

Abstract
Capacitative Ca2+ entry (CCE) activated by release/depletion of Ca2+ from internal stores represents a major Ca2+ influx mechanism in lymphocytes and other nonexcitable cells. Despite the importance of CCE in antigen-mediated lymphocyte activation, molecular components constituting this mechanism remain elusive. Here we demonstrate that genetic disruption of transient receptor potential (TRP)1 significantly attenuates both Ca2+ release-activated Ca2+ currents and inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release from endoplasmic reticulum (ER) in DT40 B cells. As a consequence, B cell antigen receptor–mediated Ca2+ oscillations and NF-AT activation are reduced in TRP1-deficient cells. Thus, our results suggest that CCE channels, whose formation involves TRP1 as an important component, modulate IP3 receptor function, thereby enhancing functional coupling between the ER and plasma membrane in transduction of intracellular Ca2+ signaling in B lymphocytes.
Keywords: B cell receptor, capacitative Ca2+ entry, store-operated Ca2+ channel, Ca2+ release, TRP
Introduction
In many cells, receptor activation evokes an increase in cytosolic Ca2+ concentration [Ca2+]i which is critical for initiating cellular responses (1–5). This increase is comprised of an initial phase that reflects Ca2+ release from intracellular stores and a second phase that is due to Ca2+ influx through Ca2+ permeable ion channels in the plasma membrane. The magnitude of the change in [Ca2+]i relies on the activation of Ca2+ entry through plasma-membrane channels induced by depletion of Ca2+ from internal stores such as endoplasmic reticulum (ER)* via inositol 1,4,5-trisphosphate (IP3) as well as other regulatory mechanisms, including modulation of Ca2+ uptake and sequestration, changes in membrane potential, and/or other as yet unknown mechanisms downstream of receptor activation.
Ca2+ entry due to store depletion is often called capacitative Ca2+ entry (CCE) and is mediated by Ca2+-permeable channels termed store-operated Ca2+ channels (SOCs). The best-studied SOCs in terms of biophysical properties are Ca2+-release-activated Ca2+ (CRAC) channels, originally described in mast cells and characterized by their inwardly rectifying current-voltage relationship as well as by their high selectivity for Ca2+ (6, 7). Other less Ca2+-selective SOCs have been also described in many tissues (8–12). Hence, the molecular identities of SOCs are still controversial and are probably not identical in the various cell types.
In lymphocytes, the importance of CCE in maintaining normal T cell function has been underscored by analysis of immunodeficiency patients, whose T cells failed to show activation of a sustained Ca2+ influx upon TCR stimulation, despite the successful release of Ca2+ from the ER stores (13–15). This defective CCE causes an insufficient T cell activation in the patient presumably through impairment of NF-AT activation, since CCE is required to maintain NF-AT activation in antigen-stimulated T cells (16, 17). Also, in B lymphocytes, the two phases of Ca2+ mobilization are known to be differentially coupled to signaling pathways (4). While the small transient Ca2+ spike mediated by release from the ER stores is sufficient to activate transcriptional regulators such as NF-κB and c-Jun NH2-terminal kinase, it is insufficient for activation of NF-AT. Instead, NF-AT activation, like in T cells, requires CCE and a sustained increase in [Ca2+]i. Collectively, these studies clearly demonstrate the importance of CCE in lymphocyte activation; however, the molecular natures of SOCs in lymphocytes are still unclear.
To address the mechanisms by which CCE is activated upon B cell receptor (BCR) engagement, we first sought to determine the molecular components of SOCs in B cells. The seven mammalian homologs of Drosophila transient receptor potential (trp) proteins, have emerged as candidate subunits of the SOCs in B cells, based on the following evidence. First, several members of TRP families, including TRP1, are indeed expressed in the spleen and B cells (18–20; see Fig. 1) . Second, once overexpressed in CHO and COS cells, TRP1 has been demonstrated to produce Ca2+-permeable cation currents that were activated after depletion of internal Ca2+ stores (21–23), while some papers proposed TRP1 as a component of receptor-mediated nonselective cation channels activated independently of depletion of stores (24, 25). These findings prompted us to examine whether TRP1 functions indeed as SOCs in the BCR signaling context. In this paper, we demonstrate by genetically disrupting TRP1 that this molecule is critical for forming functional SOCs, thereby contributing to generation of regular Ca2+ oscillations and subsequent NF-AT activation. Our results also implicate important involvement of TRP1 in coordination of elementary Ca2+ signaling events that promote functional coupling between the ER and plasma membrane in transduction of BCR-induced Ca2+ signaling.
Figure 1.
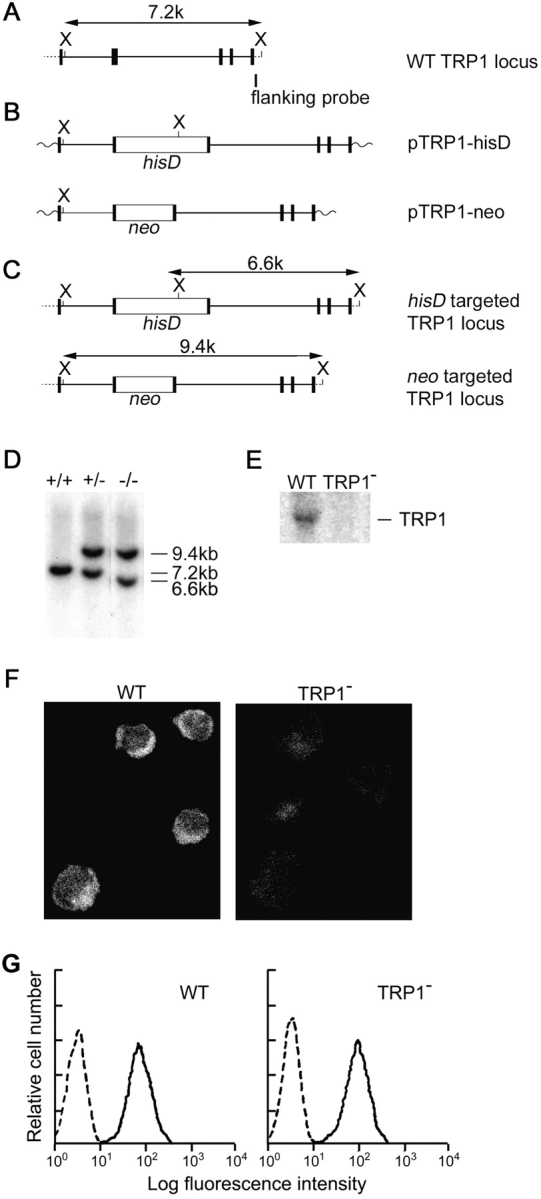
Targeted disruption of the TRP1 gene in DT40 B lymphocytes. Partial restriction map of chicken TRP1 gene (A), targeting construct (B), and expected structure of the disrupted allele (C). (D) Southern blot analysis of genomic DNAs from DT40 cells. Genomic DNAs were prepared from WT (+/+), neo-targeted (+/−), and neo/his-targeted (−/−) clones, digested with XbaI, and hybridized with a 3′-flanking probe. The restriction endonuclease cleavage site of XbaI is abbreviated as X. (E) Northern blot analysis of WT and TRP1-deficient DT40 cells (clone TRP1−-14). (F) Immunolocalization of TRP1 in WT cells, and loss of its expression in TRP1−-14 cells. The fluorescence images were acquired with a confocal laser microscope. (G) BCR expression on TRP1-deficient DT40 cells. DT40 cells were stained with FITC-conjugated anti–chicken IgM Ab.
Materials and Methods
Generation of TRP1-deficient DT40 Cells.
The chicken genomic TRP1 DNA was obtained by PCR using a pair of primers chT1–31 (sense, 5′-AGACTGTGGTATGAAGGGTTGGAAGACTTC-3′) and chT1–1 (antisense, 5′-TCGGGCGAATTTCCATTCCTTATCTTC-3′). The targeting vector of TRP1 was constructed by replacing the genomic sequence, which encodes the fifth hydrophobic segment H5 (PTLVAEGLFAFANVLSYLRLFFMY) of chicken TRP1, with a histidinol (hisD) or neomycin resistance gene (neo) cassette (26). The upstream 2.1-kb and downstream 5.2-kb genomic sequences were generated by PCR using the TRP1 genomic clone as a template. DT40 cells were cultured in RPMI 1640 supplemented with 10% FCS, 1% chicken serum, penicillin, streptomycin, and glutamine. The targeting vector was linearized and transfected sequentially into DT40 cells by electroporation (550 V, 25 μF). After isolation of several clones in the presence of 1 mg/ml histidinol or 2 mg/ml G418, genomic DNAs were prepared and analyzed by Southern blot analysis using the 3′-flanking probe. In a PCR experiment to confirm that the TRP1-deficient preparation is free from contamination of wild-type (WT) cells, a pair of primers K2723 (sense, 5′-TAGAAGAGTCACGCAATCAGCTTAGTT-3′) and K2725 (antisense, 5′-CGCAGATAACTCAGCACATTAGCAAATGC-3′) were used: K2723 and K2725 anneal with the respective sequences in the neighboring exons, of which the K2725 primer-annealing sequence in the 3′ exon is eliminated by disruption with the hisD or neo cassette in TRP1-deficient cells. Cell surface expression of BCR was analyzed by FACScan™ (Becton Dickinson) using FITC-labeled anti–chicken IgM. The two targeted mutant clones, TRP1−-14 and TRP1−-16, were analyzed extensively, although reproducibility of the results was confirmed using additional clones.
Northern Blot Analysis.
Isolation of total RNA and hybridization was performed as described previously (20). Probe used was the chicken TRP1 cDNA isolated by RT-PCR amplification from the DT40 poly(A)+ RNA using a pair of primers chT1–25 (sense, 5′-TGGATTATTGGGATGATTTGGTCAGAC-3′) and chT1–1 (antisense) mentioned above.
Immunolocalization of TRP1 Proteins.
DT40 cells on coverslips were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and incubated with anti-TRP1 polyclonal antibody (Alamone Labs, Ltd.) at 1:600 dilution, according to manufacturer's instruction. Secondary antibody was the FITC-conjugated donkey anti–rabbit IgG antisera. The fluorescence images were acquired with a confocal laser scanning microscope (LSM510; Carl Zeiss Co., Ltd.) using 505-nm long path filter for emission and an argon ion laser for excitation (488 nm).
Western Blot Analysis.
∼5 × 106 cells were lysed by 100 μl phosphate-buffered saline containing 1% NP-40. After centrifugation at 15,000 rpm for 10 min, 5 μl of supernatant was subjected to 5% SDS-PAGE and electrotransferred onto a nitrocellulose membrane. After blocking in 5% BSA, the membrane was incubated with anti-IP3 receptor (IP3R) polyclonal antibodies (27) and subsequently with horseradish peroxidase–conjugated anti–rabbit IgG antibody. The bands (>208 kD) were visualized by enhanced chemiluminescence method (Amersham Pharmacia Biotech).
cDNA Expression.
Human TRP1 (hTRP1) cDNA was cloned according to Zhu et al. (18) by RT-PCR from the human brain poly(A)+ RNA using pair of primers hTRP1–1 (5′-CATGGGCCGCGATGATGGCGGCCCTGTACC-3′) and hTRP1–2 (5′-GAAAATGGTTAATTTCTTGGATAAAACATAGC-3′). To introduce the pMXΔ-IRES-GFP plasmid (28) containing the hTRP1 cDNA to TRP1-deficient cells, vesicular somatitis virus glycoprotein pseudotyped retrovirus was used.
Measurement of Changes in [Ca2+]i.
Cells were plated onto poly-l-lysine-coated glass coverslips, and were subjected to measurements 3–6 h after plating on the coverslips. Cells on coverslips were loaded with 1 μM Fura-2/AM (Dojindo Laboratories) at 37°C for 40 min, in a physiological salt solution containing (in mM): 150 NaCl; 4 KCl; 2 CaCl2; 1 MgCl2; 5 Hepes; and 5.6 glucose, adjusted to pH 7.4 with NaOH. The coverslips were then placed in a perfusion chamber mounted on the stage of the microscope. Fluorescence images of the cells were recorded and analyzed with a video image analysis system (ARGUS-20/CA; Hamamatsu Photonics). The Fura-2 fluorescence at an emission wavelength of 510 nm (bandwidth, 20 nm) was observed at room temperature by exciting Fura-2 alternately at 340 and 380 nm (bandwidth, 11 nm). The 340/380 nm ratio images were obtained on a pixel by pixel basis, and were converted to Ca2+ concentrations by in vitro calibration. All the reagents dissolved in water, ethanol, or dimethylsulfoxide were diluted to their final concentrations in the physiological salt solution and applied to the cells by perfusion. Ca2+-free solution contains 0.5 mM EGTA but no added CaCl2. 30 mM K+-containing ‘high K+’ solution and 8 mM K+-containing solution were made by the substitution of equimolar K+ for Na+. In measuring [Ca2+]i responses under fix membrane potential, 2 μM valinomycin was added to the 8 mM K+-containing solution. Data were accumulated from three to five experiments under each experimental condition.
IP3 Generation Assay.
Cells (2 × 106) were stimulated with mAB M4 (10 μg) at 37°C for indicated time. Kinetic analysis of IP3 production was performed using BIOTRAK IP3 assay system (Amersham Pharmacia Biotech).
Evaluation of IP3R Activity.
Cells were attached to poly-l-lysine and collagen-coated coverslips, and loaded with either 20 μM Furapta-AM for 60 min in a physiological salt solution (in mM): 150 NaCl; 4 KCl; 2 CaCl2; 1 MgCl2, 5 Hepes; and 5.6 glucose adjusted to pH 7.4 with NaOH. The Furapta-loaded cells were then permeabilized by incubation with 40 μM β-escin for 2–4 min in an internal solution to wash out Furapta in the cytoplasm, which enabled measurement of the Ca2+ concentration within the organelles (27). The ratio of fluorescence intensities by excitations at 340 and 380 nm of 30–40 cells within a frame was normalized so that 1 and 0 corresponded, respectively, to the values just before the application of IP3 and after complete depletion by 10 μM at 300 nM Ca2+. The initial 20 s period for the normalized time course was fitted by single exponential function, e-rt. The rate constant, r (s−1), thus estimated was used as an index of the IP3 receptor activity.
Electrophysiology.
For electrophysiological measurements, coverslips plated with DT40 cells were transferred to the recording chamber, or DT40 cells grown in suspension were collected, resuspended in standard external, modified Ringer's solution (in mM: NaCl 145; KCl 2.8; CsCl 10; MgCl2 2; CaCl2 10; glucose 10; and Hepes 5, adjusted to pH 7.2 with NaOH), and allowed to settle in the recording chamber for 5 min. The standard pipette-filling solution contained (in mM): CsOH 145; glutamate 145; NaCl 8; MgCl2 1; EGTA 10; Mg2ATP 2; GTP 0.2; Hepes 5, adjusted to pH 7.2 with CsOH. To avoid spontaneous activation of HSOC currents, [Ca2+]i in the pipette solution was clamped to ∼100 nM by supplement with a mixture of 10 mM Cs-BAPTA and 4.3 mM CaCl2. Pipette resistance ranged from 2 to 4 MΩ when filled with the pipette solution. Patch pipettes were made from borosilicate glass capillaries (KIMAX-51; Kimble Products) using a model P-87 Flaming-Brown micropipette puller (Sutter Instrument). Patch-clamp experiments were performed in the tight-seal whole-cell configuration at 21–25°C. High-resolution current recordings were acquired by a computer-based patch-clamp amplifier system (EPC-9; HEKA). Immediately following establishment of the whole-cell configuration, 50 ms voltage ramps spanning the voltage range of –150–+50 mV were delivered from a holding potential of 0 mV at a rate of 0.5 Hz over a period of 200 s. All voltages were corrected for a liquid junction potential of 10 mV between external and internal solutions. Currents were filtered at 2.3 kHz and digitized at 100 μs intervals. Capacitative currents and series resistance were determined and corrected before each voltage ramp using the automatic capacitance compensation of the EPC-9. For analysis, the very first ramps before activation of SOC currents (usually 1–3) were digitally filtered at 2 kHz, pooled and used for leak-subtraction of all subsequent current records. The low-resolution temporal development of inward currents was extracted from the leak-corrected individual ramp current records by measuring the current amplitude at −130 mV.
NF-AT Activity.
NF-AT activity was quantitated with Lumat LB 9501 (Berthold Japan) using the NF-AT luciferase gene and the Dual-Luciferase™ Assay System (Promega) as described in Sugawara et al. (29). Luciferase activity was determined in triplicate for each experimental condition.
Online Supplemental Materials.
Supplemental figures can be accessed at http://www.jem.org/cgi/content/full/195/6/673/DC1. The data section includes the Northern blot analysis of RNA expression of TRP isoforms and measurements of TG-induced CCE in the high K+ depolarizing solution in DT40 cells.
Results
Targeted Disruption of the TRP1 Gene.
In agreement with previous reports that a trp1 transcript is detected in various human tissues including spleen, we found that the trp1 transcript was expressed in mouse spleen cells as well as chicken DT40 B cells (data not shown and Fig. 1 E). Herein, we took a genetic approach to study the physiological role of TRP1 in B cells. The trp1 gene locus in DT40 cells was disrupted by homologous recombination (26). The targeting vectors carry a neomycin or histidinol resistance gene cassette replacing the chicken genomic sequence, which contains the exon corresponding to the transmembrane region H5 of TRP1 (30) (Fig. 1 A–C). Southern and Northern blot analysis confirmed disruption of trp1 gene (Fig. 1 D and E), and confocal immunoimages demonstrated loss of TRP1 proteins in mutant cells (Fig. 1 F). The level of cell surface expression of BCR examined by staining with FITC-conjugated anti-chicken IgM antibody on TRP1-deficient clones was indistinguishable from that of parental DT40 cells (Fig. 1 G).
Defects of BCR-mediated Ca2+ Mobilization in TRP1-deficient B Cells.
We used digital video fluorescence imaging of the Ca2+-sensitive dye Fura-2 to compare BCR-induced [Ca2+]i changes in single WT and TRP1-deficient DT40 B cells. In the presence of 2 mM extracellular Ca2+, we observed the averaged [Ca2+]i increase from ∼70 to 250 nM in WT cells, whereas this [Ca2+]i increase was significantly reduced in TRP1-deficient cells (Fig. 2 A), demonstrating that TRP1 participates in BCR-mediated Ca2+ mobilization. To examine whether this Ca2+ defect in TRP1-deficient cells is solely attributable to insufficient Ca2+ influx, we also assessed the averaged [Ca2+]i rise in the absence of extracellular Ca2+. As shown in Fig. 2 B, a transient release from intracellular Ca2+ pools was also affected by loss of TRP1. Since the augmentation of [Ca2+]i rises by addition of external Ca2+ to solution, a hallmark of Ca2+ influx, was significantly potentiated in WT in comparison to that in mutant cells (Fig. 2 C), we conclude that TRP1 participates in both Ca2+ release and Ca2+ influx upon BCR cross-linking. In support of this conclusion, the defects in TRP1−-14 cells were restored by reintroduction of human TRP1 cDNA (Fig. 2 A–C).
Figure 2.

Disruption of TRP1 gene attenuates BCR-induced Ca2+ mobilization in DT40 cells. Average time courses of Ca2+ responses evoked in the presence (A, 2 mM Ca2+) and absence (B, EGTA) of extracellular Ca2+ upon BCR stimulation with anti-BCR antibody M4 (1 μg/ml) in WT cells (white box), in a TRP1-deficient clone TRP1−-14 (black box), and in TRP1−-14 cells expressing recombinant hTRP1 (TRP1/14, black triangle). Anti-BCR antibody was applied as indicated by the horizontal bars above the traces. (C) Peak BCR-induced [Ca2+]i rises in WT (n = 75), in two independent TRP1-deficient clones, TRP1−-14 (n = 31) and TRP1−-16 (n = 39), and in TRP1−-14 cells expressing recombinant hTRP1 (TRP1/14)(n = 22). The responses of the two independent TRP1-deficient clones were indistinguishable. Data points and columns are the mean ±SE. *P < 0.05; **P < 0.01; ***P < 0.001. P values are the results of Student's t test.
Ca2+ Release Defect in TRP1-deficient B Cells.
Given the evidence that BCR utilizes IP3 to release Ca2+ from the ER stores through its binding to P3Rs (31), the compromised Ca2+ release in TRP1-deficient cells could be explained by the defect in IP3 generation and/or by that in IP3R function. The former possibility is unlikely, because both levels and kinetics of IP3 generation after BCR cross-linking were comparable in mutant and WT DT40 B cells (Fig. 3 A). Then, to examine the latter possibility, we directly assessed the function of IP3Rs by monitoring the Ca2+ concentration in the lumen of the Ca2+ stores (luminal Ca2+ monitoring system) (Fig. 3 C–E; reference 27) and studying the dose–response behavior of IP3-mediated Ca2+ release in WT and TRP1-deficient cells. As shown in Fig. 3 E, TRP1-deficient cells manifested significantly lower IP3 sensitivity compared with WT DT40 cells, suggesting that the diminished activity of IP3Rs per se could at least partly account for the observed attenuation of Ca2+ release in TRP1-deficient B cells. Furthermore, since the expression levels of IP3R-1 and IP3R-2 were indistinguishable in WT and mutant cells (Fig. 3 B), it is likely that reduced activity of a normal number of IP3Rs is responsible for the observed decrease in Ca2+ release rates.
Figure 3.

IP3-induced Ca2+ release is suppressed in TRP1-deficient cells. (A) Intact BCR-induced IP3 production in TRP1-deficient cells. Cells were stimulated with anti-BCR antibody M4 (1 μg/ml) for the indicated time. Data points are the mean ±SE from four experiments. (B) Western blot analysis demonstrating the IP3R-1 or IP3R-2 expression indistinguishable in WT and mutant cells using a polyclonal antibody against either IP3R-1 or IP3R-2. (C) The ER luminal Ca2+ concentration increased with activation of the Ca2+ pump, and declined upon application of IP3. (D) The level of activation of the IP3R can be quantitatively compared by the initial rate of Ca2+ release, which we estimated by fitting an exponential curve (continuous line) to the initial part of the Ca2+ decay signal (black circles). (E) IP3-concentration dependence of Ca2+ release. Release rates were obtained by fitting a single exponential to the initial part of Ca2+ decay signal measured in luminal Ca2+ monitoring (reference 27). The continuous curve and dotted curve represent the best fit hyperbolic equations, rmax/(1 + EC50/[IP3]), where rmax is the extrapolated values of the maximal rate of Ca2+ release, for Ca2+ release rates in WT and TRP1-deficient cells, respectively. rmax and EC50 were 0.153 s-1 and 0.66 μM in WT cells, and 0.118 s−1 and 0.83 μM in mutant cells. Data obtained from TRP1−-14 and TRP1−-16 were combined. Data points are the mean ±SE from six to seven experiments. **P < 0.01.
Ca2+ Influx Defect in TRP1-deficient B Cells.
To test the hypothesis that TRP1 is part of the SOCs in B cells, we stimulated WT and TRP1-deficient DT40 B cells with thapsigargin (TG), an inhibitor of SERCA Ca2+-ATPases, that depletes intracellular Ca2+ stores and elicits CCE. As shown in Fig. 4 A, in the absence of extracellular Ca2+, TG caused a transient Ca2+ rise in both WT and mutant cells, indicating that TG-sensitive Ca2+ stores are not affected by loss of TRP1. Readmission of extracellular Ca2+ led to a steep increase of [Ca2+]i in WT B cells, whereas the [Ca2+]i increase was attenuated by ∼50% in TRP1-deficient DT40 B cells, demonstrating an important role of TRP1 in inducing CCE. Ca2+ influx through plasma membrane Ca2+ channels is driven by the electrochemical gradient across the plasma membrane. To exclude the possibility that the observed inhibition of Ca2+ influx in TRP1-deficient cells is due to decrease in electrical driving force for Ca2+ via membrane depolarization by TG treatment, we compared CCE in WT and TRP1-deficient DT40 cells in the presence of K+ ionophore valinomycin (2 μM) and 8 mM extracellular K+, previously reported to maintain the cell membrane potential at ∼–75 mV (32). Under this condition, CCE in mutant cells was still significantly less than that in WT cells (Fig. 4 B). Likewise, when high K+ (30 mM)-containing solution was employed to establish depolarizing experimental conditions, TRP1-deficient cells exhibited a reduced CCE component compared with WT cells (Fig. 4 B). Since independent TRP1-deficient clones, TRP1−-14 and TRP1−-16, showed virtually identical responses, data for the two clones were combined below.
Figure 4.

CCE is impaired in TRP1-deficient cells. TG-induced Ca2+ release and CCE. (A and B) (A) Time course of Ca2+ responses induced by external perfusion of 2 μM TG, that passively depletes internal Ca2+ stores in the Ca2+-free solution containing EGTA, and by subsequent addition of 2 mM extracellular Ca2+ to evoke CCE, in WT (white box), TRP1−-14 (black box), and hTRP1-expressing TRP1−-14 cells (TRP1/14, white triangle). The solutions contained 4 mM K+. (B) Peak [Ca2+]i rises attributable to Ca2+ release and entry induced by TG in WT, mutant clones, TRP1−-14 and TRP1−-16, and hTRP1-expressing TRP1−-14 cells (TRP1/14). Experiments were performed in the 4 mM K+-containing, physiological salt solution (n = 114, 65, 55, and 35 for WT, TRP1−-14, TRP1−-16, and TRP1/14, respectively), in high K+ (30 mM) solution (n = 153, 106, and 86 for WT, TRP1−-14, and TRP1−-16, respectively), and in 8 mM K+ plus 2 μM valinomycin-containing solution (n = 36 and 19 for WT and TRP1−-14, respectively). The responses of the two independent TRP1-deficient clones were indistinguishable. Data points and columns are the mean ±SE. **P < 0.01; ***P < 0.001. SOC currents induced by internal dialysis with 10 μM IP3 in DT40 cells (C and D). (C, left) Representative, high resolution current records at 10 s (top trace) and 200 s (lower trace) after whole cell break-in, subtracted with pooled leak currents (see Materials and Methods). The voltage ramp protocol is schematically shown on top. (Right) Average time courses of ionic currents evoked by 10 μM IP3 at –130 mV (indicated by an arrow in C in SOC-positive WT cells. Average time courses in TRP1-deficient cells were similar to WT cells. Spontaneous activation of SOC currents was not observed in the absence of intrapipette IP3, since [Ca2+]i in the pipette solution was clamped to ∼100 nM (see Materials and Methods). Data are mean ±SE. The whole-cell configuration of patch clamp recording is established at the time 0. (D) Number of WT and TRP1-deficient cells developing SOC currents in response to intracellular perfusion of 10 μM IP3. Only 20% of mutant cells show SOC current activation, while 90% of WT cells develops SOC current. (E) PCR analysis of genomic DNA demonstrating the TRP1-deficient preparations free from contamination of WT cells. PCR primers were designed as described in Materials and Methods to amplify the ∼2,080-bp genomic fragment in WT cells but not in mutant cells. Addition of 1/100 amount of genomic DNA from WT cells to that from the mutant line leads to clear PCR amplification of the WT ∼2,080-bp band, revealing that contamination of WT cells was <1% of the cell population, if at all.
To formally demonstrate that the Ca2+ influx defect by loss of TRP1 is due to loss of SOC function, we directly compared SOC currents in WT and TRP1-deficient DT40 cells in whole-cell patch clamp recordings. In the vast majority of WT DT40 cells (27 out of 30), intracellular dialysis with 10 μM IP3 via the patch pipette elicited inward currents that showed the salient features of CRAC currents (6): a positive reversal potential of >50 mV and inward rectification over the voltage range from –150–50 mV (Fig. 4 C; 200 s). The number of cells with detectable SOC currents was clearly reduced in TRP1-deficient cells in that only 6 out of 29 mutant cells showed detectable SOC currents (Fig. 4 D). Although SOCs could not be detected in ∼80% of mutant cells, the remaining ∼20% of mutant cells manifested similar characteristics in both peak current density and half-maximal activation time of SOC currents (−1.98 ± 0.35 pA/pF and 42.1 ± 14.7 s; n = 6) to those in WT cells (−1.88 ± 0.19 pA/pF and 43.0 ± 6.3 s; n = 27) (Fig. 4 D). In mutant and WT cells, the SOC currents showed similar shape of I-V curve (Fig. 4 C, left panel). Moreover, resistance to time-dependent inactivation of the SOC currents was also similar between mutant and WT cells (Fig. 4 C, right panel): 500 s after establishment of whole-cell configuration, current amplitudes remained 100 ± 7 and 98 ± 6% of the peak currents for WT (n = 5) and mutant cells (n = 4), respectively. These similarities could not be ascribed to contaminated WT cells in the TRP1-deficient preparation, because WT alleles could not be detected even though by using the PCR analysis (Fig. 4 E). Thus, genetic elimination of TRP1 impaired expression of functional SOCs activated by IP3-dependent Ca2+ release from ER stores.
Ca2+ Oscillation Defects in TRP1-Deficient B Cells.
Ca2+ signals often take the form of oscillations, i.e., periodic spiking of [Ca2+]i. Recent reports suggest that characteristics of Ca2+ oscillation, such as amplitude and frequency, contribute to the efficacy and specificity of signaling (5, 33). In WT cells and in the presence of external Ca2+, BCR stimulation evoked an initial Ca2+ transient peaking at ∼35 s (Fig. 5 B), which was followed by Ca2+ oscillations that lasted 30 min or longer (Fig. 5 A; reference 27). By contrast, and similar to responses observed in WT DT40 cells after omission of external Ca2+, the BCR-induced Ca2+ response in TRP1-deficient cells showed significant delay to reach first peak and reduced number of oscillation (Fig. 5 A–C). Strikingly, the number of oscillations in mutant cells exhibited further reduction by omission of external Ca2+, again confirming that the TRP1 deficit leads to attenuation of Ca2+ release activity through IP3R. Collectively, TRP1 plays an important role in normal BCR-mediated Ca2+ oscillations by regulating both Ca2+ release and Ca2+ influx.
Figure 5.


Reduced Ca2+ oscillation and NF-AT activity in TRP1-deficient cells. (A) Representative Ca2+ responses upon BCR ligation in Ca2+-containing and Ca2+-free external solution in single WT and mutant DT40 cells. Anti-BCR antibody was applied as indicated by the horizontal bars above the traces. (B) Delay of Ca2+ response to reach initial peak after application of anti-BCR antibody. n = 90 and 88 for WT and TRP1−, respectively, in Ca2+-containing solution, and n = 46 and 53 for WT and TRP1−, respectively, in Ca2+-free external solution. (C) Number of Ca2+ oscillations within 60 min of BCR stimulation. n = 90 and 150 for WT and TRP1−, respectively, in Ca2+-containing solution, and n = 89 and 139 for WT and TRP1−, respectively, in Ca2+-free external solution. (D) NF-AT activity in WT and mutant cells. Cells transfected with the NF-AT luciferase gene were analyzed as described in Materials and Methods. The experiment shown is representative of three independent trials. Data points and columns are the mean ±SE. **P < 0.01; ***P < 0.001.
DT40 B cells lacking TRP1 have allowed us to directly assess the effects of modulation of BCR-mediated Ca2+ oscillations on NF-AT activity. As shown in Fig. 5 D, NF-AT activation was reduced in the mutant cells. Thus, the result suggests that delayed Ca2+ onset and/or reduced number of Ca2+ oscillations, mediated by loss of TRP1, have an important impact on transcriptional regulation in B cells.
Discussion
We have demonstrated that TRP1 is critical for regulation of both IP3R Ca2+ release channels and SOCs in DT40 B lymphocytes. The functional impairment of these channels resulted in a decrease of the BCR-mediated Ca2+ mobilization, thereby leading to a reduction of NF-AT activity. Therefore, we have obtained the direct evidence that TRP1 indeed functions in the BCR signaling context, which in turn modulates gene expression in B cells.
Our data indicate that disruption of the trp1 gene significantly but incompletely eliminates SOC activity in mutant cells, which could be accounted for by at least two possibilities. First, this may imply the existence of multiple SOC subtypes in B lymphocytes. Because there are ∼20 members of the TRP family proteins including seven close mammalian relatives of the Drosophila trp gene (34), other members of these TRP family proteins could be responsible for the residual SOC activity in a TRP1-independent manner. Second, TRP proteins may assemble into a heteromultimeric complex comprising TRP1, where channel characteristics are determined by subunit compositions (35). In this regard, TRP1 could function as a regulatory subunit rather than as an indispensable subunit for the SOC activity. Although our data could not definitely distinguish these two possibilities, we speculate that the first possibility is more likely. Assuming that TRP1 could be a regulatory subunit of the SOCs in WT cells, one could envision that characteristics of SOC currents would significantly differ between WT and TRP1-deficient cells. However, our electrophysiological analysis demonstrated that despite nondetectable SOC currents in ∼80% of the mutant cells, the remaining ∼20% had SOC currents with characteristics that were indistinguishable from WT cells. Considering that a gene knock-out approach sometimes causes upregulation of other functionally redundant molecules in an attempt to cope with the deficit, this compensation extent may vary for each TRP1-deficient cell. In this regard, it has been demonstrated that activation of CRAC channel by IP3 appears to be of all-or-none type regulation (36), suggesting that opening of CRAC channels might be mediated by the small changes of key component(s). If so in the case of DT40 cells, apparently normal SOCs in ∼20% of TRP1-deficient cells may be accounted for by sufficient compensation that enables SOCs to cross the threshold for all-or-none activation.
In [Ca2+]i measurements using high K+-containing external solution, which limits Ca2+ entry only through Ca2+-selective channels such as CRAC channels, majority of WT cells (∼80%) displayed sustained TG-induced [Ca2+]i responses upon readmission of external Ca2+, whereas this sustained TG-induced [Ca2+]i responses were observed only in minority of mutant cells (∼10%) (Fig. S2). These results appear to be consistent with the electrophysiological recording data that the CRAC-like SOC current was detectable in ∼20% of mutant cells. However, in the TG-induced [Ca2+]i response, the remaining 90% of mutant cells still manifested a small transient or slowly rising tiny Ca2+ response. The straightforward explanation for why this transient rapid component detected by Ca2+ imagining might have escaped detection by patch clamp is that TG-mediated response could be composed of two components: IP3R-mediated component and other IP3R-independent one. IP3 used in electrophysiology should activate the SOC coupled to IP3R-mediated Ca2+ release from the internal Ca2+ store ER, while TG, that passively depletes internal stores by blockade of ER Ca2+-ATPase in Ca2+ imaging, may activate the SOC coupled to IP3R as well as SOCs coupled to ryanodine receptors (37). In accordance with this, Parekh et al. indeed reported that IP3 and TG are not equivalent in potency for activation of the CRAC current in rat basophilic leukemia cells (RBL-1) in electrophysiological recordings (36). It is thus possible that multiple SOC subtypes and/or multiple SOC activation mechanisms, which respond somewhat differently to store depletion signals, are expressed in DT40 cells.
Since TRP1, but not IP3R, is a plasma membrane–resident protein, our results that deletion of TRP1 perturbed the function of IP3Rs were somewhat surprising. However, given the previous observations that TRP3 directly binds to IP3Rs (37–41) and that the functional IP3R-binding sequence identified in TRP3 is conserved in TRP1 and other TRP proteins (42), it is reasonable to anticipate that TRP1, like TRP3, directly interacts with IP3Rs. If so, the straightforward explanation for our data is that the physical interaction between TRP1 and IP3Rs is required for optimal activation of both IP3Rs and SOCs. Hence, we propose that TRP1 functions not only as one component of SOCs but also as a positive regulator of IP3Rs in B cells, thereby contributing to the facilatory communication between the plasma membrane and ER. This facilitation of the coupling between the IP3Rs and SOCs by TRP1 in B cells promotes the coordinated aspect of Ca2+ signaling, Ca2+ oscillation, which could have significant impacts on the efficiency and specificity of gene expression (5, 33). Our present view concerning the role of TRP–IP3R interaction seemingly contradicts the apparently normal CCE induced by TG in DT40 cells lacking all three IP3R types (29). However, given the evidence that the ryanodine receptors RyR1 and RyR3 gates SOCs independently of IP3Rs in DT40 cells, one possibility is that the persistence of SOC is due to RyRs in the triple IP3R-deficient cells (43). Alternatively, yet unidentified Ca2+ release pathways may be involved in the induction of CCE in DT40 cells.
In the case of the Drosophila photoreceptor, phospholipase C (PLC)-β is associated with TRP through mutual interaction with a scaffolding protein, thereby making a spatially compact signaling complex between rhodopsin, PLC-β, TRP, and other regulatory proteins (44). This spatial compartmentation is thought to be important for efficient operation of the light-induced Ca2+ signaling in photoreceptor cells (45). In the case of BCR signaling, PLC-γ2, instead of PLC-β, is used to generate IP3. Given the evidence that PLC-γ2 should be targeted to subdomains of the plasma membrane known as rafts for generating IP3 (46), the compact signaling complex including PLC-γ2, TRP1, and IP3R, in close analogy to the Drosophila photoreceptor, is likely formed in rafts after BCR ligation. Indeed, TRP1 is known to be concentrated in rafts (47), wherein TRP1, with coordination of candidate scaffolding proteins such as a INAD (44) homologue or possibly SNAP-25 (48), might contribute to assembling the CCE signaling complex. Hence, TRP1 deficit may perturb an assembly of this signaling complex to decrease local concentration of functional SOCs.
Acknowledgments
This work was supported by research grants from the Ministry of Education, Science, Sports, and Culture, and by the Research for the Future program of the Japan Society for the Promotion of Science (JSPS).
Footnotes
The online version of this article contains supplemental material.
Abbreviations used in this paper: BCR, B cell receptor; CCE, capacitative Ca2+ entry; CRAC, Ca2+-released-activated Ca2+; ER, endoplasmic reticulum; IP3, inositol 1,4,5-trisphosphate; IP3R, IP3 receptor; PLC, phospholipase C; SOC, store-operated Ca2+ channel; TG, thapsigargin; TRP, transient receptor potential; WT, wild-type.
References
- 1.Clapham, D.E. 1995. Calcium signaling. Cell. 80:259–268. [DOI] [PubMed] [Google Scholar]
- 2.Berridge, M.J., M.D. Bootman, and P. Lipp. 1998. Calcium-a life and death signal. Nature. 395:645–648. [DOI] [PubMed] [Google Scholar]
- 3.Putney, J.W., and G.S.J. Bird. 1993. The signal for capacitative calcium entry. Cell. 75:199–201. [DOI] [PubMed] [Google Scholar]
- 4.Dolmetsch, R.E., R.S. Lewis, C.C. Goodnow, and J.I. Healy. 1997. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 386:855–858. [DOI] [PubMed] [Google Scholar]
- 5.Li, W.-H., J. Llopis, M. Whitney, G. Zlokarnik, and R.Y. Tsien. 1998. Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature. 392:936–941. [DOI] [PubMed] [Google Scholar]
- 6.Hoth, M., and R. Penner. 1992. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 355:353–356. [DOI] [PubMed] [Google Scholar]
- 7.Zweifach, A., and R.S. Lewis. 1993. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc. Natl. Acad. Sci. USA. 90:6295–6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fasolato, C., B. Innocenti, and T. Pozzan. 1994. Receptor-activated Ca2+ influx: how many mechanisms for how many channels? Trends Pharmacol. Sci. 15:77–83. [DOI] [PubMed] [Google Scholar]
- 9.Su, Z., P. Csutora, D. Hunton, R.L. Shoemaker, R.B. Marchase, and J.E. Blalock. 2001. A store-operated nonselective cation channel in lymphocytes is activated directly by Ca2+ influx factor and diacylglycerol. Am. J. Physiol. Cell Physiol. 280:C1284–C1292. [DOI] [PubMed] [Google Scholar]
- 10.Kaznacheyeva, E., A. Zubov, K. Gusev, I. Bezprozvanny, and G.N. Mozhayeva. 2001. Activation of calcium entry in human carcinoma A431 cells by store depletion and phospholipase C-dependent mechanisms converge on ICRAC-like calcium channels. Proc. Natl. Acad. Sci. USA. 98:148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mignen, O., and T.J. Shuttleworth. 2001. Permeation of monovalent cations through the non-capacitative arachidonate-regulated Ca2+ channels in HEK293 cells. Comparison with endogenous store-operated channels. J. Biol. Chem. 276:21365–21374. [DOI] [PubMed] [Google Scholar]
- 12.Trepakova, E.S., M. Gericke, Y. Hirakawa, R.M. Weisbrod, R.A. Cohen, and V.M.J. Bolotina. 2001. Properties of a native cation channel activated by Ca2+ store depletion in vascular smooth muscle cells. J. Biol. Chem. 276:7782–7790. [DOI] [PubMed] [Google Scholar]
- 13.Partiseti, M., F. Le Deist, C. Hivroz, A. Fischer, H. Korn, and D. Choquet. 1995. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J. Biol. Chem. 269:32327–32335. [PubMed] [Google Scholar]
- 14.Feske, S., J.M. Muller, D. Graf, R.A. Kroczek, R. Drager, C. Niemeyer, P.A. Baeuerle, H.H. Peter, and M. Schlesier. 1996. Severe combined immunodeficiency due to defective binding of the nuclear factor of activated T cells in T lymphocytes of two male siblings. Eur. J. Immunol. 26:2119–2126. [DOI] [PubMed] [Google Scholar]
- 15.Feske, S., J. Giltnane, R. Dolmetsch, L.M. Staudt, and A. Rao. 2001. Gene regulation mediated by calcium signals in T lymphocytes. Nat. Immunol. 24:316–324. [DOI] [PubMed] [Google Scholar]
- 16.Stankunas, K., I.A. Graef, J.R. Neilson, S.H. Park, and G.R. Crabtree. 1999. Signaling through calcium, calcineurin, and NF-AT in lymphocyte activation and development. Cold Spring Harb. Symp. Quant. Biol. 64:505–516. [DOI] [PubMed] [Google Scholar]
- 17.Okamura, H., and A. Rao. 2001. Transcriptional regulation in lymphocytes. Curr. Opin. Cell Biol. 13:239–243. [DOI] [PubMed] [Google Scholar]
- 18.Zhu, X., P.B. Chu, M. Peyton, and L. Birnbaumer. 1995. Molecular cloning of a widely expressed human homologue for the Drosophila trp gene. FEBS Lett. 373:193–198. [DOI] [PubMed] [Google Scholar]
- 19.Wes, P.D., J. Chevesich, A. Jeromin, C. Rosenberg, G. Stetten, and C. Montell. 1995. TRPC1, a human homolog of a Drosophila store-opereted channel. Proc. Natl. Acad. Sci. USA. 92:9652–9656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okada, T., R. Inoue, K. Yamazaki, A. Maeda, T. Kurosaki, T. Yamakuni, I. Tanaka, S. Shimizu, K. Ikenaka, K. Imoto, and Y. Mori. 1999. Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca2+-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. J. Biol. Chem. 274:27359–27370. [DOI] [PubMed] [Google Scholar]
- 21.Zitt, C., A. Zobel, A.G. Obukhov, C. Harteneck, F. Kalkbrenner, A. Lückhoff, and G. Schultz. 1996. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron. 16:1189–1196. [DOI] [PubMed] [Google Scholar]
- 22.Zhu, X., M. Jiang, M. Peyton, G. Boulay, R. Hurst, E. Stefani, and L. Birnbaumer. 1996. trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell. 85:661–671. [DOI] [PubMed] [Google Scholar]
- 23.Liu, X., W. Wang, B.B. Singh, T. Lockwich, J. Jadlowiec, B. O'Connell, R. Wellner, M.X. Zhu, and I.S. Ambudkar. 2000. Trp1, a candidate protein for the store-operated Ca2+ influx mechanism in salivary gland cells. J. Biol. Chem. 275:3403–3411. [DOI] [PubMed] [Google Scholar]
- 24.Brereton, H.M., M.L. Harland, A.M. Auld, and G.J. Barritt. 2000. Evidence that the TRP-1 protein is unlikely to account for store-operated Ca2+ inflow in Xenopus laevis oocytes. Mol. Cell. Biochem. 214:63–74. [DOI] [PubMed] [Google Scholar]
- 25.Strübing, C., G. Krapivinsky, L. Krapivinsky, and D.E. Clapham. 2001. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 29:645–655. [DOI] [PubMed] [Google Scholar]
- 26.Takata, M., H. Sabe, A. Hata, T. Inazu, Y. Homma, T. Nukada, H. Yamamura, and T. Kurosaki. 1994. Tyrosine kinases Lyn and Syk regulate B-cell receptor-coupled Ca2+ mobilization through distinct pathways. EMBO J. 13:1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyakawa, T., A. Mizushima, K. Hirose, T. Yamazawa, I. Bezprozvanny, T. Kurosaki, and M. Iino. 2001. Ca2+-sensor region of IP3 receptor controls intracellular Ca2+ signaling. EMBO J. 20:1674–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyakawa, T., A. Maeda, T. Yamazawa, K. Hirose, T. Kurosaki, and M. Iino. 1999. Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J. 18:1303–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sugawara, H., M. Kurosaki, M. Takata, and T. Kurosaki. 1997. Genetic evidence for involvement of type 1, type 2, and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B cell antigen receptor. EMBO J. 16:3078–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mori, Y., N. Takada, T. Okada, M. Wakamori, K. Imoto, H. Wanifuchi, H. Oka, A. Oba, K. Ikenaka, and T. Kurosaki. 1998. Differential distribution of TRP Ca2+ channel isoforms in mouse brain. Neuroreport. 9:507–515. [PubMed] [Google Scholar]
- 31.Kurosaki, T., A. Maeda, M. Ishiai, A. Hashimoto, K. Inabe, and M. Takata. 2000. Regulation of the phospholipase C-γ2 pathway in B cells. Immunol. Rev. 176:19–29. [DOI] [PubMed] [Google Scholar]
- 32.Hashimoto, A., K. Hirose, T. Kurosaki., and M. Iino. 2001. Negative control of store-operated Ca2+ influx by B cell receptor cross-linking. J. Immunol. 166:1003–1008. [DOI] [PubMed] [Google Scholar]
- 33.Dolmetsch, R.E., K. Xu, and R.S. Lewis. 1998. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 392:933–936. [DOI] [PubMed] [Google Scholar]
- 34.Harteneck, C., T.D. Plant, and G. Schultz. 2000. From worm to man: three subfamilies of TRP channels. Trends Neurosci. 23:159–166. [DOI] [PubMed] [Google Scholar]
- 35.Lintschinger, B., M. Balzer-Geldsetzer, T. Baskaran, W.F. Graier, C. Romanin, M.X. Zhu, and K. Groschner. 2000. Coassembly of Trp1 and Trp3 proteins generates diacylglycerol- and Ca2+-sensitive cation channels. J. Biol. Chem. 275:27799–27805. [DOI] [PubMed] [Google Scholar]
- 36.Parekh, A.B., A. Flieg, and R. Penner. 1997. The store-operated calcium current ICRAC: nonlinear activation by InsP3 and dissociation from calcium release. Cell. 89:973–980. [DOI] [PubMed] [Google Scholar]
- 37.Kiselyov, K.I., D.M. Shin, N. Shcheynikov, Y. Wang, I.M. Pessah, P.D. Allen, and S. Muallem. 2000. Gating of store-operated channels by conformation coupling to ryanodine receptors. Mol. Cell. 6:421–431. [DOI] [PubMed] [Google Scholar]
- 38.Boulay, G., D.M. Brown, N. Qin, M. Jiang, A. Dietrich, M.X. Zhu, Z. Chen, M. Birnbaumer, K. Mikoshiba, and L. Birnbaumer. 1999. Modulation of Ca2+ entry by polypeptides of the inositol 1,4,5-trisphosphate receptor (IP3R) that bind transient receptor potential (TRP): evidence for roles of TRP and IP3R in store depletion-activated Ca2+ entry. Proc. Natl. Acad. Sci. USA. 96:14955–14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiselyov, K., G.A. Mignery, M.X. Zhu, and S. Muallem. 1999. The N-terminal domain of the IP3 receptor gates store-operated hTrp3 channels. Mol. Cell. 4:423–429. [DOI] [PubMed] [Google Scholar]
- 40.Patterson, R.L., D.B. van Rossum, and D.L. Gill. 1999. Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell. 20:487–499. [DOI] [PubMed] [Google Scholar]
- 41.Ma, H.T., R.L. Patterson, D.B. van Rossum, L. Birnbaumer, K. Mikoshiba, and D.L. Gill. 2000. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 287:1647–1651. [DOI] [PubMed] [Google Scholar]
- 42.Tang, J., Y. Lin, Z. Zhang, S. Tikunova, L. Birnbaumer, and M.X. Zhu. 2001. Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of trp channels. J. Biol. Chem. 276:21303–21310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kiselyov, K., D.M. Shin, N. Shcheynikov, T. Kurosaki, and S. Muallem. 2001. Regulation of Ca2+-release-activated Ca2+ current (I CRAC) by ryanodine receptors in inositol 1,4,5-trisphosphate-receptor-deficient DT40 cells. Biochem. J. 360:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsunoda, S., J. Sierralta, Y. Sun, R. Bodner, E. Suzuki, A. Becker, M. Socolich, and C.S. Zuker. 1997. A multivalent PDZ-domain protein assembles signalling complexes in a G-protein-coupled cascade. Nature. 388:243–239. [DOI] [PubMed] [Google Scholar]
- 45.Chyb, S., P. Raghu, and R.C. Hardie. 1999. Polyunsaturated fatty acids activate the Drosophila light-sensitive channels TRP and TRPL. Nature. 397:255–259. [DOI] [PubMed] [Google Scholar]
- 46.Ishiai, M., M. Kurosaki, K. Inabe, A.C. Chan, K. Sugamura, and T. Kurosaki. 2000. Involvement of LAT, Gads, and Grb2 in compartmentation of SLP-76 to the plasma membrane. J. Exp. Med. 192:847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lockwich, T.P., X. Liu, B.B. Singh, J. Jadlowiec, S. Weiland, and I.S. Ambudkar. 2000. Assembly of trp1 in a signaling complex associated with caveolin-scaffolding lipid raft domains. J. Biol. Chem. 275:11934–11942. [DOI] [PubMed] [Google Scholar]
- 48.Yao, Y., A.V. Ferrer-Montiel, M. Montal, and R.Y. Tsien. 1999. Activation of store-operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell. 98:475–485. [DOI] [PubMed] [Google Scholar]