

Abstract
c-Jun NH2-terminal kinases (JNK) play important roles in T helper cell (Th) proliferation, differentiation, and maintenance of Th1/Th2 polarization. To determine whether JNKs are involved in antiviral T cell immunity, and whether JNK1 and JNK2 bear biological differences, we investigated the immune responses of JNK1-deficient and JNK2-deficient mice to lymphocytic choriomeningitis virus (LCMV). After LCMV infection, wild-type (JNK+/+) mice had a 5- to 10-fold increase in splenic CD8+ T cells. In contrast, infected JNK1−/− mice showed a significantly lower virus-specific CD8+ T cell expansion. However, JNK1−/− mice cleared LCMV infection with similar kinetics as JNK+/+ mice. Splenic T cells from LCMV-infected JNK1−/− animals produced interferon γ after stimulation with viral peptides. However, fewer JNK1−/− T cells acquired an activated phenotype (CD44hi) and more JNK1−/−CD8+CD44hi cells underwent apoptosis than JNK+/+ cells at the peak of the primary response. In contrast, LCMV-infected JNK2−/− mice generated more virus-specific CD8+ T cells than JNK+/+ mice. These results indicate that JNK1 and JNK2 signal pathways have distinct roles in T cell responses during a viral infection. JNK1 is involved in survival of activated T cells during immune responses, and JNK2 plays a role in control of CD8+ T cell expansion in vivo.
Keywords: viral infection, cellular activation, T lymphocytes, protein kinases, lymphocytic choriomeningitis virus
Introduction
Immune responses are mediated via cell-to-cell contact or secretion of soluble mediators. In both instances, receptor molecules transmit this information from the cell's surface to its interior, thereby transducing a cascade of intracellular events. Numerous cellular receptors on the T cell surface have been studied for their role in immune responses; however, the signaling pathways activated after ligands bind to such receptors are not completely understood. Mitogen-activated protein (MAP)* kinase signaling pathways are involved in control of immune responses (1, 2). MAP kinases link cell-surface receptors to regulatory intracellular targets leading to the control of pivotal processes such as gene expression, cell proliferation and motility, cell survival, and death (3). At least four groups of MAP kinases are expressed in mammals: extra cellular signal–related kinases (ERK) 1–2, ERK5, p38 proteins, and c-Jun NH2-terminal kinases (JNK1/2/3; reference 3).
The JNKs, also known as stress-activated protein kinases, phosphorylate the transcription factors c-Jun (part of AP-1 complex), JunD, ATF2, ATFa, Elk-1, Sap-1, nuclear factor of activated T cells (NFAT)c1, and NFAT4 (1, 2, 4, 5). Three JNK genes have been identified: JNK1, JNK2, and JNK3, and their alternative splicing results in at least 10 isoforms of JNK (6). JNK1 and JNK2 are ubiquitously expressed, whereas JNK3 is expressed mainly in brain, testis, and heart. JNK is involved in cell death, proliferation, and differentiation as well as cells' response to stress such as ultraviolet light and proinflammatory molecules. Although lymphocyte development is normal in JNK1-deficient (−/−) mice (7) and JNK2−/− mice (8), differences in lymphocyte responses have been observed. For example, when naive CD4+ T cells are stimulated in vitro with polyclonal antibodies (against CD3 and CD28) or mitogen, these cells from JNK1−/− mice preferentially differentiate into a Th2 phenotype and show a decreased activation-induced cell death (7), whereas CD4+ T cells from JNK2−/− mice show impaired IFN-γ production and a diminished Th1 response (8). One in vivo consequence of such defects has been shown to affect Leishmania major infection of JNK1−/− mice bred on a genetically resistant background. Such mice fail to resolve the infection, showing enhanced, noncurative Th2 Leishmania-specific responses (9). Hence, it is thought that JNK1 MAP kinase is required for effective polarization toward a Th1 response in vivo (9).
Because JNK has a critical role in Th activation and JNK activity is also detectable in CD8+ T cells (see Conze et al. in this issue [9a]), we evaluated the role of these MAP kinases in physiological CD8+ T cell–mediated immune responses. CD8+ T cells play a critical role in the control of many viral pathogens, including lymphocytic choriomeningitis virus (LCMV). LCMV is a natural pathogen for mice. Because the T cell response to LCMV is well characterized in mice, these mice have been used extensively to explore molecular and cellular mechanisms of viral clearance and viral persistence (for a review, see reference 10) as well as regulation of the immune response (11, 12) of both primary and memory phases. After infection with LCMV, adult immunocompetent mice mount a robust virus-specific CD8+ T cell response that clears the virus infection (13, 14). Kinetically during the LCMV infection, the virus-specific CD8+ and CD4+ T cell populations expand (15–18), followed by a contraction phase in which many of these cells die by apoptosis; a stable life-long population of memory CD8+ T cells remains (18–22), and a CD4+ T cell memory population declines slowly (18). Further, the LCMV model has been used to evaluate the physiological contributions of many soluble mediators and cell-to-cell contact molecules to the antiviral immune responses (23–30).
The role of MAP kinases in antiviral CD8+ and CD4+ T cell immune responses has not previously been established. In this paper, we evaluate the immune responses to LCMV infection as influenced by JNK1 and JNK2 MAP kinases. This was accomplished by study of mice genetically deficient for either JNK1 or JNK2, by using staining of virus-specific induced IFN-γ producing CD4+ and CD8+ T cells and tetramer staining of viral-specific CD8+ T cells (16, 17, 31). Our results reveal that JNK1 and JNK2 assume distinctly different functions in CD8+ T cell activation and expansion.
Materials and Methods
Mice.
The generation, breeding, and use of JNK1 genetically deficient (knockout; JNK1−/−), JNK2 genetically deficient (JNK2−/−), and JNK+/+ mice has been described (7, 8). Mice in each group were backcrossed to C57BL/6 for 6–8 generations. Breeding and maintenance were performed under specific pathogen-free conditions at Scripps Research Institute (La Jolla, CA) and at Yale University (New Haven, CT).
Virus, Virus Quantification, and Infection of Mice.
The Armstrong 53b strain (ARM) and the clone-13 (Cl-13) strain of LCMV were used. Virus origins, sequence, growth in baby hamster kidney cells, and titration by standard plaque assay on Vero cells have been described (32, 33). Mice were infected when 8 to 15 wk of age by intraperitoneal route using 2 × 105 PFU of LCMV ARM or by intravenous route using 2 × 106 PFU of LCMV-Cl-13 in a volume of 0.2 ml. For analyzing immune memory, mice were infected with 2 × 105 PFU of LCMV ARM intraperitoneally at least 60 d before the second infection with 2 × 106 PFU of LCMV-Cl-13 intravenously.
Peptides.
In vitro epitope-specific stimulation used MHC class I–restricted peptides (glycoprotein [GP] amino acids [aa] 33–41 [KAVYNFATC] and nucleoprotein [NP] aa 396–404 [FQPQNGQFI]) obtained from Peptidogenic, and MHC class II–restricted peptides (GP aa 61–80 [GLNGPDIYKGVYQFKSVEFD] and NP aa 309–328 [SGEGWPYIACRTSIVGRAWE]) obtained from Chiron Corp.
CTL Assay.
Primary LCMV-specific CTL activity of splenocytes obtained from LCMV-infected mice was measured in a standard 5-h 51Cr release assay as described (13). Briefly, 51Cr-labeled Balb C17 (H-2d) and MC57 (H-2b) cells either uninfected or infected with LCMV-ARM at a multiplicity of infection (MOI) of 1 were used as targets 48 h later. Effector cells harvested from spleens of LCMV-infected mice 7 d after infection were added to LCMV infected or uninfected targets at ratios of 50:1, 25:1, and 12:1. Lysis was measured after a 5-h incubation period at 37°C. Samples were set up in triplicate and variance was less than 10%. The results are expressed as the percentage of specific 51Cr release (13).
In Vitro Epitope-specific Stimulation.
Spleens were harvested from JNK1−/−, JNK2−/−, and JNK+/+ mice at different time points after infection. Cells in the spleen were obtained by teasing apart spleens, passing the cells through a nylon mesh, and lysing erythrocytes with 0.83% NH4Cl. When appropriate these cells were cultured in RMPI (GIBCO BRL/LifeTechnologies) containing 10 mM HEPES (GIBCO BRL), 50 U/ml penicillin (GIBCO BRL), 50 μg/ml streptomycin (GIBCO BRL), 1 mM sodium pyruvate (GIBCO BRL), 0.1 mM nonessential amino acids (GIBCO BRL), and 50 μM 2-mercaptoethanol. Cytokine intracellular staining was performed on 106 splenocytes stimulated 5 h with 1 μg/ml of MHC-class I peptide (NP aa 396–404 or GP aa 33–41) or 2 μg/ml of MHC class II peptide (GP aa 61–80 or NP aa 309–328) in the presence of 50 U/ml of recombinant human IL-2 (Roche) and 1 μg/ml or 5 μg/ml of brefeldin A (Sigma-Aldrich).
Flow Cytometry.
Splenocytes were stained for cell surface antigens, using anti-CD8 antibodies conjugated either to allophycocyanin (APC), PE, or FITC, and anti-CD4 antibodies conjugated either to Cychrome or FITC (BD PharMingen), for 20–30 min on ice in PBS containing 1% (vol/vol) FBS and 0.1% (wt/vol) NaN3. After, cells were washed two times, fixed, and permeabilized in 4% (wt/vol) paraformaldehyde, and 0.1% (wt/vol) saponin in HBSS. Intracellular cytokine staining was accomplished with incubation of antibodies to IFN-γ PE or IL-2-APC for 30 min on ice in 0.1% (wt/vol) saponin, 1% FBS PBS, followed by two washes and resuspension of cells in 1% (vol/vol) FBS, 0.1% (wt/vol) NaN3 PBS, as described (34). For annexin-V staining, sodium azide was omitted in all buffers and cells were first stained with surface markers (CD4, CD8, and CD44) and then exposed to annexin-V–conjugated to PE and 7-amino-actinomycin D (7-AAD; BD PharMingen) according to the manufacturer's instructions. Cells were acquired on a FACSCalibur™ flow cytometer (Becton Dickinson) and analyzed with CELLQuest™ (Becton Dickinson) and FlowJo (Treestar) software.
Statistical Analyses.
Data handling and analysis (Student's t test) were performed using Prism 3.0 (GraphPad Software).
Results
JNK1−/− and JNK2−/− Mice Clear LCMV Equivalently After an Acute Infection.
First, we determined whether a deficiency in the MAP kinases JNK1 or JNK2 would interfere in the viral clearance of LCMV. After inoculation with LCMV ARM intraperitoneally, JNK1−/−, JNK2−/−, and JNK+/+ mice cleared all detectable virus from their sera by 8 d after infection (Table I) indicating that neither JNK1 nor JNK2 deficiencies prevented the clearance of over 3–4 logs of infectious virus.
Table I.
Primary CTL Activity and Function Are Normal in JNK1−/− and JNK2−/− Animals
| Specific 51Cr releasea | ||||
|---|---|---|---|---|
| Group | Effector/ target ratio |
H-2b | H-2d | Clearance of LCMV from serab |
| JNK1−/− | 50:1 | 35 ± 4 | <1 | 4/4 |
| 25:1 | 23 ± 3 | <1 | ||
| 12:1 | 14 ± 4 | <1 | ||
| JNK2−/− | 50:1 | 40 ± 6 | <1 | 4/4 |
| 25:1 | 40 ± 2 | <1 | ||
| 12:1 | 22 ± 5 | <1 | ||
| JNK+/+ | 50:1 | 27 ± 4 | <1 | 4/4 |
| 25:1 | 22 ± 3 | <1 | ||
| 12:1 | 14 ± 2 | <1 | ||
Average of four mice per group, no statistical difference between groups.
To evaluate clearance of infectious virus, sera were harvested 8 d after LCMV inoculation while 2-3 d after inoculation with LCMV-ARM 2 × 105 PFU intraperitoneal viral titers range at 3 logs of infectious virus/ml of sera (references 23 and 34), at day 8 no detectable infectious virus was observed in any of the sera by plaque assay (reference 32).
Expansion of Virus-specific CD8+ T Cells Is Increased in JNK2−/− Mice during Acute LCMV Infection but CD8+ T Cell Immune Functions Are Normal in Both JNK1−/− and JNK2−/− Mice.
The rapid clearance of LCMV virus during acute infection is mediated by the lytic activity of CD8+ CTL (35–37). Besides this lytic activity, CTL can also secrete cytokines such as IFN−γ and IL-2 upon specific MHC-restricted TCR interaction (38). We tested for these two activities in splenocytes prepared 7 d after LCMV-ARM infection. T cells harvested from JNK1−/−, JNK2−/−, and JNK+/+ mice showed an efficient and equivalent MHC-restricted virus specific killing, which correlated directly with clearance of the virus from inoculated mice (Table I).
The expansion of LCMV-specific T cells peaks at about 8 d after infection (16), and up to 70% of total CD8+ T cells are virus specific. Immunodominant epitopes for C57BL/6 mice are GP aa 33–41 and NP aa 396–404. At day 7 after infection, the resulting percentages of LCMV-specific CD8+ T cells after in vitro stimulation with GP aa 33–41 and NP aa 396–404 were similar for all groups (Fig. 1 and Table II). Interestingly, at day 8 after infection, JNK2−/− mice showed a statistically significant increase (P < 0.05) in the relative expansion of virus specific CD8+ T cells when compared with JNK+/+ for both tested epitopes (GP33–41 shown on Fig. 1 and Table II for NP396–404 and GP 33–41). Similar results were seen in two independent experiments using 3–4 mice per group. In contrast, JNK1−/− and JNK+/+ had similar percentages of virus-specific CD8+ T cells for these peptides. T cells from naive animals of all groups failed to produce IFN-γ after LCMV-peptide stimulation.
Figure 1.

After LCMV infection, the percentage of virus specific splenic T cells is higher in JNK2−/−, but similar in JNK1−/− and JNK+/+ mice. JNK1−/−, JNK2−/−, and JNK+/+ animals were injected intraperitoneally with LCMV ARM 2 × 105 PFU. 7 (D7) or 8 (D8) d after infection, spleens were processed and stimulated for 5 h in vitro with virus specific peptide GP aa 33–41. Cells were then stained for CD8 and intra-cellular IFN-γ. Values written in the dot plots indicate the percentage of CD8+ T cells positive for IFN-γ.
Table II.
Splenocytes from 7 and 8 d after LCMV Inoculation Were Stimulated In Vitro with LCMV-specific Peptides and Cytokine Production Was Assessed by Flow Cytometry
| IFN-γ
|
IL-2
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Percentage of splenic CD8+ T cellsa , b |
Percentage of splenic CD4+ T cellsa , b |
Percentage of splenic CD8+ T cellsa , b |
Percentage of splenic CD4+ T cellsa , b |
||||||
| Group | GP 33–41 | NP 396–404 | GP 61–80 | NP 309–328 | GP 33–41 | NP 396–404 | GP 61–80 | NP 309–328 | |
| JNK1−/− | D7 | 22 ± 1 | 15 ± 1 | 5.0 ± 0.3 | 1.1 ± 0.3 | 1.8 ± 0.3 | 0.6 ± 0.1 | 2.6 ± 1 | 0.3 ± 0.05 |
| D8 | 36 ± 1 | 18 ± 1 | 4.9 ± 0.5 | 1.7 ± 0.2 | |||||
| JNK2−/− | D7 | 26 ± 2 | 18 ± 2 | 7.4 ± 1.3 | 1.5 ± 0.2 | 1.2 ± 0.1 | 0.6 ± 0.1 | 4.5 ± 0.5 | 0.3 ± 0.05 |
| D8 | 53 ± 2c | 29 ± 3c | 11.1 ± 1.7d | 1.8 ± 0.4 | |||||
| JNK+/+ | D7 | 23 ± 1 | 14 ± 1 | 6.1 ± 1.0 | 1.4 ± 0.5 | 1.4 ± 0.1 | 0.3 ± 0.03 | 3.6 ± 0.5 | 1.0 ± 0.4 |
| D8 | 30 ± 2 | 16 ± 1 | 6.3 ± 0.9 | 2.3 ± 0.3 | |||||
D7, day 7; D8, day 8.
Average of four mice per group.
Percentage of CD8+ or CD4+ positive for cytokine as measured by flow cytometry ± standard error.
P < 0.05 compared to JNK+/+.
P = 0.05.
As IL-2 plays a role in T cell proliferation, and JNK affects the stability of the IL-2 mRNA (39), we next evaluated the percentage of IL-2 producing virus-specific T cells in JNK1−/−, JNK2−/−, and JNK+/+ mice. IL-2–positive CD8+ T cells were also positive for IFN-γ. There was no significant difference in the percentage of CD8+ T cells secreting IL-2 after stimulation with either one of the two major MHC class I–restricted immunodominant peptides (see Table II). Additionally, when supernatants of splenocytes stimulated with class I–restricted peptides (GP aa 33–44 or NP aa 396–404) for 25–30 h were tested by ELISA, JNK1−/−, JNK2−/−, and JNK+/+ cells secreted similar amounts of IL-2 (data not shown). T cells from naive animals did not produce IL-2 when stimulated with LCMV-specific MHC class I–restricted peptides. Furthermore, CD25, the high affinity IL-2 receptor was similarly expressed on CD4+ and CD8+ T cells from LCMV-infected JNK1−/−, JNK2−/−, and JNK+/+ mice (data not shown).
Virus-specific CD4+ T Cell Immune Functions Are Normal in JNK1−/− and JNK2−/− Mice Infected with LCMV.
Having studied CD8+ T cell immune functions, we then analyzed CD4+ T cell functions by stimulating splenocytes independently with two MHC class II–restricted immunodominant epitopes of LCMV; GP aa 61–80 and NP aa 309–328 (40), and measuring the percentage of CD4+ T cells induced to produce IFN-γ. Previous studies showed that over 10% of CD4+ T cells can be recorded as virus-specific after LCMV peptide stimulation (19). Stimulation with GP aa 61–80 or NP aa 309–328 peptide resulted in similar percentage of IFN-γ producing CD4+ T cells from JNK1−/− and JNK+/+ mice. The percentage of CD4+ T cells secreting IFN-γ after a GP aa 61–80 was higher for JNK2−/− cells than for JNK+/+ cells.
The percentage of IL-2 secreting CD4+ T cells after in vitro stimulation with GP aa 61–80 and NP aa 309–328 MHC class II peptides was assessed (see Table II). A greater percentage of GP aa 61–80–specific CD4+ T cells produced IL-2 from JNK2−/− mice when compared with CD4+ T cells from JNK+/+ mice, similar to results obtained for IFN-γ. Furthermore, there was a dichotomy between responses to the GP (high response) and NP (low response) CD4+ T cell epitopes. When IL-2 secretion was measured by ELISA after GP aa 61–80 stimulation for 25–30 h, virus-specific JNK2−/− and JNK1−/− CD4+ T cells produced slightly more IL-2 than CD4+ T cells from JNK+/+ mice although the difference was not statistically significant (data not shown, P > 0.1). The mean fluorescence intensity was similar for IL-2 and IFN-γ producing CD4+ T cells from all groups indicating comparable levels of cytokine production.
Virus-specific T Cell Number Is Differentially Controlled in JNK1−/− and JNK2−/− Mice During Acute LCMV Infection.
During acute LCMV infection, spleens from JNK1−/− never expanded to the size observed in JNK+/+ mice. We hypothesized that the expansion of virus-specific T cells could be impaired in JNK1−/− mice. We therefore analyzed relative percentages of CD8+ T cells and total numbers of CD8+ and CD4+ T cells per spleen. 7 d after LCMV infection, JNK+/+ and JNK2−/− mice expanded their CD8+ T cell population from 16 to ∼40% (Fig. 2) , while JNK1−/− mice failed to display the degree of relative expansion of splenic CD8+ T cells. However, at 8 d after LCMV-infection, the percentage of splenic CD8+ T cells in JNK1−/− reached a similar value as that of JNK2−/− and JNK+/+ (35, 39, and 40%, respectively). However, upon analysis of the total number of splenocytes, JNK1−/− had 50% fewer splenocytes (i.e., JNK+/+ mice: 8 × 107 cells/spleen; JNK2−/− mice: 7 × 107 cells/spleen; JNK1−/−: 3 × 107 cells/spleen). From the total number of cells obtained per spleen and the percentages of CD8+ and CD4+ T cells, and the percentages of virus-specific T cells, the number of T cells per spleen specific for each immunodominant viral peptide can be calculated (Fig. 3) . Thus, JNK1−/− had lower total numbers of virus specific CD8+ and CD4+ T cells than did JNK+/+ mice for all T cell epitopes tested (Fig. 3, A and B). In contrast, JNK2−/− mice had significantly higher total numbers of virus-specific CD8+ T cells for both epitopes while the difference in the number of virus-specific CD4+ T cells was not statistically significant between JNK+/+ and JNK2−/− mice (P > 0.2). Acute VSV-infection gave similar results; smaller spleens and a significantly reduced number of VSV-specific CD8+ T cells were observed in JNK1−/− mice compared with JNK+/+, and JNK2−/− mice, thus showing that a second virus caused an equivalent phenotype to JNK1−/− mice infected with LCMV (data not shown). When LCMV spleen memory cells were quantified (60 d after inoculation), the percentage of CD8+ T cells was equivalent in all three groups.
Figure 2.
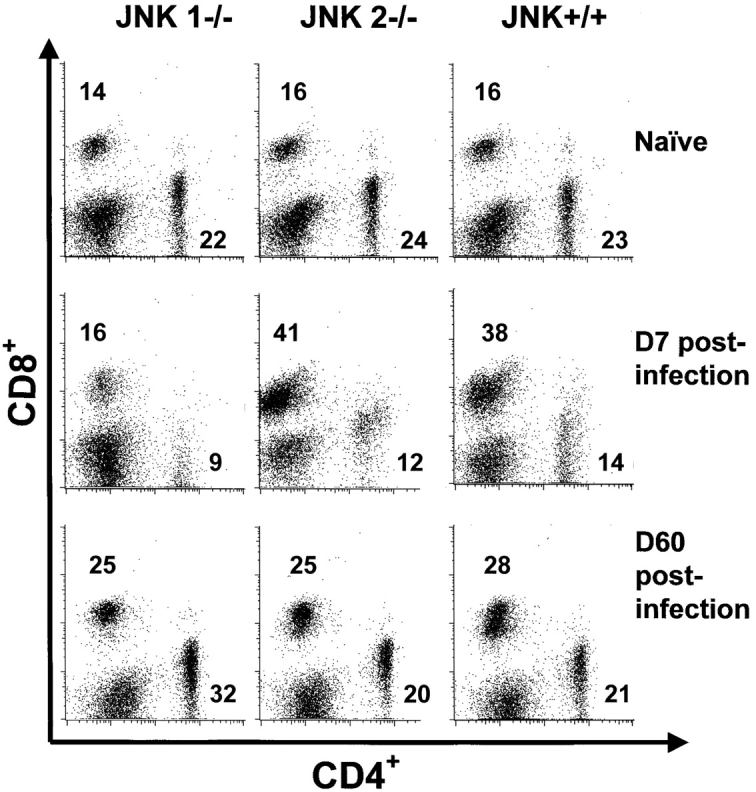
The CD8+ T cell compartment does not expand in JNK1−/− mice during the primary LCMV infection in comparison to JNK2−/− and JNK+/+ mice but equivalent number of CD8+ T cell are noted 60 d later. JNK+/+, JNK1−/−, and JNK2−/− mice were injected intraperitoneally with LCMV ARM 2 × 105 PFU. Uninfected spleen cells and spleen cells 7 (D7) or 60 d (D60) postinfection were stained for CD4 and CD8. Data represent average values obtained from three animals per group and are representative of two independent experiments. Values written in the dot plots indicate the percentage of CD8+ T cells and the percentage of CD4+ T cells
Figure 3.


The number of virus-specific CD8+ T cells and CD4+ T cells is lower in JNK1−/− mice than in either JNK2−/− or JNK+/+ mice after LCMV infection. JNK1−/−, JNK2−/−, and JNK+/+ mice were injected intraperitoneally with LCMV ARM, 2 × 105 PFU. 8 d after infection, spleen were processed and stimulated for 5 h in vitro with virus-specific peptide GP aa 33–41, NP aa 396–404, GP aa 61–80, or NP aa 309–328. Cells were then stained for surface CD4 or CD8 and intracellular IFN-γ molecules. The number of virus-specific T cells was calculated from the number of total spleen cells obtained per animal. Data represent average values obtained from three animals per group and are representative of two independent experiments. (A) CD8+ T cells; (B) CD4+ T cells. *P < 0.05 #P = 0.052 when compared with JNK+/+.
After LCMV Infection, Apoptosis Is Significantly Increased in JNK1−/− Mice.
We wished to distinguish whether the reduced number of T lymphocytes in JNK1−/− mice during acute LCMV infection was due to a defect in proliferation or to enhanced apoptosis in JNK1−/− T cells. Conze and colleagues in the accompanying paper report a decreased in vitro proliferation for purified naive CD8+ T cells in response to antibodies to CD3 and CD28 in JNK1−/− compared with JNK+/+ mice, suggesting that JNK1−/−CD8+ T cells do not proliferate as much as JNK+/+ after activation via the TCR and costimulatory molecules. However, in our hands, culture with anti-CD3 and anti-CD28 antibodies of CD8+ T cells from acutely infected mice led to apoptosis of the majority of cells owing to their enhanced state of activation (data not shown).
The percentage of activated splenic T cells dying during the peak of the immune response were identified and quantified using the CD44 marker, which is highly expressed by the expanding population of T cells (41). A lower percentage of CD8+ lymphocytes in JNK1−/− than in JNK+/+ mice had acquired a CD44hi phenotype at 8 d after LCMV infection (see Fig. 4 A). By comparison, a higher percentage of JNK2−/−CD8+ T cells were CD44hi compared with JNK+/+ cells. To evaluate apoptosis, CD8+CD44hi were gated while excluding the dead cells that were 7AAD positive, thereby allowing an evaluation of the percentage of activated cells that were annexin-V positive. As shown in Fig. 4, the percentage of cells undergoing apoptosis amongst CD8+CD44hi cells was significantly greater in JNK1−/− mice than either JNK+/+ or JNK2−/− mice. Increased apoptosis may likely account for the decreased number of virus specific CD8+ T cells found in JNK1−/− mice. We looked at CD8+CD44lo cells and found no significant difference in apoptosis between JNK1−/− and JNK+/+ cells (data not shown).
Figure 4.


During the acute infection, the number of splenic CD8+ T cells CD44hi is lower in JNK1−/− mice than in JNK2−/− and JNK+/+ mice, and a greater proportion of these cells from JNK1−/− undergo apoptosis. JNK1−/−, JNK2−/−, and JNK+/+ animals were injected intraperitoneally with LCMV ARM 2 × 105 PFU. 8 d after infection, spleen were processed to make single cell suspension devoted of red blood cells and stained directly for surface markers: CD4, CD8, and CD44 then annexin-V and 7AA-D were added. Data represent average values obtained from four animals per group and are representative of two independent experiments. The percentage of CD8+ or CD4+ that were CD44hi are shown in the left, that were annexin V+ cells among the CD8CD44hi or CD4CD44hi on the right. (A) CD8+ T cells. (B) CD4+ T cells. *P < 0.05 compared with JNK+/+.
Analysis of CD4+ T cells revealed fewer CD4+ T cells had acquired a CD44hi phenotype in JNK1−/− mice compared with JNK+/+ mice during the acute phase, but similar proportions occurred in the JNK2−/− and JNK+/+ mice. Gating on the CD4+CD44hi cells, a lower proportion of JNK2−/− cells undergoing apoptosis (i.e., annexin-V positive) was found compared with JNK+/+ and JNK1−/− cells (Fig. 4 B).
To further characterize whether apoptotic pathways were more active in JNK−/− versus JNK+/+, the expression of antiapoptotic proteins bcl-2 or bcl-XL in CD8+ T cells from all three groups was evaluated. No significant differences were observed in either bcl-2 or bcl-XL protein expression by flow cytometry for CD8+ T cells, for GP aa 33–41 or NP aa 396–404 tetramer-positive CD8+ T cells from JNK1−/−, JNK2−/−, and JNK+/+ mice (data not shown) at 8 d after LCMV infection. In addition, no significant differences in levels of mRNA expression for bcl-2 and caspase genes families as measured by Rnase Protection Assay (RPA) was observed in RNA from CD8+ T cells obtained from JNK1−/−, JNK2−/−, or JNK+/+ mice at day 8 after LCMV-inoculation (data not shown).
CD8+ T Cell Expansion Is Enhanced in JNK2−/− Mice After a Secondary Challenge with LCMV Compared with CD8+ T Cells from JNK+/+ Mice.
To evaluate whether a deficiency in JNK1 or JNK2 could affect the long-term memory of CD8+ T cells, we measured the number of virus specific T cells during the memory phase using an ex vivo stimulation with LCMV-specific peptides GP aa 33–41 and NP aa 396–404. The number of memory cells measured at 60 d or more after initial LCMV infection was higher in JNK2−/− mice but lower in JNK1−/− when compared with JNK+/+ CD8+ T memory cells. The number of splenic CD8+ T cells producing IFN-γ after an in vitro stimulation with GP aa 33–41 was 5.2 × 105 ± 2.3 × 104, 9.0 × 105 ± 9.4 × 104, 6.8 × 105 ± 9.2 × 104 for JNK1−/−, JNK2−/−, and JNK+/+ mice, respectively. This trend of lower expansion of memory CD8+ T cells in JNK1−/− but a higher expansion in JNK2−/− when compared with JNK+/+ mice was noted in all three experiments and in all three individually studied mice, although the differences were not statistically significant (P > 0.2 for JNK2−/− versus JNK+/+, and P > 0.5 for JNK1−/− versus JNK+/+).
JNK1−/−, JNK2−/−, and JNK+/+ mice were rechallenged intravenously with Cl-13, a strain of LCMV that induces persistent infection by aborting anti-LCMV–specific CD8+ T cell response after primary infection (42–44). In our studies, the immunosuppressive Cl-13 was given to mice who already had undergone primary viral infection, cleared virus, and established and maintained CD8+ and CD4+ anti-LCMV memory T cells. LCMV-specific CD8+ (Figs. 5 and 6) and CD4+ T cells increased during the secondary challenge to a level comparable to the primary response. The expansion was observed for MHC class I–restricted epitopes GP aa 33–41 and NP aa 396–404 as well as MHC class II–restricted LCMV epitopes GP aa 61–80 and NP aa 309–328 (data for GP aa 33–41 shown on Fig. 5 A and data for NP aa 396–404 shown on Fig. 5 B). Similar to the primary response, the expansion of CD8+ T cells for both GP aa 33–41 and NP aa 396–404 immunodominant epitopes was significantly greater in JNK2−/− mice than in JNK+/+ mice, suggesting that JNK2 plays a role in controlling of CD8+ T cell expansion in vivo. In contrast, despite the reduced burst size of JNK1−/− mice during the primary response, the number of virus-specific T cells after a secondary challenge was comparable to that observed for JNK+/+ mice.
Figure 5.


The number of splenic virus specific CD8+ T cells and CD4+ T cells is lower in JNK1−/− mice than in either JNK2−/− or JNK+/+ mice during the acute response but not during the secondary response. JNK1−/−, JNK2−/−, and JNK+/+ mice were injected intraperitoneally with LCMV ARM 2 × 105 PFU. Spleens were harvested at day 7, 8, 14, or 60 postinfection or 4 d after a challenge with Cl-13 (106 PFU intravenously) in memory animals. Spleen cells were stimulated for 5 h in vitro with virus specific peptides GP aa 33–41 or NP aa 396–404, then stained for CD8 and intracellular IFN-γ. The number of virus specific T cells was calculated from the number of total spleen cells obtained per animal. Data represent average values obtained from three animals per group and are representative of two independent experiments. *P < 0.05, #P = 0.057 compared with JNK+/+.
Figure 6.

The number of splenic virus specific CD8+ T cells is greater in JNK2−/− mice than in either JNK1−/− or JNK+/+ mice during the secondary response. JNK1−/−, JNK2−/−, and JNK+/+ mice were injected intraperitoneally with LCMV ARM 2 × 105 PFU, rechallenged after 60 d with Cl-13 (106 PFU intravenously), and spleens were harvested 4 d later. Spleen cells were stimulated for 5 h in vitro with virus specific peptides GP aa 33–41 or NP aa 396–404, then stained for CD8 and intracellular IFN-γ. The number of virus-specific T cells was calculated from the number of total spleen cells obtained per animal. Data represent average values obtained from three animals per group and are representative of three independent experiments. *P < 0.05 when compared with JNK+/+.
Discussion
Our results show for the first time that JNK1 and JNK2 MAP kinases play distinctly different roles in the activation of CD8+ T cells after viral infection. Although both JNK1−/− and JNK2−/− mice cleared LCMV, the magnitude of their immune responses varied markedly. After an acute LCMV infection, JNK1−/− mice did not show the characteristic 5- to 10-fold expansion of the splenic CD8+ T cells observed in immunocompetent JNK+/+ or JNK2−/− mice. JNK1−/− mice mounted LCMV-specific CTL responses, and their virus-specific CD8+ T cells produced IFN-γ and IL-2. However, at the peak of the primary response, the total number of JNK1−/− splenocytes was ∼50% less than that of JNK+/+ mice and the number of virus specific CD8+ and CD4+ T cells was significantly reduced in JNK1−/− mice. Similar results were found with VSV infection of JNK1−/− and JNK+/+ mice suggesting the generality of our finding that JNK1 and JNK2 MAP kinases play distinctive and different roles in the activation of T cells after viral infection.
After LCMV-infection, fewer JNK1−/− CD8+ T cells became activated and more of their CD8+CD44hi T cells underwent apoptosis compared with the JNK+/+. This would in part explain the impaired expansion of CD8+ T cells in these mice. Moreover, naive CD8+ T cells from JNK1−/− mice showed a decreased proliferation in response to antibodies to CD3 and CD28 compared with JNK+/+, suggesting that in addition to the enhanced apoptosis, a decreased proliferation might contribute to the decreased number of virus specific CD8+ T cells during cell activation. Interestingly, the reduced CD8+ T cell expansion in JNK1−/− mice was observed only during the primary acute viral response, as the magnitude of their secondary (immune memory) response was similar to that of the JNK+/+ mice. In contrast, JNK2-deficient mice showed an increased expansion of virus-specific CD8+ T cells during the acute infection as well as during the secondary response. The enhanced expansion of virus specific CD8+ T cells coincided with an increased proportion of CD8+CD44hi T cells compared with JNK+/+ mice.
Acute LCMV-infection triggers preferentially a Th1 response rather than a Th2 response (45). Although it has been reported that in vitro differentiation of Th0 JNK2−/− CD4+ T cells into Th1 is impaired (8), our results indicate that in vivo JNK2−/− CD4+ T cells were still capable of a similar virus-specific Th1 response during an acute viral infection, equivalent to CD4+ T cell response of JNK+/+ wild-type mice. Moreover, after acute LCMV infection, JNK2−/−CD4CD44hi T cells experienced less apoptosis, suggesting that activated CD4+ T cells may enhance the antiviral response in JNK2−/− mice by providing higher level of soluble mediators given that they were less inclined to die than CD4+ T cells from JNK+/+ mice. A second set of JNK1 and JNK2 deficient mice have been developed (46, 47). Analysis of the responses of mixed lymphocyte populations of these animals led the authors to conclude that JNK1 and 2 play a similar role in regulating IL-2 production and T cell proliferation (47). Studies with purified CD4+ T cells from the mice of our present study showed no role for JNK in these functions for CD4+ T cells. However, as the studies of Sabapathy et al. used mixed cell populations, it remained possible that CD8+ T cell effects in these cell populations influenced these results (47).
As mice deficient for either JNK1 or JNK2 showed a divergent phenotype in the expansion of CD8+ T cells during the primary response to a viral infection, we propose that these two MAP kinases have distinct roles in the activation of CD8+ T cells. JNK1−/− and JNK2−/− mice show a normal development of T and B lymphocytes (7, 8) and the differences observed here occurred during the differentiation of naive cells to fully activated cells. Many events lead to the differentiation of CD8+ T cells from a naive to an activated state including the ligation of the TCR and costimulatory molecules. Whereas naive T cells only express low levels of JNK, signals from both the TCR-CD3 complex and a costimulatory factor such as CD28 are necessary for the activation of JNKs (48–50). Additionally, many costimulatory molecules such as CD40, Ox40, 4–1BB, CTLA-4, LFA-1, and CD27 mediate their signaling through JNKs (12, 51, 52), but the results of these studies did not discriminate among different JNKs. The precise identification of signaling pathways blocked by the absence of either JNK1 or JNK2 is complicated by numerous JNK isoforms and at least a dozen upstream MAP kinase kinase kinases that can activate the JNK pathway (3). Nevertheless, we speculate that JNK1−/− mice lack one or more positive signals necessary for controlling the survival and expansion of CD8+ T cells during an immune response. Given that we detected fully functional CD8+ T cells in both JNK1−/− and JNK2−/− mice as judged by antiviral CTL activity, IL-2 production, IFN-γ production, the impaired expansion of CD8+ T cells in JNK1−/−, and the enhanced expansion in JNK2−/− suggest a role for these MAP kinases in controlling the degree (magnitude) rather than the functionality of the immune response.
Among all costimulatory molecules with a signaling pathway that could be partially or completely blocked in JNK1−/− mice, 4–1BB is a compelling candidate. First, 4–1BB enhances CD8+ T cell proliferation and IFN-γ (53–55) and is more important for the stimulation of CD8+ T cells rather than CD4+ T cells (55, 56). Moreover, a role in long-term CD8+ T cell survival has been attributed to this costimulatory molecule (56). Further, 4–1BB ligand knockout mice responded to acute LCMV infection much as JNK1−/− mice did. That is, 4–1BBL−/− mice eliminated LCMV with normal kinetics, but the expansion of their CD8+ T cells was two- to threefold lower than in wild-type mice (28). Thus, the normal signaling pathway observed after the binding of 4–1BB to 4–1BBL could be affected by the absence of the MAP kinases (e.g., JNK1). Alternatively, other costimulatory pathways could be affected by the absence of either JNK1 or JNK2 with the 4–1BB/4–1BBL pathway simply representing one possibility.
Conze et al. (9a) have assessed the in vitro functionality of CD8+ T cells in JNK1−/− and JNK2−/− mice after polyclonal or mitogenic stimulation. The CD8+ T cells from JNK1−/− proliferated less than JNK+/+ cells, possibly because of defective IL-2 production as well as decreased expression of IL-2 receptor (CD25). JNK1−/− CD8+ T cells also produced a reduced amount of IFN-γ compared with JNK+/+ cells. These in vitro observations complement our in vivo findings of a decreased ability of CD8+ T cells to expand in JNK1−/− mice infected with LCMV. However, we found no decrease in the amount of IL-2 or IFN-γ production or surface expression of CD25 by activated CD8+ T cells noted in the in vitro system of Conze et al. These discrepancies may reflect the different systems studied (i.e., in vitro polyclonal/mitogenic stimulation versus in vivo viral infection). During a viral infection, interactions implicating CD3 and CD28 and other costimulatory molecules likely occur in the activation of CD8+ T cells.
Previously, JNK was established as a key player in the polarization of CD4+ T cells (7, 8). Our study now shows that these MAP kinases are also important signaling pathways for activation of CD8+ T cells. Further, we show that individual members of the MAP kinase family have clearly distinguishable functions in vivo. The model of LCMV infection in JNK1−/− mice should prove valuable for further evaluation of the specific contributions of JNK1, JNK2, and various isoforms to the activation of T cells. These properties may allow targeting of specific immune responses to decrease CD8+ T cell expansion without affecting overall quality of the response.
Acknowledgments
We thank Dorian McGavern and Phyllis Minick for critically reviewing the manuscript.
This work was supported by a National Institutes of Health grant AI09484 to M.B.A. Oldstone; N. Arbour was supported by a postdoctoral fellowship from the Canadian Institutes of Health, D. Naniche by senior fellowship from the Leiper Trust, and D. Homann by a senior post-doctoral fellowship from the Juvenile Diabetes Foundation International (3-1999-629). R.A. Flavell and R.J. Davis are Investigators of the Howard Hughes Medical Institute.
Footnotes
Abbreviations used in this paper: aa, amino acid(s); ARM, Armstrong strain of LCMV; Cl-13, clone-13 of LCMV-ARM; GP, glycoprotein; JNK, c-Jun NH2-terminal kinase; LCMV, lymphocytic choriomeningitis virus; MAP, mitogen-activated protein.
References
- 1.Whitmarsh, A.J., and R.J. Davis. 1996. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. 74:589–607. [DOI] [PubMed] [Google Scholar]
- 2.Ip, Y.T., and R.J. Davis. 1998. Signal transduction by the c-Jun N-terminal kinase (JNK)—from inflammation to development. Curr. Opin. Cell Biol. 10:205–219. [DOI] [PubMed] [Google Scholar]
- 3.Chang, L., and M. Karin. 2001. Mammalian MAP kinase signalling cascades. Nature. 410:37–40. [DOI] [PubMed] [Google Scholar]
- 4.Chow, C.W., C. Dong, R.A. Flavell, and R.J. Davis. 2000. c-Jun NH(2)-terminal kinase inhibits targeting of the protein phosphatase calcineurin to NFATc1. Mol. Cell. Biol. 20:5227–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chow, C.W., M. Rincon, J. Cavanagh, M. Dickens, and R.J. Davis. 1997. Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science. 278:1638–1641. [DOI] [PubMed] [Google Scholar]
- 6.Gupta, S., T. Barrett, A.J. Whitmarsh, J. Cavanagh, H.K. Sluss, B. Derijard, and R.J. Davis. 1996. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 7.Dong, C., D.D. Yang, M. Wysk, A.J. Whitmarsh, R.J. Davis, and R.A. Flavell. 1998. Defective T cell differentiation in the absence of Jnk1. Science. 282:2092–2095. [DOI] [PubMed] [Google Scholar]
- 8.Yang, D.D., D. Conze, A.J. Whitmarsh, T. Barrett, R.J. Davis, M. Rincon, and R.A. Flavell. 1998. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity. 9:575–585. [DOI] [PubMed] [Google Scholar]
- 9.Constant, S.L., C. Dong, D.D. Yang, M. Wysk, R.J. Davis, and R.A. Flavell. 2000. JNK1 is required for T cell-mediated immunity against Leishmania major infection. J. Immunol. 165:2671–2676. [DOI] [PubMed] [Google Scholar]
- 9a.Conze, D., T. Krahl, N. Kennedy, L. Weiss, J. Lumsden, P. Hess, R.A. Flavell, G. Le Gros, R.J. Davis, and M. Rincon. 2002. c-Jun NH2-terminal kinase (JNK)1 and JNK2 have distinct roles in CD8+ T cell activation. J. Exp. Med. 195:811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Homann, D. 2001. Immunocytotherapy. Curr. Top. Microbiol. Immunol. In press. [DOI] [PubMed] [Google Scholar]
- 11.Slifka, M.K., and J.L. Whitton. 2000. Antigen-specific regulation of T cell-mediated cytokine production. Immunity. 12:451–457. [DOI] [PubMed] [Google Scholar]
- 12.Whitmire, J.K., and R. Ahmed. 2000. Costimulation in antiviral immunity: differential requirements for CD4(+) and CD8(+) T cell responses. Curr. Opin. Immunol. 12:448–455. [DOI] [PubMed] [Google Scholar]
- 13.Byrne, J.A., and M.B. Oldstone. 1984. Biology of cloned cytotoxic T lymphocytes specific for lymphocytic choriomeningitis virus: clearance of virus in vivo. J. Virol. 51:682–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moskophidis, D., U. Assmann-Wischer, M.M. Simon, and F. Lehmann-Grube. 1987. The immune response of the mouse to lymphocytic choriomeningitis virus. V. High numbers of cytolytic T lymphocytes are generated in the spleen during acute infection. Eur. J. Immunol. 17:937–942. [DOI] [PubMed] [Google Scholar]
- 15.Zarozinski, C.C., and R.M. Welsh. 1997. Minimal bystander activation of CD8 T cells during the virus-induced polyclonal T cell response. J. Exp. Med. 185:1629–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murali-Krishna, K., J.D. Altman, M. Suresh, D.J. Sourdive, A.J. Zajac, J.D. Miller, J. Slansky, and R. Ahmed. 1998. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 8:177–187. [DOI] [PubMed] [Google Scholar]
- 17.Butz, E.A., and M.J. Bevan. 1998. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity. 8:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Homann, D., L. Teyton, and M.B. Oldstone. 2001. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat. Med. 7:913–919. [DOI] [PubMed] [Google Scholar]
- 19.Varga, S.M., and R.M. Welsh. 1998. Detection of a high frequency of virus-specific CD4+ T cells during acute infection with lymphocytic choriomeningitis virus. J. Immunol. 161:3215–3218. [PubMed] [Google Scholar]
- 20.Selin, L.K., K. Vergilis, R.M. Welsh, and S.R. Nahill. 1996. Reduction of otherwise remarkably stable virus-specific cytotoxic T lymphocyte memory by heterologous viral infections. J. Exp. Med. 183:2489–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lau, L.L., B.D. Jamieson, T. Somasundaram, and R. Ahmed. 1994. Cytotoxic T-cell memory without antigen. Nature. 369:648–652. [DOI] [PubMed] [Google Scholar]
- 22.Asano, M.S., and R. Ahmed. 1996. CD8 T cell memory in B cell-deficient mice. J. Exp. Med. 183:2165–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berger, D.P., D. Naniche, M.T. Crowley, P.A. Koni, R.A. Flavell, and M.B. Oldstone. 1999. Lymphotoxin-beta-deficient mice show defective antiviral immunity. Virology. 260:136–147. [DOI] [PubMed] [Google Scholar]
- 24.Borrow, P., A. Tishon, S. Lee, J. Xu, I.S. Grewal, M.B. Oldstone, and R.A. Flavell. 1996. CD40L-deficient mice show deficits in antiviral immunity and have an impaired memory CD8+ CTL response. J. Exp. Med. 183:2129–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cousens, L.P., J.S. Orange, and C.A. Biron. 1995. Endogenous IL-2 contributes to T cell expansion and IFN-gamma production during lymphocytic choriomeningitis virus infection. J. Immunol. 155:5690–5699. [PubMed] [Google Scholar]
- 26.Orange, J.S., and C.A. Biron. 1996. An absolute and restricted requirement for IL-12 in natural killer cell IFN-gamma production and antiviral defense. Studies of natural killer and T cell responses in contrasting viral infections. J. Immunol. 156:1138–1142. [PubMed] [Google Scholar]
- 27.van den Broek, M.F., U. Muller, S. Huang, M. Aguet, and R.M. Zinkernagel. 1995. Antiviral defense in mice lacking both alpha/beta and gamma interferon receptors. J. Virol. 69:4792–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan, J.T., J.K. Whitmire, R. Ahmed, T.C. Pearson, and C.P. Larsen. 1999. 4-1BB ligand, a member of the TNF family, is important for the generation of antiviral CD8 T cell responses. J. Immunol. 163:4859–4868. [PubMed] [Google Scholar]
- 29.Andreasen, S.O., J.E. Christensen, O. Marker, and A.R. Thomsen. 2000. Role of CD40 ligand and CD28 in induction and maintenance of antiviral CD8+ effector T cell responses. J. Immunol. 164:3689–3697. [DOI] [PubMed] [Google Scholar]
- 30.Zimmermann, C., P. Seiler, P. Lane, and R.M. Zinkernagel. 1997. Antiviral immune responses in CTLA4 transgenic mice. J. Virol. 71:1802–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gallimore, A., A. Glithero, A. Godkin, A.C. Tissot, A. Pluckthun, T. Elliott, H. Hengartner, and R. Zinkernagel. 1998. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J. Exp. Med. 187:1383–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dutko, F.J., and M.B. Oldstone. 1983. Genomic and biological variation among commonly used lymphocytic choriomeningitis virus strains. J. Gen. Virol. 64:1689–1698. [DOI] [PubMed] [Google Scholar]
- 33.Salvato, M., E. Shimomaye, P. Southern, and M.B. Oldstone. 1988. Virus-lymphocyte interactions. IV. Molecular characterization of LCMV Armstrong (CTL+) small genomic segment and that of its variant, Clone 13 (CTL-). Virology. 164:517–522. [DOI] [PubMed] [Google Scholar]
- 34.Homann, D., A. Tishon, D.P. Berger, W.O. Weigle, M.G. von Herrath, and M.B. Oldstone. 1998. Evidence for an underlying CD4 helper and CD8 T-cell defect in B-cell-deficient mice: failure to clear persistent virus infection after adoptive immunotherapy with virus-specific memory cells from muMT/muMT mice. J. Virol. 72:9208–9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kagi, D., B. Ledermann, K. Burki, P. Seiler, B. Odermatt, K.J. Olsen, E.R. Podack, R.M. Zinkernagel, and H. Hengartner. 1994. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 369:31–37. [DOI] [PubMed] [Google Scholar]
- 36.Walsh, C.M., M. Matloubian, C.C. Liu, R. Ueda, C.G. Kurahara, J.L. Christensen, M.T. Huang, J.D. Young, R. Ahmed, and W.R. Clark. 1994. Immune function in mice lacking the perforin gene. Proc. Natl. Acad. Sci. USA. 91:10854–10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahmed, R., J.A. Byrne, and M.B. Oldstone. 1984. Virus specificity of cytotoxic T lymphocytes generated during acute lymphocytic choriomeningitis virus infection: role of the H-2 region in determining cross-reactivity for different lymphocytic choriomeningitis virus strains. J. Virol. 51:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yanagi, Y., A. Tishon, H. Lewicki, B.A. Cubitt, and M.B. Oldstone. 1992. Diversity of T-cell receptors in virus-specific cytotoxic T lymphocytes recognizing three distinct viral epitopes restricted by a single major histocompatibility complex molecule. J. Virol. 66:2527–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen, C.Y., R. Gherzi, J.S. Andersen, G. Gaietta, K. Jurchott, H.D. Royer, M. Mann, and M. Karin. 2000. Nucleolin and YB-1 are required for JNK-mediated interleukin-2 mRNA stabilization during T-cell activation. Genes Dev. 14:1236–1248. [PMC free article] [PubMed] [Google Scholar]
- 40.Oxenius, A., M.F. Bachmann, P.G. Ashton-Rickardt, S. Tonegawa, R.M. Zinkernagel, and H. Hengartner. 1995. Presentation of endogenous viral proteins in association with major histocompatibility complex class II: on the role of intracellular compartmentalization, invariant chain and the TAP transporter system. Eur. J. Immunol. 25:3402–3411. [DOI] [PubMed] [Google Scholar]
- 41.Whitmire, J.K., M.S. Asano, K. Murali-Krishna, M. Suresh, and R. Ahmed. 1998. Long-term CD4 Th1 and Th2 memory following acute lymphocytic choriomeningitis virus infection. J. Virol. 72:8281–8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sevilla, N., S. Kunz, A. Holz, H. Lewicki, D. Homann, H. Yamada, K.P. Campbell, J.C. de La Torre, and M.B. Oldstone. 2000. Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J. Exp. Med. 192:1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borrow, P., C.F. Evans, and M.B. Oldstone. 1995. Virus-induced immunosuppression: immune system-mediated destruction of virus-infected dendritic cells results in generalized immune suppression. J. Virol. 69:1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahmed, R., A. Salmi, L.D. Butler, J.M. Chiller, and M.B. Oldstone. 1984. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J. Exp. Med. 160:521–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Varga, S.M., and R.M. Welsh. 2000. High frequency of virus-specific interleukin-2-producing CD4(+) T cells and Th1 dominance during lymphocytic choriomeningitis virus infection. J. Virol. 74:4429–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sabapathy, K., Y. Hu, T. Kallunki, M. Schreiber, J.P. David, W. Jochum, E.F. Wagner, and M. Karin. 1999. JNK2 is required for efficient T-cell activation and apoptosis but not for normal lymphocyte development. Curr. Biol. 9:116–125. [DOI] [PubMed] [Google Scholar]
- 47.Sabapathy, K., T. Kallunki, J.P. David, I. Graef, M. Karin, and E.F. Wagner. 2001. c-Jun NH(2)-Terminal Kinase (JNK)1 and JNK2 have similar and stage-dependent roles in regulating T cell apoptosis and proliferation. J. Exp. Med. 193:317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su, B., E. Jacinto, M. Hibi, T. Kallunki, M. Karin, and Y. Ben-Neriah. 1994. JNK is involved in signal integration during costimulation of T lymphocytes. Cell. 77:727–736. [DOI] [PubMed] [Google Scholar]
- 49.Rincon, M., and R.A. Flavell. 1994. AP-1 transcriptional activity requires both T-cell receptor-mediated and co-stimulatory signals in primary T lymphocytes. EMBO J. 13:4370–4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weiss, L., A.J. Whitmarsh, D.D. Yang, M. Rincon, R.J. Davis, and R.A. Flavell. 2000. Regulation of c-Jun NH(2)-terminal kinase (Jnk) gene expression during T cell activation. J. Exp. Med. 191:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Calvo, C.R., D. Amsen, and A.M. Kruisbeek. 1997. Cytotoxic T lymphocyte antigen 4 (CTLA-4) interferes with extracellular signal-regulated kinase (ERK) and Jun NH2-terminal kinase (JNK) activation, but does not affect phosphorylation of T cell receptor zeta and ZAP70. J. Exp. Med. 186:1645–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Akiba, H., H. Nakano, S. Nishinaka, M. Shindo, T. Kobata, M. Atsuta, C. Morimoto, C.F. Ware, N.L. Malinin, D. Wallach, et al. 1998. CD27, a member of the tumor necrosis factor receptor superfamily, activates NF-kappaB and stress-activated protein kinase/c-Jun N-terminal kinase via TRAF2, TRAF5, and NF-kappaB-inducing kinase. J. Biol. Chem. 273:13353–13358. [DOI] [PubMed] [Google Scholar]
- 53.Kim, H.H., K. Kwack, and Z.H. Lee. 2000. Activation of c-jun N-terminal kinase by 4-1BB (CD137), a T cell co-stimulatory molecule. Mol. Cells. 10:247–252. [PubMed] [Google Scholar]
- 54.Cannons, J.L., K.P. Hoeflich, J.R. Woodgett, and T.H. Watts. 1999. Role of the stress kinase pathway in signaling via the T cell costimulatory receptor 4-1BB. J. Immunol. 163:2990–2998. [PubMed] [Google Scholar]
- 55.Shuford, W.W., K. Klussman, D.D. Tritchler, D.T. Loo, J. Chalupny, A.W. Siadak, T.J. Brown, J. Emswiler, H. Raecho, C.P. Larsen, et al. 1997. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J. Exp. Med. 186:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takahashi, C., R.S. Mittler, and A.T. Vella. 1999. Cutting edge: 4-1BB is a bona fide CD8 T cell survival signal. J. Immunol. 162:5037–5040. [PubMed] [Google Scholar]