

Abstract
Splenectomized individuals are prone to overwhelming infections with encapsulated bacteria and splenectomy of mice increases susceptibility to streptococcal infections, yet the exact mechanism by which the spleen protects against such infections is unknown. Using congenitally asplenic mice as a model, we show that the spleen is essential for the generation of B-1a cells, a B cell population that cooperates with the innate immune system to control early bacterial and viral growth. Splenectomy of wild-type mice further demonstrated that the spleen is also important for the survival of B-1a cells. Transfer experiments demonstrate that lack of these cells, as opposed to the absence of the spleen per se, is associated with an inability to mount a rapid immune response against streptococcal polysaccharides. Thus, absence of the spleen and the associated increased susceptibility to streptococcal infections is correlated with lack of B-1a B cells. These findings reveal a hitherto unknown role of the spleen in generating and maintaining the B-1a B cell pool.
Keywords: B-1a cells, asplenia, natural antibodies, Streptococcus pneumoniae, Hox11
Introduction
Absence of the spleen predisposes individuals to a risk of overwhelming infection, most often due to the encapsulated bacterium Streptococcus pneumoniae (1). Various hypotheses have been proposed to account for this increased infection risk in asplenic individuals, including lack of opsonins or marginal zone (MZ)* macrophages, altered T cell function, impaired clearance by the reticuloendothelial system, and mechanical filtration of pathogens by the spleen (2). Congenital asplenia in humans is often associated with complex heart defects and abdominal heterotaxy resulting from a laterality defect that begins to develop before spleen fate is specified (3). While cases of isolated congenital asplenia with no other obvious abnormalities have been reported (4, 5), such individuals have seldom been documented. This may reflect that congenital asplenia is a rare occurrence but, alternatively, it has been proposed that such individuals die early in childhood from overwhelming sepsis, without an autopsy being performed (6–8).
There are, broadly speaking, two major branches of the immune response to invading microorganisms. The innate immune system provides immediate protection by promptly initiating a local inflammatory reaction. The adaptive immune system expands antigen-specific T and B cells in the lymphoid organs and provides long-lasting protection against subsequent infections by the generation of memory T and B cells. In the mouse, a population of B cells, known as B-1a, cooperates with the innate immune system at the onset of bacterial and viral infections (9). B-1a cells produce natural antibodies, mostly IgM, assumedly without prior exposure to antigen. Such antibodies recognize thymus-independent antigens, such as the polysaccharides of encapsulated bacteria (10). By opsonizing invading pathogens, they play a crucial role in the activation of the complement cascade and the amplification of early innate defense mechanisms (11).
To understand the role of the spleen in the response to bacterial polysaccharides, we have used the Hox11-null mouse as a model of congenital asplenia (12, 13). We show that the spleen is indispensable for the generation and maintenance of the B-1a pool. Congenitally asplenic and splenectomized mice have reduced B-1a cells and a diminished IgM-mediated immune response against bacterial polysaccharide antigens.
Materials and Methods
Mice.
C57BL/6, Hox11-null, Rag-2-null, and SCID mice were bred and housed in specific pathogen-free conditions in the institute's animal facility. All mice used were aged 6–10 wk. The Hox11-null mice were on a mixed genetic background of MF1 (12.5%) and C57BL/6 (87.5%). For splenectomies and sham-operations, C57BL/6 mice were anaesthetized and after shaving, a small incision was made in the skin at the left flank. The peritoneum was opened and the splenic artery and vein ligated with catgut (Ethicon). The spleen was removed and the incision closed with surgical silk-thread (Ethicon). For sham-operated mice, the peritoneum was opened and surgically closed.
FACS® Analysis.
Antibodies used for lymphocyte stainings were anti–mouse CD5-Biotin (clone 53–7.3; BD PharMingen), anti–mouse B220-PE (clone RA3–6B2; Southern Biotechnology Associates), anti–mouse IgM-Cy5 (Jackson ImmunoResearch Laboratories), anti–mouse IgD-FITC (clone 11.26c2a; Southern Biotechnology Associates), anti–Mac-1 (clone M1/70, own), anti–mouse CD23-Biotin (clone B3B4; BD PharMingen), and anti–mouse CD21-FITC (clone 7G6, own). Biotinylated antibodies were detected with Streptavidin-Red670 (GIBCO BRL and Life Technologies). Mouse peritoneal cells were isolated by peritoneal lavage with 5 ml FACS® buffer, containing PBS with 3% FCS (vol/vol) and 0.01% (wt/vol) sodium azide. For the analysis of splenic lymphocytes, single-cell suspensions of total splenic tissue were prepared and depleted of erythrocytes by lysis with Gey's solution. 2 × 106 cells per sample were incubated with varying combinations of antibodies as indicated. Four-color flow cytometry was performed on a FACSCalibur™ flow cytometer (Becton Dickinson). Each analysis shown represents 200,000 events within the live lymphocyte gate. Flow cytometric profiles were analyzed using CELLQuest™ software (Becton Dickinson).
Immunization.
C57BL/6 and Hox11-null mice were immunized either subcutaneously, intramuscularly, intraperitoneally, or intravenously with 10 μl Pneumovax® 23 in 100 μl PBS. Pneumovax® 23 (Pasteur Merieux MSD GmbH) contains polysaccharides of Streptococcus pneumoniae of 23 capsular types each at a concentration of 50 μg/ml. Splenectomized and sham-operated mice were immunized directly after the operation, while still under narcosis. Mice were bled 24 h before and 6 d after immunization. The serum was isolated and used for Pneumovax® 23-specific anti–mouse IgM ELISA. Serum concentrations from 1:20 to 1:640 were tested. The 1:80 dilution is shown, as below this dilution serum IgM levels measured in Hox11-null and splenectomized mice were not above background levels.
ELISA.
ELISA plates (Nunc Maxisorb) were coated with either 5 μl Pneumovax® 23, 10 μg/ml phosphorylcholine (Sigma-Aldrich), or 10 μg/ml pneumococcal polysaccharide type 19 (American Type Culture Collection) in 50 μl PBS for 2 h at 37°C. All incubation steps were performed in a humid chamber. Blocking was achieved by addition of 200 μl 1% (wt/vol) BSA, 0.1% sodium azide in PBS followed by incubation overnight at 4°C. Plates were then washed three times with 200 μl PBS containing 0.05% (vol/vol) Tween 20 (Sigma-Aldrich) in a Skanwasher 300 (Skatron Instruments). 50 μl of diluted serum was added to each well followed by incubation for 2 h at 37°C. Plates were washed three times with PBS/Tween and 50 μl 1 μg/ml goat anti–mouse IgM alkaline phosphatase-labeled antibody (Southern Biotechnology Associates) was added. Plates were incubated for 2 h at 37°C, washed three times with PBS/Tween, and 100 μl 1 mg/ml SIGMA 104 phosphatase substrate (Sigma-Aldrich) in DEAE buffer were added. Emitted light was measured at 405 and 490 nm at various time-points afterwards. Total serum IgM antibodies were detected similarly except that plates were first coated with 50 μl 1 μg/ml goat anti–mouse IgM (Southern Biotechnology Associates) followed by addition of diluted serum. For measurements of total IgM sera were diluted between 1:500 and 1:8,000. The 1:1,000 dilution is shown. Mouse IgM (Roche Biochemicals) was used as a standard control.
Bone Marrow and Fetal Liver Transfer.
At least 3 d before irradiation, recipient mice were placed on antibiotics (0.12% (wt/vol) Borgal (Hoechst Russel) in the drinking water). The mice were maintained on antibiotics until sacrifice. 1 d before transfer, recipient mice were sublethally irradiated with 300 rads for Rag2-null and SCID mice and 600 rads for Hox11-null mice. 2 × 106 fetal liver cells of day 14.5 embryos or adult bone marrow cells in 200 μl PBS were injected intravenously into the tail vein. For immunization with Pneumovax® 23, reconstituted mice were bled 19 d after transfer and injected 24 h thereafter with 10 μl Pneumovax® 23 in 100 μl PBS, either subcutaneously, intramuscularly, intraperitoneally, or intravenously. Serum was taken 6 d after immunization and used for the detection of Pneumovax® 23-specific IgM by ELISA. Mice were then killed and bone marrow, spleen, lymph node, and peritoneal cells analyzed by FACS® to confirm complete reconstitution.
Statistical Analysis and Graphics.
Statistical analysis and graphics were done with Microsoft® Excel and GraphPad Prism®. Student's t tests with P values <0.05 were taken as significant.
Results
Hox11-Null Mice Lack B-1a Cells.
We have previously shown that the spleen plays a major role in the late phases of B cell development. Transitional 1 (T1) B cells, recently emigrated from the bone marrow, accumulate in the spleen and here differentiate to transitional 2 (T2) and mature B cells (19). To further study the function of the spleen in the development of B cells, we analyzed congenitally asplenic Hox11-null mice (12, 13). Of the major B cell and T cell populations from bone marrow, lymph node, and thymus analyzed by FACS®, we could not detect any gross phenotypic abnormalities. A minor increase in the number of T1 B cells in bone marrow and peripheral blood was observed, perhaps reflecting a shortage of space, as these cells normally collect in the spleen. Reductions were also observed in the proportion of mature B cells in blood, lymph node, and bone marrow (data not shown). This confirms the role of the spleen in the generation of normal numbers of mature B cells. The most striking finding, however, was a large reduction in the total cell number in the peritoneal cavity of Hox11-null mice (8.9 ± 2.0 × 106 in C57BL/6, 5.1 ± 1.0 × 106 in Hox11-null, Fig. 1 A). However, in the peritoneal lymphocyte population there was no significant difference in the proportion of total B cells between C57BL/6 and Hox11-null (59.5% ± 5.7% in C57BL/6, 69.1% ± 6.6% in Hox11-null). Thus, nonB cells including T cells but also myeloid cells are reduced in the peritoneal cavity of Hox11-null mice.
Figure 1.




The spleen is important for the generation of peritoneal B-1a cells in the mouse. (A) The total number of peritoneal cells is shown for 16-wk-old C57BL/6 (n = 10) and Hox11-null (n = 10) mice. Hox11 +/− and C57BL/6 mice do not differ in any of the peritoneal B cell compartments; thus C57BL/6 were used as controls throughout the paper. (B) Peritoneal cavity cells of one C57BL/6 and one Hox11-null mouse were stained with antibodies against IgM, IgD, CD5, and B220 and analyzed by flow-cytometry. The relative expression of IgM and IgD can be used to discriminate IgMposIgDdull B-1 (gate 1) from IgMdullIgDpos B-2 cells (gate 2, left panels). Based on the expression of IgM and IgD, B-1 and B-2 cells were gated as indicated and analyzed for the expression of B220 and CD5 to further distinguish between CD5posB220dull B-1a cells (top region, bottom right panels) and the CD5negB220pos B-1b population (bottom region, bottom right panels). While B-1b cells are normal in Hox11-null mice, the B220dullCD5pos B-1a population is absent. B-2 cells (gate 2) are CD5negB220pos mature B cells (top right panels). The CD5posB220pos B-1a population that remains in Hox11-null mice is indicated with an arrow. (C) The proportion of B-1a cells in the peritoneum of C57BL/6 (n = 8) and Hox11-null (n = 8) are shown. Peritoneal B cells (IgMpos and/or IgDpos) were measured by flow cytometric analysis and IgMposIgDdullCD5posB220dull were classified as B-1a cells. (D) Relative serum IgM levels in blood of C57BL/6 (n = 10) and Hox11-null mice (n = 9) were determined by anti-IgM ELISA. Each data point represents the relative serum IgM level of one mouse measured at a 1:1,000 dilution.
To identify the different populations of B cells in the peritoneal cavity we used antibodies against IgM, IgD, B220, and CD5. This antibody combination allows the discrimination of all three B cell populations within the peritoneal cavity so far described: B-1a, B-1b, and B-2 B cells (14). Peritoneal B cells of C57BL/6 and Hox11-null mice were gated into IgMposIgDdull B-1 (Fig. 1 B, gate 1, left panels) and IgMdullIgDpos B-2 cells (Fig. 1 B, gate 2, left panels) and analyzed for expression of B220 and CD5 (Fig. 1 B, right panels). B-1a cells (IgMposIgDdullCD5posB220dull) comprised 23.1% ± 5.0% of all peritoneal B cells in C57BL/6 mice, whereas these cells were only 3.3% ± 1.6% of all peritoneal B cells in Hox11-null mice (Fig. 1 C). The few CD5pos B cells in Hox11-null mice were different from those typically referred to as B-1a cells, in that they expressed high levels of B220 (Fig. 1 C, indicated by the arrow). B-1b (IgMposIgDdullCD5negB220pos) and mature B-2 cells (IgMdullIgDposCD5negB220pos) were phenotypically normal in Hox11-null mice (Fig. 1 B). B-1b comprised 13.5% ± 3.6% of all peritoneal B cells in C57BL/6 mice and 18.3% ± 4.8% in Hox11-null mice. Mature B cells comprised 24.0% ± 4.8% of all peritoneal B cells in C57BL/6 mice and 44.6% ± 7.1% in Hox11-null mice.
Several other markers can be used to discriminate between B-1 and B-2 cells: B-1 cells are Mac-1pos, CD43pos, and CD23neg, whereas B-2 cells are CD23pos, Mac-1neg, and CD43neg (15–17). Stainings with combinations of these antibodies verified that the B-1a population was reduced in the peritoneal cavity of Hox11-null mice (data not shown).
As B-1 cells are known to be the major source of natural antibodies and most of the circulating IgM is contributed by B-1a cells in the absence of previous antigen stimulation (10), serum IgM levels in the blood of mutant and C57BL/6 mice were compared, revealing significantly lower levels in Hox11-null mice (Fig. 1 D). B-1a cells are not only present within the peritoneal cavity but can also be detected in spleen and to very low amounts in lymph nodes (18). We found that B-1a cells are also present in the blood. We stained peripheral blood with antibodies against IgM, IgD, CD5, and either B220 or Mac-1. The IgMposIgDdull population includes T1 B cells, recently emigrated from the bone marrow, B-1a, and B-1b cells (Fig. 2 A and B). In the peripheral blood lymphocyte population, the proportion of total B cells was not significantly different between C57BL/6 and Hox11-null mice (Fig. 2 C).
Figure 2.



B-1a cells are absent in the blood of asplenic mice. (A) Representative flow cytometric analysis of peripheral blood cells isolated from C57BL/6 and Hox11-null mice. Cells were stained with antibodies against IgM, IgD, CD5, and B220. IgMdullIgDpos (left panels, top region) and IgMposIgDdull B lymphocytes (left panels, bottom region) were gated and analyzed for B220 versus CD5 expression (right panels). IgMdullIgDpos cells are B220posCD5neg mature B lymphocytes (top right panels). Gated IgMposIgDdull B lymphocytes (bottom right panels) comprise CD5posB220dull B-1a cells (top region) and CD5negB220pos B-1b and T1 cells (bottom region). The CD5posB220pos B-1a population that remains in Hox11-null mice is indicated with an arrow. (B) Representative flow cytometric analysis of cells stained with antibodies against IgM, IgD, CD5, and Mac-1 isolated from blood of the same C57BL/6 and Hox11-null mice analyzed in A. IgMposIgDdull B-lymphocytes were gated and analyzed for Mac-1 versus CD5 expression. Gated IgMposIgDdull B lymphocytes comprise CD5posMac-1pos B-1a cells (indicated by arrow), CD5negMac-1pos B-1b, and CD5negMac-1neg T1 cells. (C) Relative proportions of the major B cell populations in the peripheral blood of C57BL/6 and Hox11-null mice. Each value is the average of measurements obtained in five mice. For the purpose of measurement, B-1a cells are defined as IgMposIgDdullB220dullCD5pos, B-1b as IgMposIgDdullMac-1posCD5neg, and T1 as IgMposIgDdullMac-1negCD5neg. IgMposIgDdull cells constituted 26.5% ± 5.7% of total B cells (IgMpos and/or IgDpos) in the blood of C57BL/6 and 39.5% ± 6.5% in Hox11-null mice.
B-1a cells represent a significant proportion of B cells in the peripheral blood of C57BL/6 mice. B220dull B-1a cells were absent from the blood of Hox11-null mice (Fig. 2 A and C) but, as in the peritoneal cavity of Hox11-null mice, a few CD5posB220pos cells could be detected (indicated by the arrow in Fig. 2 A). To distinguish between T1 and B-1b cells we analyzed the expression of CD5 and Mac-1 on gated IgMposIgDdull cells. Transitional B cells do not express CD5 and Mac-1. In normal mice, they exit the bone marrow and collect in the spleen (19), but in asplenic mice they accumulate in the blood (Fig. 2 B and C). There was no change in the B-1b cell population (CD5negMac-1pos) but a slight reduction in the proportion of mature B cells in asplenic mice (Fig. 2 B and C).
Hox11 is expressed in the branchial arches, the developing hindbrain, the spinal cord, the pharynx, the outflow tracts of the heart, the external auditory meatus, the neurons of the developing cranial sensory ganglia, and in the spleen capsule and trabeculae, but not in hematopoietic cells during fetal life (12, 13). Extensive analysis of the spleen, bone marrow, and peritoneal cavity of adult and neonatal mice did not reveal any evidence of Hox11 expression in B cells (data not shown). To formally exclude the possibility that the reduction of the B-1a cell pool in Hox11-null mice was due to an intrinsic hematopoietic defect, rather than due to the absence of the spleen, we transferred Hox11-null fetal liver cells into SCID mice, which lack both B and T cells. This protocol allows the reconstitution of all lymphocyte populations including B-1a and B-1b cells with cells of donor origin (18). 4 wk after transfer, B-1a cells were detectable in the peritoneal cavity of SCID mice reconstituted with Hox11-null fetal liver cells (Fig. 3 A). Thus, Hox11-null progenitors retain the ability to generate B-1a cells, but are unable to do so in the absence of the spleen. A control transfer of C57BL/6 fetal liver cells into irradiated Hox11-null mice was also performed (Fig. 3 B). The majority of B-1a cells was still absent although a small number of B220dull B-1a cells was present. This indicates that there may be a limited capacity to generate some B-1a cells from wild-type precursors in the absence of the spleen.
Figure 3.


B-1a cells can be generated from Hox11-null fetal liver. (A) Fetal liver cells from C57BL/6 or Hox11-null mice were injected intravenously into irradiated SCID mice. 4 wk later peritoneal cavity cells of reconstituted mice were stained with antibodies against IgM, IgD, CD5, and B220. Data were analyzed by FACS® and compared with nonreconstituted SCID mice. B lymphocytes of reconstituted mice were gated based on IgM and/or IgD expression to remove non-B cells. (B) Fetal liver cells from C57BL/6 mice were injected intravenously into irradiated Rag2-null or Hox11-null mice. Peritoneal cells were analyzed 4 wk later as described in A.
Removal of the Spleen Depletes the B-1a Pool.
To investigate whether the spleen plays a role in the maintenance of an already established B-1a cell pool, we splenectomized normal C57BL/6 mice and determined the frequency of all peritoneal B cell populations at various time points thereafter. Cells were stained with antibodies against IgM, CD5, B220, and Mac-1 and the proportion of IgMposMac-1posCD5posB220dull B-1a cells was determined at various time points thereafter (Fig. 4). There was a similar reduction in the number of B-1a cells in splenectomized and sham-operated mice in the first 2 d. However, after day 3, the proportion of B-1a cells in sham-operated mice began to recover whereas in splenectomized mice it declined further. 6 d after the removal of the spleen, CD5posB220dull B-1a cells were ∼1/4 of those in sham-operated mice and appeared to stabilize at this level as further measurements at 10, 14 and 26 d after splenectomy revealed little change (Fig. 4 A and B). The frequency of B-1b remained stable after splenectomy (Fig. 4 C). Therefore, the spleen is indispensable for the maintenance of an already established B-1a cell pool.
Figure 4.

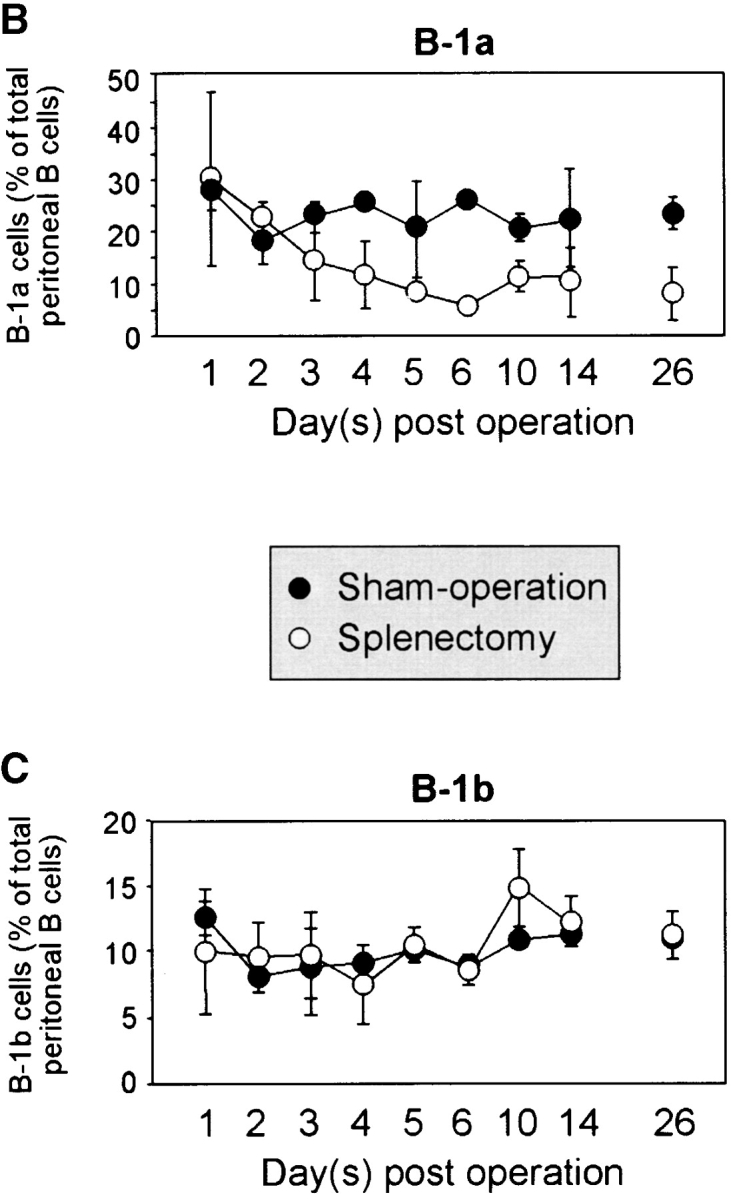
The peritoneal B-1a cell pool is rapidly reduced following splenectomy. (A) Representative FACS® analysis of peritoneal B cells isolated 10 d after splenectomy or sham operation and gated for IgMposMac-1pos cells. A reduction of the CD5posB220dull B-1a population can be seen. (B) Time course of reduction in B-1a cells upon splenectomy. C57BL/6 mice were either splenectomized or sham-operated and the frequency of B220dullCD5posIgMposMac-1pos B-1a cells determined by flow-cytometry at the indicated time points after surgery. At least three splenectomized and two sham-operated mice were examined for each time point, except for sham-operated day 6 where the cell number from only one mouse is shown. (C) Similar time course for B220posCD5negIgMposMac-1pos B-1b cells.
Despite the significant reduction, a fraction of B-1a cells survived in splenectomized mice. Our observations suggest that ∼75% of the peritoneal B-1a cells require the spleen for their generation and survival while the rest can survive without the spleen for at least 26 d. B-1a cells are considered a self-replenishing population, originally derived from fetal liver progenitors (18). The depletion of the majority of B-1a cells after splenectomy suggests that the spleen is indispensable either for their self-renewal or for their selection into the B-1a pool. We have previously shown that the spleen plays a role in the differentiation of transitional into mature B cells (19). We now demonstrate that the spleen is also required for a normal B-1a cell pool. In contrast, the spleen is not necessary for the development and survival of B-1b cells.
Asplenic Mice Do Not Respond To Vaccination with Streptococcal Polysaccharides.
B-1a cells are responsible for the production of IgM against thymus-independent antigens such as polysaccharides and phospholipids of the bacterial capsule (20–22) and are known to participate in the early response to bacterial and viral infection (9, 10, 23). The polysaccharide bacterial capsule is the major virulence factor of S. pneumoniae. Protective IgM antibodies have the function to opsonize the bacteria and facilitate their phagocytosis. Using mice challenged with a noncapsular strain of S. pneumoniae, it has been well documented that the major protective immune response is directed toward phosphorylcholine (24–26). The response against phosphorylcholine is also T cell independent. Levels of natural antibody against phosphorylcholine and pneumococcal polysaccharide were reduced in Hox11-null mice (18.2% + 17.4% in Hox11-null mice, n = 16, compared with 100.0% + 69.6% in C57BL/6, n = 14, for antiphosphorylcholine; 20.4% + 27.2% in Hox11-null mice compared with 100.0% + 108.8% in C57BL/6 for antipneumococcal polysaccharide). To determine if the absence of the spleen affected B-1a–dependent immune responses, Hox11-null, splenectomized, sham-operated, and control mice were immunized subcutaneously, intramuscularly, intraperitoneally, or intravenously with Pneumovax® 23, a vaccine composed of polysaccharides from the 23 most common types of Streptococcus pneumoniae (27). Control and sham-operated mice rapidly produced Pneumovax®-specific IgM antibodies via all four injection routes, while Hox11-null (Fig. 5 A) and splenectomized mice (Fig. 5 B) failed to respond.
Figure 5.


Hox11-null and splenectomized mice do not respond to vaccination with streptococcal polysaccharides. Pneumovax® 23 was injected subcutaneously, intramuscularly, intraperitoneally, or intravenously into either (A) C57BL/6 and Hox11-null mice or (B) sham-operated and splenectomized mice. Specific IgM antibodies in sera were measured by ELISA before and 6 d after immunization. The optical density (OD405) measured with a 1:80 serum dilution is shown. Each data point indicates the relative antibody level of a single mouse.
The Spleen, in the Absence of B-1a Cells, Is Not Sufficient for a Normal Immune Response to Bacterial Polysaccharides.
To exclude the possibility that the absence of the spleen itself, rather, or in addition to, the lack of B-1a cells, was responsible for the failure of the immune response, we generated mice lacking B-1a cells but possessing a spleen. To do this, bone marrow or fetal liver cells were transferred into Rag2-null mice (Fig. 6). It has been reported that transfer of bone marrow cells repopulates all major B cell populations with the exception of B-1a cells, whereas transfer of fetal liver cells also repopulates the B-1a population (18). 4 wk after transfer the major B cell populations in bone marrow (pro-B and pre-B, immature, transitional, and recirculating), spleen (T1, T2, and mature), and lymph nodes (mature) were analyzed. All these populations were present (data not shown). However, as expected, the peritoneal CD5pos B220dull B-1a population was lacking in the mice transferred with bone marrow but present in the mice that had received fetal liver cells (Fig. 6 A). Interestingly CD5posB220pos B cells (indicated by the arrow in Fig. 6 A) phenotypically identical to residual CD5pos B cells of Hox11-null mice (see also Fig. 1 B) were detectable after bone marrow transfer, suggesting they are not only phenotypically but also ontogenetically distinct from the CD5posB220dull B-1a cells.
Figure 6.




Mice lacking B-1a cells but possessing a spleen fail to respond to immunization with streptococcal polysaccharides. (A) Fetal liver (FL) or bone marrow (BM) cells from C57BL/6 mice were injected intraperitoneally into irradiated Rag2-null mice and four weeks later reconstituted mice were analyzed. A representative FACS® analysis of total ungated peritoneal cells stained for CD5 and B220 from a FL- and a BM-transferred mouse is shown above. The CD5posB220dull B-1a cell population is boxed. The CD5posB220pos B cells phenotypically identical to residual CD5pos B cells of Hox11-null mice are indicated by a box and arrow. (B) Representative FACS® analyses of CD23neg-gated spleen cells stained for CD21 and IgM from a FL- and a BM-transferred mouse are shown. The normal position of the MZ B cell population (MZ) is boxed. (C) Specific IgM antibodies in sera of transferred mice. Pneumovax® 23 was injected intraperitoneally 20 d after transfer. Antibodies were measured by ELISA 1 d before and 6 d after Pneumovax® 23 injection. The optical density (OD405) measured with a 1:80 serum dilution is shown. Each data point indicates the relative antibody level of a single mouse. (D) The response to Pneumovax® 23 of mice transferred with either bone marrow (BM) or dE14.5 fetal liver cells (FL) is shown as mean fold-increase of specific serum IgM as detected by ELISA. The optical density (OD405) was measured with a 1:80 serum dilution. For each route of injection, five mice were transferred with bone marrow and three with fetal liver cells.
To confirm that the B220dull B-1a cells are indeed the cells responsible for the production of IgM against bacterial polysaccharides, the reconstituted mice were immunized subcutaneously, intramuscularly, intraperitoneally, or intravenously with Pneumovax® 23 and the production of specific IgM was measured. Mice transferred with fetal liver cells, and, thus, possessing B-1a cells, produced specific IgM, regardless of the route of immunization. In contrast, mice transferred with bone marrow and, thus, lacking B-1a cells failed to respond (Fig. 6 C and D). Therefore, the presence of B-1a cells is required to induce an immune response to streptococcal polysaccharides. In contrast, the presence of the spleen, B-1b, and B-2 cells in the absence of B-1a cells is not sufficient.
The importance of MZ B cells in response to T cell–independent antigens has been recognized (28–30). Mice transferred with bone marrow cells possessed normal numbers of CD23negCD21brightIgMbright MZ B cells (reference 19; Fig. 6 B), but their inability to respond to bacterial polysaccharides indicates that in the absence of B-1a cells, MZ B cells are not sufficient for a response to Pneumovax® 23. The impaired response to streptococcal polysaccharides in mice lacking B-1a cells may indicate that B-1a and B-1b cells are not only ontogenetically (31), but also functionally, distinct. Thus, in Hox11-null, splenectomized and bone marrow reconstituted mice, there is a direct correlation between absence of B-1a cells and lack of immune response to streptococcal polysaccharides.
Discussion
Using asplenic Hox11-null mice, we have shown that the spleen is required for the normal development of the B-1a population. The origin and differentiation of B-1a cells is controversial. At one time, B-1a cells were assumed to be ontogenetically distinct from B-2 cells, based on the observation that they are derived from fetal liver but not adult bone marrow (26). The absence of N nucleotide addition and the preferential usage of proximal V gene segments in fetal precursors are possible reasons for the generation of high numbers of B-1 cells from fetal liver but not from adult bone marrow (32). More recent data show that B-1a cells may develop from conventional B cell precursors as a consequence of positive selection after encounter with antigen (33, 34). It is conceivable that this selection occurs in the spleen and the presence of B-1a precursor cells in the spleen has been described previously (35). Therefore, the spleen may offer a specialized microenvironment where ligand-dependent positive selection of B-1a precursors occurs. In addition, the spleen may function as a reservoir of fetal liver–derived B-1a precursors in adult life.
A few CD5pos B-1a cells were still present in the peritoneal cavity and the blood of Hox11-null mice and could also be detected in the peritoneal cavity of irradiated Rag2-null mice 4 wk after transfer of C57BL/6 bone marrow. However, these cells express higher levels of B220 and slightly lower levels of CD5 as compared with the CD5posB220dull B-1a cells of C57BL/6 animals. This suggests that CD5pos B cells might consist of two distinct subpopulations. B220dull CD5 cells, corresponding to B-1a B cells, are strictly dependent on the spleen for their generation, and develop rapidly in high numbers from fetal liver precursors. In contrast, B220pos CD5 cells can be generated in the absence of the spleen, can be derived from both fetal liver and adult bone marrow, and in these respects are very similar to B-1b cells. Our results demonstrate that the former population is required for natural IgM production and the response to streptococcal polysaccharide antigens.
Splenectomy of C57BL/6 mice leads to a rapid loss of ∼75% of the peritoneal B-1a cell pool. It is unknown whether the reduction in B-1a cells in splenectomized mice reflects a defect in their generation, their survival, or a combination of both. B-1a cell precursors may home to the spleen from the liver during the fetal life and B-1a B cells may be continuously generated from these precursors in the adult. Alternatively, B-1a cells may be long-lived and their rapid loss upon splenectomy would suggest that they are unable to survive in the absence of the spleen. This could reflect the requirement of a factor generated in the spleen that is essential to B-1a survival. It is possible that B-1a cells continually circulate through the spleen, where the interaction with a specialized microenvironment supports their survival.
B-1a cells were originally identified based on the expression of CD5 (36), a glycoprotein expressed by all T cells after positive selection (37, 38). In the B cell compartment, only B-1a cells are CD5pos. However, CD5 can be induced on conventional B cells after B cell antigen receptor (BCR) cross-linking (39) and low levels of CD5 are expressed by chronically stimulated, anergic B cells (40). Therefore, it has been proposed that expression of CD5 is indicative of a prior interaction of B-1a cells with antigen. The BCRs of B-1a cells comprise not only polyreactive but also autoreactive specificities and it has been demonstrated that autoantigens can be responsible for their positive selection (33). Given that CD5 is a negative regulator of BCR signaling in B-1a cells (41) and that tight regulation of the activation of B-1a cells specific for autoantigens is essential, low expression of B220 in addition to up-regulated CD5 expression might downmodulate BCR signals after positive selection by antigen and thus prevent autoimmunity (42, 43).
It has been demonstrated that fetal liver– and bone marrow–derived B cells have different signaling properties (34). The CD5posB220pos B cells present in Hox11-null, Rag2-null reconstituted, and C57BL/6 mice may be B-1a precursors that do not, for some reason, downmodulate B220 after interaction with ligand in the spleen. The reduced threshold for signaling in such cells may contribute to autoimmune phenomena attributed to CD5pos B cells. Alternatively, these cells may be ontologically distinct from typical B-1a cells. Their presence after C57BL/6 bone marrow transfer into Rag2-null mice indicates that they are a bone marrow–derived population.
At the outset, bacterial infections are often localized and are initially exposed to components of the innate immune system. Natural IgM antibodies secreted by B-1 cells are part of this system and play a crucial role in the activation of the complement cascade and the amplification of early innate defense mechanisms (11). Hox11-null mice exhibit significantly reduced levels of serum IgM and of natural antibodies. In addition, lack of B-1a cells in Hox11-null and splenectomized mice correlates with an inability to produce specific IgM in response to T cell–independent streptococcal polysaccharides. We observed that Pneumovax®-specific IgM was produced regardless of the route of inoculation. Although B-1a cells were originally described in the peritoneal cavity, we found B-1a cells in the peripheral blood. They have also been detected in the spleen and 50% of the IgA-secreting plasma cells in the mucosa of the bowel are B-1 derived (44, 45). Furthermore, B-1a cells can be detected in lungs of mice infected with influenza virus (46), suggesting that they may be able to migrate to sites of infection. Circulation through the body via the blood could be a mechanism by which B-1a cells rapidly reach infected and inflamed tissues. Therefore, this might be important to limit dissemination of invading pathogens. Thus, the presence of B-1a cells in the blood might explain the production of Pneumovax®-specific IgM in C57BL/6 mice regardless of the route of immunization while Hox11-null mice, lacking peritoneal and blood B-1a cells, did not respond.
MZ B cells of the spleen contribute to the early immune response to thymus-independent (TI)-2 antigens (28–30). However, though MZ B lymphocytes were reconstituted to normal amounts after transfer of bone marrow cells into Rag2-null mice, these animals did not produce specific IgM in response to immunization with streptococcal polysaccharides. Presence of the spleen including MZ B cells is, therefore, not sufficient for an adequate response to TI-2 antigens if B-1a cells are absent. It has recently been demonstrated that B-1 cells dominate the response to TI antigens in the peritoneum, while MZ B cells contribute to the early response to blood-borne particulate TI antigens (47). Nevertheless, mice possessing B-1a cells but lacking MZ B cells show an impaired response to the TI-2 antigen TNP-Ficoll after intraperitoneal administration (28). Hence, it is possible that both B-1a and MZ cells are required for the production of specific IgM in response to different repetitive polysaccharide antigens. While MZ B cells may be important in the late phases of infections when pathogens can be found in the blood and are filtered into the MZ of the spleen, B-1a B cells may play a role in the limitation of early bacterial growth at the site of infection (47).
In summary, we have demonstrated that the spleen is indispensable for the generation and survival of B-1a cells in the mouse. This reveals an essential role of the spleen in producing these cells, and their absence may account for the concomitant infections that occur in asplenic individuals. Future experiments will focus on determining how the spleen functions in the generation and survival of these cells.
Acknowledgments
We thank Horst Mossmann, Norbert Joswig, Uta Stauffer, and colleagues in the central animal facility for maintenance of the mouse colony and assistance with surgery. Hox11-null mice were provided by T.H. Rabbitts, Laboratory of Molecular Biology, Cambridge, UK. We thank Holger Weber for helpful discussion.
Footnotes
Abbreviations used in this paper: BCR, B cell antigen receptor; MZ, marginal zone; TI, thymus independent; T1, transitional 1; T2, transitional 2.
References
- 1.Hansen, K., and D.B. Singer. 2001. Asplenic-hyposplenic overwhelming sepsis: postsplenectomy sepsis revisited. Ped. Dev. Pathol. 4:105–121. [DOI] [PubMed] [Google Scholar]
- 2.Styrt, B. 1990. Infection associated with asplenia: risks, mechanisms, and prevention. Am. J. Med. 88:33N–42N. [PubMed] [Google Scholar]
- 3.Phoon, C.K., and C.A. Neill. 1994. Asplenia syndrome: insight into embryology through an analysis of cardiac and extracardiac anomalies. Am. J. Cardiol. 73:581–587. [DOI] [PubMed] [Google Scholar]
- 4.Feder, H.M., Jr., and H.A. Pearson. 1999. Assessment of splenic function in familial asplenia. N. Engl. J. Med. 341:210–212. [DOI] [PubMed] [Google Scholar]
- 5.Germing, U., C. Perings, S. Steiner, A.J. Peters, M.P. Heintzen, and C. Aul. 1999. Congenital asplenia detected in a 60 year old patient with septicemia. Eur. J. Med. Res. 28:283–285. [PubMed] [Google Scholar]
- 6.Dyke, M.P., R.P. Martin, and P.J. Berry. 1991. Septicemia and adrenal haemorrhage in congenital asplenia. Arch. Dis. Child. 66:636–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gillis, J., J. Harvey, D. Isaacs, M. Freelander, and B. Wyeth. 1992. Familial asplenia. Arch. Dis. Child. 67:665–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanthan, R., T. Moyana, and J. Nyssen. 1999. Asplenia as cause of sudden unexpected death in childhood. Am. J. Forensic Med. Pathol. 20:57–59. [DOI] [PubMed] [Google Scholar]
- 9.Ochsenbein, A.F., T. Fehr, C. Lutz, M. Suter, F. Brombacher, H. Hengartner, and R.M. Zinkernagel. 1999. Control of early viral and bacterial distribution and disease by natural antibodies. Science. 286:2156–2159. [DOI] [PubMed] [Google Scholar]
- 10.Hayakawa, K., and R.R. Hardy. 2000. Development and function of B-1a cells. Curr. Opin. Immunol. 12:346–353. [DOI] [PubMed] [Google Scholar]
- 11.Casali, P., and E.W. Schettino. 1996. Structure and function of natural antibodies. Curr. Top. Microbiol. Immunol. 210:167–179. [DOI] [PubMed] [Google Scholar]
- 12.Roberts, C.W., J.R. Shutter, and S.J. Korsmeyer. 1994. Development expression of Hox11 and specification of splenic cell fate. Nature. 368:747–749. 7908720 [Google Scholar]
- 13.Dear, T.N., W.H. Colledge, M.B. Carlton, I. Lavenir, T. Larson, A.J. Smith, A.J. Warren, M.J. Evans, M.V. Sofroniew, and T.H. Rabbitts. 1995. The Hox11 gene is essential for cell survival during spleen development. Development. 121:2909–2915. [DOI] [PubMed] [Google Scholar]
- 14.Herzenberg, L.A. 2000. B-1 cells: the lineage question revisited. Immunol. Rev. 175:9–22. [PubMed] [Google Scholar]
- 15.Whitmore, A.C., G. Haughton, and L.W. Arnold. 1996. Phenotype of B cells responding to the thymus-independent type-2 antigen polyvinyl pyrrolidinone. Int. Immunol. 8:533–542. [DOI] [PubMed] [Google Scholar]
- 16.Arnold, L.W., C.A. Pennell, S.K. McCray, and S.H. Clarke. 1994. Development of B-1 cells: segregation of phosphatidyl choline-specific B cells to the B-1 population occurs after immunoglobulin gene expression. J. Exp. Med. 179:1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wortis, H.H., and R. Berland. 2001. Cutting edge commentary: origins of B-1 cells. J. Immunol. 166:2163–2166. [DOI] [PubMed] [Google Scholar]
- 18.Kantor, A.B., A.M. Stall, S. Adams, K. Watanabe, and L.A. Herzenberg. 1995. De novo development and self-replenishment of B cells. Int. Immunol. 7:55–68. [DOI] [PubMed] [Google Scholar]
- 19.Loder, F., B, Mutschler, R.J. Ray, C.J. Paige, P. Sideras, R. Torres, M.C. Lamers, and R. Carsetti. 1999. B cell development takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J. Exp. Med. 190:75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lalor, P.A., and G. Morahan. 1990. The peritoneal Ly-1 (CD5) B cell repertoire is unique among murine B cell repertoires. Eur. J. Immunol. 20:485–492. [DOI] [PubMed] [Google Scholar]
- 21.Kenny, J.J., L.J. Yaffe, A. Ahmed, and E.S. Metcalf. 1983. Contribution of Lyb 5+ and Lyb 5− B cells to the primary and secondary phosphocholine-specific antibody response. J. Immunol. 130:2574–2579. [PubMed] [Google Scholar]
- 22.Masmoudi, H., T. Mota-Santos, F. Huetz, A. Coutinho, and P.A. Cazenave. 1990. All T15 Id-positive antibodies (but not the majority of VHT15+ antibodies) are produced by peritoneal CD5+ B lymphocytes. Int. Immunol. 2:515–520. [DOI] [PubMed] [Google Scholar]
- 23.Baumgarth, N., J. Chen, O.C. Herman, G.C. Jager, and L.A. Herzenberg. 2000. The role of B-1 and B-2 cells in immune protection from influenza virus infection. Curr. Top. Microbiol. Immunol. 252:163–169. [DOI] [PubMed] [Google Scholar]
- 24.Briles, D.E., M. Nahm, K. Schroer, J. Davie, P. Baker, and J. Kearney. 1981. Antiphosphocholine antibodies found in normal mouse serum are protective against intravenous infection with type 3 streptococcus pneumoniae. J. Exp. Med. 153:694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Briles, D.E., C. Forman, S. Hudak, and J.L. Claflin. 1982. Anti-phosphorylcholine antibodies of the T15 idiotype are optimally protective against Streptococcus pneumoniae. J. Exp. Med. 156:1177–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kenny, J.J., G. Guelde, R.T. Fischer, and D.L. Longo. 1994. Induction of phosphocholine-specific antibodies in X-linked immune deficient mice: in vivo protection against a Streptococcus pneumoniae challenge. Int. Immunol. 6:561–568. [DOI] [PubMed] [Google Scholar]
- 27.Barletta, R., G.A. Bruyn, and R. van Furth. 1991. Pneumococcal polysaccharide vaccines: indications, efficiency, and recommendations. Eur. J. Clin. Microbiol. Infect. Dis. 10:897–910. [DOI] [PubMed] [Google Scholar]
- 28.Guinamard, R., M. Okigaki, J. Schlessinger, and J.V. Ravetch. 2000. Absence of marginal zone B cells in Pyk-2-deficient mice defines their role in the humoral response. Nat. Immunol. 1:31–36. [DOI] [PubMed] [Google Scholar]
- 29.Martin, F., A.M. Oliver, and J.F. Kearney. 2001. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 14:617–629. [DOI] [PubMed] [Google Scholar]
- 30.Martin, F., and J.F. Kearney. 2000. B-cell subsets and the mature preimmune repertoire. Marginal zone and B1 B cells as part of a “natural immune memory”. Immunol. Rev. 175:70–79. [PubMed] [Google Scholar]
- 31.Kantor, A.B., and L.A. Herzenberg. 1993. Origin of murine B cell lineages. Annu. Rev. Immunol. 11:501–538. [DOI] [PubMed] [Google Scholar]
- 32.Benedict, C.L., S. Gilfillan, T.H. Thai, and J.F. Kearney. 2000. Terminal deoxynucleotidyl transferase and repertoire development. Immunol. Rev. 175:150–157. [PubMed] [Google Scholar]
- 33.Hayakawa, K., M. Asano, S.A. Shinton, M. Gui, D. Allman, C.L. Stewart, J. Silver, and R.R. Hardy. 1999. Positive selection of natural autoreactive B cells. Science. 285:113–116. [DOI] [PubMed] [Google Scholar]
- 34.Hardy, R.R., Y.S. Li, D. Allman, M. Asano, M. Gui, and K. Hayakawa. 2000. B-cell commitment, development and selection. Immunol. Rev. 175:23–32. [PubMed] [Google Scholar]
- 35.Arnold, L.W., S.K. McCray, C. Tatu, and S.H. Clarke. 2000. Identification of a precursor to phosphatidyl choline-specific B-1 cells suggesting that B-1 cells differentiate from splenic conventional B cells in vivo: cyclosporin A blocks differentiation to B-1. J. Immunol. 164:2924–2930. [DOI] [PubMed] [Google Scholar]
- 36.Herzenberg, L.A., A.M. Stall, P.A. Lalor, C. Sidman, W.A. Moore, D.R. Parks, and L.A. Herzenberg. 1986. The Ly-1 B cell lineage. Immunol. Rev. 93:81–102. [DOI] [PubMed] [Google Scholar]
- 37.Azzam, H.S., J.B. DeJarnette, K. Huang, R. Emmons, C.S. Park, C.L. Sommers, D. El-Khoury, E.W. Shores, and P.E. Love. 2001. Fine tuning of TCR signaling by CD5. J. Immunol. 166:5464–5472. [DOI] [PubMed] [Google Scholar]
- 38.Azzam, H.S., A. Grinberg, K. Lui, H. Shen, E.W. Shores, and P.E. Love. 1998. CD5 expression is developmentally regulated by T cell receptor (TCR) signals and TCR avidity. J. Exp. Med. 188:2301–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cong, Y.Z., E. Rabin, and H.H. Wortis. 1991. Treatment of murine CD5-B cells with anti-Ig, but not LPS, induces surface CD5: two B-cell activation pathways. Int. Immunol. 3:467–476. [DOI] [PubMed] [Google Scholar]
- 40.Hippen, K.L., L.E. Tze, and T.W. Behrens. 2000. CD5 maintains tolerance in anergic B cells. J. Exp. Med. 191:883–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sen, G., G. Bikah, C. Venkataraman, and S. Bondada. 1999. Negative regulation of antigen receptor-mediated signaling by constitutive association of CD5 with the SHP-1 protein tyrosine phosphatase in B-1 B cells. Eur. J. Immunol. 29:3319–3328. [DOI] [PubMed] [Google Scholar]
- 42.Qian, Y., C. Santiago, M. Borrero, T.F. Tedder, and S.H. Clarke. 2001. Lupus-specific antiribonucleoprotein B cell tolerance in nonautoimmune mice is maintained by differentiation to B-1 and governed by B cell receptor signaling thresholds. J. Immunol. 166:2412–2419. [DOI] [PubMed] [Google Scholar]
- 43.Berland, R., and H.H. Wortis. 2000. A model for autoantigen induction of natural antibody producing B-1a cells. Curr. Top. Microbiol. Immunol. 252:49–55. [DOI] [PubMed] [Google Scholar]
- 44.Kroese, F.G., W.A. Ammerlaan, and A.B. Kantor. 1993. Evidence that intestinal IgA plasma cells in mu, kappa transgenic mice are derived from B-1 (Ly-1 B) cells. Int. Immunol. 5:1317–1327. [DOI] [PubMed] [Google Scholar]
- 45.Bos, N.A., J.J. Cebra, and F.G. Kroese. 2000. B-1 cells and the intestinal microflora. Curr. Top. Microbiol. Immunol. 252:211–220. [DOI] [PubMed] [Google Scholar]
- 46.Baumgarth, N., O.C. Herman, G.C. Jager, L. Brown, and L.A. Herzenberg. 1999. Innate and acquired humoral immunities to influenza virus are mediated by distinct arms of the immune system. Proc. Natl. Acad. Sci. USA. 96:2250–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin, F., A.M. Oliver, and J.F. Kearney. 2001. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 14:617–629. [DOI] [PubMed] [Google Scholar]