

Abstract
Deletion, anergy, and a spectrum of functional impairments can affect virus-specific CD8 cells in chronic viral infections. Here we characterize a low frequency population of CD8 cells present in chronic hepatitis B virus (HBV) infection which survive in the face of a high quantity of viral antigen. Although they do not appear to exert immunological pressure in vivo, these CD8 cells are not classically “tolerant” since they proliferate, lyse, and produce antiviral cytokines in vitro. They are characterized by altered HLA/peptide tetramer reactivity, which is not explained by TCR down-regulation or reduced functional avidity and which can be reversed with repetitive stimulation. CD8 cells with altered tetramer binding appear to have a specificity restricted to envelope antigen and not to other HBV antigens, suggesting that mechanisms of CD8 cell dysfunction are differentially regulated according to the antigenic form and presentation of individual viral antigens.
Keywords: cytotoxic T lymphocytes, chronic hepatitis B, viral diseases, immune tolerance, viral T antigen
Introduction
During persistent virus infections antigen-specific CD8 T cells and viral antigens are able to coexist as a possible result of different mechanisms. Functionally active CD8 cells may exert selective pressure on the virus leading to the emergence of T cell escape virus mutants (1), or the virus might possess strategies to avoid recognition by CD8 T cells by altering the processing or presentation of viral antigen (2). In other cases the production of viral antigen may lead to the exhaustion of virus-specific CD8 cells (3, 4) or to the selection of virus-specific T cells with altered functions (5–10). These mechanisms are present to variable degrees in different persistent virus infections and can change in relation to the dose or strain of infecting virus, genetic background, and age of the host (11).
Hepatitis B virus (HBV)*, a noncytopathic, hepatotropic DNA virus, is one of the important pathogenic viruses able to establish a persistent infection in man, which may lead to the development of cirrhosis and hepatocellular carcinoma. Approximately 300 million individuals are chronically infected worldwide (12). Mutations in relevant CTL epitopes (13, 14) or down-regulation of MHC class I molecules (15) have been demonstrated and are likely to play a role in persistence. However, the main immunological feature that characterizes chronically infected patients is a state of relative hyporesponsiveness of HBV-specific T cells compared with that demonstrable in those patients who control the virus replication after acute infection (16). Whether this T cell hyporesponsiveness is caused by deletion of specific cells (negative selection, exhaustion) or by functional tolerance (anergy, cytokine imbalance) has not been fully tested. Clonal deletion of HBV-specific T cells as a consequence of transplacental HBV infection (in vertically infected patients) and the exhaustion of antiviral CTL by high dose of antigen (in adult infection) are likely to substantially reduce the number of HBV-specific T cells in chronically infected patients (16). Nevertheless, HBV-specific T cells are not completely deleted. Vaccination with HBsAg of neonatally infected babies is effective at inducing the clearance of HBV (17), suggesting that T cells are still present and functionally active. Similarly when adults achieve spontaneous (18) or drug-induced (19) control of HBV replication, functionally efficient HBV-specific CD8 cells in the circulation may be demonstrated and expanded. Since these data suggest that some HBV-specific CD8 cells escape deletion, whether these cells are functionally active, how they survive, and what their contribution is to the control of the virus infection, remain important unresolved questions.
In this study, to evaluate the biological properties of the HBV-specific CD8 population persisting in chronic hepatitis B patients with a high level of replicating virus, we used in parallel MHC/peptide tetramers and intracellular cytokine staining (ICCS) both directly ex vivo and after in vitro expansion. The use of more than one technique is necessary since in the presence of antigens, CD8 T cells may either lack functional activity or reactivity with the specific HLA-class I tetramers (11). We present data showing that a low frequency CD8 cell population is able to escape peripheral deletion and persist in the face of a high dose of viral antigen, displaying altered reactivity to the specific HLA-tetramer. We also explore mechanisms responsible for the reduced reactivity of these cells with HLA-peptide tetramers and their functional capacity in vitro. In addition we show that CTLs with these characteristics have reactivity restricted to the envelope antigen, suggesting that, as seen in mice (9), strategies of CD8 impairment may be differentially regulated according to the dose and presentation of individual viral antigens.
Materials and Methods
Patients.
10 HLA-A2 positive adult subjects were studied. Three had an acute self-limited HBV infection at least 1 y before this study (referred to as “immune patients”), and seven patients were chronically infected, with HBV-DNA 105–109 copies per milliliter and with elevated alanine transaminase values (normal range <40 IU/L) for >6 mo before the study. All patients were negative for antibodies to hepatitis C virus (HCV), delta virus, and to HIV-1,2. Sequencing of the core and envelope HBV genome of the immune patients at the time of acute infection showed that the core 18–27, envelope 183–91, and envelope 348–57 sequences corresponded with the index peptides used for HLA-tetramers and peptide stimulation.
Virological Assessment.
HBsAg, anti-HBs, total and IgM anti-HBc, HBeAg, anti-HBe, anti-delta, anti-HCV, anti HIV-1, and anti–HIV-2 were determined by commercial enzyme immunoassay kits (Abbott Laboratories, Ortho Diagnostic System, and Sanofi Diagnostic Pasteur). HBV-DNA level was quantified by using the Roche Amplicor Monitor assay (Roche Laboratories), with a DNA detection limit threshold of 400 copies per milliliter (0.0014 pg/ml). HBsAg in patients' serum was quantified using the Murex HBsAg Version 3 kit (Abbott Murex). The manufacturer's instructions were followed throughout with the inclusion of a standard curve made from dilutions of the 2nd British Working Standard for HBsAg (NIBSC). Serial dilutions of patients' sera were tested (usually 10−4–10−6) to ensure results within the range of the assay. All dilutions were made in normal human serum.
PCR and HBV DNA Sequencing.
DNA was extracted from serum samples taken at the time of liver biopsy using QIAamp DNA Blood minikit (QIAGEN). The HBV-DNA was amplified with primers specific for the HBV core and envelope genes, as described previously (13). The amplicons were purified and core/envelope regions were sequenced directly using ABI 377 Automated Sequencer (Applied Biosystems).
Antibodies.
For flow cytometry we used the following FITC PE or CyChrome-conjugated antibodies: anti-CD8 (RPA-T8); CD3 (HIT3a); CD27 (M-T271); CD28 (CD28.2) CD38 (HIT2); CD45RA (HI100); CD45RO (UchL1); CD56 (B159); CD62L (Dreg-56); HLA-DR (TU-36); α/β TCR (T10B9.1A-31) (BD PharMingen); CCR-3 (61828.111); CCR-5 (45549.11); CXCR-3 (49801.11); IFN-γ (25723); IL-4 (3007.11) (R&D Systems); IFN-γ (B27); and IL-10 (Jes3–9D7) (Caltag).
For TCR analysis: (Vβ1, Vβ2, Vβ3, Vβ5.1, Vβ5.2, Vβ6.1, Vβ8, BV11, Vβ12, Vβ13.1, Vβ13.6, Vβ14, Vβ16, Vβ17, Vβ20, Vβ21.3, Vβ22; Immunotech) FITC-conjugated antibodies.
Synthetic Peptides.
Peptides corresponding to the HBV genotype D sequence, and single amino acid substituted env 183–91 were purchased from Chiron Mimotopes (Clayton) or from Primm. Purity of peptides was >90% by HPLC analysis.
Staining with HLA-tetrameric Complexes.
HLA-class I tetramers were produced as described previously (20). Tetramer staining of directly purified cells or T cell lines were performed normally at 37°C for 20 min in RPMI 1640 plus 10% FCS. Cells were then washed, suspended in PBS plus 1% FCS. and mAbs against CD8 or different surface molecules were added at 4°C for 30 min. After further washings, cells were analyzed on FACSort™ (Becton Dickinson) using CELLQuest™ software immediately or after addition of 1% paraformaldehyde.
Intracellular IFN-γ Staining.
Ex vivo purified PBMCs or short-term T cell lines were stimulated at 2–3 × 106 cells per milliliter in RPMI 1640, 10% FCS with or without peptides, or with different APC pulsed with peptides for 6 h at 37°C in the presence of Brefeldin A (Sigma-Aldrich) at 10 μg/ml. Cells were washed, stained with Cy-chrome conjugated anti-CD8 antibodies (or different surface markers in selected experiments), then permeabilized and fixed using Permeafix (Ortho Diagnostic Systems) according to the manufacturer's instructions. FITC-conjugated anticytokine antibodies or isotyped-matched control were added (20 min, 4°C), washed twice and analyzed by flow cytometry. In inhibition experiments, T cell lines were incubated with HLA-A2 plus EBV-B cells or macrophages previously incubated with synthetic peptides in the presence of different concentrations of purified anti-CD8 antibody (clone RPA-T8; BD PharMingen) or with anti-IgG1 isotype control (MOPC-21; BD PharMingen). Experiments were then performed as indicated above.
Proliferation Assay using CSFE.
For cell proliferation assays, cells were labeled with CFSE (Molecular Probe) as described previously (21). Briefly, PBMCs were incubated with 1 μM CSFE in PBS at 37°C for 9 min, before stimulating with peptides. Cells were examined the same day of CSFE staining or after 8–9 and 12 d of in vitro culture for evidence of loss of CSFE intensity.
Lytic Assays.
Cytotoxic activity was assessed by incubating the T cell lines with Cr51-labeled HLA-A2 matched target cells (EBV-B cells or macrophages) for 4–5 h in round-bottomed plates. Target cells were either incubated with synthetic peptides or infected with a recombinant Modified Vaccinia Ankara (Oxxon Pharmaccines, Ltd.) carrying HBsAg or a Melanoma tumor epitope as control. Infection and expression of antigen was performed as described previously (22).
Production of T Cell Lines.
PBMCs were suspended at a concentration 2–3 × 106 cells per milliliter in complete T cell medium (MEM α medium [GIBCO BRL], supplemented with 25 mM Hepes, 2 mM l-glutamine, 0.5 mM nonessential AA, and 10% FCS). Cells were stimulated with synthetic peptides in a 96-well plate. Recombinant IL-2 (20 IU/ml) was added on day 4–5 of culture and cells were analyzed after a total of 10–12 d of culture (referred to as short term lines). Oligoclonal lines and clones were further purified by selection of IFN-γ–producing cells using MACS® Secretion Assay (Miltenyi Biotec) or separating CD8+ cells with microbeads (Dynal AS). Selected cells were then seeded in 96-well plates, at concentrations of 1–10–50 cells per well with allogenic irradiated PBMCs (1–2 × 105 cells per milliliter) in complete T cell medium plus 20 U/ml IL-2 and 10 ng/ml IL-7 (R&D Systems). Wells were restimulated with irradiated feeder cells approximately every 2 wk. Growing cells were tested for peptide specificity with ICCS.
Results
Identification of Tet/Neg Cells in Chronic HBV Patients.
The quantity and function of HBV-specific CD8 cells were analyzed in three HLA-A2+ subjects who had recovered from symptomatic acute HBV infection, and in six HLA-A2+ subjects with chronic hepatitis B (3 HBeAg+, HBV-DNA 107–109 copies per milliliter; 3 HBeAg negative, HBV-DNA 105–107 copies per milliliter). The study was focused on the following four well-described HLA-A2–restricted HBV-CTL epitopes: core 18–27; envelope 183–91; envelope 348–57; and polymerase 816–24. The frequency of these HBV-specific CD8 cells was quantified by HLA-tetramers or by ICCS, either directly ex vivo or after 10 d of in vitro expansion. Cells were stimulated once with 1 μM of peptide, a concentration that was unable either to stimulate directly or to expand in vitro HBV-specific CD8 cells in uninfected individuals (data not shown). We found that subjects who had recovered from symptomatic acute hepatitis B (hereafter referred to as “immune”) had HBV-specific frequencies of around 0.2–0.5% of total CD8. Analysis using ICCS and tetramers gave a similar pattern of response but HLA-tetramers were often able to detect more HBV-specific CD8 cells than ICCS (see for example, Fig. 1 patient 1). Cells specific for all the four epitopes expanded efficiently in vitro and produced IFN-γ (and not IL-4 or IL-10, data not shown). The frequency, hierarchy, and function of these HBV-specific CD8 responses were stable in repetitive assays performed over a follow up of >2 y. We were never able to detect the presence of peptide-induced IFN-γ positive CD8 cells in the absence of the specific tetramer positive (tet/pos) CD8 cells in this group of patients.
Figure 1.
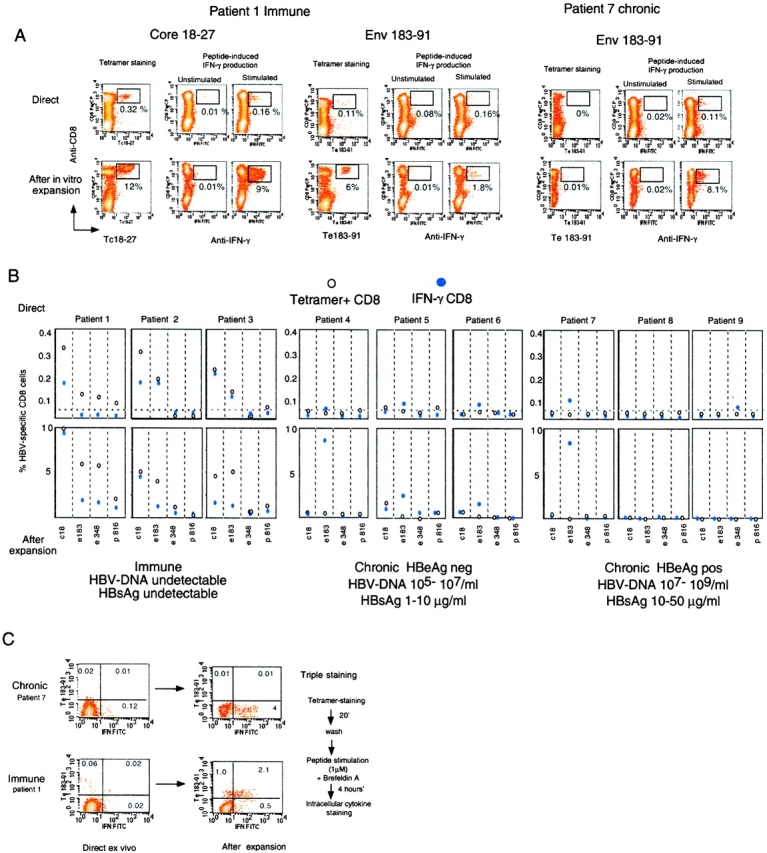
Identification of tet/neg CD8 cells. (A) Detection of epitope-specific CD8 cells with HLA-A2 tetramers and with ICCS. Dot plots are representative of results obtained in “immune” and chronic patients. Numbers indicate the frequency of double positive cells (tet/CD8+ or IFN-γ/CD8+) directly ex vivo (top dot plots) or after 10 d in vitro expansion (bottom dot plots). (B) Frequency of tet/CD8+ and IFN-γ/CD8+ cells in the indicated group of patients directly ex vivo or after 10 d expansion. Frequency of IFN-γ/CD8+ cells were calculated after subtraction of double positive cells obtained in nonstimulated cells. The background level of direct ex vivo tetramer staining (value 0.03% of CD8+ cells) has been calculated in HLA-A2+ noninfected subjects and in HLA-A2 negative HBV-infected patients; this is indicated as a dotted line. (C) Visualization of tet/pos and tet/neg cells producing IFN-γ directly ex vivo and after 10 d in vitro expansion in two representative patients. Dot plots represent live CD8 gated cells. Numbers indicate the relative frequency of the cells in the different quadrants.
The pattern of CD8 response in chronically infected patients was different. As already reported (20), direct ex vivo analysis of circulating HBV-specific CD8 cells using HLA-tetramers were around the background level (0.03%) calculated in control subjects (HLA-A2 plus nonHBV–infected and HLA-A2-HBV–infected subjects). However, direct ICCS assays using 1 μM envelope peptides were able to visualize a population of IFN-γ (and not IL-4– or IL-10–) producing CD8 cells in the absence of specific tetramer binding in 4 out of 6 patients. Although direct ex vivo frequency was low in most of the chronic patients (see patients 4–6, frequency of ∼0.04% above the nonstimulated value), in some patients (see patient 7) the env 183–91 peptide elicited IFN production by 0.11% of circulating CD8 cells. This frequency is comparable to the direct frequency of the HBV-specific CD8 cells in immune patients (Fig. 1 patients 1–3).
ICCS and tetramer staining of CD8 cells performed after peptide stimulation and 10 d of in vitro expansion confirmed the discrepancy between the two methods found directly ex vivo, with values of IFN-γ–producing CD8 cells as high as 10% (patients 4 and 7) in the absence of tetramer staining. These data suggest the presence of tetramer negative (tet/neg) antigen-specific CD8 cells. This population of cells was confirmed by triple staining performed both directly ex vivo and after expansion (Fig. 1 panel c).
Circulating tet/neg antigen-specific CD8 cells were only evident in chronically infected subjects and their specificity appeared to be restricted to envelope antigens. Whereas core 18–27 and polymerase 816–24 specific CD8 cells were visualized by both tetramer and ICCS, the same patient possessed envelope-specific CD8 cells which did not react with tetramers (Fig. 2) . The absence of circulating tet/pos envelope-specific cells prompted us to investigate whether these cells were preferentially sequestrated within the liver. Analysis of intrahepatic T cells in patients with chronic hepatitis B undergoing liver biopsy failed to reveal intrahepatic compartmentalization of Te183–91 positive CD8 cells (Table I). However, the paucity of T cells obtained from liver biopsies did not allow a parallel analysis of IFN-γ–producing cells, precluding testing whether tet/neg-envelope specific cells were present in the liver of these patients. Lack of reactivity of envelope-specific CD8 cells in chronic patients toward the specific tetramers was confirmed using two different preparations, a range of tetramer concentrations (from 1, 2, and 5 μg/ml) and performing the staining at 4 and 37°C (data not shown). The inability of HLA-A2 tetramers to bind to envelope-specific cells is not due to expression of different HLA-A2 subtypes since all the chronic patients (4–7) carried the common HLA-A201 allele subtype. The HLA-restriction of the envelope peptide recognition was investigated by using HLA-A2 positive and negative target cells. Recognition of only HLA-A2 positive pulsed target cells excluded the possibility that the IFN+ tet/neg cells were activated by presentation of envelope peptides through different HLA-class I molecules. Envelope epitopes were recognized not only as synthetic peptides, but also after the processing of endogenously synthesized envelope antigen by a vaccinia virus (data not shown).
Figure 2.

Tet/neg CD8 cells are specific only for envelope epitopes. Dot plots obtained with tetramer staining and ICCS of CD8 lines specific for the indicated core, envelope and polymerase epitopes. Lines were selected by stimulating PBMCs of patient 5 (chronic HBV) in vitro with the corresponding peptides for 10 d.
Table I.
| PatientsHLA-A2+ | HBeAg | HBV-DNA106 cells/ml | ALT U/L | Tc18-27 | Te183-91 | Tp575-83 | T HIV gag77-85 |
|---|---|---|---|---|---|---|---|
| Patient 5 | Neg | 0.8 × 106 | 74 | 1.1% | 0.02% | ND | ND |
| Patient 7 | Pos | 210 × 106 | 227 | 0.3% | 0.08% | 0.1% | ND |
| Patient 8 | Pos | 40 × 106 | 505 | 0.1% | 0.2% | 0.3% | 0.2% |
| Patient 9 | Pos | 20 × 106 | 120 | 0.04% | 0.02% | 0.2% | 0.1% |
| Patient 10a | Pos | 15 × 106 | 120 | 0.01% | 0.02% | 0.01% | ND |
Background level staining of CD8 cells in biopsies from HLA-A2 negative patients with chronic hepatitis B was 0.1% (data from four patients not shown).
Patient 10 had a detectable circulating population of tet/neg env 183-91 specific CD8 cells (5% of total CD8 after 10 d in vitro expansion, data not shown).
Functional T Cell Avidity and TCR Expression of Tet/Neg CD8 Cells.
To investigate the mechanism underlying the lack of tetramer reactivity, we first examined whether tet/neg cells represented a population of T cells with lower TCR functional avidity than the tet/pos cells. Tet/neg (patient 7, chronic) and tet/pos (patient 1, immune) envelope 183–91-specific CD8 cells were either stimulated directly with different concentrations of peptide, or using HLA-A2+ macrophages from an uninfected individual pulsed with the indicated concentrations of peptides (Fig. 3 a). Equivalent concentrations of envelope peptide (10 nM/1 nM) were necessary to activate the tet/neg or the tet/pos envelope specific CD8 cells. Identical results were obtained when CD8 lines from the other chronic patients were compared with lines of immune subjects (data not shown).
Figure 3.

Characterization of tet/neg CD8 cells. (A) Functional TCR avidity of env183 specific CD8 lines: env 183-specific CD8 line of immune patient 1 (tet/pos) and chronic patient 7 (tet/neg) were expanded in parallel in vitro. 8 d after the second round of in vitro stimulation, IFN-γ production, and tetramer staining of CD8 cells was examined. Lines were stimulated with peptide alone or with peptide-pulsed irradiated HLA-A2+ APC. (B) TCR expression of tet/neg CD8 cells. A representative short-term line (10 d in vitro expansion) of patient 4 is shown. The line was tested for Te183–91 staining (left dot plots) and in parallel stained with anti-CD3 and analyzed for IFN-γ production after specific peptide stimulation. Dot plots of ICCS represent gated CD8 cells and show CD3 expression of nonstimulated and peptide-stimulated CD8 cells. Percentage of CD8 TCRlow is shown in the bottom left quadrant. (C) The expression of CD3 and CD8 molecules by env 183–91 specific CD8 cells of patient 2 (immune) and patient 5 (chronic) was determined in parallel by gating on IFN-γ plus CD8 cells after stimulation with env 183–91 peptide (1 μM). At the time of experiments, short term lines were also tested in parallel for tetramer-specific binding and the env 183–91 specific line of patient 5 did not have Te183–91/CD8+ cells. Similar results were obtained with lines of other two chronic patients (patients 4 and 7). (D) Anti-CD8 equally inhibits tet/pos and tet/neg env-specific CD8 lines. Short-term CD8 lines (10 d in vitro expansion) of chronic patient 7 and immune patient 1 were incubated with the indicated concentrations of anti-CD8 and then stimulated with different doses of peptides. Data are presented as percentage of inhibition calculated by dividing the numbers of IFN-γ–producing cells in the presence of anti-CD8 with the numbers observed in the absence of antibody. Original magnification: ×100.
Then, we evaluated whether reduced TCR or CD8 expression could explain the inefficient tetramer binding (23). A tet/neg CD8 line (short term line after 10 d in vitro expansion) containing a population of envelope-specific CD8 cells showed a homogeneous pattern of CD3 staining before restimulation, with no subpopulation of down-regulated cells visible (Fig. 3 B). Furthermore, the level of CD3 expression of envelope 183–91 specific IFN+ tet/neg CD8 cells after peptide-specific stimulation was equivalent to the level found in IFN+ tet/pos cells (Fig. 3 C).
Likewise, in these short-term lines, CD8 molecule expression levels were not reduced in tet/neg cells, but instead were consistently higher in tet/neg than in tet/pos cells (Fig. 3 C). This suggests that the CD8 molecule could contribute to the overall functional avidity observed in tet/neg cells. The importance of the CD8 molecule in the activation of envelope-specific CD8 cells was confirmed by inhibition experiments with anti-CD8 antibody, showing a comparable level of CD8 dependence in both envelope-specific CD8 populations (Fig. 3 D).
Repetitive In Vitro Stimulation Enhances Tetramer-binding Ability.
Then, we investigated whether repetitive in vitro stimulation could convert tet/neg cells to a tet/pos phenotype. This conversion has been shown in a mouse model of influenza virus infection and attributed to the ability of in vitro restimulation to reorganize the display of TCR (21, 24). In agreement with this work, tetramer env 183–91-positive cells became detectable after repetitive in vitro stimulation of IFN+ tet/neg CD8 cells. 2–3 rounds of in vitro stimulation were necessary in chronic patients 3–5 (Fig. 4 A), whereas we were unable to convert cells of patient 7 even after 3 rounds of stimulation (40 d of in vitro expansion (data not shown).
Figure 4.


Effect of antigenic stimulation on tetramer staining by env-183 specific CD8 cells. (A). PBMCs were stimulated directly in vitro with peptide (1 μM) and at 10–12 d intervals expanded once (1×) and twice (2×) in vitro with irradiated APC (EBV-B cells) pulsed with peptide (1 μM). Dot plots show Te183–91 staining directly ex vivo and after 20 d (1×) and 30 d (2×) of in vitro expansion. Numbers in the top right dot plots indicate the percentage of total CD8 cells tet/pos. Numbers in boxes indicated the percentage of total CD8 that produce IFN-γ after peptide-specific stimulation. Binding of Tc18–27 was always negative. (B) Tetramer-binding assay in different populations of HBV-specific CD8 cells. Tet/pos converted cells specific for envelope 183–91 and conventional tet/pos CD8 cells specific for core 18–27 (patients 1 and 5) and envelope 183–91 of patient 1 were incubated with graded concentration of tetramers for 20 min at the indicated temperatures before adding anti-CD8. Cells were washed, fixed, and analyzed immediately by flow cytometry. Data are shown as mean fluorescence intensity of double positive tet/CD8 population. (C) Tetramer dissociation assay. Tet/pos envelope 183 specific CD8 cells of chronic (converted tet/pos cells) and immune patients (tet/pos) were stained with saturating concentrations of Te183 (1 μg/ml) at 37°C for 20 min before addition of anti-CD8 antibody. Cells were then washed (three times), kept at 4°C, and analyzed by FACS® at the indicated time points. Results are expressed as a percentage of tet/pos CD8+ present at time 0.
Although repetitive in vitro stimulation may reorganize the TCR display to allow HLA-tetramer binding (24, 25), the “converted” tet/pos envelope-specific CD8 cells retain unique characteristics. This was evident when tetramer staining of these cells and of conventional tet/pos CD8 cells was performed at different temperatures. Whereas specific staining intensity was increased at 37°C in all the classical tet/pos cells, the numbers (data not shown) and intensity (Fig. 4 B) of converted tet/pos-CD8 was less temperature dependent.
This confirmed that the envelope-specific CD8 cells of chronic patients are not classical “low affinity” tetramer-binding cells, since in this case staining performed at 37°C should eliminate any binding seen at 4°C (26). At the same time, the temperature independent binding of converted tet/pos cells suggests a lack of active participation in the staining process, since redistribution of the TCR and HLA-tetramer internalization would be maximal at the physiological temperature (26). In line with this interpretation, converted tet/pos cells also had an altered pattern of decay (Fig. 4 C). Envelope-specific CD8 cells of chronic and immune patients were stained with tetramers and after washing the tetramer-dissociation rate was analyzed. The intensity of tetramer-binding in envelope-specific CD8 cells from chronic patients had a more pronounced decay compared with envelope-specific CD8 cells of acute patients.
Taken together, these data indicate that even though the envelope-specific CD8 cells, which persist in chronically infected patients, can be converted to a tet/pos phenotype, they seem to maintain a different functional identity. To further characterize the maturation state of these ‘converted’ cells we compared their phenotypic markers (Table II) with those expressed by conventional tet/pos cells, which had been expanded in parallel. Despite the same numbers of rounds of in vitro restimulation, converted tet/pos CD8 cells maintain a less terminally differentiated phenotype (CD38lo, CD56lo, and CD28h) than classical tet/pos CD8 cells (CD38h, CD56h, and CD28lo) (27). Of note, the differences described in staining pattern and phenotype between “converted” and “classical” tet/pos cells argue against the possibility of converted/tet pos cells being derived in culture from a contaminating population of classical tet/pos cells, but confirm their distinct functional identities.
Table II.
Phenotypic Analysis of CD8 Lines
| Markers | CD8 linechronic env 183 | CD8 line immunecore 18-27 |
|---|---|---|
| HLA-DR | ++ | ++ |
| CD69 | +/− | +/− |
| CD56 | − | ++ |
| CD38 | − | ++ |
| CD62L | − | −/+ |
| CCR5 | + | + |
| CCR3 | − | − |
| CXCR3 | ++ | ++ |
| CD45 R0 | ++ | ++ |
| CD45 RA | − | − |
| CD27 | − | − |
| CD28 | ++ | − |
| CD95 | ++ | ++ |
++, high expression; +, intermediate; −/+, low expression; −, absent.
Contribution of Tet/Neg CD8 Cells to HBV Control.
Having demonstrated the persistence of envelope-specific CD8 cells in the circulation of chronically HBV-infected patients we wanted to determine whether these cells exert any immunological pressure on the virus or whether they present defects in antiviral functions. The high level of HBV replication present in these chronic patients (105–109 copies per milliliter) suggests that envelope-specific CD8 cells have a negligible effect on HBV replication, but specific mutations in envelope epitopes could allow virus to escape CD8 control. Functionally active core 18–27 specific CD8 cells are demonstrable only in chronically infected patients with virus mutations in the core epitope (13, 20). Consistent with these previous findings, core 18–27 specific CD8 were only efficiently expanded from patient 5, in whom the core sequence was mutated in position 27, a mutation that reduces HLA-binding affinity (Fig. 1 and Table III). In contrast, expansion of envelope-specific CD8 cells was not attributable to mutations within the corresponding envelope epitopes (Table III).
Table III.
Expansion of Epitope-specific CD8 Cells and Viral Sequence
| Frequency of specific CD8 cells
|
Epitope sequence | ||
|---|---|---|---|
| Patients | Direct | After expansion | |
| Patient 4 | |||
| Env 183–91 | 0.03% | 9% | WT |
| Core 18–27 | 0.02% | 0% | WT |
| Patient 5 | |||
| Env 183–91 | 0.06% | 2.8% | WT |
| Core 18–27 | 0.04% | 1.8% | 127 |
| Patient 6 | |||
| Env 183–91 | 0.04% | 2% | WT |
| Core 18–27 | 0.02% | 0.4% | WT |
| Patient 7 | |||
| Env 183–91 | 0.08% | 9.4% | WT |
| Core 18–27 | 0.02% | 0% | WT |
The lack of selective pressure exerted by the envelope-specific cells prompted us to investigate whether these cells are in a tolerant state or display alterations in antiviral functions. Production of antiviral cytokines did not appear to be impaired, since tet/neg CD8 cells produce IFN-γ, directly ex vivo and after in vitro culture (Fig. 1 B). Lytic ability was tested in a conventional Cr51 release assay, showing that envelope-specific tet/neg CD8 cells expanded in vitro were able to lyse target cells pulsed with the specific peptide (Fig. 5 B). Cells were also perforin positive by ICCS (data not shown). Ideally, cytolytic ability should be tested directly ex vivo, since this function could be altered by in vitro stimulation (7). However, the frequency of HBV envelope-specific CD8 cells was too low to allow detection of cytolytic ability ex vivo even in immune subjects (data not shown).
Figure 5.


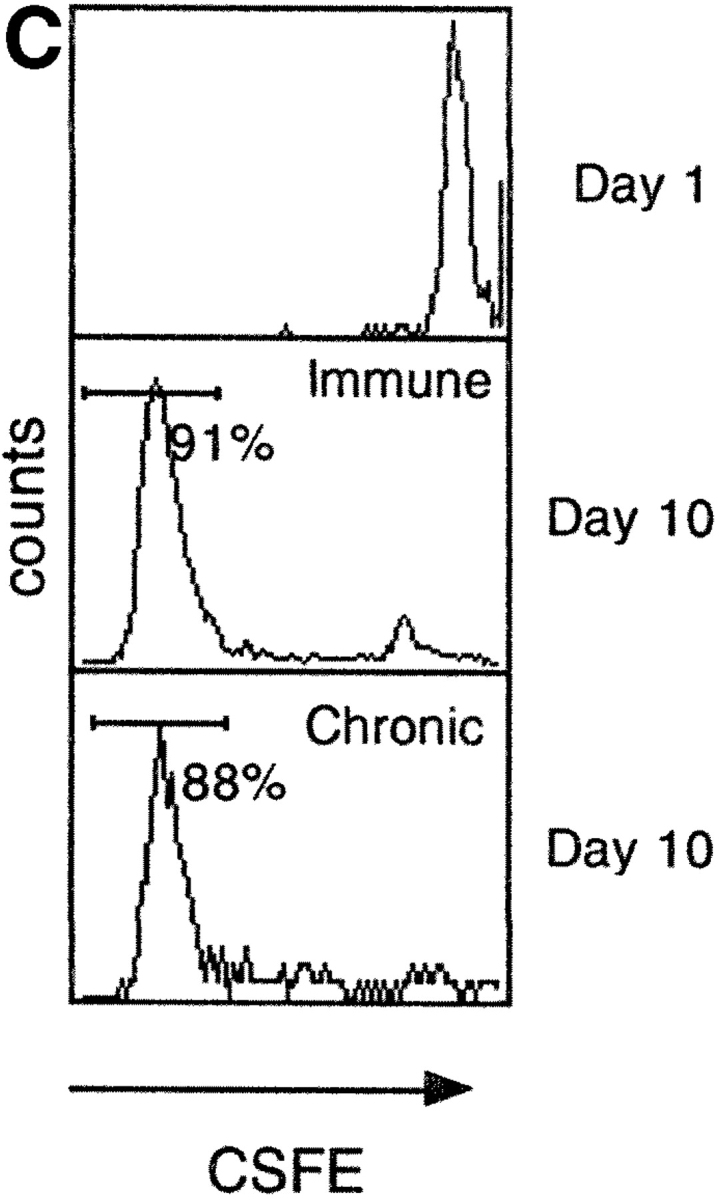
Functional characterization of tet/neg CD8 cells. (A) Functional TCR affinity of env183–91 CD8 cells of chronic and immune subjects. PBMCs stimulated with different concentrations of peptides were tested after 10 d of in vitro expansion for HLA-tetramer binding and peptide specific IFN-γ production. Bars represent the frequency of IFN-γ–producing CD8 cells after stimulation with different doses of env 183 peptide. Numbers in boxes represent the percentage of tet/pos CD8 cells found in the same line. (B) Lysis of peptide coated target cells (EBV-B cells) by tet/pos and tet/neg envelope specific short-term CD8 lines. Lines examined with ICCS showed 4–5% of peptide specific IFN-γ–producing CD8 cells and experiments were performed with an E/T ratio of 10:1. (C) Proliferation of tet/neg envelope 183-specific CD8 cells. PBMCs were labeled with CSFE before peptide specific stimulation. Dye intensity was tested in total CD8 cells at day 1 and after 10 d of in vitro expansion for peptide-specific IFN-y–producing CD8 cells. Histograms show representative results obtained from patient 2 (immune/core 18–27 specific CD8 cells) and patient 6 (chronic/env 183–91 specific CD8 cells).
A defect in clonal expansion could impair the ability of virus-specific CD8 cells to expand and control viral replication (28), but the high frequency of envelope-specific CD8 cells in PBMCs of chronic patients after 10 d of culture suggested that such an expansion defect was not operative. To better analyze whether the envelope-specific CD8 cells of chronic patients present a different activation threshold for CD8 expansion, PBMCs of different immune and chronic patients were stimulated directly after ex vivo purification with varying doses of envelope 183–91 peptide. The quantity of envelope-specific CD8 cells expanded by 10 d of in vitro stimulation was tested with tetramer and ICCS (Fig. 5 A). Similar quantities of envelope peptide (1 uM–100 nM) were necessary to expand envelope 183–91 specific cells from PBMCs of chronic and immune patients. The only difference between the cells expanded from chronic patients and the ones present in immune patients is in the ability to bind tetramers and not in the dose of peptide required for CD8 expansion. These results show that tet/neg cells are not in an anergic state and further support the finding that these cells have comparable functional T cell avidity to tet/pos cells. Interestingly, the expansion potential of envelope-specific cells present in chronic patients is similar to that of resting CD8 cells present in immune patients. This was tested by comparing the rate of division of core 18–27 specific CD8 cells of immune patients with the envelope 183–91 specific CD8 cells of chronic patients. Circulating PBMCs were labeled with the fluorescent dye CSFE and stimulated with the envelope 183–91 (chronic) or core 18–27 (immune) peptide. CSFE staining of CD8 cells was measured (Fig. 5 C) at the time of in vitro stimulation and after 10 or 12 d of in vitro culture. The results showed that the efficiency of cell division reached after in vitro expansion was identical for both populations (Fig. 5 C). Thus, tet/neg cells of chronic patients have a potential capacity for in vitro expansion similar to the immunodominant response seen in patients who are able to control HBV infection.
Different Activation Requirements of Tet/Neg CD8 Cells.
Although we were unable to demonstrate any functional deficits of tet/neg CD8 cells, this was difficult to reconcile with the demonstrated absence of a selective pressure on the virus, which suggests that envelope-specific CD8 cells can ignore HBV in vivo. Ideally, the activation of envelope-specific CD8 cells should be tested using infected hepatocytes (29, 30) or liver endothelial cells (31) to mimic physiological in vivo presentation. In the absence of such a system, we compared the presentation of HLA–peptide complexes by circulating APC and tetramers. MHC/peptide oligomers have been used in other studies to dissect the minimum requirements for T cell activation (32, 33). Fig. 6 A shows the experiments in which IFN-γ production by the different populations of HBV-specific CD8 cells found in short term lines (10 d of in vitro expansion) was tested after pulsing cells with a tetramer concentration that fully stained specific CD8 cells (1 μg/ml). In parallel, the same CD8 cells were stimulated with APCs (B cells or macrophages) pulsed with peptides (1 μM). Tet/pos CD8 cells (core 18–27 and envelope 183–91 of immune (patient 1) and core 18–27 of chronic (patient 5) patients could be stimulated by tetramers alone to produce IFN-γ. In contrast, tetramers did not stimulate the production of IFN-γ by tet/neg CD8 cells (Fig. 6 A). Increasing the quantity of tetramer or the time of stimulation (up to a maximum of 4 h) did not change the results (data not shown). Thus, the lack of tetramer binding correlated with an inability to activate CD8 cells for IFN-γ production.
Figure 6.

Analysis of CD8 activation with HLA-tetramers. (A) Short-term CD8 lines (10 d in vitro expansion) were stimulated with tetramers (1 μg/ml) or with HLA-A2+ APC pulsed with 1 μM of peptide. IFN-γ–producing CD8 cells were examined with ICCS. Bars represent the percentage of total CD8 cells producing IFN-γ. Representative experiments of chronic patient 5 and immune patient 1 are shown. A similar pattern was observed in chronic patient 4 and 7 and in immune patient 2. (B) Converted tet/pos cells are not activated by HLA tetramer. Top dot plots show the phenotypic characterisation (tetramer staining only) of a tet/pos core 18–27 specific CD8 oligoclonal line derived from an immune patient (patient 1) and of a converted tet/pos env 183–91 CD8 line of chronic patient 6. Oligoclonal lines were produced after selection and expansion (four rounds of in vitro stimulation) of IFN-γ–producing cells. Bottom dot blots examined the same lines stained with tetramers and then stimulated or not stimulated (tetramer only) with peptide pulsed HLA-A2+ APC. Lines were then tested with ICCS for production of IFN-γ. Dot blots of gated CD8 cells are shown. Note that the decreased staining intensity of Te 183–91 in the ICCS dot plots is explained by the faster decay of tetramer binding present in converted tet/pos cells. Numbers in the top corners represent the percentage of total CD8 cells producing IFN-γ.
More surprisingly, the envelope-specific CD8 cells (from oligoclonal lines of chronic patients obtained after four rounds of in vitro stimulation of antigen-specific selected cells) were not activated by tetramers even after conversion to a tet/pos phenotype (Fig. 6 B). Even when tet/pos ‘converted’ CD8 cells acquired an intensity of fluorescent staining identical to classical tet/pos cells, these cells were still not activated by tetramers, but only by peptide-loaded APC (Fig. 6 B). Since activation of CD8 cells by tetramers requires TCR clustering (32, 33), the inability of HLA-tetramers to fully activate envelope-specific CD8 cells of chronic patients suggests that tetramers alone are insufficient to cluster the TCRs in a way that permits full stimulation. Alternatively, the TCRs may aggregate, but need costimuli provided by target cells (34). These data show that even after conversion to a tet/pos phenotype, the envelope-specific CD8 cells of chronic patients maintain different requirements for T cell activation in comparison with “classical” tet/pos HBV-specific CD8 cells.
Selective Persistence of Tet/Neg CD8 Cells.
During persistent viral infection the TCR repertoire of virus-specific CD8 cells is continuously selected (35). To determine whether this feature might also apply to the altered tetramer-binding CD8 cells persisting in chronic hepatitis B, the Vβ profile and fine-specificity of the envelope-specific CD8 cells generated in immune or chronic individuals was tested. Envelope specific IFN-γ–producing CD8 cells present in short-term lines were stained with a panel of 17 different Vβ-specific antibodies. This Vβ antibody panel covers approximately half of the available TCR repertoire, but allows an assessment of oligo/polyclonality of the specific CD8 cells present in immune or chronically infected subjects to be made.
Fig. 7 A shows that the Vβ usage of tet/neg envelope-specific CD8 cells of chronic patients 4 and 7 was restricted to Vβ17, whereas a more heterogeneous Vβ usage was displayed by envelope-specific CD8 cells of immune subjects. Results from functional fingerprint experiments were consistent with the broader T cell usage of immune subjects. The production of IFN-γ by envelope-specific CD8 cells of immune (tet/pos) and chronic patients (tet/neg) was stimulated with single alanine substituted env 183–91 analogue peptides. Tet/pos envelope-specific CD8 cells of immune patients displayed a degree of flexibility in the recognition of the substituted peptides, whereas tet/neg cells of chronic patients were activated only by the WT (nonmutated) env 183–91 sequence (Fig. 7 B). Therefore, altered tetramer binding is present only in a selected oligoclonal envelope-specific CD8 population.
Figure 7.

Narrow TCR usage of envelope-specific CD8 cells of chronic patients. (A) Analysis of Vβ usage of envelope specific CD8 cells of indicated patients. PBMCs were stimulated with envelope peptide and after 10 d of in vitro expansion, cell lines were analyzed with ICCS for presence of peptide-specific IFN-γ–producing CD8 cells. The percentage of IFN-γ CD8 cells expressing each Vβ chain was calculated by analysis of at least 1,000 IFN-γ CD8+ cells, indicated by bars. (B) Functional fingerprinting of envelope-specific CD8 cells of chronic and immune patients. PBMCs were stimulated with 1 μM of env 183–91 peptide (wt) and with analogue single alanine substituted peptides. Percentage of IFN-γ CD8 cells specific for the wt peptide was calculated after 10 d of in vitro expansion.
Discussion
CD8 T cells in chronic viral infections (11) and tumors (36) are prone to deletion or functional inactivation. A particular characteristic of chronic HBV infection is the production of large quantities of subviral particles containing only envelope antigens and associated host-derived lipid (37, 38). These particles are present in a 103–106-fold excess over virions, reaching exceptional quantities of ∼3 × 1013 particles per milliliter (1–300 μg/ml; reference 39). HBV subviral particles are noninfectious and the reason for evolving this level of synthetic effort is not well understood. A possible explanation is that this constitutes a way of subverting the antiviral response. In this work we demonstrate that a population of envelope-specific CD8 cells persist in chronic hepatitis B, apparently escaping exhaustion mediated by high concentrations of antigen. These virus-specific CD8 cells are not the classical tolerant cells, since they can be stimulated in vitro to produce IFN-γ, as well as being able to lyse and expand efficiently. However, a population of specific CD8 cells with altered HLA/peptide tetramer binding and ignoring HBV in vivo appear to have been selected by envelope antigen.
Until now CD8 cells unable to efficiently bind MHC class I tetramers have been observed in mouse models of viral infections (21, 23, 40) and in humans, in selected CTL clones from a patient with melanoma (41). They have been found in situations of high doses of viral antigen or with partial/complete homology to self-peptide, reinforcing the idea that tet/neg CD8 cells are a population of cells able to persist in the presence of large amounts of cognate antigen.
It has been proposed that the mechanisms underlying this altered tetramer binding may include TCR down-regulation and the selection of a low avidity T cell population (11). The former is precluded by our data showing no TCR downmodulation on tet/neg CD8 cells of chronic patients. As regards the question of “low avidity T cells,” this has been reported as an explanation for lack of tetramer binding and increased tetramer dissociation rates (40, 42). However, it is becoming increasingly clear that HLA-tetramers cannot measure overall functional T cell avidity (41, 43, 44), since this is often highly dependent on other factors such as costimulatory molecules and signal transduction pathways (45). Tetramer binding may reflect the intrinsic TCR/MHC affinity (46), but even the accuracy of this is questionable due to the multivalent engagement of tetramers (45) and their requirement for correct TCR organization at the immunological synapse (24). Thus, we cannot formerly exclude that tet/neg CD8 cells have an intrinsic low TCR affinity, but data from three different functional assays (IFN-γ production, T cell expansion, and cytotoxicity) clearly show that they do not represent low avidity T cells.
Our data are more in line with the explanation for altered tetramer-binding proposed by Braciale et al. (21) as representing a state of “incomplete T cell maturation.” In their model of influenza-infected mice, they suggest that tetramer-negativity may result from a defect in TCR organization (21), an hypothesis further supported by recent experiments where T cells could be rendered “tet/neg” by chemical disruption of lipid rafts and consequent loss of normal TCR topology (24). The influenza-specific tet/neg cells reverted to a tet/pos phenotype after repetitive in vitro stimulation. In agreement with these data in mice, we found that envelope-specific CD8 cells in the majority of patients with chronic hepatitis B revert to a tet/pos phenotype after repetitive in vitro stimulation. This phenotypic change can be explained by the demonstration that in vitro T cell activation can modulate the plasma membrane cholesterol content and redistribute the TCR to enhance MHC dimer binding (25). Even though tetramer binding of these envelope-specific CD8 cells can be promoted, the cells maintain peculiar features that still differentiate them from the “classical” HBV-specific CD8 cells visualized by tetramers directly ex vivo. In this study we found that the reverted tet/pos envelope-specific CD8 cells are insensitive to temperature-induced up-regulation of tetramer-binding. The fact that staining is not augmented at physiological temperatures further reinforces the possibility that processes such as redistribution of TCR, or tetramer internalization, are defective in these cells. Future studies of membrane mobility, raft organization, and TCR display will be needed to clarify these issues.
In addition to demonstrating the presence of virus-specific CD8 cells with altered tetramer binding, we also found that the behavior of CD8 cells in patients with chronic HBV infection differed according to their antigen specificity. Here, and in previous work (13, 20), an efficient core 18–27 specific CD8 response was only demonstrated in the circulation of patients with chronic hepatitis B when mutations in the core epitope were present. High levels of replication (HBV-DNA >107 copies per milliliter) of nonmutated HBV are associated with an inability to expand core 18–27 CD8 cells in the circulation. In contrast, env 183–91–specific CD8 cells are able to expand, displaying the same proliferative potential, activation threshold, and IFN-γ production ability as memory CD8 cells present in immune individuals. This CD8 responsiveness is found in patients that show high levels of virus replication without mutations in the relevant epitopes, suggesting that the functional activity of these cells in vitro is not mirrored by the exertion of immunological pressure in vivo. The tet/neg CD8 cells that persist in patients with chronic HBV infection are therefore not ‘anergic’ but seem more similar to the ‘ignorant’ cells present in mice which express high quantities of antigen localized to peripheral sites (47–50) or the liver (51–53). Of note, the tet/neg CD8 cells present in influenza-transgenic mice display similar behavior, in that they ignore the product of the transgene in vivo, but are not anergic, since tolerance can be broken by viral infection (40).
The demonstration that different mechanisms can operate in the same host to silence antiviral CD8 responses has already been reported in LCMV infection (9). In this murine infection, Zajac et al. have suggested that the important parameter influencing the fate of CD8 cells specific for different antigens could be the degree of activation. Our findings may be in line with this interpretation. Despite presenting similar HLA-A2–binding affinity, core 18–27 specific CD8 cells are numerically dominant over envelope specificities in acute and resolved HBV infection, suggesting that the core 18–27 epitope may be more immunogenic than the envelope specificities. This could depend on the different quantity of envelope or core peptides presented at the surface of the infected cells. A further explanation for the peculiar features of envelope-specific CD8 cells may be the fact that core and envelope antigens are present in the patient in different forms and concentrations (12). Core is preferentially (but not exclusively) a cell-associated antigen (54), whereas envelope is present in large quantity in a soluble form, making it the ideal substrate for presentation by liver endothelial cells. These cells are a population of nonmyeloid APCs, which are present in the liver, and are specialized in the presentation of exogenous soluble antigen, and in the induction of antigen-specific CD8 cell tolerance (31).
Envelope-specific tet/neg CD8 cells might therefore escape deletion by high dose of antigen because they are kept in a state of “partial tolerance” by liver endothelial cells. This interpretation could explain why, in vivo, these CD8 cells appear to ignore the infecting virus. This state of tolerance is clearly only partial. Tet/neg envelope-specific CD8 cells can be activated in vitro and the TCR repertoire of tet/neg envelope-specific CD8 cells is narrower in chronic patients than in immune, suggesting that only an oligoclonal population of CD8 cells is able to persist. Interestingly, we have been unable to detect envelope-specific CD8 cells in three patients with minimal liver injury, high levels of HBV DNA, and a concentration of HBsAg >80 μg/ml (unpublished data), further suggesting that there is a concentration threshold of envelope antigen above which envelope-specific CD8 cells are deleted.
Whatever the explanations for the persistence of envelope-specific CD8 cells in patients with chronic hepatitis B, the demonstration that these cells are not fully anergic may have important therapeutic implications. Thus, their presence might explain the efficacy of envelope-based therapeutic vaccination in patients (55, 56) and animals with chronic hepadnavirus infections (57, 58). In addition, the demonstration that tumor or virus-specific CD8 cells which ignore high doses of antigen can acquire protective properties in vivo (59), might support the idea that indifferent CD8 cells may represent a population of cells that can be expanded for therapeutic use. It will also be interesting to see whether the persistence of tet/neg CD8 cells is a general phenomenon, occurring in other human chronic infections and tumors, or is mainly dependent on the HBV liver tropism.
Acknowledgments
The authors wish to thank Oxxon Pharmaccines, Ltd (Oxford, UK) for recombinant Vaccinia Ankara expressing HBsAg and melanoma antigen. Helpful suggestions and/or critical evaluation of this manuscript were provided by Thomas Cameron, Francis Chisari, and Enza Piccolella.
This work was mainly supported by a National Lottery Board grant awarded through the Digestive Disorders Foundation. G.J.M. Webster is supported by Glaxo SmithKline. M.K. Maini is supported by a collaborative grant from The Edward Jenner Institute of Vaccine Research.
Footnotes
Abbreviations used in this paper: HBV, hepatitis B virus; ICCS, intracellular cytokine staining; tet/neg, tetramer negative; tet/pos, tetramer positive.
References
- 1.Pircher, H., A. Moskophidis, U. Rohrer, K. Burki, H. Hengartner, and R. Zinkernagel. 1990. Viral escape by selection of cytotoxic T cell-resistant virus variants in vivo. Nature. 346:629–633. [DOI] [PubMed] [Google Scholar]
- 2.Ploegh, H.L. 1998. Viral strategies of immune evasion. Science. 280:248–253. [DOI] [PubMed] [Google Scholar]
- 3.Moskophidis, D., F. Lechner, H. Pircher, and R.M. Zinkernagel. 1993. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 362:758–761. [DOI] [PubMed] [Google Scholar]
- 4.Gallimore, A., A. Glithero, A. Godkin, A.C. Tissot, A. Pluckthun, T. Elliott, H. Hengartner, and R. Zinkernagel. 1998. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J. Exp. Med. 187:1383–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greten, T., J. Slansky, R. Kubota, S. Soldan, E. Jaffee, T. Leist, D. Pardoll, S. Jacobson, and J. Schneck. 1998. Direct visualisation of antigen-specific T cells: HTLV-1 tax 11-19 specific CD8+ T cells are activated in peripheral blood and accumulate in cerebrospinal fluid from HAM/TSP patients. Proc. Natl. Acad. Sci. USA. 95:7568–7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lechner, F., D. Wong, P. Dunbar, R. Chapman, R. Chung, P. Dohrenwend, G. Robbins, R. Phillips, P. Klenerman, and B. Walker. 2000. Analysis of succesful immune responses in person infected with hepatitis C virus. J. Exp. Med. 191:1499–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiong, Y., J. Luscher, J. Altman, M. Hulsey, H. Robinson, M. Ostrowski, B. Barber, and K. MacDonald. 2001. Simian immunodeficiency virus (SIV) infection of rhesus macaque induces SIV-specific CD8+ T cells with a defect in effector function that is reversible on extented IL-2 incubation. J. Virol. 75:3028–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Appay, V., D. Nixon, S. Donohoe, G. Gillespie, T. Dong, A. King, G. Ogg, H. Spiegel, C. Conlon, C. Spina, et al. 2000. HIV-specific CD8+ T cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 192:63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zajac, A.J., J.N. Blattman, K. Murali-Krishna, D.J.D. Sourdive, M. Suresh, J.D. Altman, and R. Ahmed. 1998. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 188:2205–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ou, R., S. Zhou, L. Huang, and D. Moskophidis. 2001. Critical role for α/β and γ interferons in persistence of lymphocytic choriomeningitis virus by clonal exhaustion of cytotoxic T cells. J. Virol. 75:8407–8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Welsh, R. 2001. Assessing CD8 T cell number and dysfunction in the presence of antigen. J. Exp. Med. 193:19–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seeger, C., and W.S. Mason. 2000. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64:51–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertoletti, A., A. Costanzo, F.V. Chisari, M. Levrero, M. Artini, A. Sette, A. Penna, T. Giuberti, F. Fiaccadori, and C. Ferrari. 1994. Cytotoxic T lymphocyte response to a wild type hepatitis B virus epitope in patients chronically infected by variant viruses carrying substitutions within the epitope. J. Exp. Med. 180:933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertoletti, A., A. Sette, F.V. Chisari, A. Penna, M. Levrero, M. De Carli, F. Fiaccadori, and C. Ferrari. 1994. Natural variants of cytotoxic epitopes are T-cell receptor antagonists for antiviral cytotoxic T cells. Nature. 369:407–410. [DOI] [PubMed] [Google Scholar]
- 15.Michalak, T., P. Hodgson, and N. Churchill. 2000. Posttranscriptional inhibition of class I major histocompatibility complex presentation on hepatocytes and lymphoid cells in chronic woodchuck hepatitis virus infection. J. Virol. 74:4483–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chisari, F., and C. Ferrari. 1995. Hepatitis B virus immunopathogenesis. Annu. Rev. Immunol. 13:29–60. [DOI] [PubMed] [Google Scholar]
- 17.Lee, G., L. Hwang, R. Beasley, S. Chen, and T. Lee. 1983. Immunogenicity of hepatitis B virus vaccine in healthy Chinese neonates. J. Infect. Dis. 148:526–529. [DOI] [PubMed] [Google Scholar]
- 18.Rehermann, B., D. Lau, J.H. Hoofnagle, and F.V. Chisari. 1996. Cytotoxic T lymphocyte responsiveness after resolution of chronic hepatitis B virus infection. J. Clin. Invest. 97:1655–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boni, C., A. Penna, G. Ogg, A. Bertoletti, M. Pilli, A. Cavalli, S. Urbani, R. Boheme, R. Panebianco, F. Fiaccadori, and C. Ferrari. 2001. Lamivudine treatment can overcome cytotoxic T cell hyporesponsiveness in chronic hepatitis B: new perspective for immune therapy. Hepatology. 33:963–971. [DOI] [PubMed] [Google Scholar]
- 20.Maini, M.K., C. Boni, C.K. Lee, J.R. Larrubia, S. Reignat, G.S. Ogg, A.S. King, J. Herberg, R. Gilson, A. Alisa, et al. 2000. The role of virus-specific CD8+ cells in viral control and liver damage during persistent hepatitis B virus (HBV) infection. J. Exp. Med. 191:1269–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spencer, J., and T. Braciale. 2000. Incomplete CD8+ T lymphocyte differentiation as a mechanism for subdominant cytotoxic T lymphocyte responses to a viral antigen. J. Exp. Med. 191:1687–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dorrell, L., C. O'Callaghan, W. Britton, S. Hambleton, A. McMichael, G. Smith, S. Rowland-Jones, and T. Blanchard. 2000. Recombinant modified vaccinia Ankara efficiently restimulates human cytotoxic T lymphocytes in vitro. Vaccine. 15:327–336. [DOI] [PubMed] [Google Scholar]
- 23.Moser, J., J. Altman, and A. Lukacher. 2001. Antiviral CD8+ T cell responses in neonatal mice: susceptibility to polyoma virus-induced tumors is associated with lack of cytotoxic function by viral antigen-specific T cells. J. Exp. Med. 193:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drake, D., III, and T. Braciale. 2001. Cutting edge: lipid raft integrity affects the efficiency of MHC Class I tetramer binding and cell surface TCR arrangement on CD8+ T cells. J. Immunol. 166:7009–7013. [DOI] [PubMed] [Google Scholar]
- 25.Fahmy, T., J. Bieler, M. Edidin, and J. Schneck. 2001. Increased TCR avidity after T cell activation: a mechanism for sensing low-density antigen. Immunity. 14:135–143. [PubMed] [Google Scholar]
- 26.Whelan, J., P. Dunbar, D. Price, M. Purbhoo, F. Lechner, G. Ogg, G. Griffiths, R. Phillips, V. Cerundolo, and A. Sewell. 1999. Specificity of CTL interactions with peptide-MHC class I tetrameric complexes is temperature dependent. J. Immunol. 163:4342–4348. [PubMed] [Google Scholar]
- 27.Pittet, M., D. Speiser, D. Valmori, J. Cerottini, and P. Romero. 2000. Cutting edge: cytolytic effector function in human circulating CD8+ T cells closely correlates with CD56 surface expression. J. Immunol. 164:1148–1152. [DOI] [PubMed] [Google Scholar]
- 28.Ehl, S., P. Klenerman, P. Aichele, H. Hengartner, and R.M. Zinkernagel. 1997. A functional and kinetic comparison of antiviral effector and memory cytotoxic T lymphocyte populations in vivo and in vitro. Eur. J. Immunol. 27:3404–3413. [DOI] [PubMed] [Google Scholar]
- 29.Bertolino, P., M.-C. Trescol-Biemont, and C. Rabourdin-Combe. 1998. Hepatocytes induce functional activation of naive CD8+ T lymphocytes but fail to promote survival. Eur. J. Immunol. 28:221–236. [DOI] [PubMed] [Google Scholar]
- 30.Bertolino, P., D. Bowen, G. McCaughan, and B. Fazekas de St. Groth. 2001. Antigen-specific primary activation of CD8+ T cells within the liver. J. Immunol. 166:5430–5438. [DOI] [PubMed] [Google Scholar]
- 31.Limmer, A., J. Ohl, C. Kurts, H.-G. Ljunggren, Y. Reiss, M. Groettrup, F. Momburg, B. Arnold, and P. Knolle. 2000. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T cell tolerance. Nat. Med. 6:1348–1354. [DOI] [PubMed] [Google Scholar]
- 32.Boniface, J., J. Rabinowitz, C. Wulfing, J. Hampl, Z. Reich, J. Altman, R. Kantor, C. Beeson, H. McConnell, and M. Davis. 1998. Initiation of signal transduction through the T cell receptor requires the peptide multivalent engagement of MHC ligands. Immunity. 998:459–466. [DOI] [PubMed] [Google Scholar]
- 33.Cochran, J., T. Cameron, and L. Stern. 2000. The relationship of MHC-peptide binding and T cell activation probed using chemically defined MHC class II oligomers. Immunity. 12:241–250. [DOI] [PubMed] [Google Scholar]
- 34.Delon, J., C. Gregoire, B. Malissen, S. Darche, F. Lemaitre, P. Kourilsky, J.-P. Abastado, and A. Trautmann. 1998. CD8 expression allows T cell signaling by monomeric peptide-MHC complexes. Immunity. 9:467–473. [DOI] [PubMed] [Google Scholar]
- 35.Lin, M., and R. Welsh. 1998. Stability and diversity of T cell receptor repertoire usage during lymphocytic choriomeningitis virus infection of mice. J. Exp. Med. 188:1993–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee, P., C. Yee, P. Savage, L. Fong, D. Brockstedt, J. Weber, D. Johnson, S. Sweeter, J. Thompson, P. Greenberg, et al. 1999. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 5:677–685. [DOI] [PubMed] [Google Scholar]
- 37.Prince, A. 1968. An antigen detected in the blood during the incubation period of serum hepatitis. Proc. Natl. Acad. Sci. USA. 968:814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Millman, I., V. Zavotone, B. Gerstley, et al. 1969. Australia antigen detected in nuclei of liver cells of patients with viral hepatitis by fluorescent antibody technique. Nature. 222:181–184. [DOI] [PubMed] [Google Scholar]
- 39.Kim, C., and J. Tilles. 1973. Purification and biophysical characterization of hepatitis B antigen. J. Clin. Invest. 52:1176–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Visser, K., T. Cordaro, D. Kioussis, J. Haanen, N. Schumacher, and A. Kruisbeek. 2000. Tracing and characterisation of the low-avidity self-specific T cell repertoire. Eur. J. Immunol. 30:1458–1468. [DOI] [PubMed] [Google Scholar]
- 41.Rubio-Godoy, V., V. Dutoit, D. Rimoldi, D. Lienard, F. Lejeune, D. Speiser, P. Guillaume, J.C. Cerottini, P. Romero, and D. Valmori. 2001. Discrepancy between ELISPOT IFN-γ secretion and binding of A2/peptide multimers to TCR reveals interclonal dissociation of CTL effector function from TCR-peptide/MHC complexes half-life. Proc. Natl. Acad. Sci. USA. 98:10302–10307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nugent, C.T., D.J. Morgan, J.A. Biggs, A. Ko, I.M. Pilip, E.G. Pamer, and L.A. Sherman. 2000. Characterization of CD8+ T lymphocytes that persist after peripheral tolerance to a self antigen expressed in the pancreas. J. Immunol. 164:191–200. [DOI] [PubMed] [Google Scholar]
- 43.Slifka, M., and J. Whitton. 2001. Functional avidity maturation of CD8+ T cells without selection of higher affinity TCR. Nat. Immunol. 2:711–717. [DOI] [PubMed] [Google Scholar]
- 44.Derby, M.A., J. Wang, D.H. Margulies, and J.A. Berzofsky. 2001. Two intermediate-avidity cytotoxic T lymphocyte clones with a disparity between functional avidity and MHC tetramer staining. Int. Immunol. 13:817–824. [DOI] [PubMed] [Google Scholar]
- 45.Margulies, D. 2001. TCR avidity: it's not how strong you make it, it's how you make it strong. Nat. Immunol. 2:669–670. [DOI] [PubMed] [Google Scholar]
- 46.Busch, D.H., and E.G. Pamer. 1999. T cell affinity maturation by selective expansion during infection. J. Exp. Med. 189:701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oldstone, M., M. Nerenberg, P. Southern, J. Price, and H. Lewicki. 1991. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: role of anti-self (virus) immune response. Cell. 65:319–326. [DOI] [PubMed] [Google Scholar]
- 48.Ohashi, P., S. Ohen, K. Burki, H. Pircher, C. Ohashi, B. Odermatt, B. Malissen, R. Zinkernagel, and H. Hengartner. 1991. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 65:305–318. [DOI] [PubMed] [Google Scholar]
- 49.Ochsenbein, A., P. Klenerman, U. Karrer, B. Ludewig, M. Pericin, H. Hengartner, and R. Zinkernagel. 1999. Immune surveillance against a solid tumor fails because of immunological ignorance. Proc. Natl. Acad. Sci. USA. 96:2233–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wick, M., P. Dubey, H. Koeppen, C. Siegel, P. Fields, L. Chem, J. Bluestone, and H. Schreiber. 1997. Antigenic cancer cells grow progressively in immune hosts without evidence of T cell exhaustion or systemic anergy. J. Exp. Med. 186:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Voehringer, D., C. Blaser, A. Grawitz, F. Chisari, K. Buerki, and H. Pircher. 2000. Break of T cell ignorance to a viral antigen in the liver induces hepatitis. J. Immunol. 165:2415–2422. [DOI] [PubMed] [Google Scholar]
- 52.Sette, A., C. Oseroff, J. Sidney, J. Alexander, R. Chesnut, K. Kakimi, L. Guidotti, and F. Chisari. 2001. Overcoming T cell tolerance to the hepatitis B virus surface antigen in hepatitis B virus-transgenic mice. J. Immunol. 166:1389–1397. [DOI] [PubMed] [Google Scholar]
- 53.Mancini, M., M. Hadchouel, H. Davis, R. Whalen, P. Tiollais, and M. Michel. 1996. DNA-mediated immunization in a transgenic mouse model for the hepatitis B surface antigen chronic carrier state. Proc. Natl. Acad. Sci. USA. 93:12496–12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Naoumov, N., B. Portmann, R. Tedder, B. Ferns, A. Eddleston, G. Alexander, and R. Williams. 1990. Detection of hepatitis B virus antigens in liver tissue. Gastroenterology. 99:1248–1253. [DOI] [PubMed] [Google Scholar]
- 55.Couillin, I., S. Pol, M. Mancini, F. Driss, C. Brechot, P. Tiollais, and M.L. Michel. 1999. Specific vaccine therapy in chronic hepatitis B: induction of T cell proliferative responses specific for envelope antigens. J. Infect. Dis. 180:15–26. [DOI] [PubMed] [Google Scholar]
- 56.Pol, S., B. Nalpas, F. Driss, M.L. Michel, P. Tiollais, J. Denis, and C. Brecho. 2001. Efficacy and limitations of a specific immunotherapy in chronic hepatitis B. J. Hepatol. 34:917–921. [DOI] [PubMed] [Google Scholar]
- 57.Pancholi, P., D. Lee, Q. Liu, C. Tackney, P. Taylor, M. Perkus, L. Andrus, B. Brotman, and A. Prince. 2001. DNA prime/canarypox boost-based immunotherapy of chronic hepatitis B virus infection in a chimpanzee. Hepatology. 33:448–454. [DOI] [PubMed] [Google Scholar]
- 58.Rollier, C., C. Sunyach, L. Barraud, N. Madani, C. Jamard, C. Trepo, and L. Cova. 1999. Protective and therapeutic effect of DNA-based immunization against hepadnavirus large envelope protein. Gastroenterology. 116:658–665. [DOI] [PubMed] [Google Scholar]
- 59.Speiser, D., R. Miranda, A. Zakarian, M. Bachmann, K. McKall Faienza, B. Odermatt, D. Hanahan, R. Zinkernagel, and P. Ohashi. 1997. Self antigens expressed by solid tumors do not efficiently stimulate naive or activated T cells: implications for immunotherapy. J. Exp. Med. 186:645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]