

Abstract
The balance between pro and antiinflammatory cytokines secreted by T cells regulates both the initiation and perpetuation of inflammatory bowel diseases (IBD). In particular, the balance between interferon (IFN)-γ/interleukin (IL)-4 and transforming growth factor (TGF)-β activity controls chronic intestinal inflammation. However, the molecular pathways that evoke these responses are not well understood. Here, we describe a critical role for the transcription factor T-bet in controlling the mucosal cytokine balance and clinical disease. We studied the expression and function of T-bet in patients with IBD and in mucosal T cells in various T helper (Th)1- and Th2-mediated animal models of chronic intestinal inflammation by taking advantage of mice that lack T-bet and retroviral transduction techniques, respectively. Whereas retroviral transduction of T-bet in CD62L+ CD4+ T cells exacerbated colitis in reconstituted SCID mice, T-bet–deficient T cells failed to induce colitis in adoptive transfer experiments suggesting that overexpression of T-bet is essential and sufficient to promote Th1-mediated colitis in vivo. Furthermore, T-bet–deficient CD62L− CD4+ T cells showed enhanced protective functions in Th1-mediated colitis and exhibited increased TGF-β signaling suggesting that a T-bet driven pathway of T cell activation controls the intestinal balance between IFN-γ/IL-4 and TGF-β responses and the development of chronic intestinal inflammation in T cell–mediated colitis. Furthermore, TGF-β was found to suppress T-bet expression suggesting a reciprocal relationship between TGF-β and T-bet in mucosal T cells. In summary, our data suggest a key regulatory role of T-bet in the pathogenesis of T cell–mediated colitis. Specific targeting of this pathway may be a promising novel approach for the treatment of patients with Crohn's disease and other autoimmune diseases mediated by Th1 T lymphocytes.
Keywords: T-bet, GATA-3, cytokines, colitis, IFN-γ
Introduction
Crohn's disease and ulcerative colitis are the two major forms of inflammatory bowel diseases (IBD)* in humans (1–4). Whereas Crohn's disease is characterized by a transmural, granulomatous inflammation that can occur anywhere in the gastrointestinal tract, ulcerative colitis causes a more superficial, continuous inflammation that is restricted to the large bowel (5). Although the etiology of the diseases is unknown, it has been suggested that an activation of the mucosal immune system in response to bacterial antigens with consecutive pathologic cytokine production and activation of matrix metalloproteinases plays a key pathogenic role (6–8). In particular, cytokines produced by T lymphocytes appear to initiate and perpetuate chronic intestinal inflammation (9–15). Interestingly, cytokine production by lamina propria (LP) CD4+ T lymphocytes differs between Crohn's disease and ulcerative colitis. Whereas the former disease is associated with increased production of Th1 type cytokines such as IFN-γ and TNF, the latter disease is associated with T cells producing large amounts of the Th2 type cytokine IL-5 while IFN-γ production is unaffected (10, 13, 16). The proinflammatory function of both Th1 and Th2 cytokines in experimental models of IBD is counteracted by the immunosuppressive cytokine TGF-β, mainly secreted by Th3 cells and a unique population of regulatory T cells (17–19).
Recent studies have focused on the molecular mechanisms underlying Th1 and Th2 T cell development (20, 21). T lymphocytes transit through sequential stages of cytokine activation, commitment, silencing, and physical stabilization during polarization into differentiated effector subsets, a process tightly controlled by regulatory transcription factors (21–24). Various transcription factors such as c-maf, GATA-3, NFATc1, NIP45, JunB, and signal transducer and activator of transcription (STAT)-6 have been shown to induce or augment Th2 cytokine production, although only c-maf and GATA-3 are expressed selectively in Th2 cells (25–30). In particular, GATA-3 has been shown to promote expression of several Th2 cytokines, including IL-4, IL-5, and IL-13, either by transactivation of cytokine gene promoters and enhancers or the induction of chromatin remodelling (24, 27, 31–34). On the other hand, STAT-1 and STAT-4 transcription factors are specifically associated with IFN-γ and IL-12/IL-23 signaling in T lymphocytes, respectively, and play a key role in regulating cytokine production of Th1 cells at the transcriptional level (21, 35, 36).
However, the STAT proteins are expressed in both Th1 and Th2 subsets and may not have a major role in directly regulating the transcription of the IFN-γ gene. Indeed, some IFN-γ production is retained by STAT-4– and STAT-1–deficient T cells (37, 38). Thus, it appears that alternative regulatory pathways exist that control IFN-γ gene expression. Recently a novel transcription factor of the T-box family, denoted T-bet (39), has been cloned that has shed light on the Th1-restricted expression of IFN-γ. T-bet has been found to be expressed by IFN-γ–producing Th1 but not Th2 cells and increased transcripts for T-bet have been reported to occur within 72 h after stimulation of T cells under Th1-inducing conditions (22, 39). Retroviral transduction of T-bet into primary developing T cells or even fully polarized Th2 cells induces high levels of IFN-γ production and simultaneously represses production of IL-4 and IL-5 (39). T-bet thus appears to be a master switch for Th1 T cell development and the regulation of T cell effector function. Based on this observation, we analyzed in the present study the role of T-bet in regulating cytokine production and clinical disease in chronic intestinal inflammation. We demonstrate a critical role for this factor in controlling IBD by regulating the cytokine balance in T cells.
Materials and Methods
Patients.
Gut specimens obtained from patients with Crohn's disease or control patients were studied. The Crohn's disease group (n = 21) consisted of 10 men and 11 women, ranging from 19 to 57 y of age. The diagnosis for each patient was made using clinical parameters, radiographic studies, and histologic criteria. At the time of sample collection, 12 patients of the Crohn's disease group were receiving corticosteroids and three patients were receiving an oral sulfasalazine preparation. The control group consisted of colonic specimens from 23 patients. There were 12 male and 11 female patients in the control group ranging from 41 to 78 y of age. These patients were not receiving sulfasalazine or corticosteroids at the time of the resection. In addition, colonic sections from four patients with ulcerative colitis were obtained. Collection of surgical samples was approved by the ethical committee and the institutional review board of the University of Mainz.
Immunohistochemistry.
Immunohistochemistry was performed on 7-μm cryosections from gut specimens of both control and Crohn's disease/ulcerative colitis patients, as described previously (11, 40). Briefly, tissues were fixed in 4% paraformaldehyde/PBS and washed in 0.01 M PBS. Samples were then pretreated with 10% of serum (corresponding to the secondary antibody) in PBS/0.1% Triton-X and incubated overnight at 4°C with the primary antibody (5–10 μg/ml monoclonal or polyclonal rabbit anti–T-bet (39) or goat anti–GATA-3; obtained from Santa Cruz Biotechnology, Inc.) in PBS/0.5% BSA/0.2% saponin. Samples without primary antibody served as negative control. The following day, samples were rinsed in PBS and incubated with a biotinylated secondary IgG antibody (1:200 dilution; obtained from Pierce Chemical Co.) for 1 h at room temperature followed by incubation with streptavidin conjugated Cy2 or Cy3 (Dianova) (1:500–1:1,000 dilution) for 1 h at room temperature. Samples were rinsed with PBS and, in some experiments, subjected to a second cycle of staining by using anti-CD3 as primary antibody (monoclonal mouse anti–human CD3; BD PharMingen) and streptavidin-conjugated Cy3 as chromogen (40). Slides were mounted with DAPI-containing mounting medium for fluorescence (Vector Laboratories) and analyzed with a Olympus Microscope. 6–10 high power fields (HPF) were finally counted in all patients per condition. No staining was detected in samples without primary antibody.
Isolation of Human LP T Cells.
LP mononuclear cells (LPMCs) were isolated using a previously described technique (10). LP T cells were prepared from the resultant cell population as described previously (11). The resulting cells were >90% T cells, as assessed by FACS® analysis (FACStar™; Becton Dickinson). The T cells were either analyzed directly or cultured in complete media with antibodies to human CD2 and CD28 (1 μg/ml; obtained from BD PharMingen). Complete medium consisted of RPMI 1640 supplemented with 3 mM l-glutamine, 10 mM Hepes buffer, 10 μg/ml gentamycin (Whittaker), 100 U/ml each of penicillin and streptomycin (Whittaker), 0.05 mM 2 ME (Sigma-Aldrich), and 10% heat-inactivated FCS.
Electrophoretic Mobility Shift Assays.
Oligonucleotides for electrophoretic mobility shift assays (EMSAs) were synthesized, annealed, and end-labeled with γ-[32P]-ATP (>6,000 Ci/mmol; NEN Life Sciences Labs) using bacteriophage T4 polynucleotide kinase (New England Biolabs Inc.). Synthetic, double-stranded oligonucleotides containing binding sites for SP-1, AP-2, μE5, and GATA-3 for EMSA were obtained from Santa Cruz Biotechnology, Inc. and MWG Biotech, respectively. The sequences of T-bet binding sites have been described elsewhere (39). Binding reactions (15 μl) for EMSA contained 2 μg synthetic DNA duplex of poly (dI-dC) (Amersham Pharmacia Biotech), 25,000 cpm (Cerenkov) of end-labeled DNA probe, and incubation buffer (10 mM Hepes, pH 7.9, 100 mM NaCl, 10% glycerol, 0.5 mM MgCl2, 1 mM DTT). After preincubation for 15 min at room temperature, radiolabeled DNA was added to the reaction for an additional 15 min. 2 μg of the monoclonal anti–mouse T-bet IgG1 (4B10; reference 39), a c-jun antibody (Santa Cruz Biotechnology, Inc.) or a polypeptide T-bet antibody (H2N-CSDSGLGEGDTKRRRI-CONH2; obtained from Eurogentec) were added as indicated. Finally, the complexes were separated from unbound specific probe by electrophoresis on native 4% polyacrylamide gels. After electrophoresis, the gels were dried and exposed to Kodak films on intensifying screens at −80°C.
Western Blot Analysis.
For Western blot analysis, equal amounts of extract (30 μg) were added to 10 μl electrophoresis sample buffer. After boiling, the extracts were loaded on 10% SDS-PAGE gels and electrophoretically separated for 2 h at 100 V. Proteins were transferred to nitrocellulose membranes and detection was performed with a polyclonal rabbit or monoclonal mouse anti–T-bet (39), rabbit anti–mouse GATA-3, polyclonal goat or rabbit anti-Smad3, goat anti-Smad7 (all obtained from Santa Cruz Biotechnology, Inc.), and β-actin antibodies (Sigma-Aldrich), HRP-conjugated secondary antibodies (donkey anti–rabbit Ig, obtained from Sigma-Aldrich; anti–goat IgG, obtained from Santa Cruz Biotechnology, Inc.; anti–rat IgG, obtained from Pierce Chemical Co.) and the ECL Plus or ECL Western blotting analysis system (Amersham Pharmacia Biotech).
Animals.
2–4-mo-old Balb/c and B6 mice were obtained from the central breeding facility at the University of Mainz or from Charles River Laboratories; S129/B6 wild-type and STAT-1–deficient mice (38) were obtained from Taconic; T-bet knockout mice and transgenic mice expressing T-bet in T cells under the control of the CD2 promoter/enhancer construct have been described previously (41, 42). B cell–deficient TCR-α−/−/μ−/− mice and IL-10–deficient mice (4–12 wk old) with chronic enterocolitis have been described previously (43, 44). C.B.-17 SCID or recombination activation gene (RAG)-deficient mice were obtained from Charles River Laboratories or Taconic.
Screening of T-bet Knockout Mice.
Tail DNA from mice was isolated and subjected to polymerase chain reaction (PTC-100 Thermal Cycler; MJ Research Inc.) using three synthetic primers (T-bet1 5′-GCG CGA AGG GGC CAC CAA AGA ACG GAG-3′, T-bet2 5′-GAC TGA AGC CCC GAC CCC CAG TCC TAA G-3′, T-bet3 5′-TGG GCA TAC AGG AGG CAG CAA CAA ATA-3′; obtained from GIBCO BRL) and the Advantage PCR kit (CLONTECH).
Isolation of LPMCs.
LPMCs were isolated from freshly obtained colonic specimens using a modification of the technique described by van der Heijden and Stok, as described previously (45). After removal of the Peyer's patches, the colon was washed in HBSS free of calcium and magnesium, cut in 3-mm pieces and incubated twice in HBSS containing EDTA (0.35 mg/ml) and DTT (0.145 mg/ml; both obtained from Sigma-Aldrich) at 37°C for 15 min. Next, the tissue was digested further in RPMI 1640 containing collagenase D (400 U/ml) and DNase I (0.1 mg/ml) (Boehringer Mannheim) in a shaking incubator at 37°C. LP cells were then layered on a 40–100% Percoll gradient (Amersham Pharmacia Biotech) and lymphocyte-enriched populations were isolated from the cells at the 40–100% interface. In some experiments, LPMCs were further enriched for T cells using cell adherance to plastic dishes for 1 h at 37°C.
Cytokine Assays.
To measure cytokine production, 106 splenic T cells or 0.5–2 × 106 T cell enriched LPMC per ml were activated with 10 μg/ml purified hamster anti–mouse CD3ε (clone 145–2C11) and 1 μg/ml soluble hamster anti–mouse CD28 (clone 37.51) with or without IFN-γ and cultured in complete medium (RPMI 1640 supplemented with 3 mM l-glutamine, 10 mM Hepes buffer, 100 U/ml penicillin/streptomycin, 0.05 mM 2 ME, 10% heat inactivated FCS) or serum-free medium at 37°C in a humidified atmosphere containing 5% CO2. After 48 h (96 h for TGF-β), culture supernatants were removed and assayed for cytokine concentration. Cytokine concentrations were determined by specific ELISA using commercially available recombinant cytokines and antibodies (BD PharMingen, Genzyme Corp., R&D Systems, and Boehringer Mannheim).
Retroviral Gene Transfer in Primary Splenic CD4+ T Lymphocytes.
For retroviral gene transfer of primary T cells the GFP-RV vector and its derivates were used. The GFP-RV bicistronic vector was constructed by inserting the encephalomyocarditis virus ribosomal entry sequence (IRES) and the GFP allele into the MSCV2.2 retroviral vector (31, 32). The GFP-RV vector and the GATA-3-GFP vector were donated by K. Murphy and the Phoenix-Eco packaging cell line was obtained from G. Nolan (46). The pGCIRES and pGCIRES.T-bet retroviral constructs have been described previously (39).
Splenic CD4+ T cells from healthy Balb/c mice were isolated, as described previously (11). In brief, spleens were aseptically removed and washed thoroughly in HBSS free of calcium and magnesium. The tissue was cut into small pieces and pushed through a 40-μm nylon cell strainer (Falcon; Becton Dickinson). Erythrocytes were removed from the resulting cell suspension by hypotonic lysis in ACK buffer. Spleen cells were then washed in PBS, centrifuged at 1,600 rpm for 10 min at 4°C and CD4+ T cells were isolated using immunomagnetic beads and MACS® (Miltenyi Biotec). Cell purity was >97%, as assessed by FACS® analysis. Retroviral infection of primary splenic CD4+ T cells with polybrene (Sigma-Aldrich) and retroviral supernatants was performed as described previously (32). The following day cells were incubated with PE-labeled CD62L antibodies (BD PharMingen) and sorted by FACS® (Becton Dickinson) to obtain CD62L+ GFP+ double positive CD4+ T lymphocytes. Finally, 1–5 × 105 CD62L+ GFP+ CD4+ T cells were injected in CB-17 SCID mice. SCID mice were maintained in isolated cages under specific pathogen free conditions.
Adoptive Transfer of CD4+ T Cell Subsets in Immunocompromised Hosts.
To induce colitis by adoptive transfer of CD4+ T cells, a modification of a previously described protocol was used (11, 17). In brief, CD4+ T cells were purified from spleen mononuclear cells of healthy mice using FITC-conjugated mAbs, anti-FITC immunomagnetic beads, and MACS® (Miltenyi Biotech), or anti-CD4 beads (Dynal) followed by enzymatic removal of the beads. The resulting CD4+ T cells (purity >97%) were further separated by immunomagnetic beads into CD62L+ and CD62L− T cells. The former cells (purity: >95%) showed high expression of CD45RB by FACS® analysis. 0.5–1 × 106 CD62L+ CD45RBhigh CD4+ T cells or 106 CD62L+ CD45RBhigh and CD62L− (ratio 1:4) CD4+ T cells were finally transferred into C.B.-17 SCID or RAG-1–deficient mice. Colitis activity was monitored by weight curves, endoscopy and histologic analysis, as specified below. Mice were maintained in isolated cages under specific pathogen free conditions. No evidence of graft versus host disease, such as skin inflammation and histopathologic evidence of small bowel inflammation, was observed in the reconstituted animals under our experimental conditions.
Induction of Colitis by Haptenizing Agents.
Oxazolone-induced colitis in S129/B6 mice was induced using a modification of previously described method (47), as this mouse strain exhibits increased resistance to inducible colitis by hapten reagents (6). In brief, mice were sensitized by epicutaneous application of 3% oxazolone (4-ethoxymethylene-2-phenyl-2-oxazolin-5-one; obtained from Sigma-Aldrich) in 100% ethanol (150 μl) on day 0 followed by intrarectal administration of 1% oxazolone in 50% ethanol (100 μl) to anesthetized mice on day 7.
Treatment of Mice with Neutralizing Antibodies to IL-4.
In some experiments, colitic mice were treated with neutralizing antibodies to IL-4 (11B.11; 7 mg/wk; obtained from the Biological Resources Branch, DCTD, Frederick, MD, as specified in Results).
Assessment of Contact Hypersensitivity.
Contact hypersensitivity after oxazolone sensitization was assessed on day 7, essentially as described by Xu et al. (48). In brief, ear swelling before and 24 h after ear challenge with 1% oxazolone in 100% ethanol (vol: 20 μl) was determined using a dial thickness gauge (Mitutoyo). The DTH responses were expressed as percentage increase in ear thickness after oxazolone challenge over the baseline values (100%).
Histologic Analysis of Colon Cross Sections.
Tissues were removed from colitic mice at indicated time points, and cryosections or paraffin sections were made and stained with hematoxylin and eosin. For colitis induced by CD62L+ CD4+ T cell transfer, the degree of inflammation and epithelial injury on microscopic cross sections of the colon was graded semiquantitatively from 0 to 4: inflammation score 0 = no evidence for inflammation; 1 = low level of inflammation with scattered infiltrating mononuclear cells (1–2 foci only); 2 = moderate inflammation with multiple foci; 3 = high level of inflammation with increased vascular density and marked wall thickening; and 4 = maximal severity of inflammation with transmural leukocyte infiltration and loss of goblet cells. Injury score 0 = no epithelial injury; 1 = occasional epithelial lesion; 2 = 1–2 foci of ulcerations; and 3 = extensive ulcerations. Grading of colitis activity was done in a blinded fashion by the same pathologist (J. Glickman). Small bowel sections were taken from some animals as an additional control and showed no evidence for inflammation.
For histopathologic grading of oxazolone-induced colitis five criteria (hypervascularization, presence of mononuclear cells, epithelial hyperplasia, epithelial injury, and presence of granulocytes) were scored from 0 to 3 yielding an additive score between 0 (no colitis) and 15 (maximal colitis activity).
In Vivo Endoscopic Analysis of the Colon.
A novel method was developed to perform endoscopy in mice using a mini-endoscope (length: 30 or 60 mm, diameter: 0.89 mm) with an intralux vision light source (Volpi AG, Switzerland and Insight Instruments) (unpublished data). In brief, mice were anesthetized with avertine and the colon was flushed with PBS. Prominent endoscopic signs of inflammation in SCID mice were masking of the normal vascular pattern, the presence of mucosal granularity, and the appearance of ulcers. Based on our data in >100 endoscopies in colitic SCID/RAG mice a murine endoscopic index of colitis severity (MEICS) was developed that consists of the following four criteria: ulcerations, none: 0, 1–2: 1, 3–5: 2, >5: 3 points × 2; masking of the normal vascular pattern: none: 0, moderate: 1, marked: 2, complete or almost complete: 3 points; mucosal granularity: none: 0, moderate: 1, marked: 2, extreme: 3 points; and surface involved in cm: none: 0, 1–2: 1, 3–4: 2, >4: 3 points. The cumulative MEICS score was between 0 and 15 points. This index allows monitoring of colitis activity in individual SCID/RAG mice over several months, as the endoscopic procedure can be repeated without problems for up to eight times. To determine colitis activity in reconstituted SCID or RAG mice the mice were monitored by endoscopy at indicated time points after the cell transfer.
FACS® Analysis and Staining for T-bet and STAT-1.
FACS® analysis of splenic and LP cells was performed as described previously (11). Antibodies against murine CD4 (Cychrome) and CD25 (FITC) were obtained from BD PharMingen. Intracellular staining for T-bet was performed as described previously (39). Intracellular staining for STAT-1 was done using the same method and anti–human STAT-1 antibodies (Santa Cruz Biotechnology, Inc.).
Statistical Analysis.
Statistical analysis was made using the Student's t test and the program Statworks for Macintosh and MS Office (Excel).
Results
Reciprocal Expression of GATA-3 and T-bet in LP T Cells from Patients with Crohn's Disease.
Changes in cytokine production by LP T cells have been implicated as a key phenomenon in the pathogenesis of IBD. In particular, an increased production of Th1 (such as IFN-γ and TNF) cytokines is present in patients with Crohn's disease, while ulcerative colitis is associated with production of high levels of the Th2 cytokine IL-5 (9–11, 13). Interestingly, a novel transcription factor of the T box family, denoted T-bet, has been recently shown to control both IFN-γ and IL-5 production (39). Therefore, we focused in an initial series of experiments on the expression of T-bet by purified LP T cells in IBD patients. Immunofluorescence double staining studies showed an accumulation of T-bet expressing LP T cells in patients with Crohn's disease (Fig. 1 A–D). In addition, it was found that T-bet was strongly expressed in both the cytoplasm and the nucleus of LPMCs in patients with Crohn's disease, while only a weak staining in perinuclear areas or no staining was observed in control patients and patients with ulcerative colitis (Fig. 1 E). To verify these data on increased expression of T-bet in patients with Crohn's disease, we isolated nuclear extracts of purified LP T cells from patients with Crohn's disease and control patients and analyzed expression of T-bet by EMSA and Western blot analysis. We observed that patients with Crohn's disease expressed higher amounts of nuclear T-bet compared with control patients (Fig. 2 A–C). This observation was supported by intracellular FACS® staining of cells for T-bet demonstrating strongly increased T-bet levels in Crohn's disease patients compared with controls (Fig. 2 D). Similarly, the expression of the IFN-γ associated transcription factor STAT-1 was upregulated in Crohn's disease compared with control patients, although to a much lesser extent (Fig. 2 D).
Figure 1.
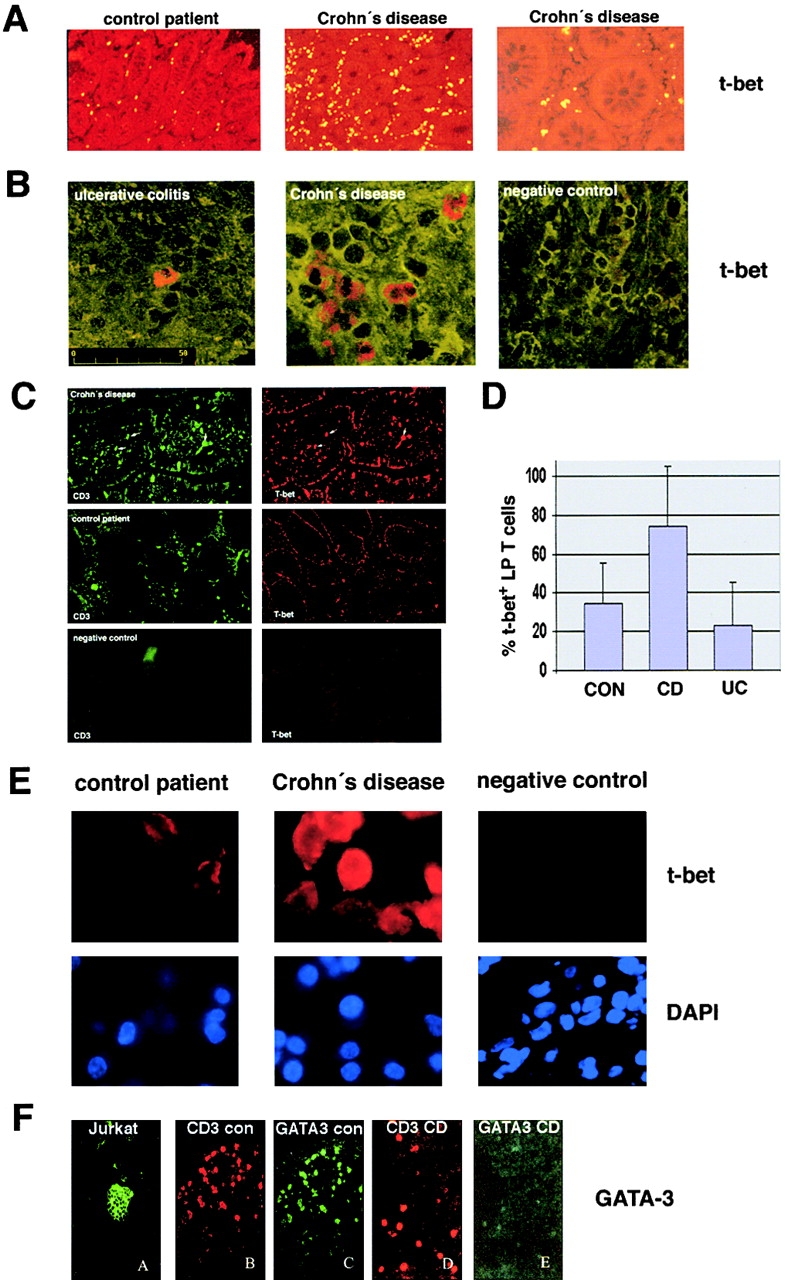
Accumulation of T-bet–expressing T lymphocytes in the LP of patients with Crohn's disease. (A and B) Analysis of T-bet expression in the LP from patients with IBD and control patients. Colon cryosections were stained with a T-bet antibody and sections were analyzed by immunofluorescense microscopy and confocal laser microscopy, respectively. Many T-bet positive cells were seen in patients with Crohn's disease but not ulcerative colitis. Data are representative of four to five patients per group. (C) Immunohistochemical double staining analysis of the cellular expression of T-bet plus CD3 in consecutive cross sections. Cryosections from patients with Crohn's disease (n = 10), ulcerative colitis (n = 4) and control patients (n = 8) were stained with antibodies to CD3 and T-bet, as indicated. One representative experiment is shown. Double positive cells are indicated by arrows. (D) Quantitative analysis of T-bet-positive CD3+ LP T cells in patients with Crohn's disease, ulcerative colitis, and control patients. The percentage of double-positive T cells in four to six patients per group was quantified as specified in Materials and Methods. Data represents mean values ± SD. (E) Analysis of T-bet expression by LPMCs from patients with Crohn's disease and control patients. Nuclei were counterstained with DAPI, as specified in Materials and Methods. Most T cells from control patients showed weak expression of T-bet in perinuclear areas, whereas high cytoplasmic and nuclear expression was noted in patients with Crohn's disease. (F) Double staining analysis for GATA-3 and CD3 in the LP of patients with Crohn's disease (CD) and control patients (con), as indicated. Colon cryosection were stained with a polyclonal anti–GATA-3 antibody and anti-CD3 antibodies. There was a marked reduction of GATA-3 expression in LP T cells from patients with Crohn's disease compared with control patients. Data are representative of four to six patients per group. Cytospins from PMA- plus ionomycin-stimulated peripheral blood T cells served as a positive control.
Figure 2.



Increased nuclear expression of T-bet in LP T cells from patients with Crohn's disease. (A) EMSA using nuclear extracts of purified LP T cells from Crohn's disease and control patients and a radiolabeled T-bet DNA binding site. The location of the T-bet signal is indicated. (B) Auto- and cross-competition assay. Specificity of the EMSA signal obtained with the T-bet–binding site was shown by autocompetition with unlabeled T-bet binding site, whereas cross-competition with unlabeled SP-1, μE5, and AP-2 sites did not affect the signal. (C) Analysis of T-bet expression in nuclear extracts of LP T cells (Crohn's disease: n = 3; controls: n = 2) by Western blot analysis. A high expression of T-bet was observed in patients with Crohn's disease but not control patients. A second independent experiments with four patients per group showed similar results (data not shown). (D) Intracellular staining for STAT-1 and T-bet by FACS® using LP cells from patients with Crohn's disease or control patients, as indicated. One representative experiment out of three is shown.
Next, we assessed the expression of GATA-3 in LP T cells from Crohn's disease patients, since this transcription factor has been shown to directly transactivate the IL-5 promoter and to mediate Th2 effector function (27, 32). We observed that GATA-3 expression was downregulated in LP T cells from Crohn's disease patients compared with control patients, as assessed by immunohistochemical double staining analysis for CD3 and GATA-3 on colon cryosections (Fig. 1 F). Taken together, these data suggested a reciprocal expression pattern of GATA-3 and T-bet in Crohn's disease LP T cells that is associated with increased IFN-γ but decreased IL-4 and IL-5 production in this disease.
Induction of T-bet Expression in Th1- but not Th2-mediated Animal Models of Chronic Intestinal Inflammation.
Since the above data were consistent with the possibility of a regulatory role for T-bet in controlling cytokine production in the inflamed intestine, we focused our attention on the expression of T-bet in several different animal models of T cell–mediated animal models of chronic intestinal inflammation previously shown to be associated with Th1 or Th2 subsets. Accordingly, we isolated nuclear proteins from T cell enriched LPMCs in these colitis models and assessed T-bet expression by EMSA and Western blot analysis (Fig. 3) . T-bet was found to be strongly expressed in T cell enriched LP cells in the Th1-mediated colitis model observed in SCID or RAG mice reconstituted with CD62L+ CD4+ T cells. Time course studies showed that increased T-bet expression in the colon occurred as early as 3 wk after cell transfer before the onset of colitis (Fig. 3 A–C). Maximum expression was noted 6 wk after cell transfer when the mice started to develop colitis, although increased T-bet expression was also observed in the full-blown colonic inflammation seen at 12 wk after T cell transfer. Furthermore, increased T-bet expression was consistently noted in two additional Th1-mediated animal models of chronic intestinal inflammation, namely colitis in IL-10–deficient mice and colitis induced by the hapten reagent 2,4,6,-trinitrobenzene sulfonic acid (TNBS) (Fig. 3 D). In contrast, unchanged or lower levels of T-bet were detected in T cell enriched LP cells in oxazolone colitis and TCR-α−/− μ−/− associated colitis; two colitis models that are believed to be mediated by IL-4–producing T cells and Th2 cells, respectively (43, 47). These findings suggested that T-bet is potentially a key regulator of the mucosal Th1/Th2 cytokine balance in experimental colitis in vivo.
Figure 3.
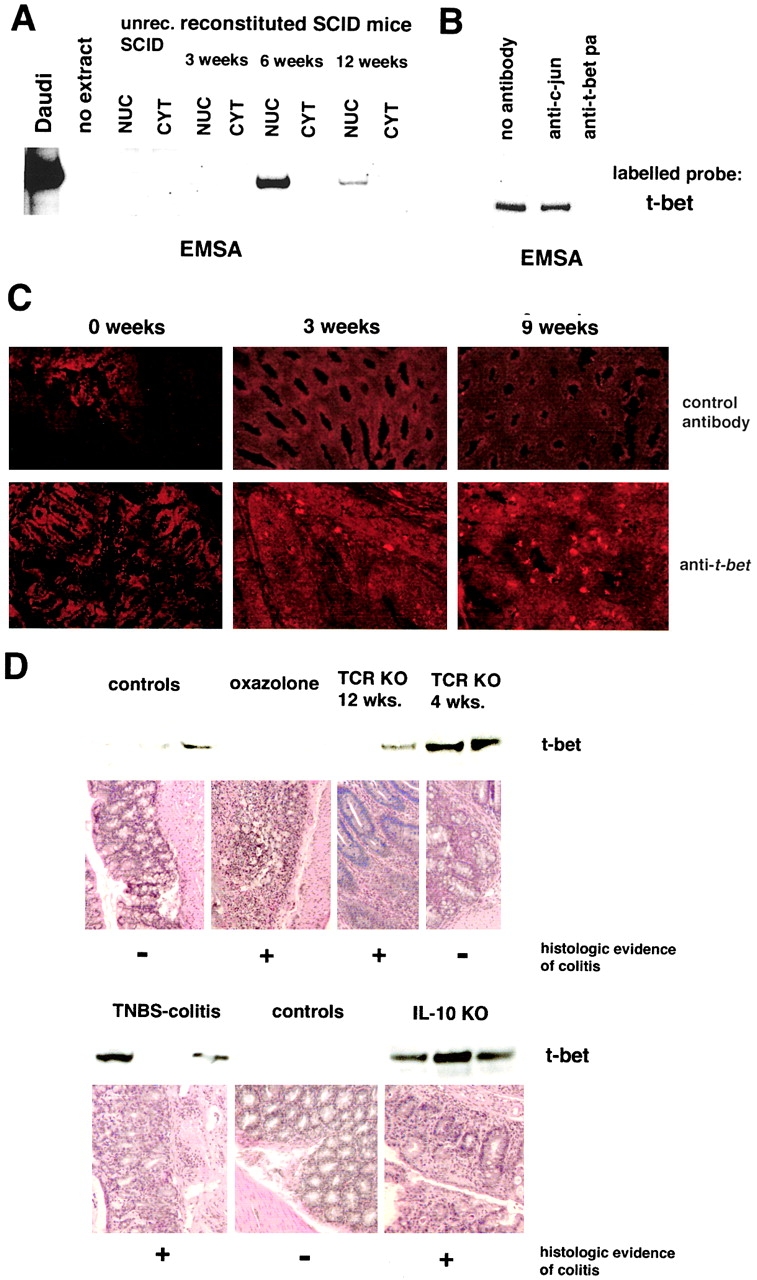
Increased expression of T-bet in Th1- but not Th2-mediated animal models of chronic intestinal inflammation A. Analysis of T-bet expression in C.B.-17 SCID mice reconstituted with 106 CD62L+ CD4+ T cells from Balb/c mice. EMSA for T-bet using nuclear (NUC) and cytoplasmic (CYT) extracts from spleen cells of unreconstituted and CD62L+ CD4+ T cell reconstituted SCID mice at indicated time points. SCID mice showed clinical signs of colitis 8–9 wk after T cell transfer. (B) Supershift assay for T-bet using nuclear extract from spleen cells of SCID mice 6 wk after reconstitution with CD62L+ CD4+ T cells from Balb/c mice. (C) Immunohistochemical analysis of the colon of SCID mice for T-bet expression at indicated time points after reconstitution with CD62L+ CD4+ T cells. T-bet expressing cells in the LP were noted as soon as 3 wk after T cell transfer. (D) Expression of T-bet in extracts from LPMCs in Th1- and Th2-mediated animal models of chronic intestinal inflammation. Whereas increased T-bet expression by Western blot analysis was noted in Th1-mediated models such as TNBS-induced colitis in Balb/c mice and colitis in 4–12-wk-old IL-10 knockout mice, both oxazolone-induced colitis and TCR-α−/− μ−/− colitis were associated with normal or reduced T-bet expression in LPMCs.
Retroviral Overexpression of T-bet Induces an Early Onset of Severe CD62L+ CD4+ Th1 T Cell-mediated Colitis in SCID Mice.
We next determined the potential regulatory role of T-bet in Th1-mediated colitis in vivo by retroviral overexpression techniques. Accordingly, we analyzed the colitogenic potential of T cells after infection with a T-bet retrovirus (Fig. 4 A). We observed in two independent experiments that FACS®-sorted GFP+ CD62L+ double positive CD4+ T cells that were retrovirally transduced with T-bet induced an earlier onset of severe colitis in SCID mice compared with SCID mice reconstituted with control transduced CD62L+ T cells, as assessed by weight loss curves (Fig. 4 A). Furthermore, SCID mice reconstituted with T-bet transduced T cells developed a massive rectal prolapse as well as macroscopic signs of severe colitis (Fig. 4 B and C). This phenotype suggested that the overexpression of T-bet induced by retroviral gene transfer (Fig. 4 D) accelerates development of Th1-mediated chronic intestinal inflammation.
Figure 4.


Overexpression of T-bet accelerates development of Th1-mediated colitis. (A) Transfer of T-bet transduced CD62L+ CD4+ T cells causes rapid onset of colitis in SCID mice. Splenic CD4+ T cells from Balb/c mice were isolated and infected with the T-bet retrovirus followed by FACS® sorting for GFP/CD62L double positive T cells. GFP+ CD62L+ CD4+ T cells were then injected into C.B.-17 SCID mice followed by monitoring of the body weight at indicated time points after cell transfer. Data represent mean values ± SEM. Two independent experiments are shown (right and left). (B) Rectal prolapse upon reconstitution of SCID mice with T-bet transduced (T-bet RV – lower panel) and control transduced (CON-RV, top panel) CD62L+ T cells. The development of a marked rectal prolapse was noted much earlier in the former than in the latter group. (C) Macroscopic evidence of colitis in SCID mice reconstituted with T-bet transduced (T-bet RV, bottom panel) and control transduced (CON-RV, top panel) CD62L+ T cells as compared with an unreconstituted SCID mouse. (D) Western blot analysis for T-bet expression in T-bet transduced versus control transduced T cells before the T cell transfer. As expected, retroviral infection with the T-bet virus led to much higher T-bet levels in the T cells as compared with control infected T cells. The β-actin staining served as loading control.
Mice Lacking T-bet (T-bet Knockout) Are More Susceptible to Th2-mediated Colitis.
Since the above data strongly suggested that overexpression of T-bet plays an important role in Th1-mediated colitis, we next analyzed the susceptibility of mice in which the T-bet gene has been inactivated by homologous recombination for T cell–mediated colitis. In an initial series of studies, we determined whether T-bet–deficient mice exhibited an altered susceptibility to Th2-mediated colitis using the oxazolone-induced colitis model that has previously been shown to be dependent on IL-4 production by T cells (47). As shown in Fig. 5, T -bet knockout mice showed enhanced susceptibility to oxazolone-induced colitis compared with both wild-type littermates and heterozygous T-bet mice, as shown by weight curves (Fig. 5 A), macroscopic (Fig. 5 B), and histopathologic (Fig. 5 C and D) criteria. This was accompanied by a marked increase in IL-4 production by splenic CD3+ T cells, while IFN-γ production by these cells was not significantly changed (Fig. 5 E).
Figure 5.

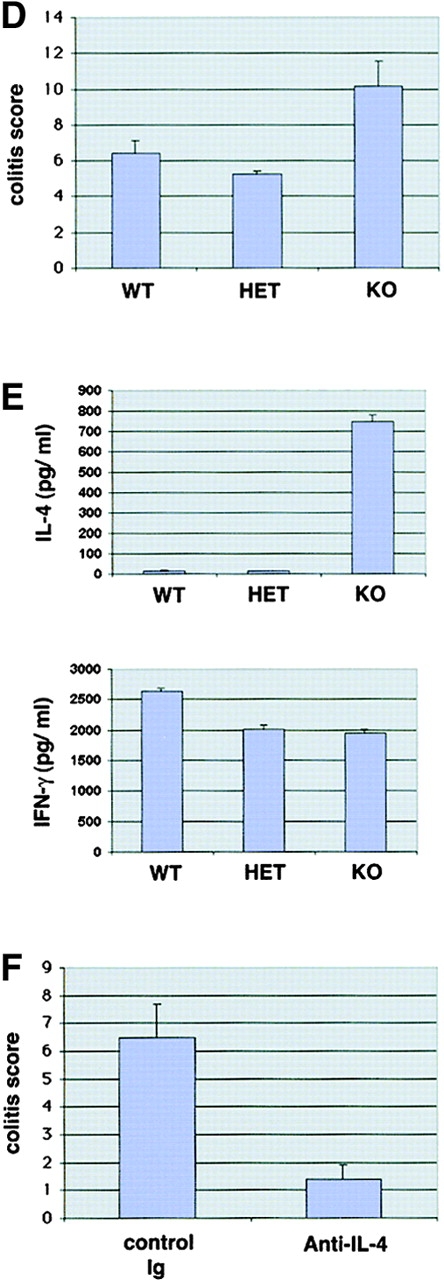
T-bet–deficient mice show enhanced susceptibility to oxazolone-induced colitis. (A) T-bet wild-type (WT), heterozygous (HET) and knockout mice (KO) were sensitized to oxazolone followed by intrarectal treatment with 1% oxazolone in 50% ethanol after 1 wk and monitoring of body weight (left). Data are given as mean values ± SEM and represent six to eight mice per group. Significant differences are indicated (*P < 0.05). In addition, ear thickness after local oxazolone challenge was determined in sensitized animals, as specified in Materials and Methods (right). Data are given as mean values ± SEM and represent six to eight mice per group. No significant differences were noted. (B and C) Macroscopic and histologic analysis of the colon of T-bet wild-type and knockout mice 7 d after intrarectal administration of oxazolone. One representative colon per group is shown. There was a marked inflammation of the mucosa and submucosa in T-bet–deficient mice associated with edema, hypervascularization, and ulcer formation (arrowhead in bottom left panel) that was more pronounced compared with wild-type animals. (D) Histopathologic assessment of colitis activity in wild-type and T-bet–deficient mice given oxazolone. Paraffin sections were scored in a blinded fashion, as described in Materials and Methods. Data are given mean values ± SEM and represent six to eight mice per group. (E) Cytokine production by splenic CD3+ T cells from oxazolone-treated mice. T cells were stimulated with antibodies to CD3 and CD28 followed by analysis of culture supernatants by ELISA. Data represent four to eight mice per group and are given as mean values ± SEM. A marked induction of IL-4 production was seen in T-bet–deficient mice. No marked changes in IFN-γ production by CD3+ T cells were detected probably due to the fact that IFN-γ production from T-bet–deficient CD8+ T cells is unimpaired (reference 47). (F) Anti–IL-4 treatment suppresses colitis activity in T-bet knockout mice. T-bet knockout mice were sensitized to oxazolone followed by intraperitoneal administration of neutralizing anti–IL-4 antibodies and intrarectal oxazolone treatment. Colon samples were obtained after 7 d followed by histopathologic analysis. Data represent three to four mice per group and are given as mean values ± SEM.
To determine whether the observed increase in IL-4 production was responsible for the differences between wild-type and T-bet knockout mice, we next administered antibodies to IL-4 or control rat Ig to T-bet knockout mice after oxazolone sensitization. It was found that antibodies to IL-4 suppressed histologic colitis activity in oxazolone-treated T-bet knockout mice (Fig. 5 F) strongly suggesting that the protective role of T-bet in Th2-mediated colitis is due to its direct or indirect regulation of IL-4 production.
T-bet Deficiency Protects from Th1-mediated Experimental Colitis in an Adoptive Transfer Model Using CD62L+ CD4+ T Cells.
We next assessed the effects of T-bet deficiency in Th1-mediated colitis induced by transfer of CD4+ CD62L+ CD45Rbhigh T cells in SCID and RAG knockout mice (11, 17, 49). Whereas transfer of T-bet–expressing CD4+ CD62L+ T cells from wild-type mice resulted in clinical and endoscopic signs of severe colitis, transfer of T-bet−/− CD4+ CD62L+ T cells failed to induce chronic diarrhea, weight loss, rectal prolapse, and endoscopic signs of colitis (Fig. 6 A, B, and D). Furthermore, transfer of T-bet–deficient T cells resulted in a markedly reduced colitis activity in mice reconstituted with CD62L+ CD45Rbhigh CD4+ T cells in three independent experiments, as assessed by macroscopic and histologic criteria (Fig. 6 C, E, and F). This protective effect of T-bet deficiency on CD62L+ CD4+ T cell–induced colitis was at least as pronounced as that seen upon transfer of STAT-1-deficient CD62L+ CD4+ T cells (histopathologic score: STAT-1−/− reconstituted mice: 1.25 ± 0.9 vs. T-bet−/− reconstituted mice: 0.8 ± 0.2). T cells from T-bet knockout mice produced more IL-4 but less IFN-γ before the T cell transfer and, consistently, T-bet–deficient cells isolated from reconstituted mice produced less IFN-γ compared with cells from both wild-type and T-bet heterozygous T cell reconstituted mice (Fig. 6 G) suggesting that T-bet deficiency suppresses proinflammatory cytokine production by CD4+ T cells in colitis in vivo. Interestingly, CD4+ CD62L+ T cells from heterozygous T-bet mice showed a marked variability to induce colitis in three independent experiments (Fig. 6 A-F), possibly due to a threshold effect of T-bet expression on the colitogenic potential of T cells.
Figure 6.
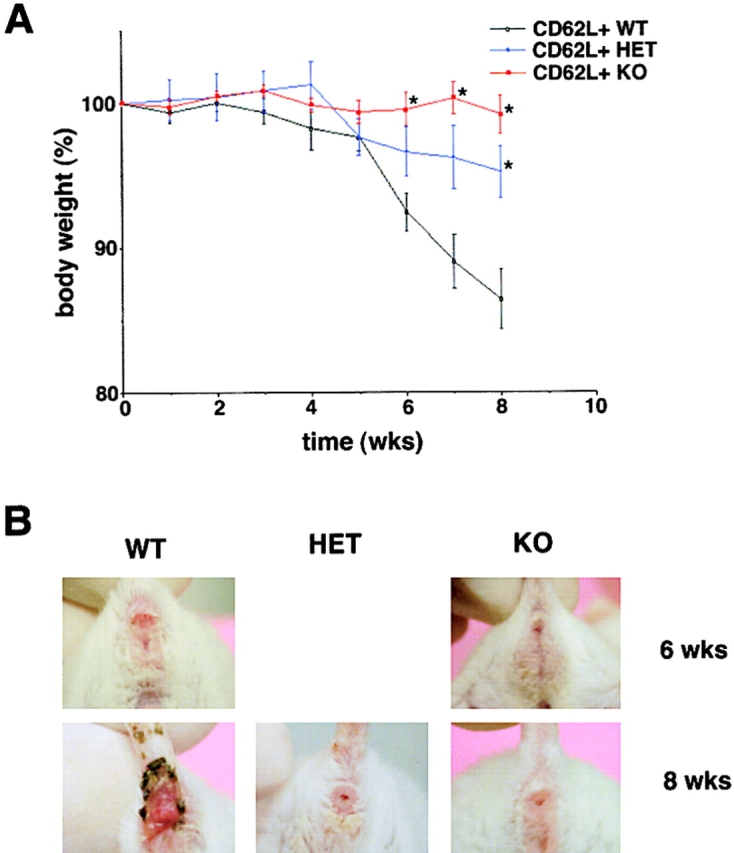

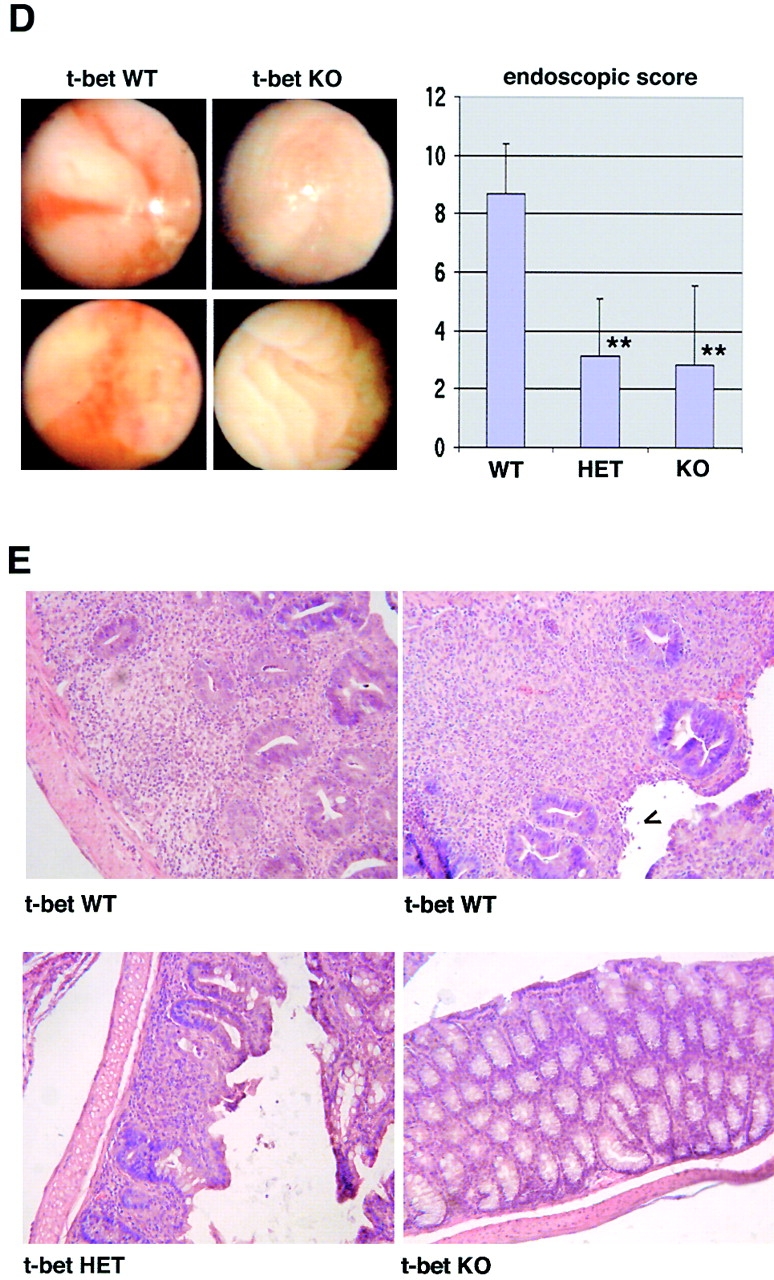
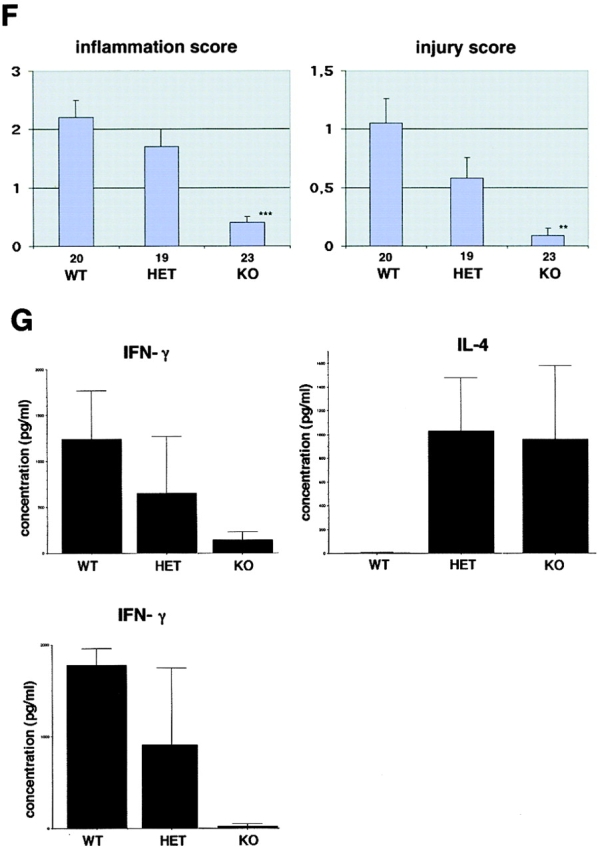
T-bet deficiency protects from Th1-mediated colitis induced by CD62L+ CD4+ T cell transfer in immunocompromised mice. Immunocompromised mice were reconstituted with CD62L+ CD4+ T cells from wild-type and T-bet–deficient mice. In contrast to mice reconstituted with cells from wild-type animals, mice injected with CD62L+ CD4+ T cells from T-bet knockout mice showed neither wasting disease (A) nor rectal prolaps (B) and macroscopic (C) or endoscopic (D) signs of inflammation. A shows weight data (mean values ± SEM) from one representative experiment out of three and represents data from five to six mice per group. C shows one representative colon and spleen per group 8 wk after the T cell transfer. A significant reduction in endoscopic severity of colitis was noted in mice reconstituted with CD62L+ CD4+ T cells lacking T-bet compared with cells from wild-type mice (D; P < 0.05). In particular, no bleeding ulcers and erosions were noted in the former mice that were frequently present after transfer of CD62L+ CD4+ T cells from wild-type mice (left panels in D). Furthermore, there was a striking reduction of cell infiltration in the colon in mice reconstituted with T-bet–deficient T cells compared with mice reconstituted with T cells from wild-type mice (E). Quantitative histopathologic assessment of colitis activity showed a highly significant (**P < 0.01; ***P < 0.0001) suppression of both the inflammation and injury score in mice reconstituted with CD62L+ CD4+ T cells lacking T-bet compared with cells from wild-type mice (F). Data were pooled from three independent experiments with at least five mice per group (n = 19–23 per group) and are stated as mean values ± SEM. In addition, cytokine production by splenic CD4+ T cells was assessed before and 8 wk after the T cell transfer. T cells were stimulated with anti-CD3 plus CD28 antibodies and cytokine concentrations were determined by specific ELISA after 2 d. Data are shown as mean values ± SEM (G).
T-bet Controls the Mucosal Balance between IFN-γ and IL-4 Production by T Cell–enriched LP Cells in the Absence of Colitogenic Stimuli.
Since the above data suggested that T-bet controls the balance between Th1 and Th2 cytokine production by mucosal T lymphocytes in vivo, we next assessed the structure of the LP and cytokine production by LPMCs in mice lacking T-bet. We observed no macroscopic or histologic abnormalities in the small and large bowel of T-bet heterozygous and T-bet knockout mice in the absence of colitogenic stimuli (Fig. 7 A). To analyze cytokine production by T cell enriched LPMCs from T-bet–deficient mice and wild-type littermates, cells were stimulated by anti-CD3 plus anti-CD28 for 48 h and cytokine production in culture supernatants was determined by ELISA (Fig. 7 B). It was found in two independent experiments (purple and red bars) that T cell–enriched LPMCs from T-bet knockout mice secreted lower levels of IFN-γ than cells from wild-type littermates in the absence of colitogenic stimuli. In contrast, production of the Th2 type cytokines IL-4, IL-6, and IL-10 by T cell–enriched LPMCs was augmented in T-bet–deficient animals compared with wild-type mice. In particular, IL-4 production by T-bet−/− and T-bet+/− LPMC was increased compared with T-bet–expressing LPMCs from wild-type littermates (Fig. 7 B). These changes in cytokine production were seen using LPMCs from both the small and large bowel suggesting that T-bet is a regulator of the mucosal Th1/Th2 cytokine balance in the entire gut immune system. Next, we assessed whether increased Th2 cytokine production by LPMCs in T-bet knockout mice was associated with evidence for activated IL-4 signaling in LP T cells. There was increased GATA-3 expression in nuclear extracts from LPMC of T-bet–deficient mice, as shown by both gel retardation assays and Western blot analysis (Fig. 7 C), consistent with an increased presence of Th2 effector T cells in the mucosa of T-bet knockout mice.
Figure 7.
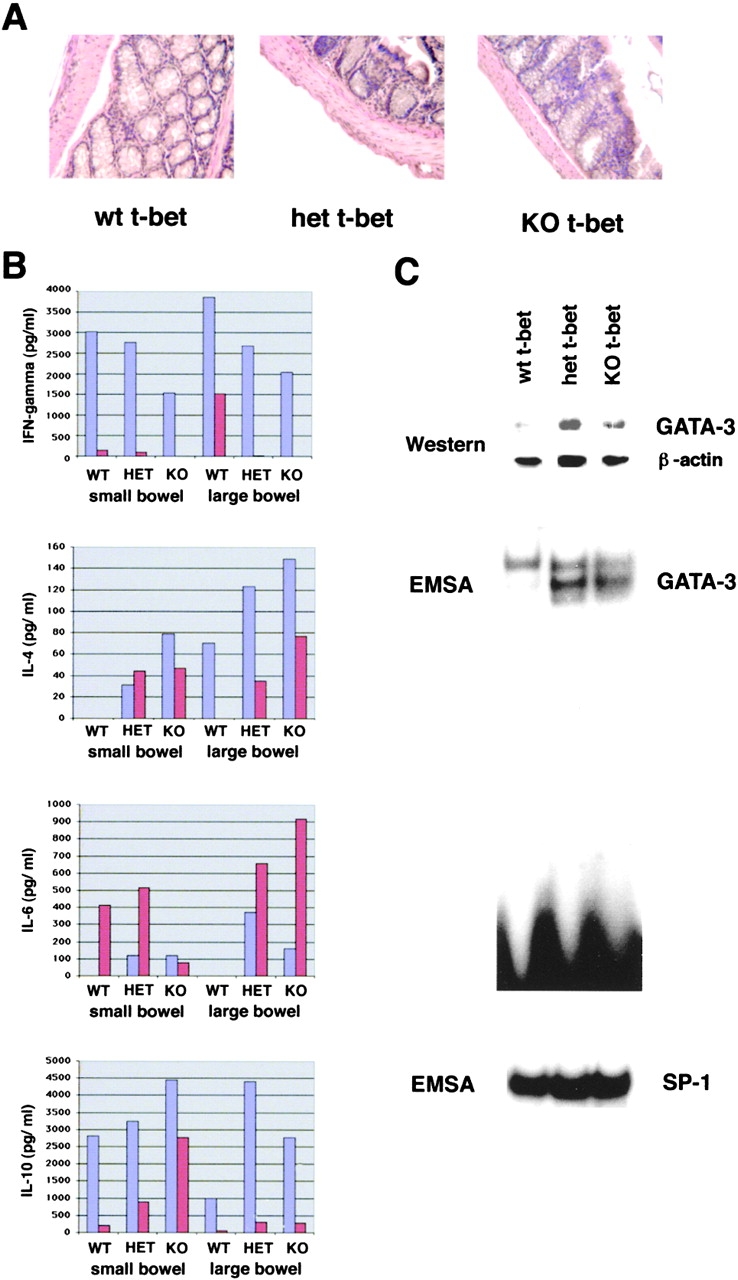
Changes in Th1 and Th2 cytokine production by LP cells in T-bet–deficient mice. (A) Histologic analysis of the large bowel of T-bet knockout mice (KO), heterozygous mice (HET), and wild-type littermates (WT). There was no evidence for colitis in T-bet–deficient animals. Similarly, no evidence of intestinal inflammation was observed in the small bowel (data not shown). (B) T cell–enriched LPMCs were isolated from the small and large bowel of wild-type (WT) and T-bet–deficient (KO) mice as well as from heterozygous (HET) mice and stimulated with antibodies to CD3 and CD28. Supernatants were taken after 48 h and analyzed for cytokine content by specific ELISA. Two representative experiments (experiment 1: red bars; experiment 2: purple bars) out of four are shown. (C) Western blot analysis and EMSA analysis for GATA-3 expression in nuclear extracts of LPMC from wild-type and T-bet–deficient mice. There was an increased expression of GATA-3 in LPMCs from T-bet knockout mice. The staining for β-actin and the EMSA for SP1 served as loading controls.
T-bet Deficient Regulatory CD62L− CD4+ T Cells Show Enhanced Protective Functions in Th1-mediated Colitis and Exhibit Increased TGF-β Production and Signaling.
Since the balance between IFN-γ and TGF-β appears to regulate chronic intestinal inflammation (50), we focused in a final series of studies on the effects of T-bet deficiency on TGF-β production and signaling in T cells and the effect of TGF-β on T-bet levels. We observed that treatment of activated T cell–enriched LPMCs with TGF-β but not IL-4 suppressed T-bet expression suggesting a reciprocal relationship between T-bet and TGF-β levels (Fig. 8 A). Furthermore, we found that T cell enriched LPMC in T-bet knockout mice produced increased amounts of TGF-β compared with cells from wild-type littermates (Fig. 8 B). This increased production of TGF-β in the absence of T-bet could be important for the regulatory function of T-bet in colitis, since TGF-β production by T cells has been recently suggested to play a key role in suppressing chronic intestinal inflammation. In intestinal inflammation, TGF-β is mainly produced by a unique population of regulatory CD25+ CD45RBlow CD62L− CD4+ T cells that have been shown to suppress colitis activity in SCID mice when cotransferred with CD4+ CD62L+ T cells (17). Furthermore, at least in the spleen IL-4–producing T cells have been shown to produce high amounts of TGF-β in secondary cultures (51).
Figure 8.



The enhanced protective capacity of CD62L− CD4+ T cells from T-bet knockout mice is associated with increased production of TGF-β and TGF-β signaling. (A) TGF-β downregulates T-bet expression in T cell–enriched LPMCs. Cells were cultured in the presence of antibodies to CD3 and CD28 with or without recombinant IL-4 and TGF-β (1 ng/ml). Cellular extracts were made after 48 h and analyzed for the expression of T-bet and β-actin by Western blot analysis. (B) TGF-β production by T cell–enriched LPMCs from wild-type (WT), T-bet heterozygous (HET), and T-bet knockout (KO) mice in the absence of colitogenic stimuli. Cells were stimulated with antibodies to CD3 plus CD28 and supernatants were analyzed by ELISA. (C) T-bet expression in splenic CD25+, CD62L+, CD4+, and CD62L− CD4+ T cells from healthy wild-type mice. Cytoplasmic (CYT) and nuclear (NUC) extracts from these cells were isolated and analyzed for T-bet expression by Western blotting. (D) Evidence for increased TGF-β–mediated signaling in T-bet–deficient CD4+ T cells. CD4+ T cells from wild-type and T-bet knockout mice were stimulated with anti-CD3 plus anti-CD28 and rIFN-γ for 12 h followed by protein extraction and Western blot analysis. Cellular extracts were analyzed for Smad7 expression whereas nuclear and cytoplasmic extracts from CD4+ T cells were analyzed for Smad3 levels. (E) Inflammation score of mice reconstituted with CD62L+ CD4+ T cells from wild-type mice and CD62L− CD4+ T cells from T-bet knockout mice (KO) and wild-type (WT) control mice. Data represent three to four mice per group and are given as mean values ± SEM. (F) FACS® analysis of splenic CD4+ T cells from mice reconstituted with CD62L+ CD4+ T cells from wild-type mice plus CD62L− CD4+ T cells from T-bet knockout mice (KO) or wild-type (WT) control mice. One representative experiment is shown. (G) TGF-β production by LPMCs from the above reconstituted mice. Cells were stimulated with antibodies to CD3 plus CD28 in serum free medium and supernatants were collected after 3 d followed by analysis of supernatants by ELISA. Data represent mean values ± SEM.
Interestingly, we observed that both splenic CD62L+ and CD62L− CD4+ T cells from healthy mice expressed large amounts of nuclear T-bet by Western blot analysis, whereas splenic CD25+ cells showed lower amounts of nuclear T-bet expression (Fig. 8 C). Furthermore, CD62L− CD45RBlow CD4+ T cells from T-bet knockout mice showed decreased expression of Smad7, an inhibitory Smad protein that is induced by IFN-γ and suppresses TGF-β signaling in T cells, compared with cells from wild-type mice. Consistently, splenic T-bet−/− CD4+ T cells exhibited increased nuclear Smad3 expression compared with T-bet–expressing CD62L− CD4+ T cells indicative of enhanced TGF-β signaling (Fig. 8 D). Based on these findings on the enhanced production of TGF-β and increased TGF-β signaling in T cells in the absence of T-bet, we then analyzed the potential regulatory capacity of CD62L− CD4+ T lymphocytes from wild-type, T-bet heterozygous, and T-bet knockout mice to suppress colitis induced by T-bet–expressing CD62L+ CD4+ T cells. Cotransfer of regulatory CD62L− CD4+ T cells plus naive CD62L+ CD4+ T cells from T-bet–expressing wild-type mice led to less severe colitis compared with mice reconstituted with CD62L+ CD4+ T cells confirming a protective role for this T cell subset in vivo (Fig. 8 E), as described previously by others (17). Moreover, cotransfer of T-bet–deficient regulatory CD62L− CD4+ T cells caused a more pronounced protective effect on CD62L+ CD4+ T cell–mediated colitis compared with regulatory CD62L− cells from wild-type mice (Fig. 8 E). This finding was associated with an increased production of TGF-β by LP T cells from reconstituted mice and by a relative expansion of the number of CD4+ CD25+ T cells in the spleen of reconstituted mice (Fig. 8 F and G).
Discussion
The balance between pro and antiinflammatory cytokines secreted by T cells has been recently suggested to be a major regulatory factor for both the initiation and perpetuation of IBD. In particular, the balance between IFN-γ/IL-4 and TGF-β activity appears to regulate chronic intestinal inflammation (43, 50, 52). In this study, we have identified T-bet as a key transcription factor that determines the mucosal cytokine balance in experimental colitis in vivo. In an initial series of studies, we demonstrated that T-bet is expressed in the nuclei of LP T cells in Crohn's disease but not in ulcerative colitis and control patients. We then took advantage of genetically altered strains of mice which overexpress or lack T-bet to uncover a critical role for this factor in murine IBD. We studied both Th1- and Th2-mediated models of IBD using both T-bet overexpressor transgenic and T-bet–deficient mice as well as adoptive transfer of T-bet–transduced and T-bet–deficient T cells. These studies revealed that T-bet–deficient T cells fail to induce Th1-mediated colitis in an adoptive transfer model of colitis while IL-4–mediated colitis is enhanced in T-bet–deficient mice. Conversely, overexpression of T-bet resulted in an earlier onset of severe Th1-mediated IBD. Finally, we showed that regulatory T cells from T-bet–deficient mice exhibit an enhanced protective capacity in the context of Th1-mediated chronic intestinal inflammation most likely caused by increased TGF-β production and signaling. Thus, these data unequivocally identify T-bet as a master switch for the regulation of T cell–mediated colitis in vivo.
The Functions of T-bet in T Cell–dependent Experimental Colitis In Vivo Correlate with T-bet–induced Changes in the Mucosal Th1/Th2 Cytokine Balance.
Previous studies have shown that both Th1 and Th2 type cytokines can cause chronic intestinal inflammation (17, 43, 45). With regard to Th1-mediated colitis both IFN-γ and TNF appear to be the major driving forces that imprint a Th1 phenotype onto the mucosal immune response, while IL-4 has been shown to mediate Th2-dependent colitis (49). In this study, we have analyzed the expression and role of T-bet in several Th1- (TNBS-colitis, IL-10−/− colitis, CD62L+ adoptive transfer system) and Th2- (oxazolone-colitis, TCR−/− colitis) mediated animal models of chronic intestinal inflammation. We observed that T-bet is a critical factor for both the initiation and perpetuation of Th1-mediated colitis in an adoptive transfer model, as shown by the fact that T-bet–deficient CD62L+ CD4+ T cells failed to induce Th1-mediated colitis in immunodeficient hosts, whereas T-bet–overexpressing CD4+ T cells induced a more rapid onset of colitis and T-bet heterozygous T cells resulted in an intermediate severity of colitis. This finding could be due to T-bet–dependent induction of Th1 cytokine production, as LP T cells from mice reconstituted with T-bet–deficient CD62L+ CD4+ T cells produced low amounts of IFN-γ. However, the protective effects of T-bet deficiency in T cells on Th1-mediated colitis were at least as good as those observed in STAT-1–deficient mice that have been shown to elicit poor biological responses to IFN-α and IFN-γ but not IL-10 (38, 53). Interestingly, T cells from IFN-γ knockout mice are known to induce Th1-mediated colitis upon transfer in immunocompromised mice (54) suggesting that T-bet has important regulatory functions in Th1 cells outside its capacity to control IFN-γ production that are essential for the development of Th1-mediated colitis. In any case, our data strongly suggest that T-bet is a master control element in the development of Th1-mediated disease.
In the absence of colitogenic stimuli T cell–enriched LP cells from T-bet–deficient mice produced decreased amounts of IFN-γ but substantially increased amounts of IL-4 compared with cells from wild-type littermates. This effect of T-bet deficiency on IFN-γ production by LP cells is likely to be due to reduced IFN-γ gene transcription, since potential regulatory binding sites for T-bet have been identified both in intronic regions of the IFN-γ gene and the IFN-γ promoter and T-bet has been shown to transactivate the IFN-γ promotor in the EL4 T cell line (39). Recent studies from Reiner and colleagues further reveal that T-bet controls Th1 effector fate by regulating IL-12β2 chain expression and directing chromatin remodeling to individual IFN-γ alleles (55). Although T-bet inhibits IL-4 and IL-5 production by an IFN-γ–independent mechanism (39), the effect on IL-4 production could also be in part due to the observed reduction of IFN-γ in T-bet–deficient LP cells, since IFN-γ has been shown to inhibit the IL-4/STAT-6 signaling pathway (56, 57) via STAT-1–dependent induction of the silencer of cytokine signaling 1 (58). Consequently, IL-4–dependent or possibly T-bet–cued activation of GATA-3 may result in Th2 T cell cytokine production (27). In any case, the observed increased production of IL-4 by LP T cells lacking T-bet appears to be relevant for chronic intestinal inflammation, as T-bet knockout mice exhibited an increase in IL-4 production during oxazolone-induced colitis and antibodies to IL-4 were capable of suppressing such an effect. Thus, the lack of T-bet expression in T lymphocytes is associated with an enhanced susceptibility to IL-4–mediated chronic intestinal inflammation. On the other hand, overexpression of T-bet in T cells results in a strikingly enhanced susceptibility for Th1-mediated colitis. Since Crohn's disease in humans appears to be mediated by IL-12 driven mucosal effector Th1 cells producing IFN-γ and TNF-5, (45, 59) and since we observed increased expression of T-bet in LP T cells from patients with this disease, these data suggest that a T-bet driven signaling pathway may be responsible for the overproduction of proinflammatory Th1 cytokines in this disease (10, 14, 16). Furthermore, the observed reduction in GATA-3 expression in this context presumably leads to further changes in the balance between T-bet and GATA-3 in Crohn's disease and may account for the reduced IL-4 production by LP T cells in this disease (10).
T-bet and TGF-β Production and Signaling in T Cells.
Regulatory T cells producing IL-10 or TGF-β have been previously shown to mediate protective effects in Th1-mediated colitis by suppressing the activity of T lymphocytes (17–19, 60). Interestingly, we observed that CD62L− CD4+ T cells from T-bet knockout mice exhibit a stronger protective effect on CD62L+ CD4+ T cell–induced colitis than the corresponding cell population from wild-type mice. This observation appears to be related to differences in TGF-β production and signaling, as CD4+ T cells from T-bet–deficient mice exhibited increased nuclear Smad3 expression. After binding of TGF-β to its receptor on T cells, Smad3 is known to interact with the TGF-β receptor I followed by importin-1β– and RanGTPase-mediated import of Smad3 into the nucleus where it controls expression of TGF-β target genes (61). Interestingly, IFN-γ has been previously shown to inhibit TGF-β signaling by a Jak1/STAT-1–mediated rapid activation of the synthesis of the inhibitory Smad-7 protein, which in turn can prevent the interaction of Smad3 with the TGF-β type I receptor (60–64). Furthermore, Smad7 can form a complex with the ubiquitin-ligase Smurf2 that targets the TGF-β receptor for degradation (65). Thus, the reduced production of IFN-γ by splenic CD4+ T cells and T cell–enriched LP cells in T-bet–deficient animals may cause reduced expression of Smad7 followed by increased TGF-β signaling via Smad3/4. Consistent with this hypothesis, we observed that CD62L− CD4+ T cells from T-bet–deficient mice express reduced levels of Smad7 compared with T cells from wild-type mice. Since administration of neutralizing antibodies to TGF-β is known to suppress the protective capacity of CD62L− CD4+ T cells on Th1-mediated colitis (17), these data suggest that the enhanced regulatory capacity of T-bet–deficient CD62L− CD4+ cells is due to increased TGF-β production and signaling. The enhanced TGF-β production of T-bet–deficient T cells may even be augmented after T cell transfer, as TGF-β has been demonstrated to positively regulate its own production (51). The relevance of a defect in TGF-β1 expression or TGF-β–mediated signaling via Smad3 for the mucosal immune system has been shown by the observation that knockout mice for these proteins develop T cell–mediated chronic intestinal inflammation (52, 66–68). Finally, we observed that TGF-β in turn downregulates T-bet expression in mucosal cells suggesting a reciprocal relationship between TGF-β and T-bet levels in T cells.
In summary, the present study identifies T-bet as a master switch for T cell-mediated chronic intestinal inflammation and the regulation of protective immune responses by TGF-β (Fig. 9) . Thus, modulation of T-bet function appears to be an attractive target for local therapeutic intervention in Th1-mediated chronic intestinal inflammation such as is observed in Crohn's disease.
Figure 9.

A regulatory role for T-bet in mucosal cytokine production. T-bet controls Th1 and Th2 cytokine production in colitis and its levels are downregulated by TGF-β. On the other hand, downregulation of T-bet is associated with increased TGF-β levels possibly due to failure of T-bet–mediated activation of Smad7.
Acknowledgments
The authors would like to thank Claudia Stemmann for help with the mouse genotyping, Dr. Müller-Lobeck (DKD Wiesbaden, Germany) and Prof. Dr. Schürmann (Iserlohn, Germany) for providing gut specimens, B. Bartsch for technical help, and Warren Strober (Mucosal Immunity Section, NIAID, Bethesda, MD) for critical reading of the manuscript.
The research of M.F. Neurath was supported by grants from the Innovationsstiftung Rheinland-Pfalz, the SFB 458, and the Gerhard Hess program of the Deutsche Forschungsgemeinschaft. M.F. Neurath is a recipient of the J. William Fulbright scholarship for senior scientists. R.S. Blumberg was supported by National Institutes of Health (NIH) grants (DK44319, DK53056, DK51362) and the Harvard Digestive Diseases Center. L.H. Glimcher was supported by NIH grant AI37833, a grant from the Juvenile Diabetes Foundation, and a gift from The G. Harold and Leila Y. Mathers Charitable Foundation. S.J. Szabo is a Fellow of the Leukemia Society of America and the recipient of a grant from the Burroughs Wellcome Foundation. E.F. Nieuwenhuis was supported by the Ter Meulen Fund, Netherlands.
L.H. Glimcher and R.S. Blumberg share senior authorship of this manuscript.
Footnotes
Abbreviations used in this paper: EMSA, electrophoretic mobility shift assay; IBD, inflammatory bowel disease; IRES, ribosomal entry sequence; LP, lamina propria; LPMC, LP mononuclear cell; RAG, recombination activation gene.
References
- 1.Podolsky, D.K. 1991. Inflammatory bowel disease. N. Engl. J. Med. 325:928–937. [DOI] [PubMed] [Google Scholar]
- 2.Neurath, M.F., S. Finotto, and W. Strober. 2001. Immunology of inflammatory bowel disease. Clinical Immunology. 2nd edition. In press. [DOI] [PubMed] [Google Scholar]
- 3.Blumberg, R.S., L.J. Saubermann, and W. Strober. 1999. Animal models of mucosal inflammation and their relation to human inflammatory bowel disease. Curr. Opin. Immunol. 11:648–656. [DOI] [PubMed] [Google Scholar]
- 4.Friedman, S., and R.S. Blumberg. 1999. Inflammatory bowel disease. Harrison's Principles of Internal Medicine. 15th edition. McGraw-Hill, New York. 1679–1691.
- 5.Duchmann, R., and M. Zeitz. 1998. Crohn's disease. Handbook of Mucosal Immunology. P. Ogra and W. Strober, editors. Academic Press, NY. 1055–1080.
- 6.Elson, C.O., R.B. Sartor, G.S. Tennyson, and R.H. Riddell. 1995. Experimental models of inflammatory bowel disease. Gastroenterology. 109:1344–1367. [DOI] [PubMed] [Google Scholar]
- 7.Sartor, R. 1998. Postoperative recurrence of Crohn's disease: the enemy is within the fecal stream. Gastroenterology. 114:398–400. [DOI] [PubMed] [Google Scholar]
- 8.Shanahan, F. 2001. Inflammatory bowel disease: immunodiagnostics, immunotherapeutics, and ecotherapeutics. Gastroenterology. 120:622–635. [DOI] [PubMed] [Google Scholar]
- 9.Breese, E., C.P. Braegger, C.J. Corrigan, J.A. Walker-Smith, and T.T. MacDonald. 1993. Interleukin-2 and interferon-γ secreting T cells in normal and diseased human intestinal mucosa. Immunology. 78:127–131. [PMC free article] [PubMed] [Google Scholar]
- 10.Fuss, I., M.F. Neurath, M. Boirivant, J.S. Klein, C.D. Motte, S.A. Strong, C. Fiocchi, and W. Strober. 1996. Disparate CD4+ lamina propria (LP) lymphocyte secretion profiles in inflammatory bowel disease. J. Immunol. 157:1261–1270. [PubMed] [Google Scholar]
- 11.Atreya, R., J. Mudter, S. Finotto, J. Müllberg, T. Jostock, S. Wirtz, M. Schütz, B. Bartsch, M. Holtmann, C. Becker, et al. 2000. Blockade of IL-6 trans-signaling suppresses T cell resistance against apoptosis in chronic intestinal inflammation: Evidence in Crohn's disease and experimental colitis in vivo. Nat. Med. 6:583–588. [DOI] [PubMed] [Google Scholar]
- 12.Fuss, I.J., T. Marth, M.F. Neurath, G.R. Pearlstein, A. Jain, and W. Strober. 1999. Anti-interleukin 12 treatment regulates apoptosis of Th1 T cells in experimental colitis in mice. Gastroenterology. 117:1078–1088. [DOI] [PubMed] [Google Scholar]
- 13.Plevy, S.E., C.J. Landers, J. Prehn, N.M. Carramanzana, R.L. Deem, D. Shealy, and S.R. Targan. 1997. A role for TNF-α and mucosal T helper-1 cytokines in the pathogenesis of Crohn's disease. J. Immunol. 159:6276–6282. [PubMed] [Google Scholar]
- 14.Targan, S.R., S.B. Hanauer, S.J.V. Deventer, L. Mayer, D.H. Present, T. Braakman, K.L.D. Woody, T.F. Schaible, and P.J. Rutgeerts. 1997. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor α for Crohn's disease. N. Engl. J. Med. 337:1029–1035. [DOI] [PubMed] [Google Scholar]
- 15.Pender, S.L., J.M. Fell, S.M. Chamow, A. Ashkenazi, and T.T. MacDonald. 1998. A p55 TNF receptor immunoadhesin prevents T cell-mediated intestinal injury by inhibiting matrix metalloproteinase production. J. Immunol. 160:4098–4103. [PubMed] [Google Scholar]
- 16.Targan, S.R., R.L. Deem, M. Liu, S. Wang, and A. Nel. 1995. Definition of a lamina propria T cell responsive state. Enhanced cytokine responsiveness of T cells stimulated through the CD2 pathway. J. Immunol. 154:664–675. [PubMed] [Google Scholar]
- 17.Powrie, F., J. Carlino, M.W. Leach, S. Mauze, and R.L. Coffman. 1996. A critical role for transforming growth factor-beta but not interleukin-4 in the suppression of T helper type 1-mediated colitis by CD45Rb(low) CD4+ T cells. J. Exp. Med. 183:2669–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neurath, M.F., I. Fuss, B.L. Kelsall, D.H. Presky, W. Waegell, and W. Strober. 1996. Experimental granulomatous colitis in mice is abrogated by induction of TGF-β-mediated oral tolerance. J. Exp. Med. 183:2515–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitani, A., I.J. Fuss, K. Nakamura, O.M. Schwartz, T. Usui, and W. Strober. 2000. Treatment of experimental (TNBS)-colitis by intranasal administration of TGF-β1 plasmid: TGF-β1–mediated suppression of Th1 response occurs by IL-10 induction and IL-12Rb2 chain down-regulation. J. Exp. Med. 192:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leonard, W.J. 1996. STATs and cytokine specificity. Nat. Med. 2:968–969. [DOI] [PubMed] [Google Scholar]
- 21.Rengarajan, J., and S.J. Szabo. 2000. Transcriptional regulation of Th1/Th2 polarization. Immunol. Today. 21:479–483. [DOI] [PubMed] [Google Scholar]
- 22.Grogan, J.L., M. Mohrs, B. Harmon, D.A. Lacy, J.W. Sedat, and R.M. Locksley. 2001. Early transcription and silencing of cytokine genes underlie polarization of T helper cell subsets. Immunity. 14:205–215. [DOI] [PubMed] [Google Scholar]
- 23.Rao, A., C. Luo, and P.G. Hogan. 1997. Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol. 15:707–747. [DOI] [PubMed] [Google Scholar]
- 24.Asnagli, H., and K.M. Murphy. 2001. Stability and commitment in T helper cell development. Curr. Opin. Immunol. 13:242–247. [DOI] [PubMed] [Google Scholar]
- 25.Li, B., C. Tournier, R.J. Davis, and R.A. Flavell. 1999. Regulation of IL-4 expression by the transcription factor JunB during T helper cell differentiation. EMBO J. 18:420–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim, J.I., I.C. Ho, M.J. Grusby, and L.H. Glimcher. 1999. The transcription factor c-maf controls the production of interleukin-4 but not other Th2 cytokines. Immunity. 10:745–751. [DOI] [PubMed] [Google Scholar]
- 27.Zheng, W., and R.A. Flavell. 1997. The transcription factor GATA-3 is necessary and sufficent for Th2 cytokine gene expression in CD4+ T cells. Cell. 89:587–596. [DOI] [PubMed] [Google Scholar]
- 28.Glimcher, L.H., and K.M. Murphy. 2000. Lineage commitment in the immune system: the T helper lymphocyte grows up. Genes Dev. 14:1693–1711. [PubMed] [Google Scholar]
- 29.Kurata, H., H.J. Lee, A. O' Garra, and N. Arai. 1999. Ectopic expression of activated STAT-6 induces the expression of Th2-specific cytokines and transcription factors in developing Th1 cells. Immunity. 11:677–688. [DOI] [PubMed] [Google Scholar]
- 30.Ranger, A.M., M.R. Hodge, E.M. Gravallese, M. Oukka, L. Davidson, F.W. Alt, F.C. Brousse, T. Hoey, M. Grusby, and L.H. Glimcher. 1998. Delayed lymphoid repopulation with defects in IL-4-driven responses produced by inactivation of NF-ATc. Immunity. 8:125–134. [DOI] [PubMed] [Google Scholar]
- 31.Ouyang, W., M. Löhning, Z. Gao, M. Assenmacher, S. Ranganath, A. Radbruch, and K.M. Murphy. 2000. Stat-6 independent GATA-3 autoactivation directs IL-4 independent Th2 development and commitment. Immunity. 12:27–37. [DOI] [PubMed] [Google Scholar]
- 32.Ouyang, W., S.H. Ranganath, K. Weindel, D. Bhattacharya, T.L. Murphy, W.C. Sha, and K.M. Murphy. 1998. Inhibition of Th1 development mediated by GATA-3 through an IL-4 independent mechanism. Immunity. 9:745–755. [DOI] [PubMed] [Google Scholar]
- 33.Lee, G.R., P.E. Fields, and R.A. Flavell. 2001. Regulation of IL-4 gene expression by distal regulatory elements and GATA-3 at the chromatin level. Immunity. 14:447–459. [DOI] [PubMed] [Google Scholar]
- 34.Loots, G.G., R.M. Locksley, C.M. Blakespoor, Z.E. Wang, W. Miller, E.M. Rubin, and K.A. Frazer. 2000. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science. 288:136–140. [DOI] [PubMed] [Google Scholar]
- 35.O'Shea, J.J. 1997. Jaks, STATs, cytokine signal transduction, and immunoregualtion: are we there yet? Immunity. 7:1–11. [DOI] [PubMed] [Google Scholar]
- 36.Oppmann, B., R. Lesley, B. Blom, J.C. Timans, Y. Xu, B. Hunte, F. Vega, N. Yu, J. Wang, K. Singh, et al. 2000. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biologic activities similar as well as distinct from IL-12. Immunity. 13:715–725. [DOI] [PubMed] [Google Scholar]
- 37.Carter, L.L., and K.M. Murphy. 1999. Lineage-specific requirement for signal transducer and activator of transcription Stat4 in interferon-γ production from CD4+ versus CD8+ T cells. J. Exp. Med. 189:1355–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Durbin, J.E., R. Hackenmiller, M.C. Simon, and D.E. Levy. 1996. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 84:443–450. [DOI] [PubMed] [Google Scholar]
- 39.Szabo, S.J., S.T. Kim, G.L. Costa, X. Zhang, C.G. Fathman, and L.H. Glimcher. 2000. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 100:655–669. [DOI] [PubMed] [Google Scholar]
- 40.Finotto, S., G.T.D. Sanctis, H.A. Lehr, U. Herz, M. Buerke, M. Schipp, B. Barsch, R. Atreya, E. Schmitt, P.R. Galle, H. Renz, and M.F. Neurath. 2001. Treatment of allergic airway inflammation and hyperresponsiveness by local antisense-induced blockade of GATA-3 expression. J. Exp. Med. 193:1247–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szabo, S.J., B.M. Sullivan, C. Stemmann, A.R. Satoskar, B.P. Sleckman, and L.H. Glimcher. 2002. T-bet is essential for Th1 lineage commitment and IFN-γ production in CD4 but not CD8 T cells. Science. 295:338–342. [DOI] [PubMed] [Google Scholar]
- 42.Finotto, S., M.F. Neurath, J.N. Glickman, S. Qin, H.A. Lehr, F.H. Green, K. Ackerman, K. Haley, P.R. Galle, S.J. Szabo, J.M. Drazen, G.T. de Sanctis, and L.H. Glimcher. 2002. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science. 295:336–338. [DOI] [PubMed] [Google Scholar]
- 43.Mizoguchi, A., E. Mizoguchi, and A.K. Bhan. 1999. The critical role for interleukin-4 but not interferon-γ in the pathogenesis of colitis in T-cell receptor α mutant mice. Gastroenterology. 116:320–326. [DOI] [PubMed] [Google Scholar]
- 44.Kuhn, R., J. Lohler, D. Rennick, K. Rajewsky, and W. Mueller. 1993. Interleukin-10 deficient mice develop chronic enterocolitis. Cell. 75:263–274. [DOI] [PubMed] [Google Scholar]
- 45.Neurath, M.F., I. Fuss, B.L. Kelsall, E. Stuber, and W. Strober. 1995. Antibodies to IL-12 abrogate established experimental colitis in mice. J. Exp. Med. 182:1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hofmann, A., G.P. Nolan, and H.M. Blau. 1996. Rapid retroviral delivery of tetracycline-inducible genes in a single autoregulatory cassette. Proc. Natl. Acad. Sci. USA. 93:5185–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boirivant, M., I.J. Fuss, A. Chu, and W. Strober. 1998. Oxazolone colitis: a murine model of T helper cell type 2 colitis treatable with antibodies to interleukin-4. J. Exp. Med. 188:1929–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu, H., N.A. DiIulio, and R.L. Fairchild. 1996. T cell populations primed by hapten sensitization in contact sensitivity are distinguished by polarized patterns of cytokine production: interferon γ-producing (Tc1) effector CD8+ T cells and interleukin-4/-10 producing (Th2) negative regulatory CD4+ T cells. J. Exp. Med. 183:1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Claesson, M.H., S. Bregenholt, K. Bonhagen, S. Thoma, P. Moeller, M.J. Grusby, F. Leithauser, M.H. Nissen, and J. Reimann. 1999. Colitis-inducing potency of CD4+ T cells in immunodeficient, adoptive hosts depends on their state of activation, IL-12 responsiveness, and CD45RB surface phenotype. J. Immunol. 162:3702–3710. [PubMed] [Google Scholar]
- 50.Strober, W., B. Kelsall, I. Fuss, T. Marth, B. Ludviksson, R. Ehrhardt, and M.F. Neurath. 1997. Reciprocal IFN-γ and TGF-β responses regulate the occurrence of mucosal inflammation. Immunol. Today. 18:61–64. [DOI] [PubMed] [Google Scholar]
- 51.Seder, R.A., T. Marth, M.C. Sieve, W. Strober, J.J. Letterio, A.B. Roberts, and B.L. Kelsall. 1998. Factors involved in the differentiation of TGF-β-producing cells from naive CD4+ T cells: IL-4 and IFN-γ have opposing effects, while TGF-β positively regulates its own production. J. Immunol. 160:5719–5728. [PubMed] [Google Scholar]
- 52.Yang, X., J.J. Letterio, R.J. Lechleider, L. Chen, R. Hayman, H. Gu, A.B. Roberts, and C. Deng. 1999. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 18:1280–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meraz, M.A., J.M. White, K.C. Sheehan, E.A. Bach, S.J. Rodig, A.S. Dighe, D.H. Kaplan, J.K. Riley, A.C. Greenlund, D. Campbell, et al. 1996. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 84:431–442. [DOI] [PubMed] [Google Scholar]
- 54.Simpson, S.J., S. Shah, M. Comiskey, V.P. de-Jong, B. Wang, E. Mizoguchi, A.K. Bhan, and C. Terhorst. 1998. T cell-mediated pathology in two models of experimental colitis depends predominantly on the interleukin 12/Signal transducer and activator of transcription (Stat)-4 pathway, but is not conditional on interferon γ expression by T cells. J. Exp. Med. 187:1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mullen, A.C., F.A. High, A.S. Hutchins, H.W. Lee, A.V. Villarino, D.M. Livingston, A.L. Kung, N. Cereb, T.P. Yao, S.Y. Yang, and S.L. Reiner. 2001. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science. 292:1907–1910. [DOI] [PubMed] [Google Scholar]
- 56.Venkataraman, C., S. Leung, A. Salvekar, H. Mano, and U. Schindler. 1999. Repression of IL-4 induced gene expression by IFN-γ requires STAT-1 activation. J. Immunol. 162:4053–4061. [PubMed] [Google Scholar]
- 57.Dickensheets, H.L., C. Venkataraman, U. Schindler, and R.P. Donnelly. 1999. Interferons inhibit activation of STAT-6 by interleukin-4 in human monocytes by inducing SOCS-1 gene expression. Proc. Natl. Acad. Sci. USA. 96:10800–10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen, P.X., J.A. Losman, and P. Rothman. 2000. SOCS proteins, regulators of intracellular signaling. Immunity. 13:287–290. [DOI] [PubMed] [Google Scholar]
- 59.Monteleone, G., L. Biancone, R. Marasco, G. Morrone, O. Marasco, F. Luzza, and F. Pallone. 1997. Interleukin-12 is expressed and actively released by Crohn's disease intestinal lamina propria mononuclear cells. Gastroenterology. 112:1169–1178. [DOI] [PubMed] [Google Scholar]
- 60.Monteleone, G., A. Kumberova, N.M. Croft, C. McKenzie, H.W. Steer, and T.T. MacDonald. 2001. Blocking Smad7 restores TGF-β1 signaling in chronic inflammatory bowel disease. J. Clin. Invest. 108:601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kurisaki, A., S. Kose, Y. Yoneda, C.H. Heldin, and A. Moustakis. 2001. Transforming growth factor β induces nuclear import of Smad3 in an importin-β1 and Ran-dependent manner. Mol. Cell. Biol. 12:1079–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ulloa, L., J. Doody, and J. Massague. 1999. Inhibition of transforming growth factor-β/SMAD signalling by the interferon-γ/STAT pathway. Nature. 397:710–713. [DOI] [PubMed] [Google Scholar]
- 63.Nakao, A., M. Afrakhte, A. Moren, T. Nakayama, J.L. Christian, R. Heuchel, S. Itoh, M. Kawabata, N.E. Heldin, C.H. Heldin, and P.T. Dijke. 1997. Identification of Smad7, a TGF-β-inducible antagonist of TGF-β signalling. Nature. 389:631–635. [DOI] [PubMed] [Google Scholar]
- 64.Hayashi, H., S. Abdollah, Y. Qiu, J. Cai, Y. Xu, B.W. Grinnell, M.A. Richardson, J.N. Topper, M.A. Gimbrone, J.L. Wrana, and D. Falb. 1997. The MAD-related protein Smad7 associates with the TGF-β receptor and functions as an antagonist of TGF-β signaling. Cell. 89:1165–1173. [DOI] [PubMed] [Google Scholar]
- 65.Kavsak, P., R.K. Rasmussen, C.G. Causing, S. Bonni, H. Zhu, G.H. Thomsen, and J.L. Wrana. 2000. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF-β receptor for degradation. Mol. Cell. 6:1365–1375. [DOI] [PubMed] [Google Scholar]
- 66.Shull, M.M., I. Ormsby, A.B. Kier, S. Pawlowski, R.J. Diebold, M. Yin, R. Allen, C. Sidman, G. Proetzel, D. Calvin, N. Annunziata, and T. Doetschmann. 1992. Targeted disruption of the mouse transforming growth factor β-1 gene results in multifocal inflammatory disease. Nature. 359:693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kulkarni, A.B., C.G. Huh, D. Becker, A. Geiser, M. Lyght, K.C. Flanders, A.B. Roberts, M.B. Sporn, J.M. Ward, and S. Karlsson. 1993. Transforming growth factor β1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA. 90:770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Diebold, R.J., M.J. Eis, M. Yin, I. Ormsby, G.P. Boivin, B.J. Darrow, J.E. Saffitz, and T. Doetschman. 1995. Early-onset multifocal inflammation in the transforming growth factor β1-null mouse is lymphocyte mediated. Proc. Natl. Acad. Sci. USA. 92:12215–12219. [DOI] [PMC free article] [PubMed] [Google Scholar]