

Abstract
Antigen-specific B cells express major histocompatibility complex class II and can present antigen directly to T cells. Adoptive transfer experiments using transgenic B and T cells demonstrated that antigen-specific B cells can also efficiently transfer antigen to another cell for presentation to T cells in vivo. To identify the antigen-presenting cell that receives antigens from B cells, a strategy was developed to follow the traffic of B cell–derived proteins in vivo. B cells were labeled with the fluorescent dye CFSE and loaded with antigen, before adoptive transfer into recipient mice. Populations of splenocytes from the recipient mice were later assayed for the presence of fluorescent proteins and for the ability to activate T cells. A small number of CD8α+CD4−CD11blo dendritic cells (DCs) contain proteins transferred from B cells and these DCs effectively present antigens derived from the B cells to T cells. The results suggest that CD8α+ DCs sample the cells and membranes in their environment for presentation to T cells circulating through the T cell zone. This function of CD8α+ DCs may be relevant to the priming of an immune response or the induction of T cell tolerance.
Keywords: T cells, B cells, dendritic cells, antigen-presenting cells, flow cytometry
Introduction
Intercellular communication is key to immune system function and it has become clear that “conversations” between multiple cells are common and important (1–3). Much focus has been placed on the roles of receptor–ligand interactions in dialogs between pairs of cells, however these are not the only mechanisms of information transfer between cells. Cellular communication includes the exchange of cell components, such as the secretion of exosomes (membranous vesicles; references 4–6), the phagocytosis of dead cells (7, 8), and the direct uptake of proteins from membranes (3, 9). The work described here demonstrates that cell-associated antigen traffics between antigen-containing cells in the blood and CD8α+CD11blo dendritic cells (DCs)*in the spleen for presentation to naive CD4+ T cells.
Antigen-specific B cells function as uniquely effective antigen concentrating particles due to their ability to bind antigen to their specific Ig receptors (10–12). Acutely activated B cells present this antigen complexed with MHC class II to T cells to solicit help for proliferation and antibody production (13). We recently demonstrated that antigen-specific B cell–T cell interactions in vivo occur both directly between T and B cells, and indirectly, via an intermediary APC. In this novel pathway of antigen-specific lymphocyte regulation, antigen-specific B cells efficiently transfer antigen to another APC in vivo for presentation to T cells (2).
In this study, we set out to further characterize this process and to identify the presenting APC. Our strategy was to perform adoptive transfer of dye-labeled, HEL-loaded B cells alone, with no T cells, then isolate the APC to which the B cell transferred antigen from the recipient spleen. Splenocytes from recipient mice which contained dye-labeled proteins from the B cells were fractionated and characterized by flow cytometry. The isolated populations of cells were then tested for the ability to present antigen to T cells in vitro. We show here that the presenting APC is a CD8α+ DCs which expresses high levels of MHC class II and CD86. We propose that this subset of DCs constitutively samples the cells and membranes in its environment, the T cell zone, and presents those cell-associated antigens to T cells.
Materials and Methods
Mice.
IgHEL transgenic mice (13a) on the C57Bl/6 background, 3A9 TCR transgenic mice (13b) on the B10.BR background (>10× backcrossed), B10.BR and B6 stock mice were bred and maintained at the Biomedical Research Centre according to University standards. Bcl-2 transgenic mice in the B6 background were purchased from The Jackson Laboratory. Bcl-xL on the B6 background was a gift of Dr. Jeffrey Rathnell (University of Pennsylvania, Philadelphia, PA). The transgenic phenotypes of all donor cells were verified by flow cytometry before transfer into the recipient mice.
CFSE Labeling of T or B Cells.
T cells from spleen and lymph nodes of transgenic TCR or B cells from spleens of IgHEL mice, were labeled with 5-(and 6-) carboxyfluorescein diacetate succinimidyl ester (CFSE) modified as follows (14). The cells were washed once with complete medium RPMI 1640, 10% FCS, 10 mM Hepes, resuspended at a concentration of 107 cells per milliliter, incubated with 5 μM CFSE (Molecular Probes) for 7 min at room temperature then washed with complete medium.
B Cell Purification and Antigen Loading.
CFSE-labeled transgenic B cells were purified using B220+ microbeads (Miltenyi Biotec) as per the manufacturer's instructions. Purified B cells, as verified by FACS® (92–98%) were loaded in vitro with 10 ng/ml of HEL for 1 h on ice. The cells were washed in complete medium and then in HBSS.
Adoptive Transfer.
CFSE-labeled transgenic T cells were washed, counted, and resuspended in HBSS for injections. Cell suspensions containing 1–3 × 106 CD4+ cells were injected intravenously via the lateral tail vein. Several hours later or the next day, cell suspensions containing 1–3 × 106 HEL-specific transgenic B cells were intravenously injected. Recipient mice were killed 14–16 h after the transfer, the spleens were collected, and cell suspensions prepared for flow cytometry analysis.
In Vivo Protein Trafficking Experiments.
Transgenic B cells were CFSE-labeled, then purified and HEL loaded as described above. Then, the cells were washed, counted, and resuspended in HBSS. The B220+ HEL-loaded transgenic B cells (3–6 × 106) were injected intravenously into B10.BR recipients. 12–14 h after the injections the recipient mice were killed and the spleens were collected for APC isolation. Each spleen was collected in 1 ml of RPMI 1640, 10 mM Hepes, and 0.01 mg/ml of Liberase Blenzyme 3 (Roche Diagnostics), the spleens were minced with razor blades and incubated for 1 h at 37°C. Fragments were dissociated by passing five times through 21-g needle in order to dissociate undigested material then washed twice with complete medium.
In Vivo Protein Trafficking Experiments Using Bcl-2 and Bcl-xL Mice.
Bead purified, CFSE labeled B cells from both transgenic or nontransgenic mice were adoptively transferred into B10.BR recipients by intravenous injection. 16 h after the transfer, the recipients were killed and the spleens collected into 1 ml of RPMI 1640, 10 mM Hepes, and 0.01 mg/ml of Liberase and prepared as above. The cell suspension then were stained and analyzed by flow cytometry. To test the survival ability of the cells, both transgenic or nontransgenic purified B cells were cultured in vitro for 16–18 h in complete media. Then the cells were stained and analyzed by flow cytometry.
In Vitro T Cell Activation.
Populations of APC isolated from splenocytes of recipient mice were cultured in round-bottomed 96-well plates with 3A9 transgenic CD4+ T cells (3 × 105 cells per well) in complete medium with 10% FCS for 16 h at 37°C. The cells were then stained and analyzed by flow cytometry.
Flow Cytometry.
Analytical flow cytometry was conducted on a FACSCalibur™ and analyzed using CELLQuest™ (Becton Dickinson) and FSCPress (Ray Hicks, University of Cambridge, Cambridge, UK) software. Cell sorting was performed using FACSVantage™ (Becton Dickinson). Antibodies specific for CD69, CD11c, CD11b, and CD86 were purchased from BD PharMingen. B220, CD4, CD8a, I-Ap were purchased from Cederlane, and 7-AAD was purchased from Molecular Probes. The hybridoma secreting the clonotypic anti-3A9 TCR antibody, 1G12, was a gift of D. Peterson, Washington University, St. Louis, MO (15).
Biotinylation of Thymocytes.
3 × 107 thymocytes were resuspended in HBSS and chilled on ice. Then the cells were incubated for 30 min on ice with sulfo-NHS-biotin (Pierce Chemical Co.) at a final concentration of 0.4 mg/ml. The cells were washed and resuspended in HBSS plus 2% BSA. The effectiveness of the biotinylation was measured by flow cytometry staining with streptavidin-conjugated with allophycocyanin. The biotinylated thymocytes were incubated with streptavidin (33 μg/ml; Sigma-Aldrich) for 30 min on ice, then washed and incubated with HEL-biotin for 1 h on ice and washed twice with HBSS.
Anti–Mouse IgM (α-IgM)–HEL Conjugation.
Purified HEL was incubated with SMCC (succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate; Pierce Chemical Co.) at 0.1 mg of SMCC per mg of HEL, for 1 h at room temperature, under rotation, and in the dark. Anti–mouse IgM (α-mIgM) was reduced in 20 mM DTT per ml of antibody at room temperature for 30 min. HEL in SMCC and reduced α-mIgM were passed over individual PD-10 columns previously equilibrated with 50 mM of MES (2-[N-morpholino] ethanosulfunic acid, 2 mM EDTA), pH 6.0. Derivitized HEL and reduced α-mIgM were incubated for 1 h at room temperature under rotation in the dark and the reaction was stopped adding 34 ug of N-ethylmaleimide. The conjugate was isolated from free HEL by affinity chromatography on protein G and finally dialyzed into PBS, pH 7.8.
Results
We originally observed the presentation of antigen transferred from B cells in parental-into-F1 adoptive transfers (H-2bb→H-2kb; reference 2). This adoptive transfer system permits the study of the trafficking of antigens associated with cells present in the blood in which the donor cells cannot directly present antigen to T cells. We subsequently observed that the 3A9 HEL-specific TCR develops differently on the H-2kb and the H-2kk backgrounds, in that positive selection occurs more efficiently on the latter background (reference 16; data not shown). In light of these results, we modified our transfer system to be fully allogeneic, H-2bb donor B cells into H-2kk recipient mice seeded with H-2kk 3A9 T cells. Allogeneic CTL activity in the recipient is not expected in the short time frame of the experiments (16 h), but NK cell activity is expected. This low level of acute allogeneic reactivity between donor cells and recipients may amplify the trafficking of cell-associated antigens in the blood (discussed further below).
Splenocytes from 3A9 TCR transgenic mice on the B10.BR (H-2kk) background were adoptively transferred intravenously into B10.BR recipients. The TCR transgenic splenocytes were labeled with the fluorescent dye, CFSE, to aid in tracking the cells. MHC-mismatched (H-2bb) transgenic IgHEL B cells were loaded in vitro with HEL, at a dose of antigen that loads only the antigen-specific B cells and no other APC (unpublished data), or sham-loaded. The MHC-mismatched, loaded transgenic B cells were transferred intravenously into the same B10.BR recipients previously seeded with HEL-specific T cells. 16 h after transfer, recipient mice were killed and the splenocytes prepared for flow cytometry. HEL-specific T cells from recipient spleens were analyzed for the expression of the early activation marker, CD69, as a measure of T cell encounter with HEL peptide/I-Ak complexes.
Few CD4+ T cells from the recipients (CFSE−) expressed CD69, demonstrating that transfer of allogeneic B cell did not induce measurable activation of the recipient's endogenous CD4+ T cells (Fig. 1 a and c). Similarly, HEL-specific CD4+ T cells (CFSE+) from recipients which received sham-loaded HEL-specific B cells were not activated (Fig. 1 b). In contrast, HEL-specific CD4+ T cells (CFSE+) from recipient mice which received HEL-loaded B cells were fully activated (Fig. 1 d). The activation of these T cells was antigen-specific, as it required the presence of both the antigen, and the antigen-specific TCR (Fig. 1 d). The experiment was also performed using purified CD4+ TCR transgenic cells, with similar results (data not shown). The MHC class II–expressing APC that induced this activation is resident in the recipient, as the transferred B cells did not express the restricting MHC haplotype. Accordingly, the antigen must have been transferred from the antigen-specific B cells to another APC in the recipient for presentation to the T cells. The transfer of antigen from B cells to endogenous APC in this fully allogeneic transfer system induces activation of naive, H-2kk T cells, similar to our original observation using the parental into F1 transfer system (H-2bb B cells into H-2kk; reference 2). The transfer of antigen for presentation to T cells does not require the allogeneic difference, however, as T cell activation was also efficiently induced after the transfer of HEL-loaded H-2bb B cells into tolerant recipients (H-2kk→H-2bb bone marrow chimeras; Fig. 2) .
Figure 1.

HEL-loaded, HEL-specific H-2bb B cells mediate the activation of H-2kk-restricted HEL-specific T cells in vivo. HEL-specific 3A9, transgenic T cells from the B10.BR(H-2kk) background were labeled with CFSE and adoptively transferred intravenously into intact B10.BR recipients. Equal number of HEL-specific IgHEL transgenic B cells on the C57Bl/6 (H-2bb) background were loaded with HEL in vitro or sham loaded, then adoptively transferred into the recipients containing transferred T cells. 16 h later, the recipient mice were killed and the splenocytes analyzed by flow cytometry. Files containing data for total lymphocytes as well as live-gated files containing data only for CFSE+ cells were collected for each sample. Data were gated for lymphocytes in FSC, SSC, and B220+ cells were eliminated from the analysis by negative gating. T cell activation was measured by the expression of the early activation marker CD69 by nontransgenic CD4+ T cells (a and c) or by transgenic CFSE+ CD4+ (b and d). Data are representative of three individuals from each of more than eight experiments.
Figure 2.

Presentation of transferred antigen does not require an allogeneic response. HEL-specific IgHEL(H-2bb) transgenic splenocytes were loaded with PBS or HEL ex vivo, and injected intravenously into B10.BR (H-2kk) mice which had been previously seeded with CFSE-labeled 3A9 HEL-specific T cells (H-2kk) by adoptive transfer. 20 h after transfer, mice were killed and the spleens prepared and analyzed by flow cytometry. Data were gated to include lymphocytes and exclude dead cells, and for CFSE+ CD4+ HEL-specific T cells. CD69 expression was used as a measure of T cell activation. Each point represent a single mouse.
We tested whether nontransgenic B cells have the ability to transfer antigen as donor cells in vivo. Nontransgenic B cells were loaded with HEL by incubating B6 splenocytes with an anti-IgM antibody conjugated with HEL, to target HEL to the BCR. Thymocytes were also incubated with the antibody-HEL conjugate, to test the specificity of the conjugate for B cells. As shown in Fig. 3 A, the anti–IgM-HEL conjugate effectively loaded HEL on to the surface of nontransgenic B cells, but not onto thymocytes. HEL-loaded donor cells were adoptively transferred into B10.BR recipients which had been previously seeded with HEL-specific transgenic T cells. T cell activation was assayed by flow cytometry 14 h after transfer. MHC-mismatched, HEL-loaded nontransgenic B cells mediated the activation of HEL-specific T cells as effectively as HEL-loaded transgenic B cells (Fig. 3 B). This result demonstrates that the ability to transfer antigen in vivo is not peculiar to transgenic B cells.
Figure 3.


Antigen concentrated by nontransgenic B cells is effectively transferred for presentation to T cells in vivo. HEL was loaded onto nontransgenic B6 B cells (H-2bb) using an anti-IgM antibody conjugated with HEL to target HEL to the BCR. (A) HEL loading was measured by flow cytometry using the HEL-binding antibody, HyHEL-9. The histograms represent surface HEL on: IgHEL transgenic B cells after loading with HEL (HEL), nontransgenic B cells after loading with anti–IgM-HEL conjugate (αIgM-HEL), nontransgenic thymocytes after loading with anti-IgM-HEL conjugate (thymus), and IgHEL transgenic B cells after sham loading (Sham). Data were gated for live lymphocytes and for B220+ cells. (B) Loaded donor cells were transferred into B10.BR recipients which had been previously seeded with CFSE-labeled 3A9 HEL-specific transgenic T cells. 14 h after transfer, mice were killed and the spleens prepared for flow cytometry. Data were gated for live lymphocytes and either CFSE+CD4+ responder T cells (HEL-specific) or CFSE−CD4+ responder T cells (endogenous), and activation was measured using CD69. Each point represents a single mouse. Data are representative of two experiments.
Intriguingly, the ability to transfer antigen is not a B cell–specific phenomenon; any cell with membrane-associated antigen can transfer that antigen in vivo. HEL was concentrated on the cell surface of thymocytes by first surface biotinylating the cells, then incubating with streptavidin, and finally with HEL-biotin. These HEL-loaded thymocytes were as effective as HEL-loaded IgHEL B cells in transferring antigen in vivo (Fig. 4) . We obtained similar results using biotinylated L cells (data not shown). These data demonstrate that B cell-specific molecules are not required for antigen transfer, and suggest that the antigen transfer and presentation process is a general mechanism for handling cell-associated antigen. B cells, however, may represent a special case of this general mechanism, in that they function here as antigen concentrating particles by binding antigen via their antigen-specific Ig receptor.
Figure 4.

HEL-loaded thymocytes transfer antigen for presentation to T cells in vivo. HEL was loaded onto the surface of thymocytes by first surface biotinylating the thymocytes, then incubating with streptavidin, then finally incubating with HEL-biotin. Effective biotinylation and HEL-loading were confirmed by flow cytometry. HEL-loaded B6 thymocytes or HEL-loaded or sham-loaded B6 IgHEL B cells were injected intravenously into B10.BR recipient mice previously seeded with CFSE-labeled HEL-specific 3A9 transgenic T cells. 12–16 h later, mice were killed and spleens prepared for flow cytometry. Data were gated on live lymphocytes and CFSE+CD4+ responder T cells. CD69 expression was used as a measure of T cell activation. The percentage of CFSE+CD4+ cells expressing CD69 in mice receiving HEL-loaded IgHEL cells was considered maximal T cell activation and was >50% in both experiments. Data are presented from single individuals in two separate experiments. Similar results were obtained in a third experiment, except that the transfer of HEL-loaded thymocytes induced 30% more activation than HEL-loaded IgHEL cells. Each point represents an individual mouse.
We established the sensitivity of the T cell activation assay by transferring diminishing numbers of HEL-loaded donor B cells. Transfer of 3 × 106 donor cells induced high T cell activation and transfer of 3 × 105 cells induced substantial T cell activation (Fig. 5 A). In contrast, transfer of 3 × 104 donor cells did not activate T cells. We showed previously by Western analysis that 3 × 106 HEL-transgenic B cells bind ∼2 ng of HEL (2). By extrapolation, we estimate that >20 pg of antigen transferred by donor B cells is required to induce T cell activation in this system.
Figure 5.


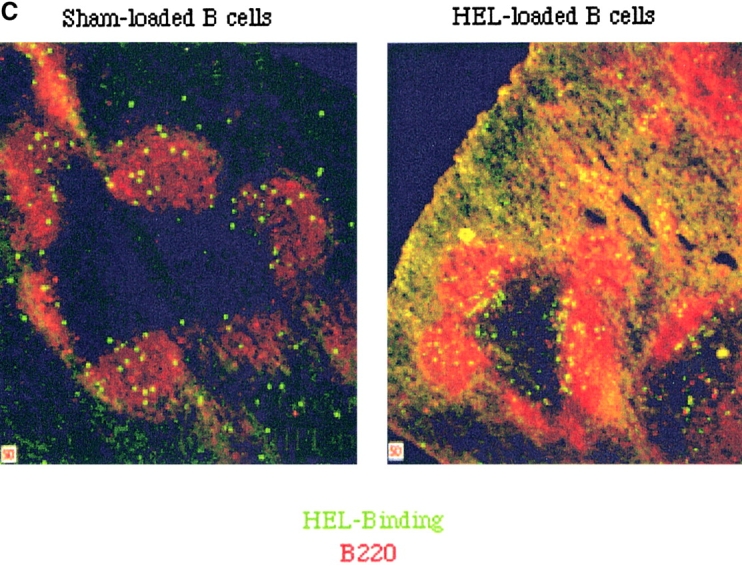
The percentage of HEL-specific T cells activation increases with the number of cells and the time after adoptive transfer. (A) Varying numbers of HEL or sham-loaded, HEL-specific transgenic B cells (H-2bb) were adoptively transferred into intact B10.BR mice (H-2kk), which had been previously seeded with CFSE-labeled, HEL-specific transgenic T cells (H-2kk). 16 h later, the recipient mice were killed and the splenocytes analyzed by flow cytometry. Squares represent sham-loaded and circles represent HEL-loaded transgenic B cells. (B) 106 HEL-specific transgenic B cells were adoptively transferred into intact B10.BR mice which had been previously seeded with 106 CFSE-labeled, HEL-specific transgenic T cells. The recipient mice were killed at different time point after the transfer and the spleens were prepared and analyzed by flow cytometry. Data were gated as in Fig. 1. (C) HEL-specific B cells pulsed with HEL ex vivo and injected intravenously are excluded to the margins of the B cell follicles in the spleen. Splenocytes from IgHEL transgenic mice were pulsed with HEL or PBS alone, washed and transferred into intact syngenic recipients by intravenous injections. Mice were killed after 12 h and the spleens flash frozen. Cryosections were stained and analyzed by fluorescent confocal microscopy.
Antigen transfer, processing, and presentation are required before T cell activation. The kinetics of T cell activation thus reflect the successful completion of these processes. To determine the time frame in which antigen transfer, processing and presentation occur in vivo, adoptive transfers were performed as described above and then mice were killed at varying time points after B cell transfer. T cell activation is detectable by 3 h after transfer and >30% of the T cells are activated by 6 h after transfer (Fig. 5 B). These results demonstrate rapid antigen transfer from B cells injected into the blood to another APC for presentation in vivo. T cell activation due to presentation of antigen transferred from intravenously injected B cells was detected in T cells in the spleen and not the lymph nodes (2). B cells are preferentially excluded from the B cell follicles in the presence of systemic antigen (17). B cells loaded with antigen ex vivo then adoptively transferred by intravenous injection are also excluded from the B cell follicles into the T cell zone (Fig. 5 C). The excluded antigen-loaded B cells are preferentially retained in the spleen (unpublished data), which, together with the rapid kinetics demonstrated here, strongly suggests that the APC which receives and presents antigen is resident in the spleen.
We set out to isolate and characterize the APC by tracking proteins derived from the transferred B cells. In vitro experiments using selectively depleted and enriched populations of APC suggested a role for CD11c+ DCs as the dominant presenting APC (unpublished data). To track the transfer of proteins from the B cells to other cells in the recipient mice, the B cells were labeled with CFSE before transfer. CFSE is a fluorescent dye that binds covalently to proteins within the cells, and so can be used to track the proteins of labeled cells (14). CFSE-labeled HEL-loaded B cells were transferred into recipient mice without HEL-specific T cells. The spleens of the recipient mice were gently dissociated, stained, and analyzed by flow cytometry.
The transferred CFSE+ B cells were easily detectable among the splenocytes (Fig. 6 A) as a small, very bright population. Another distinct population of CFSEmed cells was detected which was dimmer for CFSE than the transferred B cells, but easily distinguishable from CFSE− cells. Most of these CFSEmed cells express the DC marker, CD11c (Fig. 6 A and B). The CD11c, CFSEmed population represent, on average, 0.04% of total splenocytes and 1.4% of CD11c+ cells. The diminished fluorescence in this small population suggests that fragments of the B cells are associated with the cells. These fragments could be derived either from whole cells that have been engulfed by DCs and are being degraded, from membrane fragments, or from exosomes secreted by the B cells. The CD11c+, CFSEmed population does not express the B cell marker B220, suggesting that B cell–derived fragments are within the DCs rather than adhered to the DC cell surface (Fig. 6 B). The CD11c−, CFSEmed cells express B220, do not express the macrophage marker, F4/80, and are present in the CFSE-labeled B cell population before transfer (data not shown) and are likely to be poorly labeled transferred B cells.
Figure 6.


A small subpopulation of CD11c1 cells acquires proteins from fluorescent-labeled B cells. Purified HEL-specific transgenic B cells on the C57Bl/6 (H-2bb) background were labeled with CFSE, loaded with HEL, and adoptively transferred into unmanipulated B10.BR (H-2kk) recipients. 12 h later the mice were killed and the spleens were gently prepared, stained, and analyzed by flow cytometry. The very bright CFSE+, CD11c− population represents the transferred B cells (0.08%). Data were gated using a wide FSC, SSC gate which included lymphocytes, large cells, and granular cells. Dead cells were eliminated from the analysis by negative gating using 7-AAD. Data are representative of 20 independent experiments. (B) CFSE-labeled B cells are not adhered to the surface of the DCs. The CFSEhi transferred B cells and the CD11c+, CFSEmed populations identified in Fig. 2 were analyzed for the expression of the B cell marker, B220, by flow cytometry. Data were gated as in Fig. 2. The data are representative of three independent experiments.
Subpopulations of cells were sorted by flow cytometry and tested for the ability to activate T cells in vitro. The in vitro presenting activity of the total splenocytes from these preparations varied between experiments, from undetectable to 15% of maximum T cell activation (Fig. 7) , suggesting that the presenting cell is rare and difficult to extract. The majority of DCs, the CD11c+, CSFE− population, were very inefficient in activating T cells (Fig. 4). Similarly, the CD11c−CFSEmed population was ineffective in activating T cells. In contrast, the small subpopulation of DCs that contain proteins from the transferred B cells, which are CD11c+CFSEmed, were very efficient in activating T cells. These data demonstrate that a CD11c+ cell receives antigen from B cells in vivo, and that the cells effectively present that antigen with MHC class II to T cells.
Figure 7.

Splenic DCs which receive transferred proteins in vivo (CD11c+, CFSEmed) directly activate HEL-specific T cells. Purified HEL-specific transgenic B cells on the C57Bl/6 (H-2bb) background were labeled with CFSE, loaded with HEL ex vivo and adoptively transferred into unmanipulated B10.BR (H-2kk) recipients. Populations of DCs were tested for the ability to directly activate T cells in vitro. The CD11c+ CFSE−, CD11c+ CFSEmed, and CD11c− CFSEmed populations identified in the Figure were sorted by flow cytometry and cultured overnight in vitro with HEL-specific T cells. The purity of the sorted CD11c+, CFSEmed population was 89%. Data were gated for live lymphocytes. In the presence of exogenous HEL, 70.8% of HEL-specific T cells expressed CD69, in the absence of antigen, 5.6% of HEL-specific T cells expressed CD69. All points are data for three culture wells, except for one point, without error bars in the total APC which represents the mean of two wells. Error bars represent the SD. The functional data are representative of six independent experiments.
The CD11c+CFSEmed presenting population was characterized further by analyzing the expression of markers for different known DC subsets (Fig. 8 A). The CD11c+CFSEmed population expresses moderate CD11b, no CD4 and high levels of CD8α chain (Fig. 8 B). The CD11c+ CFSEmed CD8α+ population represents 5% (± 1.9%) of the CD11c+CD8α+ population, 1.4% (± 0.72%) of the total CD11c+ population, and 0.04% (± 0.002%) of the total splenocyte population (excluding red cells and dead cells). The CD11c+ CFSEmed cells express MHC class II and CD86 (Fig. 8 B) at levels which are high, but which overlap with the levels expressed by the total CD11c+ population.
Figure 8.
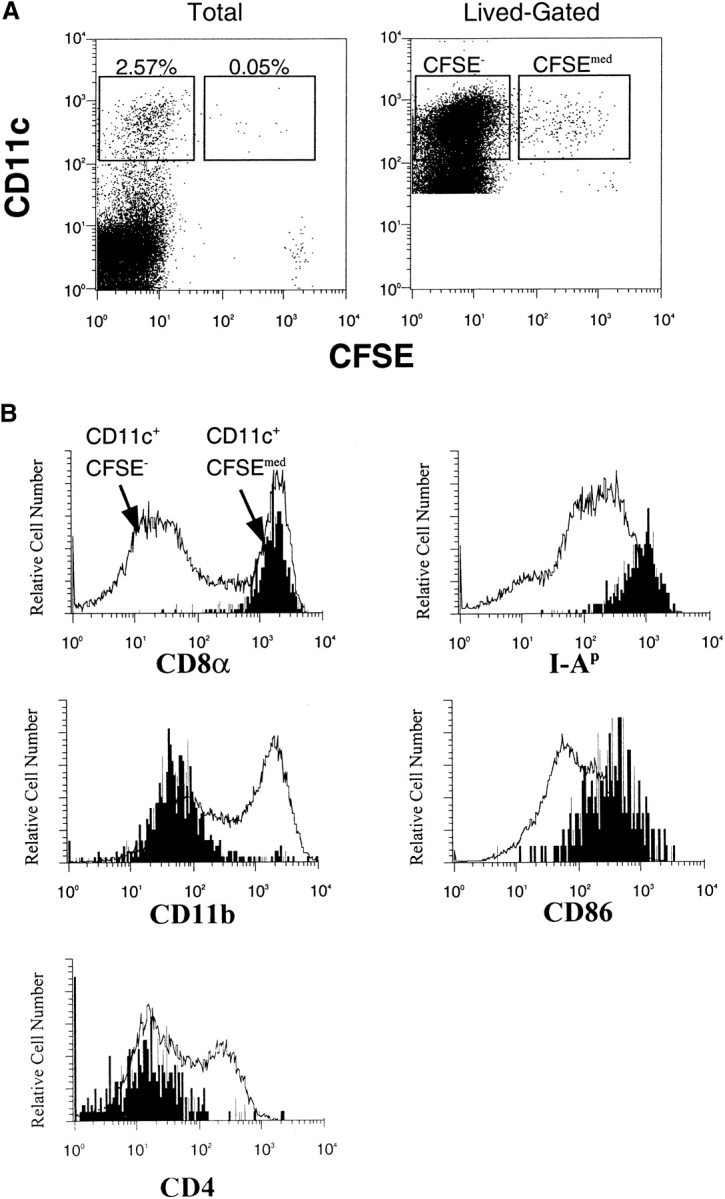

Splenic DCs containing transferred proteins express high levels of CD8α, I-A, and CD86. Purified HEL-specific transgenic B cells on the C57Bl/6 (H-2bb) background were labeled with CFSE, loaded with HEL ex vivo, and adoptively transferred into unmanipulated B10.BR (H-2kk) recipients. (A) Data were first collected for total live cells, and then subsequent data were collected using lived-gating on CD11c+ to collect sufficient events for analysis. (B) Lived-gated files were gated in the CD11c+, CFSE− and CD11c+, CFSEmed populations identified in the Figure and were analyzed for the expression of surface markers by flow cytometry. Data were gated as in Fig. 6. The data are representative of three independent experiments. (C) Stable surface phenotype of CD11c+CFSEmed early after donor cells injections. B6 splenocytes (H-2bb) were labeled with the dye CFSE and adoptively transferred into B10.BR recipients (H-2kk) by intravenous injection. The recipient mice were killed 2–6 h after the injections. Spleens were prepared and analyzed by flow cytometry. The CD11c+ CFSE− and CD11c+ CFSEmed populations were identified, then analyzed for the expression of surface markers. Data were gated as in the Figure.
To determine whether uptake of transferred material had induced increased MHC class II and CD86 expression in the CD11c+ CFSEmed population, this population was analyzed 2 and 6 h after donor cell transfer. As shown in Fig. 8 C, levels of these activation markers do not differ between the early and later time points. These data suggest that the levels of MHC class II and CD86 are constitutively high on the DCs which receive transferred antigen.
CFSEmedCD8α+CD11blo DCs are present after syngenic transfers of H-2kk B cells into H-2kk recipients, though at frequencies approximately fourfold lower than after allogeneic transfer (Fig. 9) . The observation that target DCs are more numerous after an allogeneic transfer than after a syngenic transfer suggests that the allogeneic response promotes transfer, probably via cell death. The allogeneic transfers are useful because direct T cell–DC interactions are genetically isolated interactions from T cell–B cell interactions. The syngenic transfers are useful because they are more physiological, though direct T cell–DC interactions cannot be distinguished from interactions from T cell–B cell interactions. The transfer of antigen may occur by different mechanisms in each case (discussed further below), and it is interesting that the CFSEmedCD8α+CD11blo DCs receive antigen in both cases.
Figure 9.

The number of DCs containing transferred proteins increases linearly with the number of transferred donor cells. Varying numbers of CFSE-labeled B6 (H-2bb) or B10.BR (H-2kk) splenocytes were adoptively transferred into B10.BR recipients by intravenous injection. 18 h after the transfer, the mice were killed and the spleens were prepared and analyzed by flow cytometry for the presence of CD11c+CFSEmed cells. Data were gated using a wide FCS, SSC gate that includes lymphocytes and larger granular cells. Dead cells were eliminated by negative gating using 7-AAD. Each point represents a single mouse. The data is representative of five independent experiments.
We addressed the possibility that cell death contributes to antigen transfer by testing whether transgenic overexpression of the antiapoptotic molecule bcl-2 by the donor B cells changes the acquisition of transferred protein by DCs (18, 19). Overexpression of bcl-2 by the donor B cells decreases passive B cell death in vitro (Fig. 10 A and B) but does not change the number of target DCs which receive transferred protein in allogeneic transfers. Similar results were obtaining using transgenic mice in which the B cells overexpress bclXL (data not shown). These data suggest that the differences we observe between the syngenic and allogeneic transfers are not due to death that can be rescued by bcl-2, and that antigen transfer in both cases is unlikely to depend on passive B cell death. In particular, these data suggest that our ex vivo manipulations of the donor cells (pulsing B cells with antigen) do not damage the cells and induce passive cell death before or after transfer.
Figure 10.


Overexpression of Bcl-2 by B cells decreases passive death in vitro but does not affect DC acquisition of transferred proteins. (A) Purified B cells from C57BL/6 stock mice and from C57BL/6-TgN(22Wehi) transgenic mice, which overexpress Bcl-2 were labeled with CFSE and cultured in complete media for 16–18 h. The cells were then washed and stained with 7-AAD and B220 and analyzed by flow cytometry. Data were gated for B220+ cells and 7-AAD was used as a measure of cell death. Data represent the mean of triplicate cultures and error bars represent standard deviation. The data are representative of two independent experiments. (B) Purified B cells were labeled with CFSE and injected intravenously into B10BR (H-2kk) recipients. 16 h later, mice were killed and the splenocytes were prepared and analyzed by flow cytometry. Data were gated to exclude dead cells and red blood cells. CD11c+ and CFSE+ were gated and counted. Each point represent a single mouse. The data represent three independent experiments.
Discussion
CD8α+CD11blo DCs loaded in vivo with cell-associated antigen were directly identified and characterized for phenotype and function. Antigen-specific B cells efficiently transfer antigen to another cell for presentation to T cells (2). Nontransgenic B cells also transfer antigen, as do nonB cells surface-loaded with antigen, which suggests that such transfer is a general mechanism of antigen trafficking. Here we show that this presenting cell is a CD8α-expressing DC, which also expresses low CD11b and low CD4. These DCs express higher levels of MHC class II and CD86 than the total population of DCs, and these high levels are constitutive rather than induced. DCs which receive transferred proteins effectively present antigen to T cells ex vivo. Strikingly, bcl-2–mediated inhibition of apoptosis had no effect on the transfer process. Together, these results suggest that the CD8α CD11blo DCs constitutively sample the cells and membranes in their environment and present these cell-associated antigens to T cells.
Proteins derived from transferred HEL-loaded B cells were visualized within a small subpopulation of DCs, and this subpopulation efficiently presented B cell–derived HEL to T cells ex vivo. The B cell proteins are likely to be located within the DCs, as B220 was not detectable on the surface of the DCs and the antigen accessed the MHC class II pathway for presentation to T cells. The antigen is likely transferred together with many other proteins derived from the B cell, as presenting activity (demonstrating the presence of the antigen) corresponded with CFSE-labeling (demonstrating the presence of B cell proteins). In other recipient mice, proteins from B cells that were not loaded with antigen (sham) were also detected within DCs. This result demonstrates that B cell activation is not required for the transfer of protein and suggests that the uptake of proteins from other cells is a constitutive activity of these DCs.
Multiple subsets of DC have been identified in recent years (20), including three subsets in the mouse spleen: CD4+CD8α−CD11bhi, CD4−CD8α+CD11blo, and CD4− CD8α−CD11bhi. The CD4−CD8α+CD11blo DC is located in the T cell zones of the spleen and LNs and is thought to be important in tolerance induction (21). It has been known for some time that DCs can be either immunogenic or tolerogenic (22–24), and immature DCs have recently been shown to be tolerogenic in vivo in humans (25). The relationship between the tolerogenic activity of in vitro–generated immature DCs, and the activities of different DCs populations in vivo is not yet clear.
Different functions have been ascribed to the CD8α+ and CD8α2 subsets, including the induction of IFN-γ–producing Th1 cells via IL-12 by the former and the induction of Th2 cells by the latter (26, 27). Liu et al. (29) have proposed a corresponding nomenclature: DC1 and DC2, with the caveat that there are substantial differences between the mouse and the human DC systems (28, 29). Our understanding of the function of these different subsets is complicated by the plasticity of the phenotypes (29). For example, both CD8α and DEC-205 expression can be up-regulated upon activation in vitro by culture of DCs in the presence of inflammatory mediators (30). It has been proposed that CD8α+CD11blo DCs are tolerogenic to T cells, whereas CD4+CD8α−CD11bhi DCs are immunogenic (references 24 and 31; discussed further below), but much of the work cited was performed in vitro.
Cross-presentation involves the acquisition of antigen from genetically disparate cells for presentation to T cells (24, 32, 33). MHC class I–mediated cross-presentation has been described in a variety of murine models and has been shown to be tolerogenic for CD8+ T cells (32, 34). A recent report identified the presenting cell in an MHC class I cross-presentation system as a CD8α+ DC (35). In this work, DCs from mice primed with antigen-loaded splenocytes were sorted by flow cytometry and this bulk population CD8α+CD11c+ DCs of cells was shown to present antigen to MHC class I–restricted T cells. We show here that the same, or very similar, subset of DCs is responsible for MHC class II presentation of antigen received from B cells. We directly identified and characterized the individual CD8α+CD11c+ cells that received transferred antigen and presented that antigen with MHC class II to T cells, and showed that they have an activated phenotype.
MHC class I and II pathways are generally distinct. MHC class I presents primarily endogenous antigens and MHC class II presents primarily exogenous antigens (36). In CD8α+ DCs, however, the MHC class I pathway has access to exogenous antigens. The observation that CD8α+ DCs mediate both MHC class I cross-presentation and MHC class II presentation of antigen transferred from B cells suggests that these cells have a specialized function in vivo. CD8α+ DCs may be specialized for sampling the cells and membranes in their environment for presentation to T cells circulating through the T cell zone (37). It was recently shown that CD8α+ DCs do not present soluble antigen to MHC class II–restricted T cells (38), which supports the notion that this subset of DCs is specialized to present cell-associated antigens. Such antigen presentation in the absence of additional inflammatory stimuli may be tolerogenic to the T cells by inducing abortive proliferation (2, 24). Indeed, this subset of DCs has recently been shown to mediate tolerance of T cells in vivo when antigen was delivered to DCs via the DEC-205 receptor (39). Furthermore, delivery of a signal to CD40 switched the activity from tolerance to priming.
The CFSEmedCD11c+ express CD8α and low levels of CD11b (CFSEmedCD8α+CD11blo), which suggests that these cells are interdigitating cells resident in the T cell zone of the spleen (20, 21). The CFSEmedCD11c+ DCs also express elevated levels of CD86 and MHC class II, phenotypes which are associated with activated or mature DCs. The levels of activation markers in this subpopulation, however, overlap significantly with those of the larger DC population, so the extent of induced activation is unclear. A subset of cells may be constitutively activated, or recent uptake of cellular material may activate the cells. Intriguingly, this activated or mature phenotype of the CFSEmedCD8α+CD11blo cells is consistent with the phenotype observed in pulmonary DCs in mice treated with antigen delivered nasally (40). DCs isolated from treated mice express increased levels of MHC II and CD86, but were tolerogenic upon transfer, and induced Tr1 cells by an IL-10–dependent mechanism.
DCs have been shown to phagocytose both apoptotic and necrotic cells and to present the antigens from those cells in the context of MHC class I and II (7, 8, 41). The phagocytosis of dead cells can induce activation of in vitro–generated DCs, and this activation is associated with necrotic rather than apoptotic cells. The activated phenotype of CD8α+ DCs that contain B cell–derived proteins supports the possibility that phagocytosis of dead B cells is the mechanism of antigen transfer in the experiments described here. However, the vast majority of B cells transferred were alive at the time of transfer, and enriching for live cells did not improve presentation of antigen in vivo (data not shown). The possibility remains that the transferred B cells died in situ, and were then phagocytosed by CD8α+ DCs. Inaba et al. demonstrated that fragments of dead, antigen-loaded B cells can be phagocytosed by in vitro–generated DCs and that antigen from the B cells can be efficiently transferred in vitro to DCs for presentation on MHC class II molecules (8). They also showed that upon adoptive transfer these loaded DCs migrate to the LNs in vivo, and transfer the antigen to host DCs. Here we have demonstrated that antigen transfer from B cells to DCs occurs efficiently in vivo, with the functional consequence of T cell activation.
Both self-tolerant and acutely activated B cells are excluded from B cell follicles into the T cell zones of lymphoid tissues (17, 42). In our system, antigen-pulsed B cells are also excluded from the follicle into the margin with the T cell zone. Accordingly, B cells which have bound antigen preferentially colocalize with CD8α+ DCs, which are also located in the T cell zone. In particular, excluded tolerant B cells, which have a short half-life of 1–2 d (17), may be in the proper location to transfer antigen to CD8α+ DCs upon their death. The self-antigens concentrated by the B cell could then be presented by the DCs via MHC class I and class II to T cells. Although the transfer of protein from B cells to CD8α+ DCs is not dependent on the activation of B cells in our transfer model, it is possible that activation, and thus the location of B cell death, may be important at lower frequencies in vivo.
We are currently investigating the mechanism of antigen transfer. In addition to the possibility that dead B cells are phagocytosed by DCs, it is also possible that exosomes secreted by live B cells are internalized and processed by DCs, or that membrane is extracted from B cells by DCs. B cells have been shown to secrete in vitro small membranous vesicles, or exosomes, which resemble MIIC compartments (4). Indeed, reticulocytes, platelets, B lymphoblastoid cells, and DCs all secrete exosomes in vitro, which raises the possibility that vesicle-mediated transfer of molecules between cells may be a common form of information transfer in vivo (4, 43, 44). Contact-dependent direct membrane transfer between DCs was recently demonstrated in vitro and represents a possible mechanism for the transfer of antigen from B cells to DCs (37).
Our working model is that two concurrent mechanisms of antigen transfer are operating in our system. The first mechanism of antigen transfer is the DCs phagocytosis of allogeneic donor cells killed by NK cells. It is likely that the fully allogeneic system, the transfer of H-2bb donor cells into H-2kk recipients, has allowed us to amplify what would otherwise be a barely detectable process for handling antigens derived from dead cells in the blood.
The second mechanism of antigen transfer is the transfer of antigen from live B cells. Transfer of antigen from syngenic donor cells occurs and is not changed by the overexpression of bcl-2 (data not shown). In addition, we and others have shown that live B cells produce particles that transfer antigen (unpublished data). Accordingly, the transfer of antigen from live donor cells to DCs is possible. Both of these mechanisms, the phagocytosis of dead cells and transfer of antigen from live cells, could contribute to normal antigen trafficking in vivo.
The MHC class II–mediated presentation by CD8α+ cells that we describe here is unusual in that the antigens are derived from antigen-specific B cells, which are themselves effective APC. The antigens concentrated by B cells can be presented to T cells either directly by B–T interaction, or indirectly by presentation by CD8α+ cells. Antigen-specific B cells may concentrate circulating antigen too dilute to be presented by other APC, then transfer this antigen to CD8α+ DCs resident in the T cell zone of the spleen for presentation to T cells. The important role for B cells early in an immune response or in tolerance induction may be to concentrate a unique subset of antigens. In this way, the B cells would confer the unique properties of their antigen-specific receptor onto another cell type. The recipient cell, the CD8α+ DCs, may be specialized to receive this concentrated, cell-associated antigen for presentation to T cells.
Acknowledgments
The authors would like to thank the UBC Multiuser Flow Cytometry Facility (G. Osborne, R.Wee, and A. Johnson) and the BRC Animal Unit (L. Wong and S. Kleczkowski) for excellent technical advice and assistance, Dr. J. Rathmell for the gift of bcl-XL mice, and Drs. B. Weintraub, K. McNagny, and D. Carlow for comments on the paper. The author also acknowledges the support of Dr. C. Goodnow and the Stanford Cell Imaging Facility.
This work was supported by MRC of Canada grant MT15480. S. Townsend is a scholar of the Canadian Arthritis Network.
Footnotes
Abbreviation used in this paper: DC, dendritic cell.
References
- 1.Bennett, S.R., F.R. Carbone, F. Karamalis, R.A. Flavell, J.F. Miller, and W.R. Heath. 1998. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 393:478–480. [DOI] [PubMed] [Google Scholar]
- 2.Townsend, S.E., and C.C. Goodnow. 1998. Abortive proliferation of rare T cells induced by direct or indirect antigen presentation by rare B cells in vivo. J. Exp. Med. 187:1611–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batista, F.D., D. Iber, and M.S. Neuberger. 2001. B cells acquire antigen from target cells after synapse formation. Nature. 411:489–494. [DOI] [PubMed] [Google Scholar]
- 4.Raposo, G., H.W. Nijman, W. Stoorvogel, R. Liejendekker, C.V. Harding, C.J. Melief, and H.J. Geuze. 1996. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 183:1161–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thery, C., A. Regnault, J. Garin, J. Wolfers, L. Zitvogel, P. Ricciardi-Castagnoli, G. Raposo, and S. Amigorena. 1999. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J. Cell Biol. 147:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zitvogel, L., A. Regnault, A. Lozier, J. Wolfers, C. Flament, D. Tenza, P. Ricciardi-Castagnoli, G. Raposo, and S. Amigorena. 1998. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat. Med. 4:594–600. [DOI] [PubMed] [Google Scholar]
- 7.Albert, M.L., B. Sauter, and N. Bhardwaj. 1998. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 392:86–88. [DOI] [PubMed] [Google Scholar]
- 8.Inaba, K., S. Turley, F. Yamaide, T. Iyoda, K. Mahnke, M. Inaba, M. Pack, M. Subklewe, B. Sauter, D. Sheff, et al. 1998. Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J. Exp. Med. 188:2163–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang, J.F., Y. Yang, H. Sepulveda, W. Shi, I. Hwang, P.A. Peterson, M.R. Jackson, J. Sprent, and Z. Cai. 1999. TCR-mediated internalization of peptide-MHC complexes acquired by T cells. Science. 286:952–954. [DOI] [PubMed] [Google Scholar]
- 10.Rock, K.L., B. Benacerraf, and A.K. Abbas. 1984. Antigen presentation by hapten-specific B lymphocytes: role of surface immunoglobulin receptors. J. Exp. Med. 160:1102–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aluvihare, V.R., A.A. Khamlichi, G.T. Williams, L. Adorini, and M.S. Neuberger. 1997. Acceleration of intracellular targeting of antigen by the B-cell antigen receptor: importance depends on the nature of the antigen-antibody interaction. EMBO J. 1:3553–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siemasko, K., B.J. Eisfelder, C. Stebbins, S. Kabak, A.J. Sant, W. Song, and M.R. Clark. 1999. Igα a and Igβ are required for efficient trafficking to late endosomes and to enhance antigen presentation. J. Immunol. 162:6518–6525. [PubMed] [Google Scholar]
- 13.Cooke, M.P., A.W. Heath, K.M. Shokat, Y. Zeng, F.D. Finkelman, P.S. Linsley, M. Howard, and C.C. Goodnow. 1994. Immunoglobulin signal transduction guides the specificity of B cell-T cell interactions and is blocked in tolerant self-reactive B cells. J. Exp. Med. 179:425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13a.Goodnow, C.C., J. Crosbie, S. Adelstein, T.B. Lavoie, S.J. Smith-Gill, R.A. Brink, H. Pritchard-Briscoe, J.S. Wotherspoon, R.H. Loblay, K. Raphael, et al. 1988. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 334:676–682. [DOI] [PubMed] [Google Scholar]
- 13b.Ho, W.Y., M.P. Cooke, C.C. Goodnow, and M.M. Davis. 1994. Resting and anergic B cells are defective in CD28-dependent costimulation of naive CD4+ T cells. J. Exp. Med. 179:1539–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyons, A.B., and C.R. Parish. 1994. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods. 171:131–137. [DOI] [PubMed] [Google Scholar]
- 15.Peterson, D.A., R.J. DiPaolo, O. Kanagawa, and E.R. Unanue. 1999. Quantitative analysis of the T cell repertoire that escapes negative selection. Immunity. 11:453–462. [DOI] [PubMed] [Google Scholar]
- 16.Tourne, S., V. Kouskoff, W. Ho, M. Davis, C. Benoist, and D. Mathis. 1999. Inhibition of thymocyte positive selection by natural MHC: peptide ligands. Eur. J. Immunol. 29:394–402. [DOI] [PubMed] [Google Scholar]
- 17.Cyster, J.G., S.B. Hartley, and C.C. Goodnow. 1994. Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire. Nature. 371:389–395. [DOI] [PubMed] [Google Scholar]
- 18.Strasser, A., S. Whittingham, D.L. Vaux, M.L. Bath, J.M. Adams, S. Cory, and A.W. Harris. 1991. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc. Natl. Acad. Sci. USA. 88:8661–8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Reilly, L.A., A.W. Harris, D.M. Tarlinton, L.M. Corcoran, and A. Strasser. 1997. Expression of a bcl-2 transgene reduces proliferation and slows turnover of developing B lymphocytes in vivo. J. Immunol. 159:2301–2311. [PubMed] [Google Scholar]
- 20.Kamath, A.T., J. Pooley, M.A. O'Keeffe, D. Vremec, Y. Zhan, A.M. Lew, A. D'Amico, L. Wu, D.F. Tough, and K. Shortman. 2000. The development, maturation, and turnover rate of mouse spleen dendritic cell populations. J. Immunol. 165:6762–6770. [DOI] [PubMed] [Google Scholar]
- 21.Steinman, R.M., M. Pack, and K. Inaba. 1997. Dendritic cells in the T-cell areas of lymphoid organs. Immunol. Rev. 156:25–37. [DOI] [PubMed] [Google Scholar]
- 22.Finkelman, F.D., A. Lees, R. Birnbaum, W.C. Gause, and S.C. Morris. 1996. Dendritic cells can present antigen in vivo in a tolerogenic or immunogenic fashion. JI. 157:1406–1414. [PubMed] [Google Scholar]
- 23.Banchereau, J., and R.M. Steinman. 1998. Dendritic cells and the control of immunity. Nature. 392:245–252. [DOI] [PubMed] [Google Scholar]
- 24.Steinman, R.M., S. Turley, I. Mellman, and K. Inaba. 2000. The induction of tolerance by dendritic cells that have captured apoptotic cells. J. Exp. Med. 191:411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dhodapkar, M.V., R.M. Steinman, J. Krasovsky, C. Munz, and N. Bhardwaj. 2001. Antigen-specific Inhibition of effector T cell function in humans after injection of immature dendritic cells. J. Exp. Med. 193:233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hochrein, H., K. Shortman, D. Vremec, B. Scott, P. Hertzog, and M. O'Keeffe. 2001. Differential production of IL-12, IFN-α, and IFN-γ by mouse dendritic cell subsets. J. Immunol. 166:5448–5455. [DOI] [PubMed] [Google Scholar]
- 27.Maldonado-Lopez, R., T. De Smedt, P. Michel, J. Godfroid, B. Pajak, C. Heirman, K. Thielemans, O. Leo, J. Urbain, and M. Moser. 1999. CD8α1 and CD8α-subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J. Exp. Med. 189:587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rissoan, M.C., V. Soumelis, N. Kadowaki, G. Grouard, F. Briere, R. de Waal Malefyt, and Y.J. Liu. 1999. Reciprocal control of T helper cell and dendritic cell differentiation. Science. 283:1183–1186. [DOI] [PubMed] [Google Scholar]
- 29.Liu, Y.J., H. Kanzler, V. Soumelis, and M. Gilliet. 2001. Dendritic cell lineage, plasticity and cross-regulation. Nat. Immunol. 2:585–589. [DOI] [PubMed] [Google Scholar]
- 30.Brasel, K., T. De Smedt, J.L. Smith, and C.R. Maliszewski. 2000. Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood. 96:3029–3039. [PubMed] [Google Scholar]
- 31.Suss, G., and K. Shortman. 1996. A subclass of dendritic cells kill CD4 T cells via Fas/Fas-ligand-induced apoptosis. J. Exp. Med. 183:1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bevan, M.J. 1976. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 143:1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carbone, F.R., C. Kurts, S.R. Bennett, J.F. Miller, and W.R. Heath. 1998. Cross-presentation: a general mechanism for CTL immunity and tolerance. Immunol. Today. 19:368–373. [DOI] [PubMed] [Google Scholar]
- 34.Miller, J.F., C. Kurts, J. Allison, H. Kosaka, F. Carbone, and W.R. Heath. 1998. Induction of peripheral CD8+ T-cell tolerance by cross-presentation of self antigens. Immunol. Rev. 165:267–277. [DOI] [PubMed] [Google Scholar]
- 35.den Haan, J.M., S.M. Lehar, and M.J. Bevan. 2000. CD8+ but not CD8− dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192:1685–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Germain, R.N., and D.H. Margulies. 1993. The biochemistry and cell biology of antigen processing and presentation. Annu. Rev. Immunol. 11:403–450. [DOI] [PubMed] [Google Scholar]
- 37.Harshyne, L.A., S.C. Watkins, A. Gambotto, and S.M. Barratt-Boyes. 2001. Dendritic cells acquire antigens from live cells for cross-presentation to CTL. J. Immunol. 166:3717–3723. [DOI] [PubMed] [Google Scholar]
- 38.Pooley, J.L., W.R. Heath, and K. Shortman. 2001. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8− dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J. Immunol. 166:5327–5330. [DOI] [PubMed] [Google Scholar]
- 39.Hawiger, D., K. Inaba, Y. Dorsett, M. Guo, K. Mahnke, M. Rivera, J.V. Ravetch, R.M. Steinman, and M.C. Nussenzweig. 2001. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akbari, O., R.H. DeKruyff, and D.T. Umetsu. 2001. Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen. Nat. Immunol. 2:725–731. [DOI] [PubMed] [Google Scholar]
- 41.Gallucci, S., M. Lolkema, and P. Matzinger. 1999. Natural adjuvants: endogenous activators of dendritic cells. Nat. Med. 5:1249–1255. [DOI] [PubMed] [Google Scholar]
- 42.Cyster, J.G., and C.C. Goodnow. 1995. Antigen-induced exclusion from follicles and anergy are separate and complementary processes that influence peripheral B cell fate. Immunity. 3:691–701. [DOI] [PubMed] [Google Scholar]
- 43.Rabesandratana, H., J.P. Toutant, H. Reggio, and M. Vidal. 1998. Decay-accelerating factor (CD55) and membrane inhibitor of reactive lysis (CD59) are released within exosomes during In vitro maturation of reticulocytes. Blood. 91:2573–2580. [PubMed] [Google Scholar]
- 44.Heijnen, H.F., A.E. Schiel, R. Fijnheer, H.J. Geuze, and J.J. Sixma. 1999. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and α-granules. Blood. 94:3791–3799. [PubMed] [Google Scholar]