Dr. C. Bogdan for providing antisera against iNOS, iNOS−/− mice, and for stimulating discussions. We are indebted to Cedric Cheminay for the preparation of primary macrophages and members of the Hensel lab for critical reading of the manuscript.
This work was supported by the Deutsche Forschungsgemeinschaft by grants HE 1964/2-3 and HE1964/4-2.
Footnotes
Abbreviations used in this paper: aa, amino acid(s); DPI, diphenyliodonium chloride; GFP, green fluorescent protein; iNOS, inducible nitric oxide synthase; L-NMMA, l-monomethylarginine; MOI, multiplicity of infection; RNI, reactive nitrogen intermediates; ROI, reactive oxygen intermediates; SCV, Salmonella-containing vesicle; SPI, Salmonella pathogenicity island; STE, Salmonella-translocated effector; TTSS, type III secretion system.
References
- 1.Shiloh, M.U., J.D. MacMicking, S. Nicholson, J.E. Brause, S. Potter, M. Marino, F. Fang, M. Dinauer, and C. Nathan. 1999. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity. 10:29–38. [DOI] [PubMed] [Google Scholar]
- 2.Nathan, C., and M.U. Shiloh. 2000. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. USA. 97:8841–8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hueck, C.J. 1998. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev. 62:379–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galan, J.E. 2001. Salmonella interactions with host cells: type III secretion at work. Annu. Rev. Cell Dev. Biol. 17:53–86. [DOI] [PubMed] [Google Scholar]
- 5.Wallis, T.S., and E.E. Galyov. 2000. Molecular basis of Salmonella-induced enteritidis. Mol. Microbiol. 36:997–1005. [DOI] [PubMed] [Google Scholar]
- 6.Richter Dahlfors, A., A.M.J. Buchan, and B.B. Finlay. 1997. Murine salmonellosis studied by confocal microscopy: Salmonella typhimurium resides intracellularly inside macrophages and exerts a cytotoxic effect on phagocytes in vivo. J. Exp. Med. 186:569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finlay, B.B., and S. Falkow. 1989. Salmonella as an intracellular parasite. Mol. Microbiol. 3:1833–1841. [DOI] [PubMed] [Google Scholar]
- 8.Fields, P.I., R.V. Swanson, C.G. Haidaris, and F. Heffron. 1986. Mutants of Salmonella typhimurium that cannot survive within the macrophage are avirulent. Proc. Natl. Acad. Sci. USA. 83:5189–5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foster, J.W., and M.P. Spector. 1995. How Salmonella survive against the odds. Annu. Rev. Microbiol. 49:145–174. [DOI] [PubMed] [Google Scholar]
- 10.Groisman, E.A., and H. Ochman. 1997. How Salmonella became a pathogen. Trends Microbiol. 5:343–349. [DOI] [PubMed] [Google Scholar]
- 11.Hensel, M. 2000. Salmonella Pathogenicity Island 2. Mol. Microbiol. 36:1015–1023. [DOI] [PubMed] [Google Scholar]
- 12.Shea, J.E., M. Hensel, C. Gleeson, and D.W. Holden. 1996. Identification of a virulence locus encoding a second type III secretion system in Salmonella typhimurium. Proc. Natl. Acad. Sci. USA. 93:2593–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shea, J.E., C.R. Beuzon, C. Gleeson, R. Mundy, and D.W. Holden. 1999. Influence of the Salmonella typhimurium pathogenicity island 2 type III secretion system on bacterial growth in the mouse. Infect. Immun. 67:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ochman, H., F.C. Soncini, F. Solomon, and E.A. Groisman. 1996. Identification of a pathogenicity island required for Salmonella survival in host cells. Proc. Natl. Acad. Sci. USA. 93:7800–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hensel, M., J.E. Shea, S.R. Waterman, R. Mundy, T. Nikolaus, G. Banks, A. Vazquez-Torres, C. Gleeson, F. Fang, and D.W. Holden. 1998. Genes encoding putative effector proteins of the type III secretion system of Salmonella pathogenicity island 2 are required for bacterial virulence and proliferation in macrophages. Mol. Microbiol. 30:163–174. [DOI] [PubMed] [Google Scholar]
- 16.Cirillo, D.M., R.H. Valdivia, D.M. Monack, and S. Falkow. 1998. Macrophage-dependent induction of the Salmonella pathogenicity island 2 type III secretion system and its role in intracellular survival. Mol. Microbiol. 30:175–188. [DOI] [PubMed] [Google Scholar]
- 17.Uchiya, K., M.A. Barbieri, K. Funato, A.H. Shah, P.D. Stahl, and E.A. Groisman. 1999. A Salmonella virulence protein that inhibits cellular trafficking. EMBO J. 18:3924–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vazquez-Torres, A., Y. Xu, J. Jones-Carson, D.W. Holden, S.M. Lucia, M.C. Dinauer, P. Mastroeni, and F.C. Fang. 2000. Salmonella pathogenicity island 2-dependent evasion of the phagocyte NADPH oxidase. Science. 287:1655–1658. [DOI] [PubMed] [Google Scholar]
- 19.Gallois, A., J.R. Klein, L.A. Allen, B.D. Jones, and W.M. Nauseef. 2001. Salmonella pathogenicity island 2-encoded type III secretion system mediates exclusion of NADPH oxidase assembly from the phagosomal membrane. J. Immunol. 166:5741–5748. [DOI] [PubMed] [Google Scholar]
- 20.Cherayil, B.J., and D. Antos. 2001. Inducible nitric oxide synthase and Salmonella infection. Microbes Infect. 3:771–776. [DOI] [PubMed] [Google Scholar]
- 21.Mastroeni, P., A. Vazquez-Torres, F.C. Fang, Y. Xu, S. Khan, C.E. Hormaeche, and G. Dougan. 2000. Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. II. Effects on microbial proliferation and host survival in vivo. J. Exp. Med. 192:237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vazquez-Torres, A., J. Jones-Carson, P. Mastroeni, H. Ischiropoulos, and F.C. Fang. 2000. Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. I. Effects on microbial killing by activated peritoneal macrophages in vitro. J. Exp. Med. 192:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valdivia, R.H., and S. Falkow. 1997. Fluorescence-based isolation of bacterial genes expressed within host cells. Science. 277:2007–2011. [DOI] [PubMed] [Google Scholar]
- 24.Datsenko, K.A., and B.L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA. 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maloy, S.R., V.L. Stewart, and R.K. Taylor. 1996. Genetic Analysis of Pathogenic Bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- 26.Bowe, F., and F. Heffron. 1994. Isolation of Salmonella mutants defective for intracellular survival. Methods Enzymol. 236:509–526. [DOI] [PubMed] [Google Scholar]
- 27.Green, L.C., D.A. Wagner, J. Glogowski, P.L. Skipper, J.S. Wishnok, and S.R. Tannenbaum. 1982. Analysis of nitrate, nitrite, and nitrate in biological fluids. Anal. Biochem. 126:131–138. [DOI] [PubMed] [Google Scholar]
- 28.Raetz, C.R., R.J. Ulevitch, S.D. Wright, C.H. Sibley, A. Ding, and C.F. Nathan. 1991. Gram-negative endotoxin: an extraordinary lipid with profound effects on eukaryotic signal transduction. FASEB J. 5:2652–2660. [DOI] [PubMed] [Google Scholar]
- 29.Cherayil, B.J., B.A. McCormick, and J. Bosley. 2000. Salmonella enterica serovar typhimurium-dependent regulation of inducible nitric oxide synthase expression in macrophages by invasins SipB, SipC, and SipD and effector SopE2. Infect. Immun. 68:5567–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monack, D.M., W.W. Navarre, and S. Falkow. 2001. Salmonella-induced macrophage death: the role of caspase-1 in death and inflammation. Microbes Infect. 3:1201–1212. [DOI] [PubMed] [Google Scholar]
- 31.Hensel, M., J.E. Shea, C. Gleeson, M.D. Jones, E. Dalton, and D.W. Holden. 1995. Simultaneous identification of bacterial virulence genes by negative selection. Science. 269:400–403. [DOI] [PubMed] [Google Scholar]
- 32.Webb, J.L., M.W. Harvey, D.W. Holden, and T.J. Evans. 2001. Macrophage nitric oxide synthase associates with cortical actin but is not recruited to phagosomes. Infect. Immun. 69:6391–6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miao, E.A., and S.I. Miller. 2000. A conserved amino acid sequence directing intracellular type III secretion by Salmonella typhimurium. Proc. Natl. Acad. Sci. USA. 97:7539–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bogdan, C., M. Röllinghoff, and A. Diefenbach. 2000. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr. Opin. Immunol. 12:64–76. [DOI] [PubMed] [Google Scholar]
- 35.Umezawa, K., T. Akaike, S. Fujii, M. Suga, K. Setoguchi, A. Ozawa, and H. Maeda. 1997. Induction of nitric oxide synthesis and xanthine oxidase and their roles in the antimicrobial mechanism against Salmonella typhimurium infection in mice. Infect. Immun. 65:2932–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eriksson, S., J. Bjorkman, S. Borg, A. Syk, S. Pettersson, D.I. Andersson, and M. Rhen. 2000. Salmonella typhimurium mutants that downregulate phagocyte nitric oxide production. Cell. Microbiol. 2:239–250. [DOI] [PubMed] [Google Scholar]
- 37.Vazquez-Torres, A., and F.C. Fang. 2001. Oxygen-dependent anti-Salmonella activity of macrophages. Trends Microbiol. 9:29–33. [DOI] [PubMed] [Google Scholar]
- 38.Vodovotz, Y., D. Russell, Q.W. Xie, C. Bogdan, and C. Nathan. 1995. Vesicle membrane association of nitric oxide synthase in primary mouse macrophages. J. Immunol. 154:2914–2925. [PubMed] [Google Scholar]
- 39.Meresse, S., K.E. Unsworth, A. Habermann, G. Griffiths, F. Fang, M.J. Martinez-Lorenzo, S.R. Waterman, J.-P. Gorvel, and D.W. Holden. 2001. Remodelling of the actin cytoskeleton is essential for replication of intravacuolar Salmonella. Cell. Microbiol. 3:567–577. [DOI] [PubMed] [Google Scholar]
- 40.Ferrari, G., H. Langen, M. Naito, and J. Pieters. 1999. A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell. 97:435–447. [DOI] [PubMed] [Google Scholar]
- 41.De Groote, M.A., U.A. Ochsner, M.U. Shiloh, C. Nathan, J.M. McCord, M.C. Dinauer, S.J. Libby, A. Vazquez-Torres, Y. Xu, and F.C. Fang. 1997. Periplasmic superoxide dismutase protects Salmonella from products of phagocyte NADPH-oxidase and nitric oxide synthase. Proc. Natl. Acad. Sci. USA. 94:13997–14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crawford, M.J., and D.E. Goldberg. 1998. Role for the Salmonella flavohemoglobin in protection from nitric oxide. J. Biol. Chem. 273:12543–12547. [DOI] [PubMed] [Google Scholar]
- 43.Bryk, R., P. Griffin, and C. Nathan. 2000. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature. 407:211–215. [DOI] [PubMed] [Google Scholar]
- 44.Beuzon, C.R., S. Meresse, K.E. Unsworth, J. Ruiz-Albert, S. Garvis, S.R. Waterman, T.A. Ryder, E. Boucrot, and D.W. Holden. 2000. Salmonella maintains the integrity of its intracellular vacuole through the action of sifA. EMBO J. 19:3235–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deiwick, J., T. Nikolaus, J.E. Shea, C. Gleeson, D.W. Holden, and M. Hensel. 1998. Mutations in Salmonella Pathogenicity Island 2 (SPI2) genes affecting transcription of SPI1 genes and resistance to antimicrobial agents. J. Bacteriol. 180:4775–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]