

Abstract
Recombinant porcine parvovirus virus-like particles (PPV-VLPs) are particulate exogenous antigens that induce a strong, specific cytotoxic T lymphocyte (CTL) response in the absence of adjuvant. In the present report, we demonstrate in vivo that dendritic cells (DCs) present PPV-VLPs to CD8+ T cells after intracellular processing. PPV-VLPs are captured by DCs with a high efficacy, which results in the delivery of these exogenous antigens to 50% of the whole spleen DC population. In vivo, a few hours after injection, PPV-VLPs are presented exclusively to CD8+ T cells by CD8α− DCs, whereas 15 hours later they are presented mainly by CD8α+ DCs. After PPV-VLPs processing, a fraction of CD11b+ DCs undergo phenotypic changes, i.e., the up-regulation of CD8α and CD205 and the loss of CD4 molecules on their surface. The failure to detect mRNA coding for CD8α in CD11b+ DCs suggests that CD8α expression by these cells is not due to de novo synthesis. In recombination-activating gene knockout mice (Rag−/−), CD11b+ DCs did not express CD8α and PPV-VLPs presentation by CD8α+ DCs was severely diminished. These results indicate that both CD8α− and CD8α+ DCs play an important role in the induction of CTL responses by exogenous antigens, such as VLP.
Keywords: virus-like particles, dendritic cells, cross-priming, CTL, exogenous antigens
Introduction
The induction of CTL responses requires the presentation of antigen-derived peptides associated to MHC class I molecules on the surface of APCs to specific CD8+ T cells. These peptides essentially derive from antigens processed in the cytosol of APCs. Thus, antigens that do not reach the cytosol of APCs should not elicit a CTL response. However, it is now well established that host APCs can process exogenous cell-associated antigens and present them associated to MHC class I molecules through a process called cross-priming (1, 2). In this process, cell-associated antigens gain access to the MHC class I pathway by the transfer of these antigens from cells that expressed or carried them to APCs. Soluble exogenous antigens can also gain access to the cytosol of APCs through an alternative pathway exclusive for macrophage and dendritic cells (DCs)*(3), although cell-associated antigens are much more efficiently presented than soluble antigens (4, 5). Both macrophages (6) and DCs (7) have been reported to cross-present antigens, but only DCs are able to stimulate naive CD8+ T cells (8). Two routes of cross-priming have been proposed. One route involves the passage of antigens from endosomes to cytosol and in the other route the antigens do not escape from endosomes and are processed inside these vesicles (9). The first route seems to be mostly used by DCs whereas the second one is specific to macrophages (10).
DCs do not constitute a homogeneous cell population. On the basis of the expression of CD8α homodimer and CD4 molecules, these three subpopulations of DCs have been described in murine spleen: CD4−CD8α−, CD4+ CD8α− (both are CD11bhigh), and CD8α+CD11c+ (CD11blow) (11, 12). However, so far the attention has been focused mainly on the study of CD8α− and CD8α+ DCs, which until recently have been considered to be derived from myeloid and lymphoid progenitors, respectively (13, 14). It has recently been reported that CD8α+ but not CD8α− DCs can cross-prime CD8+ T cells in vivo excluding a role for CD8α− DCs in CTL induction (15).
Virus-like particles (VLPs) have clearly revealed an exceptional capacity to trigger CTL responses (3, 16–19). However, the mechanisms of uptake, processing, and presentation of these exogenous particles remain unclear. In particular, the APC involved in the induction of CTL response by VLPs is still unknown. Indeed, DCs and macrophages have been shown to be involved in the processing of VLPs (16, 20–22), but no direct in vivo evidence has been obtained identifying the APC that can uptake, process, and present VLPs.
We have developed an antigen delivery system based on nonreplicative, recombinant porcine parvovirus (PPV)-VLPs formed by the self-assembly of the viral protein (VP)2 of PPV (19, 23). The VP2 protein, the most abundant structural VP of the PPV capsid (24) and carrying foreign CD8+ T cell epitopes, self-assembles into 25-nM pseudo viral particles after expression in insect cells (19). Mice immunized with these PPV-VLPs, carrying a CD8+ T cell epitope from the lymphocytic choriomeningitis virus (LCMV) nucleoprotein and in the absence of adjuvant, developed a CTL response against LCMV that protected mice against a lethal intracerebral injection of LCMV based on the induction of high frequency of CTLs of high avidity (19, 25). This cytotoxic response was restricted to MHC class I molecules and mediated by CD8+ T cells (19).
We have studied the mechanisms of in vivo presentation of particulate exogenous antigens using PPV-VLPs as a model to determine whether particulate antigens can be captured and processed directly by DCs or if they induce CTL response by cross-priming after capture by other cells. In this report, we demonstrate that PPV-VLPs target DCs with a very high efficiency and directly induce a specific CTL response without cross-priming. CD8α− and CD8α+ DCs capture and process these particles. We also establish that CD8α− DCs play an important role in CTL induction by these exogenous antigens. Furthermore, this study demonstrates for the first time that stimulation by VLPs induces phenotypic changes on CD8α− DCs, which leads to the acquisition of several surface molecules and the loss of others.
Materials and Methods
Mice.
6–8-wk-old female C57BL/6 (H-2b) mice were purchased from Janvier. Transporter associated with antigen processing (TAP)1 female knockout mice (TAP−/−) were a gift from A. Bandeira (Institut Pasteur, Paris, France). Recombination-activating gene (Rag)2 knockout mice (Rag2−/−) and β2-microglobulin (β2M−/−) knockout mice were obtained from the Centre de Développement des Techniques Avancées pour l'Expérimentation Animale (Orléans, France). All animals were bred onto a C57BL/6 background. The mice were maintained under specific pathogen-free conditions.
PPV-VLPs.
The construction, characterization, and purification of recombinant and control PPV-VLPs were previously described in detail (19). In brief, the VP2 gene was expressed with the 257–264 peptide plus natural flanking sequences (LEQLESIINFEKLTE) from chicken egg ovalbumin (OVA257–264) in its 5′ end (PPV-VLPs carrying the OVA257–264 epitope [PPV-VLPs-OVA]) or without this sequence (PPV-VLPs) using a baculovirus vector system. After the infection of Sf9 insect cells, the recombinant VLPs were purified by salt precipitation with 20% ammonium sulfate followed by dialysis. Characterization of PPV-VLPs-OVA and PPV-VLPs by CsCl sedimentation analysis and electron microscopy revealed identical properties to those of native PPV virions. In some experiments, PPV-VLPs-OVA were labeled with the fluorochrome Alexa 488, using the Alexa Fluor™ 488 Protein Labeling Kit (Molecular Probes) according to the manufacturer's instructions.
The concentration of PPV-VLPs-OVA was determined by densitometry and by double-antibody sandwich ELISA. The densitometric assay was performed with 1D Image Analysis Software 2.0.1 (Eastman Kodak Co.) using BSA as reference. The double-antibody sandwich ELISA was performed as previously described (26), using as capture antibody the anti-PPV mAb 15C5 and as detection antibody the anti-PPV biotinylated mAb 13C6 (27). Highly purified PPV-VLPs from size exclusion chromatography were used as standard reference. PPV-VLPs are 25-nM particles formed by 60 copies of VP2 (64 kD), and therefore the molecular mass of the particles is 3,840 kD.
Endotoxin values were determined in each sample of VLPs, using the Limulus amebocyte lysate test (BioWhittaker Inc.). For PPV-VLPs, endotoxin values were <500 pg/mg of protein and for PPV-VLPs-OVA, <10 ng/mg.
Peptides and Cell Lines.
The OVA257–264 peptide (SIINFEKL) that corresponds to an immunodominant H-2b–restricted CTL epitope of OVA was purchased from Neosystem. B3Z, a CD8+ T cell hybridoma (28), specific for OVA257–264 epitope in the context of Kb, was a gift from N. Shastri (University of California, Berkeley, CA). The thymoma EL-4 was obtained from American Type Culture Collection.
Preparation of DCs and Other APCs.
Spleens from either normal or immunized mice were removed and treated for 45 min at 37°C with 400 U/ml collagenase type IV and 50 μg/ml DNase I (Boehringer) in RPMI 1640. After inhibition of collagenase activity with 6 mM EDTA in PBS, spleens were dissociated in Ca2+- and Mg2+-free PBS in the presence of 2.5 mM EDTA and 0.5% FCS (GIBCO BRL) for in vitro and ex vivo assays or BSA (Sigma-Aldrich) for immunization with DCs. In all assays involving DCs, the same lot of endotoxin-free FCS (as determined by Limulus amebocyte lysate test) was used (batch 3013260S). All solutions were also tested for endotoxins with the same test. Single spleen cell suspensions were prepared and blocked with anti-CD16/32 (2.4G2 clone; BD PharMingen) and with colloidal super-paramagnetic microbeads, conjugated to anti-CD11c mAb (magnetic-activated cell sorting [MACS]–anti-CD11c, N418 clone; Miltenyi Biotec), according to the manufacturer's instructions. CD11c+ cells were positively selected with high speed MACS (AutoMACS; Miltenyi Biotec). The purified DC preparations contained 3–10% autofluorescent cells (defined as double positive cells in a FL2 vs. FL3 dot plot without antibody labeling). The purity of DC preparations (excluding autofluorescent cells) was always 95–99%. CD11c+ cells were H-2 Kb+, I-Ab low, CD40low, CD80low, and CD86−. 25–30% were CD8α+ and 60–70% were CD8α− CD11b+.
B220+ spleen cells were obtained according to the same protocol (but without collagenase/DNase treatment) using rat anti–mouse B220 mAb (MACS–anti-B220, clone RA3-6B2). CD11b+ CD11c− spleen cells were obtained from the CD11c− fraction after CD11c+ cell purification by magnetic sorting. The CD11c− cells were stained with anti–mouse CD11b mAb (Mac-1α, MACS–anti-CD11b, clone M1/70) and purified with AutoMACS as well.
For the purification of CD8α− and CD8α+ DCs or of CD11bhigh DCs, spleen cells were stained with MACS–anti-CD11c, PE–anti-CD11c (HL-3 clone) and anti–CD16-32 antibodies and FITC–anti-CD8α (53-6.7 clone) or FITC–anti-CD11b (M1/70 clone; all three clones from BD PharMingen). After sorting by AutoMACS, CD11c+ cells were immediately sorted out in a FACScan™ Plus (BD Biosciences) or in a MoFlo® (Cytomation, Inc.) cell sorter. Autofluorescent cells were gated out during cell sorting. In all cases, the purity of both subpopulations was between 96–99%.
Antigen Presentation Assay.
For in vitro assays, CD11c+ spleen cells (105 cells/well) were first pulsed with antigen (PPV-VLPs-OVA, PPV-VLPs, or OVA257–264 peptide) for 4 h in 96-well culture microplates in a final volume of 0.2 ml of RPMI 1640 Glutamax-I, plus 5 × 10−5 M 2-ME, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 10% FCS (10% RPMI; all from GIBCO BRL). Subsequently, APCs were washed with 10% RPMI and incubated overnight with 105 B3Z cells/well in a final volume of 0.2 ml 10% RPMI at 37°C. The stimulation of B3Z cells was monitored by IL-2 release in supernatants, which was measured using the classic CTLL-2 bioassay. 104 cells/well of the CTLL-2 cell line were cultured with 100-μl supernatant in a final volume of 0.2 ml. 2 d later, [3H]thymidine (ICN Biomedicals) was added and the cells were harvested 6 h later with an automated cell harvester (Skatron). Incorporated thymidine was detected by cell scintillation counting. All experiments were done in duplicate. Results are expressed in counts per minute.
For ex vivo assays, PPV-VLPs-OVA or PPV-VLPs were injected into the retro-orbital venous sinus of mice. APCs were isolated and incubated with B3Z hybridoma overnight in the same conditions as previously described.
CTL Assay.
Spleen cells of immunized or control mice were obtained 7 d after immunization and were stimulated in vitro for 5 d with 1 μM OVA257–264 peptide in the presence of syngeneic irradiated naive spleen cells. The cytotoxic activity of these effector cells was tested on 51Cr-labeled EL-4 target cells pulsed with 50 μM OVA257–264 at different effector/target ratios. The released radioactivity was measured in the supernatant. The percentage of specific lysis was calculated as 100 × (experimental release − spontaneous release)/(maximum release − spontaneous release). Maximum release was determined by adding 1% Triton X-100 to EL-4 cells. Spontaneous release was obtained with target cells incubated without effector cells. Nonpulsed EL-4 cells were used as control of specificity.
Flow Cytometry.
Cells were preincubated with a rat anti–CD16/32 mAb (2.4G2 clone; BD PharMingen) for 15 min to block unspecific binding of primary antibody and then stained with the primary antibodies for 30 min. Cells were washed twice and propidium iodide was added to label dead cells. A minimum of 2 × 104 events were acquired for each sample on FACScan™ or FACSCalibur® cytometers and analyzed using CELLQuest™ software (all from BD Biosciences). The following mAbs were used: anti-CD3ε (145-2C11 clone), anti-CD4 (L3T4, RM4-5 clone), anti-CD8α (Ly-2, 53-6.7 clone), anti-CD8β (Ly-3.2, 53-5.8 clone), anti-CD11b (Mac-1α, M1/70 clone), anti-CD11c (HL-3 clone), anti-CD45R (B220, RA3-6B2 clone), anti-CD86 (B7.2, GL1 clone), and CD90 (Thy1.2, 30-H12), all purchased from BD PharMingen. Anti-CD8α (CT-CD8a clone; Caltag) and anti-CD205 (NLDC-145 clone; Cedarlane Laboratories Ltd.) were also used.
Reverse Transcription (RT)-PCR.
Total RNA was extracted using RNA Plus solution (Quantum Appligene Société Anonyme), from 5−10 × 105 purified DCs subpopulations (before or after overnight culture). cDNA was synthesized from total RNA in the presence of random primer p(dN)6 (Boehringer) using Moloney murine leukemia virus reverse transcriptase SuperScript™ (GIBCO BRL). For all samples, synthesis of cDNA was controlled by RT-PCR using β-actin primers for 30 cycles. CD8α chain mRNA was analyzed using these primers: CAC GAA TAA TAA GTA CGT TCT CAC C (sense) and ATG TAA ATA TCA CAG GCG AAG TCC A (antisense). PCR were performed using 1 IU of Goldstar DNA Taq polymerase (Advanced Biotechnologies), 50 pmol of appropriate primers, 250 μM of each dNTP except for dCTP (3,000 Ci/mmol; NEN Life Science Products) in 96-well polycarbonate Costar Thermowell™ strips (Corning) in a PTC-100™ programmable thermal controller (MJ Research, Inc.). 40 cycles of amplifications were performed as follows: 1 min at 94°C, 45 s at 48°C, 1 min at 72°C, followed by 10 min of elongation at 72°C. Samples were separated in 2% agarose gels and stained with ethidium bromide.
Results
In Vivo Induction of OVA257–264-specific Cytotoxic Response by PPV-VLPs-OVA.
In a previous report (19), we established that PPV-VLPs carrying a CD8+ T cell epitope of LCMV (PPV-VLPs-LCMV) induced a strong LCMV-specific CTL response when injected without adjuvant. In the present study, PPV-VLPs carrying an H-2b–restricted CD8+ T cell epitope from OVA257–264 (PPV-VLPs-OVA) were used. We first tested the ability of PPV-VLPs-OVA to induce a CTL response against OVA257–264 peptide–coated cells. C57BL/6 mice were immunized intraperitoneally with a single injection of 10 μg PPV-VLPs-OVA or control PPV-VLPs in PBS. As shown in Fig. 1 A, 7 d after immunization the mice immunized with PPV-VLPs-OVA developed a strong and specific CTL response against the OVA257–264 epitope, whereas, as expected, mice injected with control PPV-VLPs did not show a significant CTL response. Similar CTL responses were obtained after two intraperitoneal injections (with a 21-d interval) or one intravenous injection of PPV-VLPs-OVA (unpublished data). This result confirmed our earlier demonstration of the high immunogenicity of PPV particles in the absence of adjuvant. As previously demonstrated (19), the CTL response induced by PPV-VLPs was MHC class I restricted and mediated by CD8+ T cells. Furthermore, inhibition by lactacystin and N-acetyl-leucinyl-leucinyl-norleucinal of PPV-VLPs-OVA presentation by purified DCs demonstrated that processing PPV-VLPs-OVA requires proteasome (unpublished data). These results confirm that PPV-VLPs can effectively deliver an exogenous peptide into the MHC class I pathway.
Figure 1.

PPV-VLPs-OVA induce specific CTL responses and are presented in vitro and in vivo by DCs in a TAP-dependent way. (A) C57BL/6 mice were intraperitoneally immunized with 10 μg PPV-VLPs-OVA (•, ○) or 10 μg control PPV-VLPs (▪, □). 7 d later, their spleen cells were stimulated in vitro with the OVA257–264 peptide and irradiated syngeneic cells for 5 d. Cytotoxic activity of the effector cells was measured on 51Cr-labeled EL-4 cells loaded with the OVA257–264 peptide (•, ▪) or incubated with medium alone (○, □). Data are expressed as mean ± SEM of the percent lysis from two mice per group. One representative experiment out of three is depicted. (B) CD11c+ spleen cells (105 cells/well) were incubated in vitro with PPV-VLPs-OVA (•), control PPV-VLPs (○), or OVA257–264 peptide (□) for 4 h and then washed and cultured overnight with 105 cells/well of B3Z cells. (C) Representative FACS® diagrams of purified CD11c+ (left), CD11b+ CD11c− (center), or B220+ (right) spleen cells. C57BL/6 mouse spleens were removed and after collagenase/DNase I digestion cells were stained with MACS beads–anti-CD11c or MACS beads–anti-B220 mAbs and passed through a high speed magnetic-activated cell sorter (AutoMACS). The CD11c− fraction was then labeled with MACS beads–anti-CD11b and resorted by AutoMACS. (D) C57BL/6 mice were intravenously injected with 100 μg PPV-VLPs-OVA and 90 min later various numbers of CD11c+ (•), CD11b+ CD11c− (♦), and B220+ (▪) cells purified from their spleens were cocultured overnight with 105 B3Z cells/well. (E) TAP1−/− (○) and TAP1+/+ (•) C57BL/6 mice were intravenously injected with 100 μg PPV-VLPs-OVA and 90 min later CD11c+ cells purified from their spleens were cultured overnight at various cell numbers with 105 B3Z cells/well. The presentation of OVA257–264 peptide to B3Z cells was monitored by IL-2 production, measured by a CTLL proliferation assay, and expressed as mean ± SEM counts per minute (cpm) of duplicate wells. One representative experiment out of three or four is depicted in each case (two or three mice pooled per group).
In Vitro Processing of PPV-VLPs-OVA by DCs.
DCs have been clearly recognized as being the only APC capable of stimulating naive T cells. Therefore, we wondered whether DCs could process PPV-VLPs-OVA and present the OVA257–264–Kb complex to a specific hybridoma, as the first step of CTL induction. Dendritic spleen cells were highly purified from naive mice using the CD11c molecule, which is considered the most generalized marker of DCs, with >96% purity (Fig. 1 C). CD11c+ cells were incubated with PPV-VLPs-OVA, PPV-VLPs, or OVA257–264 peptide. The presence of OVA257–264–Kb complexes on DCs was monitored by the stimulation of B3Z cells. DCs incubated with PPV-VLPs-OVA or OVA257–264 peptide efficiently presented the OVA257–264 epitope, whereas DCs incubated with PPV-VLPs did not stimulate B3Z cells (Fig. 1 B). Moreover, PPV-VLPs-OVA were highly efficient in delivering the OVA257–264 epitope into the MHC class I pathway, as compared in an equimolar ratio to the OVA257–264 peptide (Fig. 1 B). CD11c+ bone marrow–derived DCs were also able to stimulate B3Z cells when incubated with PPV-VLPs-OVA (unpublished data). Therefore, DCs are able to process PPV-VLPs-OVA in vitro and present the OVA257–264 epitope to MHC class I–restricted T cells.
In Vivo Processing of PPV-VLPs-OVA by APCs.
We examined whether DCs can capture and process PPV-VLPs in vivo using an ex vivo antigen-presenting assay. C57BL/6 mice were intravenously injected with 50 μg (13 pmol/mouse) PPV-VLPs-OVA or with PPV-VLPs. 90 min later, CD11c+, CD11b+ CD11c− (essentially macrophages and granulocytes), and B220+ cells (B cells) were purified and cocultured with B3Z hybridoma. The purity of B cells was always >99%, whereas that of CD11b+ CD11c− was >90% (Fig. 1 C). When incubated with B3Z cells, only DCs purified from PPV-VLPs-OVA–injected mice were able to present the OVA257–264 epitope, whereas CD11b+ CD11c− and B220+ spleen cells were inefficient (Fig. 1 D). The same APC populations purified from control PPV-VLPs–injected mice failed to stimulate B3Z cells, although after overnight in vitro incubation with OVA257–264 peptide, they were fully able to stimulate B3Z cells (unpublished data). Thus, in vivo, DCs are the only APC capable of efficiently processing PPV-VLPs-OVA. DCs from mice injected with 10 μg (2.6 pmol/mouse) PPV-VLPs-OVA can also present the OVA257–264 peptide, although to a lower extent (unpublished data).
Cytosolic antigens have to be processed in the cytosol of APCs and then transported to the Golgi complex to be presented by MHC class I molecules. TAP molecules are required as an essential step in this transport. Therefore, we analyzed the ex vivo PPV-VLPs-OVA presentation by DCs in TAP1−/− syngeneic mice. 90 min after PPV-VLPs-OVA intravenous injection, spleen DCs from TAP1−/− mice failed to present the OVA257–264 epitope, whereas spleen DCs from TAP1+/+ mice efficiently stimulated B3Z cells (Fig. 1 E). Although TAP1−/− cells have a diminished expression of class I MHC molecules (29), their capacity to present peptide–Kb complexes was not essentially affected. Indeed, TAP1+/+ and TAP1−/− DCs incubated in vitro with the OVA257–264 peptide equally presented this epitope (unpublished data). Therefore, in vivo, the processing of PPV-VLPs-OVA required TAP molecules, i.e., a cytosolic processing in DCs.
Induction of CTL Response by DCs In Vivo Loaded with PPV-VLPs-OVA.
To establish whether DCs can induce CTL response against the epitope delivered by PPV-VLPs, naive syngeneic mice were immunized with CD11c+, CD11b+ CD11c−, or B220+ spleen cells purified from mice intravenously injected with PPV-VLPs-OVA 90 min before purification. 7 d later, the CTL response against OVA257–264-coated target cells was analyzed. As shown in Fig. 2 , only mice that received CD11c+ cells, purified from PPV-VLPs-OVA–injected mice, developed a CTL response against the OVA257–264 epitope. In contrast, mice that received CD11b+ CD11c− or B220+ cells purified from the same mice did not develop such a specific CTL response. Therefore, DCs are the only APC involved in the in vivo induction of CTL response by PPV-VLPs-OVA.
Figure 2.

CD11c+ spleen cells, pulsed in vivo with PPV-VLPs-OVA and injected into naive mice, induce a CTL response against OVA257–264 peptide. C57BL/6 mice were intravenously injected with 10 μg PPV-VLPs-OVA (•, ♦, and ▪) or of control PPV-VLPs (○). 90 min later, CD11c+ (○, •), CD11b+ CD11c− (♦), and B220+ (▪) spleen cells were purified and intravenously injected into naive syngeneic mice (1.5 × 105 cells/mice). 7 d later, spleen cells from injected mice were stimulated in vitro with OVA257–264 peptide for 5 d in the presence of irradiated syngeneic cells. The cytotoxic activity of effector cells was measured on 51Cr-labeled EL-4 cells loaded with OVA257–264. In the inset the CTL activity on unloaded 51Cr-labeled EL-4 cells is shown. Data are expressed as the mean ± SEM of the percent lysis from duplicate samples of three mice per group. One representative experiment out of three is represented.
Induction of CTL Response by PPV-VLPs-OVA Does Not Require Cross-Priming.
It is currently assumed that exogenous antigens induce CTL response through the cross-priming of DCs (1, 2) in a process that involves the transfer of cell-associated antigens to DCs. Thus, we next determined whether cross-priming is involved in CTL activation by PPV-VLPs through the capture of PPV-VLPs associated to cells by DCs. β2M−/− and β2M+/+ C57BL/6 mice were intravenously injected with PPV-VLPs-OVA and 90 min later their spleen DCs were purified. In vivo–loaded DCs from both groups of mice were then injected into syngeneic naive β2M+/+ mice and 1 wk later the CTL response against OVA257–264 was analyzed (Fig. 3) . We reasoned that because β2M−/− APCs are unable to present MHC class I–restricted epitopes, a CTL response against the OVA257–264 epitope could be induced only by transfer of cell-associated antigens from β2M−/− to β2M+/+ DCs. Fig. 3 shows that CTLs against OVA257–264-coated target cells were only observed after the injection of DCs from β2M+/+ mice injected with PPV-VLPs-OVA, but not after the administration of DCs purified from β2M−/− mice. These results clearly show that DCs capture exogenous PPV-VLPs-OVA and directly induce CTL response, and exclude that the observed CTL response is due to cross-priming.
Figure 3.

DCs pulsed with PPV-VLPs-OVA induce a CTL response without the requirement of cross-priming. β2M−/− (○) and β2M+/+ (•) C57BL/6 mice were intravenously injected with 10 μg PPV-VLPs-OVA, whereas β2M+/+ C57BL/6 mice were injected with PPV-VLPs as control (□). 90 min later, CD11c+ spleen cells were purified from each group and intravenously injected into β2M+/+ naive syngeneic mice (1.5 × 105 cells/mice). 7 d later, spleen cells from injected mice were stimulated in vitro with the OVA257–264 peptide for 5 d in the presence of irradiated syngeneic cells. The cytotoxic activity of effector cells was measured on 51Cr-labeled EL-4 cells loaded with OVA257–264 or incubated with medium alone. In the inset the CTL activity on unloaded 51Cr-labeled EL-4 cells is shown. Data are expressed as the mean ± SEM of the percent lysis from duplicate samples of two mice per group. One representative experiment out of two is shown.
Processing of PPV-VLPs-OVA by Subpopulations of DCs.
In mice, spleen DCs do not constitute a homogeneous cell population. On the basis of the CD8α chain expression, it is now accepted that two major subsets of DCs can be distinguished: CD8α− and CD8α+ DCs. To evaluate the capacity of these DC populations to process and present PPV-VLPs, CD8α− CD11c+ and CD8α+ CD11c+ cells were purified. A two-step method was used: first, an enrichment step of DCs by MACS anti-CD11c mAb and then a fluorescent-activated cell sorting, using PE–anti-CD11c and FITC–anti-CD8α. This proceeding allows the recovery of a high number of purified DCs in a short time with few steps and minimal manipulation. As shown in a representative experiment (Fig. 4 A), the purity of both cell populations was always >96%. Both DC subpopulations were equally able to stimulate B3Z cells after in vitro incubation with OVA257–264 peptide (Fig. 4 B, left), which shows that CD8α− and CD8α+ DCs have the same presentation capacity. These two DC subpopulations were incubated with PPV-VLPs-OVA and as shown in Fig. 4 B (right), CD8α− CD11c+ cells effectively presented the OVA257–264 epitope whereas CD8α+ CD11c+ cells weakly stimulated B3Z cells.
Figure 4.

Differential kinetics of in vivo PPV-VLPs-OVA presentation by CD8α− and CD8α+ DCs. (A) Purification of CD8α− and CD8α+ CD11c+ spleen cells. Spleens from C57BL/6 mice were removed and after collagenase/DNase I digestion, cells were stained with MACS beads–anti-CD11c, PE–anti-CD11c, and FITC–anti-CD8α antibodies and passed through an AutoMACS and then immediately through a FACScan™ or MOFLO®. The percentages of the different cell populations obtained after each step of purification are indicated and correspond to naive mice. (B) In vitro antigen presentation assays. Spleen CD8α− (•) and CD8α+ (○) CD11c+ cells were incubated in vitro with OVA257–264 peptide (left) or PPV-VLPs-OVA (right) for 4 h. They were then washed and cultured overnight with 105 B3Z cells/well. (C) Ex vivo PPV-VLPs-OVA presentation assays. Mice were intravenously injected with 50 μg PPV-VLPs-OVA at 90 min (left) or 15 h (right) before DC purification. CD8α− (•) and CD8α+ (○) CD11c+ cells as well as CD11c− (⋄) spleen cells were purified and cultured overnight with 105 B3Z cells/well. In the insets, CD8α− (•) and CD8α+ (○) CD11c+ cells purified from PPV-VLPs-OVA–injected mice were cultured overnight with 105 B3Z cells/well in the presence of 10−1 nM OVA257–264 peptide. The presentation of the SIINFEKL peptide to B3Z cells was monitored by IL-2 production, measured by a CTLL proliferation assay, and expressed as mean ± SEM counts per minute (cpm) of duplicate wells. One representative experiment out of two (for B) or seven (for C) is depicted (three to five pooled mice per group).
To see if a similar difference could also be observed in vivo, we performed an ex vivo PPV-VLPs-OVA presentation assay. Mice were intravenously injected with 50 μg PPV-VLPs-OVA 90 min and 15 h before DC purification. Splenic CD8α− and CD8α+ DCs from both groups of mice were simultaneously purified and incubated with B3Z. When DCs were purified 90 min after injection, the B3Z hybridoma was selectively stimulated by CD8α− DCs, whereas CD8α+ DCs had a weak stimulatory activity. In contrast, 15 h after injection, although CD8α− DCs were still able to stimulate B3Z cells, a very high stimulation was observed with CD8α+ DCs (Fig. 4 C). Similar results were obtained in 11 independent experiments confirming that CD8α− DCs are capable of presenting PPV-VLPs at early times, but 15 h later CD8α+ DCs are the main DC subset capable of presenting this exogenous antigen. Similar results were obtained with DCs from mice injected with 10 μg PPV-VLPs-OVA (unpublished data), although the level of B3Z stimulation was lower. Interestingly enough, under these sub-optimal conditions the presentation by CD8α− DCs at 90 min was rather low compared with the presentation of CD8α+ DCs at 15 h. Both DC subpopulations, purified 90 min or 15 h after PPV-VLPs-OVA injection and cultured overnight with the OVA257–264 peptide (10−1 nM), efficiently stimulated B3Z cells, which shows that these DCs retained the capacity to present antigens at both assayed times (Fig. 4 C, insets). Therefore, these two subpopulations seem to differ in their capacity to take up/process PPV-VLPs, but not in their capacity to present the OVA257–264 epitope delivered by such exogenous antigens.
PPV-VLPs Are Taken by DCs In Vivo.
To characterize the in vivo uptake of PPV-VLPs, we labeled PPV-VLPs-OVA with Alexa 488, a strong green fluorescent dye that fluorescence does not extinguish at low pH. After labeling, Alexa 488–PPV-VLPs-OVA maintained their capacity to be processed and presented by DCs to B3Z cells (unpublished data), showing the preservation of their biological activity. We injected Alexa 488–PPV-VLPs-OVA into naive C57BL/6 mice. 90 min later, their spleen cells were sorted out into CD11c+, CD11b+, and B220+ subpopulations by magnetic sorting and were labeled with several mAbs. A representative experiment is depicted in Fig. 5 A and summarized in Fig. 5 B. 90 min after injection, only a small fraction of B220+ spleen cells captured Alexa 488–PPV-VLPs-OVA (Fig. 5 A, a). In contrast, half of the CD11c+ cells were Alexa 488+ (Fig. 5 A, b). When CD11b+ cells were isolated and labeled with an anti-CD11c antibody, these two main cell populations were evidenced: one that was strongly Alexa 488+ CD11c+ and another that was Alexa 488low CD11c− (Fig. 5 A, c). These results demonstrated that in the pool of CD11b+ cells, PPV-VLPs are mainly captured by CD11c+ cells (i.e., DCs). Granulocytes, labeled with anti-GR1 mAb, did not show any uptake of Alexa 488–PPV-VLPs-OVA (unpublished data), which suggests that the weak uptake observed in the CD11c− CD11b+ population could be attributed to spleen macrophages. 15 h after injection, DCs remained strongly positive for Alexa 488, whereas B cells were still negative and macrophages were weakly positive for fluorescent VLPs (unpublished data).
Figure 5.
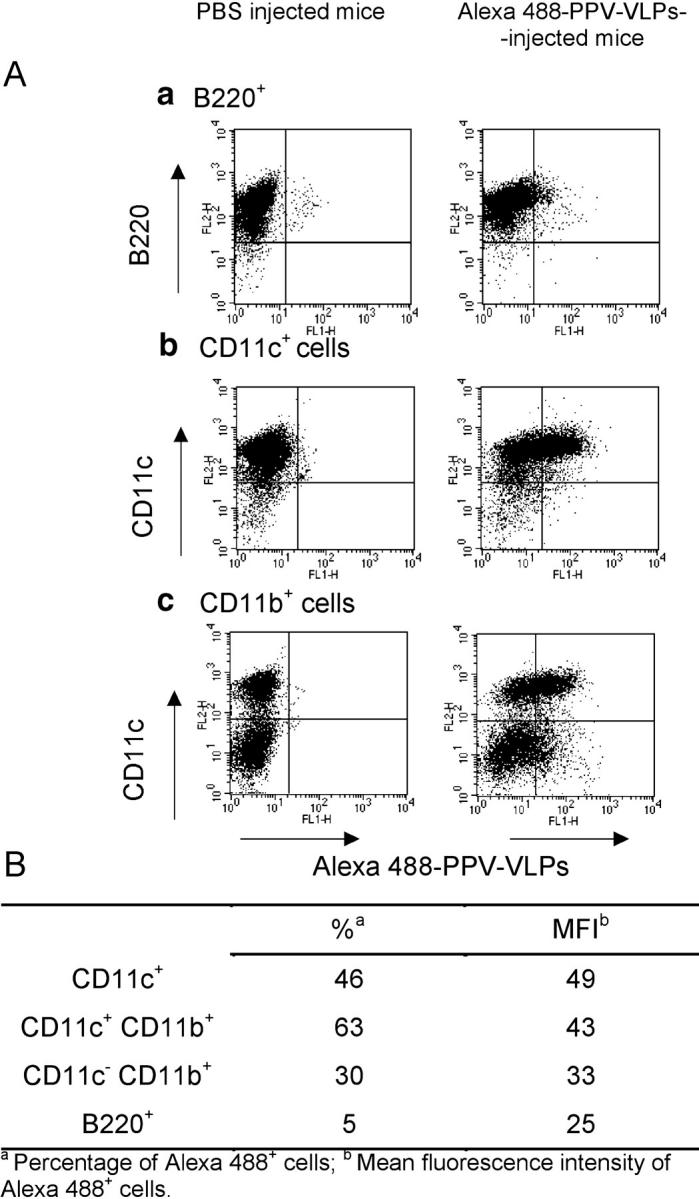
PPV-VLPs-OVA are uptaken in vivo by DCs. (A) One C57BL/6 mouse was intravenously injected with 50 μg Alexa 488–PPV-VLPs. One PBS-injected mouse was used as a control. 90 min later, their spleen cells were obtained and sorted out into B220+ (a), CD11c+ (b), or CD11b+ (c) subpopulations by magnetic sorting. All cell populations obtained were then stained with PE–anti-B220, PE–anti-CD11b, and PE–anti-CD11c and analyzed on a FACStar™ cytometer. Dot plot analysis of B220+ (a), CD11c+ (b), and CD11b+ (c) cells purified from PPV-VLPs–injected or control mice and labeled with the indicated PE-coupled mAbs are shown. B220+ cells were negative for CD11c and CD11b. (B) The percentage and mean fluorescence intensity of Alexa 488+ cells in CD11c+, CD11b+, and B220+ cells in the spleen of Alexa 488–PPV-VLPs-OVA–injected mice, as detailed in A. One representative experiment out of two is depicted.
Considering the different capacity of CD8α− and CD8α+ DCs to present PPV-VLPs-OVA, we investigated whether a differential uptake of PPV-VLPs-OVA by these two DC populations exists. Mice were injected with Alexa 488–PPV-VLPs-OVA and 90 min or 15 h later their spleen DCs were purified and labeled with anti-CD8α and anti-CD4 mAbs to distinguish the three main spleen DC subpopulations: CD4− CD8α+, CD4+ CD8α−, and CD4− CD8α− DCs (Fig. 6 A). 90 min after Alexa 488–PPV-VLPs-OVA injection, all DC subpopulations in the spleen captured PPV-VLPs and kept the fluorescence for at least 15 h (Fig. 6 B). However, CD4− CD8α+ DCs took slightly more PPV-VLPs than CD8α− DCs. Furthermore, 15 h later we observed a decrease in the percentage and mean fluorescence intensity FL1 of Alexa 488+ CD4+ CD8α− DCs (Fig. 6 C), which was not observed with other DC subpopulations. This decrease correlated with a diminution in the proportion of this subpopulation in the whole spleen DC pool.
Figure 6.

In vivo uptake of PPV-VLPs by DC subpopulations. C57BL/6 mice were intravenously injected with 50 μg Alexa 488–PPV-VLPs-OVA. 90 min or 15 h later their CD11c+ spleen cells were purified and stained with PE and APC-coupled mAbs for analysis on a FACScalibur® cytometer. Noninjected mice were used as control. (A) CD11c+ spleen cells from control mice were stained with APC–anti-CD8α and either PE–anti-CD11c (left) or PE–anti-CD4 (right). In this representative experiment, CD11c+ purified spleen cells contained 53% CD4+ CD8α−, 17% CD4− CD8α+, and 20% CD4− CD8α−. Similar values were obtained from injected mice. (B) FL1 (Alexa 488) histograms, gated on regions marked on the right panel in A, corresponding to CD4− CD8α+, CD4+ CD8α−, and CD4− CD8α− CD11c+ cells purified 90 min (thick line histograms) or 15 h (gray histograms) after injection or from control (thin line histograms). (C) Percentage and mean fluorescence intensity of Alexa 488+ cells, corresponding to the histograms depicted in B. One representative experiment out of two is depicted.
In conclusion, although CD8α+ DCs were unable to present PPV-VLPs-OVA 90 min after injection, they had already captured them. These results demonstrate that the differences observed in PPV-VLPs-OVA presentation between CD8α− and CD8α+ DCs could not be attributed to a difference in the uptake of PPV-VLPs-OVA.
CD8α− CD11b+ CD11c+ Cells, In Vivo Pulsed with PPV-VLPs-OVA, Express the CD8α Molecule, Up-regulate CD205 and Down-regulate CD4 in Rag2+/+ but Not in Rag2−/− Mice.
Considering the different capacity of DC subpopulations to present epitopes delivered by PPV-VLPs at different times, and the decrease of the proportion of CD4+ CD8α− DCs observed 15 h after the uptake of Alexa 488–PPV-VLPs-OVA, we analyzed whether these differences could be related to changes in the composition of spleen DC populations. Therefore, we injected 50 μg PPV-VLPs to Rag2+/+ mice and 15 h later we recovered the CD11c+ spleen cells for FACS® analysis. After PPV-VLPs injection, no major changes were observed in the percentages of DC subpopulations in a CD11c versus CD8α dot plot, except for a little shift toward the right of the CD8α− subpopulation (Fig. 7 A). However, when CD8α expression was analyzed against CD11b expression in CD11c+ cells, a significant number of CD11b+ cells expressed CD8α after PPV-VLPs injection. Furthermore, these CD8α+ CD11b+ CD11c+ cells represented an important percentage of total CD8α+ DCs (Table I). The same analysis performed with another anti-CD8α antibody showed similar results (Fig. 7 A). This CD8α+ CD11b+ DC population was observed until 24 h after PPV-VLPs injection and then disappeared (unpublished data). One possible explanation for this result could be that CD8+ T cells interacting with DCs remained attached after sorting, giving a false CD8+ staining on DCs. However, anti-CD90 antibody (unpublished data) as well as an anti-CD8β antibody did not label DCs (Fig. 7 A), making this hypothesis unlikely. At the same time a strong decrease in the percentage of CD4+ CD11b+ DCs was observed. This modification was accompanied by a diminution in the intensity of CD4 expression. Furthermore, the expression of CD205 was up-regulated after PPV-VLPs injection in CD11b+ DCs. The same analysis performed 90 min after PPV-VLPs-OVA injection showed no change in the expression of CD8α, CD4, or CD205 on CD11b− or CD11b+ DCs compared with noninjected mice (unpublished data). These phenotypic changes were accompanied by the up-regulation or expression of several molecules related to the maturation of DCs, such as CD86 (Fig. 7 A), MHC class I and II, CD40, and CD80 (unpublished data) molecules. The injection of 5 and 25 μg (1.3 and 6.5 pmol, respectively) PPV-VLPs induced similar changes, although to a lower extent (unpublished data).
Figure 7.

CD11b+ DCs from Rag2+/+ but not from Rag2−/− mice express CD8α, up-regulate CD205, and down-regulate CD4 molecules after PPV-VLPs injection. (A) C57BL/6 Rag2+/+ or Rag2−/− mice were intravenously injected with 50 μg PPV-VLPs. Noninjected mice from each strain were used as a control. 15 h later, CD11c+ spleen cells were purified and stained with anti-CD11b and anti-CD8α antibodies plus one of the following antibodies: anti-CD11c, anti-CD4, anti-CD205, anti-CD8β, or anti-CD86. Cells were analyzed on a FACScalibur® cytometer. One representative experiment out of two (for Rag2+/+) or three (for Rag2−/−) is depicted. (B) Expression of CD8α mRNA by DC subsets. Mice were injected with PPV-VLPs and 90 min later, CD8α− CD11c+ spleen cells were purified and cultured for 15 h (Exp. 1) or for different times (0, 3, 5, 8, and 15 h, Exp. 2). Then, the mRNA was extracted for RT-PCR (two rounds of 40 cycles per round) using primers for CD8α. mRNA from CD8α+ DCs purified 90 min after injection was used as a positive control.
Table I.
CD11b+ DCs Express CD8α in Rag2+/+ Mice Injected with PPV-VLPs-OVA
| CD11b+ CD8α1 cells percentage in total CD8α1 DCs |
CD11b+ CD8α1 cells percentage in total CD11b+ DCs |
|||||||
|---|---|---|---|---|---|---|---|---|
| Rag2+/+
mice |
Rag2−/−
mice |
Rag2+/+
mice |
Rag2−/−
mice |
|||||
| Injection of PPV-VLPs |
− | + | − | + | − | + | − | + |
| Exp. 1 | 19 | 43 | 5 | 12 | 7 | 15 | 4 | 4 |
| Exp. 2 | 30 | 45 | 17 | 28 | 11 | 23 | 9 | 11 |
| Exp. 3 | ND | ND | 19 | 22 | ND | ND | 9 | 8 |
C57BL/6 Rag2+/+ or Rag2−/− mice were intravenously injected with 50 μm PPV-VLPs. Noninjected mice from each strain were used as a control. 15 h later, CD11c+ spleen cells were labeled with CD11b and anti-CD8α antibodies and analyzed on a FACScalibur® cytometer. At least 2 × 104 events were analyzed.
To examine whether T cells play a role in these events, we repeated these experiments using Rag2−/− mice. In Rag2−/− mice, CD11b+ DCs showed no expression of CD8α, but there was a decrease in the percentage of CD8α+ CD11b− DCs after PPV-VLPs injection (Fig. 7 A and Table I). The percentage of CD4+ DCs was only slightly decreased whereas the expression of CD205 showed a small increase. However, CD8α− and CD8α+ DCs from Rag−/− mice showed an up-regulation of CD86 (Fig. 7 A) and MHC class I and II, CD40, and CD80 molecules (unpublished data) showed that they are indeed targeted by PPV-VLPs. The capture of Alexa 488–PPV-VLPs-OVA by CD4+ CD8α−, CD4− CD8α−, and CD4− CD8α+ DCs from Rag−/− mice, 90 min and 15 h after injection, was comparable to that observed with normal mice (unpublished data).
The expression of the CD8α molecule could be attributed to these two different origins: the translation of CD8α mRNA by CD11b+ DCs or the uptake of CD8α molecules from other cells, such as CD8+ T cells or CD8α+ CD11b− DCs. To address this question, we analyzed the expression of CD8α mRNA by two rounds of RT-PCR (40 cycles/round) of spleen DC subsets purified 90 min after PPV-VLPs injection and cultivated for different times (0, 3, 5, 8, and 15 h). CD8α mRNA was found only in CD8α+ CD11b− DCs but not in CD11b+ DCs (Fig. 7 B), which shows that CD8α molecules present at the surface of CD11b+ DCs after PPV-VLPs injection did not come from de novo synthesis of this molecule.
Comparable Presentation of the OVA257–264 Epitope by CD11bhigh DCs 90 min and 15 h after Injection of PPV-VLPs-OVA.
Because some CD11b+ DCs express the CD8α molecule 15 h after PPV-VLPs injection, we analyzed the presentation of the OVA257–264 epitope by these CD8α+ CD11b+ DCs. Because DCs express variable levels of CD11b, it is difficult to define clear-cut subpopulations based on this marker. Therefore, only CD11bhigh DCs were purified from mice injected with PPV-VLPs-OVA 90 min or 15 h earlier (CD11blow DCs always contained both CD8α− and CD8α+ cells). CD11bhigh DCs did not express CD8α 90 min after PPV-VLPs-OVA injection, but they expressed this molecule 15 h later (Fig. 8 A). Moreover, CD11bhigh were able to present the OVA257–264 epitope 90 min as well as 15 h after PPV-VLPs-OVA injection (Fig. 8 B). These results show that the CD11b+ DCs that acquired the CD8α molecule were able to present the OVA257–264 epitope.
Figure 8.

CD11b+ DCs present the OVA257–264 epitope 90 min as well as 15 h after the injection of PPV-VLPs-OVA. Mice were intravenously injected with 50 μg PPV-VLPs-OVA and 90 min or 15 h later, CD11bhigh CD11c+ spleen cells were purified by AutoMACS and a MOFLO® cell sorter. (A) Dot plot analysis of CD11bhigh DCs stained with an anti-CD8α antibody after sorting. (B) Splenic CD11c− (□) and CD11bhigh CD11c+ cells purified 90 min (○) or 15 h (•) after PPV-VLPs-OVA injection were cultured overnight with 105 B3Z cells/well. (C) Ex vivo PPV-VLPs-OVA presentation by DC subsets in Rag−/− mice. Naive Rag+/+ (•, ○) or Rag−/− (▪, □) mice were intravenously injected with 50 μg PPV-VLPs-OVA, 90 min (left) or 15 h (right) before DC purification. CD8α− (•, ▪) and CD8α+ (○, □) CD11c+ as well as CD11c− (▴) cells were simultaneously purified from these mice and cultured overnight with 105 B3Z cells/well. The presentation of the SIINFEKL peptide to B3Z cells was monitored by IL-2 production, measured by a CTLL proliferation assay, and expressed as mean ± SEM counts per minute (cpm) of duplicate wells. One representative experiment out of two is presented (three pooled mice per group).
Presentation of the OVA257–264 Epitope by CD8α− and CD8α+ DCs from Rag−/− Mice.
Because we did not detect the phenotypic changes in Rag−/− mice injected with PPV-VLPs that we observed in Rag+/+ mice, we analyzed the OVA257–264 epitope presentation by CD8α− and CD8α+ DCs from Rag−/− mice 90 min and 15 h after PPV-VLPs-OVA injection. 90 min after PPV-VLPs-OVA injection, CD8α+ DCs from Rag−/− and Rag+/+ mice were unable to present the OVA257–264 epitope, whereas CD8α− DCs from both strains stimulated the B3Z hybridoma. When the antigen presentation was performed 15 h after PPV-VLPs-OVA injection, the CD8α− DCs from both strains presented the OVA257–264 epitope with an efficacy comparable to the stimulation obtained 90 min after injection (Fig. 8 C). In contrast, CD8α+ DCs from Rag+/+ mice strongly stimulated B3Z cells whereas CD8α+ DCs from Rag−/− mice did not exhibit such a dramatic increase in their capacity to stimulate B3Z cells and presented the OVA257–264 epitope with an efficacy similar to CD8α− DCs. These results could suggest that T cells are required for CD8α+ DCs to present the OVA257–264 epitope. Thus, to verify this hypothesis we transferred CD90+ cells (98% T cells, 43% CD4+, 33% CD8+, B220−, CD11b−, CD45RB+, and CD69−; data not shown) purified from Rag+/+ mice to naive Rag−/− mice. 2 d later, we intravenously injected PPV-VLPs-OVA to these mice and then tested the ability of CD8α− and CD8α+ DCs to present the OVA257–264 epitope 15 h after injection. Surprisingly, under these conditions, neither CD8α− nor CD8α+ DCs were able to stimulate B3Z cells (unpublished data).
Discussion
VLPs clearly demonstrated their potential as vector for vaccination (3, 16–19, 21, 30) and have proven to be a potent CTL inducer when compared with other vectors (31). We have developed a VLPs system from PPV that has shown a powerful capacity to elicit both CD4+ (32) and CD8+ (19, 25, 33) T cell responses in the absence of any adjuvant. In this study, we analyzed the in vivo processing and presentation of PPV-VLPs carrying a CD8+ T cell epitope. In vitro, PPV-VLPs-OVA were as efficient as the synthetic OVA257–264 peptide in stimulating specific T cell hybridoma. In vivo, after intravenous injection of PPV-VLPs-OVA, only DCs were capable of presenting the OVA257–264 epitope to CD8+ T cells. Additional experiments revealed that PPV-VLPs were efficiently captured by DCs. Finally, only DCs purified from PPV-VLPs-OVA–injected mice could elicit a CTL response. These data clearly demonstrate that in vivo, processing and presentation of PPV-VLPs are performed by DCs.
Several mechanisms responsible for the processing of exogenous antigens in the MHC class I pathway have been described (34). Some are TAP- and proteasome-independent pathways, based on the regurgitation of antigen (35) or the recycling of MHC class I molecules (36) in which antigens are most likely degraded in endosomes and bind to MHC class I molecules without transfer to the cytosol. An alternative pathway, TAP and proteasome dependent, involves the transfer of antigens from phagosomes/macropinosomes to the cytosol, processing by the proteasome complex, and translocation into endoplasmic reticulum/Golgi network using TAP molecules, following the classic MHC class I pathway (37). This last pathway has been shown to be much more efficient than the TAP-independent pathways (38). Some antigens could generate CTL response using simultaneously multiple MHC class I processing pathways (39). In the present study, we demonstrated that although PPV-VLPs are exogenous antigens, they enter into the MHC class I pathway, gain access to cytosol, and are processed by a classic pathway as evidenced by the absence of stimulation of hybridoma cells by PPV-VLPs-OVA–pulsed DCs from TAP1−/− mice and by the requirement for proteasome processing (unpublished data).
Bohm et al. (16) have reported that hepatitis B surface antigen particles are processed by macrophages as well as DCs and that both cell types can prime a CTL response in vivo. However, the processing pathway of these VLPs are clearly different from that of PPV-VLPs. Hepatitis B surface antigen particles bind to recycling rather than new synthesized MHC class I molecules (40). In contrast, PPV-VLPs–derived peptides bind to new, nascent MHC class I molecules, as evidenced by the absence of PPV-VLPs-OVA presentation in TAP−/− mice. DCs, but not macrophages, have the ability to transport the antigens from endosomes to cytosol and then use the cytosolic machinery of processing (41). This may explain why PPV-VLPs, which need cytosolic processing, can only be presented by DCs in vivo.
Cross-priming, a process first described by Bevan (1, 2; for review see reference 42), allows exogenous antigens to elicit a CTL response. The TAP transporter is required for in vivo cross-priming of MHC class I–restricted antigens (43). Hence, considering that PPV-VLPs processing is TAP dependent, we analyzed whether the CTL response induced by PPV-VLPs-OVA is mediated by cross-priming or by direct priming, i.e., by the DC that capture PPV-VLPs-OVA. The transfer of β2M−/− DCs from PPV-VLPs-OVA–injected mice to naive β2M+/+ mice did not induce a CTL response, whereas the PPV-VLPs-OVA–pulsed β2M+/+ DCs induced such a response. This result clearly shows that DCs initially targeted by PPV-VLPs-OVA are the APCs that induce the CTL response. It should also be noted that the CTL response induced by PPV-VLPs is CD4 independent (19), whereas so far, the CTL responses induced by cross-priming are described to be CD4 dependent (42, 44). Therefore, our data demonstrate that DCs capture exogenous PPV-VLPs-OVA and are able to directly induce a CTL response without cross-priming.
The role of dendritic subsets in the induction of T cell responses is still a matter of intense debate. It has been shown that in mice, splenic CD8α+ DCs have the ability to produce large amounts of IL-12 and preferentially induce Th1 responses. By contrast, CD8α− DCs do not produce large amounts of IL-12 and preferentially induce Th2 responses (45, 46). However, polarization depends upon the site of injection, because the intravenous injection of CD8α− or CD8α+ DCs pulsed with peptide induces nonpolarized Th response (47). Concerning the induction of CTL response, one recent study (15) reported that only CD8α+ but not CD8α− DCs from mice injected with OVA-loaded splenocytes can cross-prime CD8+ T cells, although both DC subsets were shown to capture the same amounts of antigen. They suggested that this differential ability could be associated with a different in vivo processing due to a selective capacity of CD8α+ DCs to transport antigen from the endosome to cytosol. Pooley et al. (48) also found that CD8α+ DCs were the principal APC 18 h after an intravenous injection of a high amount of OVA protein. The injection of mice with peptide-pulsed CD8α− and CD8α+ DCs has clearly demonstrated that both DC subsets are equally able to induce a CTL response (47, 49).
In this study we have shown that all DC subsets can capture Alexa 488–PPV-VLPs-OVA to a similar extent, although CD8α+ DCs showed a little higher mean fluorescence intensity than CD8α− DC populations, perhaps because they are slightly larger than CD8α− DCs (49). However, we also showed that shortly after PPV-VLPs-OVA injection, only CD8α− DCs are responsible for the OVA257–264 epitope presentation whereas 15 h later, the presentation is mainly performed by CD8α+ DCs. Thus, these results demonstrated that CD8α− DCs have the capacity to present the OVA257–264 epitope very early after PPV-VLPs capture, whereas CD8α+ DCs presented the OVA257–264 epitope only after a lag time. Because PPV-VLPs are captured efficiently by both subsets, the difference in the accessibility of PPV-VLPs to DCs cannot explain these results. Interestingly, CD8α− from Rag−/− and Rag+/+ mice exhibited, at 90 min as well as at 15 h, comparable efficacy to capture and present PPV-VLPs-OVA, which shows that the processing by this DC subset is T cell independent. In contrast, 15 h after PPV-VLPs-OVA injection, CD8α+ DCs from Rag−/− mice were unable to present the OVA257–264 epitope with the high efficacy of CD8α+ DCs from Rag+/+ mice. This could suggest that T cells play a role in the licensing of CD8α+ DCs for exogenous antigen presentation. Therefore, our results could suggest that CD8α− and CD8α+ DCs have differential requirements for MHC class I–restricted presentation of exogenous antigens.
Using two different anti-CD8α antibodies, we demonstrated that 15 h after the injection of PPV-VLPs, a significant percentage of CD11b+ DCs expressed CD8α. These cells also expressed CD205. Moreover, the proportion of CD4+ CD8α− DCs showed an important decrease at that time, associated with a diminution in the intensity of expression of the CD4 molecule on those cells. Our study is the first in vivo report showing the apparition of a CD8α+ and CD205+ CD11b+ DC population in the spleen after the injection of an antigen. A very recent report suggests that CD8α+ DCs could originate from the CD8α− DC subset by a maturation process involving CD8α, DEC-205, and CD24 up-regulation 18 h after CD8α+ DC transfer (50). However, in that study, the phenotypic change was observed after cell transfer without any stimulation, whereas in our case it was induced by the injection of an antigen. An association of some T cells with DCs, which could eventually explains our results, is excluded because: (a) all purification steps included EDTA, which disrupts interactions between cells, (b) no CD8α mRNA was detected in sorted CD8α− DCs, and (c) anti-CD3ε, anti-CD8β, and anti-CD90 antibodies did not specifically bind to purified CD11b+ DCs. A population of CD11bdullCD11c+ that expressed CD8α in mice treated with Flt3L was described in two previous reports (51, 52). However, in this study the CD8α+ population is CD11b+, indicating that these populations are different.
The absence of CD8α mRNA in CD11b+ DCs harvested at various times after PPV-VLPs injection suggests that CD8α expression on these cells is not due to de novo synthesis. The lack of expression of the CD8α molecule on CD11b+ DCs from Rag2−/− mice injected with PPV-VLPs, as well as the slight reduction in CD4 expression and the small increase of CD205, also suggests that T cells may play a role in these phenotypic changes. Indeed, the similar uptake of Alexa 488–PPV-VLPs-OVA in Rag+/+ and Rag−/− mice (unpublished data), as well as the up-regulation of CD86 in DCs of Rag−/− mice, excluded the possibility that these effects were due to a lack of accessibility of VLPs to DCs from Rag−/− mice. It remains to be determined if the dramatic increase of CD8α+ presentation observed 15 h after PPV-VLPs-OVA injection was due to the CD11b+ CD8α+ or to the CD11b− CD8α+ population, which was inefficient at 90 min and therefore required longer times than CD8α− DCs for VLPs processing.
This study clearly demonstrates that CD8α− DCs have the capacity to transfer the VLPs to the cytosolic pathway and present these exogenous antigens without cross-priming almost immediately after antigen uptake and independently of T cells. In contrast, CD8α+ DCs cannot present PPV-VLPs immediately after capture, but exhibited a very strong capacity to present the OVA257–264 epitope carried by VLPs at longer times after VLPs capture. The fact that in Rag−/− mice the lack of expression of CD8α by CD11b+ DCs was accompanied by the inability of CD8α+ DCs to acquire the same antigen-presenting capacity than in Rag+/+ mice suggests that both events are closely linked.
In conclusion, this study highlighted the specialization of the various DC subsets. Our results, which show that CD8α− DCs can acquire molecules such as CD8α and CD205 after activation, strongly support the view that in vivo studies addressing the functions of DC subsets must define DC subsets carefully, based on the analysis of various markers.
Acknowledgments
We are grateful to Laleh Majlessi for her help in PCR analysis, Catherine Fayolle for endotoxin dosage, Anne Louise, Anne-Marie Balazuc, and Javier Sarraseca for their technical assistance, and Laleh Majlessi and Richard Loman for their critical reading of the manuscript.
G. Morón was supported by a Postdoctoral Fellowship of the Consejo Nacional de Investigaciones Científicas y Tecnológicas and by the European Economic Community grant QLK2-CT-1999-00429. This project is a collaborative work between the Pasteur Institute and INGENASA and is supported by the European Commission grant QLK2-CT-1999-00318.
I. Casal's present address is Programa de Biotecnología, Centro Nacional de Investigaciones Oncológicas, 28029 Madrid, Spain.
Footnotes
Abbreviations used in this paper: β2M, β2-microglobulin; DC, dendritic cell; LCMV, lymphocytic choriomeningitis virus; MACS, magnetic-activated cell sorting; OVA257–264, peptide corresponding to the sequence of amino acids 257–264 of the chicken egg albumin; PPV, porcine parvovirus; PPV-VLPss-OVA, PPV virus-like particles carrying OVA257–264; Rag, recombination-activating gene; RT, reverse transcription; TAP, transporter associated with antigen processing; VLP, virus-like particle; VP, viral protein.
References
- 1.Bevan, M.J. 1976. Minor H antigens introduced on H-2 different stimulating cells cross-react at the cytotoxic T cell level during in vivo priming. J. Immunol. 117:2233–2238. [PubMed] [Google Scholar]
- 2.Bevan, M.J. 1976. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 143:1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reimann, J., and R. Schirmbeck. 1999. Alternative pathways for processing exogenous and endogenous antigens that can generate peptides for MHC class I-restricted presentation. Immunol. Rev. 172:131–152. [DOI] [PubMed] [Google Scholar]
- 4.Li, M., G.M. Davey, R.M. Sutherland, C. Kurts, A.M. Lew, C. Hirst, F.R. Carbone, and W.R. Heath. 2001. Cell-associated ovalbumin is cross-presented much more efficiently than soluble ovalbumin in vivo. J. Immunol. 166:6099–6103. [DOI] [PubMed] [Google Scholar]
- 5.Carbone, F.R., and M.J. Bevan. 1990. Class I–restricted processing and presentation of exogenous cell-associated antigen in vivo. J. Exp. Med. 171:377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellone, M., G. Iezzi, P. Rovere, G. Galati, A. Ronchetti, M.P. Protti, J. Davoust, C. Rugarli, and A.A. Manfredi. 1997. Processing of engulfed apoptotic bodies yields T cell epitopes. J. Immunol. 159:5391–5399. [PubMed] [Google Scholar]
- 7.Albert, M.L., B. Sauter, and N. Bhardwaj. 1998. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 392:86–89. [DOI] [PubMed] [Google Scholar]
- 8.Ronchetti, A., P. Rovere, G. Iezzi, G. Galati, S. Heltai, M.P. Protti, M.P. Garancini, A.A. Manfredi, C. Rugarli, and M. Bellone. 1999. Immunogenicity of apoptotic cells in vivo: role of antigen load, antigen-presenting cells, and cytokines. J. Immunol. 163:130–136. [PubMed] [Google Scholar]
- 9.den Haan, J.M., and M.J. Bevan. 2001. Antigen presentation to CD8(+) T cells: cross-priming in infectious diseases. Curr. Opin. Immunol. 13:437–441. [DOI] [PubMed] [Google Scholar]
- 10.Yrlid, U., M. Svensson, C. Johansson, and M.J. Wick. 2000. Salmonella infection of bone marrow-derived macrophages and dendritic cells: influence on antigen presentation and initiating an immune response. FEMS Immunol. Med. Microbiol. 27:313–320. [DOI] [PubMed] [Google Scholar]
- 11.Grabbe, S., E. Kampgen, and G. Schuler. 2000. Dendritic cells: multi-lineal and multi-functional. Immunol Today. 21:431-433. [DOI] [PubMed] [Google Scholar]
- 12.Vremec, D., J. Pooley, H. Hochrein, L. Wu, and K. Shortman. 2000. CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J. Immunol. 164:2978–2986. [DOI] [PubMed] [Google Scholar]
- 13.Anjuere, F., P. Martin, I. Ferrero, M.L. Fraga, G.M. del Hoyo, N. Wright, and C. Ardavin. 1999. Definition of dendritic cell subpopulations present in the spleen, Peyer's patches, lymph nodes, and skin of the mouse. Blood. 93:590–598. [PubMed] [Google Scholar]
- 14.Vremec, D., and K. Shortman. 1997. Dendritic cell subtypes in mouse lymphoid organs: cross-correlation of surface markers, changes with incubation, and differences among thymus, spleen, and lymph nodes. J. Immunol. 159:565–573. [PubMed] [Google Scholar]
- 15.den Haan, J.M., S.M. Lehar, and M.J. Bevan. 2000. CD8+ but not CD8− dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192:1685–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bohm, W., R. Schirmbeck, A. Elbe, K. Melber, D. Diminky, G. Kraal, N. van Rooijen, Y. Barenholz, and J. Reimann. 1995. Exogenous hepatitis B surface antigen particles processed by dendritic cells or macrophages prime murine MHC class I-restricted cytotoxic T lymphocytes in vivo. J. Immunol. 155:3313–3321. [PubMed] [Google Scholar]
- 17.Oliveira-Ferreira, J., Y. Miyahira, G.T. Layton, N. Savage, M. Esteban, D. Rodriguez, J.R. Rodriguez, R.S. Nussenzweig, F. Zavala, and Y. Myahira. 2000. Immunogenicity of Ty-VLP bearing a CD8(+) T cell epitope of the CS protein of P. yoelii: enhanced memory response by boosting with recombinant vaccinia virus. Vaccine. 18:1863–1869. [DOI] [PubMed] [Google Scholar]
- 18.Chackerian, B., D.R. Lowy, and J.T. Schiller. 2001. Conjugation of a self-antigen to papillomavirus-like particles allows for efficient induction of protective autoantibodies. J. Clin. Invest. 3:415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sedlik, C., M. Saron, J. Sarraseca, I. Casal, and C. Leclerc. 1997. Recombinant parvovirus-like particles as an antigen carrier: a novel nonreplicative exogenous antigen to elicit protective antiviral cytotoxic T cells. Proc. Natl. Acad. Sci. USA. 94:7503–7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bachmann, M.F., M.B. Lutz, G.T. Layton, S.J. Harris, T. Fehr, M. Rescigno, and P. Ricciardi-Castagnoli. 1996. Dendritic cells process exogenous viral proteins and virus-like particles for class I presentation to CD8+ cytotoxic T lymphocytes. Eur. J. Immunol. 26:2595–2600. [DOI] [PubMed] [Google Scholar]
- 21.Rudolf, M.P., S.C. Fausch, D.M. Da Silva, and W.M. Kast. 2001. Human dendritic cells are activated by chimeric human papillomavirus type-16 virus-like particles and induce epitope-specific human T cell responses in vitro. J. Immunol. 166:5917–5924. [DOI] [PubMed] [Google Scholar]
- 22.Lenz, P., P.M. Day, Y.Y. Pang, S.A. Frye, P.N. Jensen, D.R. Lowy, and J.T. Schiller. 2001. Papillomavirus-like particles induce acute activation of dendritic cells. J. Immunol. 166:5346–5355. [DOI] [PubMed] [Google Scholar]
- 23.Sedlik, C., J. Sarraseca, P. Rueda, C. Leclerc, and I. Casal. 1995. Immunogenicity of poliovirus B and T cell epitopes presented by hybrid porcine parvovirus particles. J. Gen. Virol. 76:2361–2368. [DOI] [PubMed] [Google Scholar]
- 24.Ranz, A.I., J.J. Manclus, E. Diaz-Aroca, and J.I. Casal. 1989. Porcine parvovirus: DNA sequence and genome organization. J. Gen. Virol. 70:2541–2553. [DOI] [PubMed] [Google Scholar]
- 25.Sedlik, C., G. Dadaglio, M.F. Saron, E. Deriaud, M. Rojas, S.I. Casal, and C. Leclerc. 2000. In vivo induction of a high-avidity, high-frequency cytotoxic T-lymphocyte response is associated with antiviral protective immunity. J. Virol. 74:5769–5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rueda, P., J. Fominaya, J.P. Langeveld, C. Bruschke, C. Vela, and J.I. Casal. 2000. Effect of different baculovirus inactivation procedures on the integrity and immunogenicity of porcine parvovirus-like particles. Vaccine. 19:726–734. [DOI] [PubMed] [Google Scholar]
- 27.Casal, J.I., E. Cortés, J.A. López de Turiso, and C. Vela. 1992. Production of porcine and canine parvovirus-like particles using recombinant baculovirus. Baculovirus and Recombinant Protein Production Processes. J.M. Vlak, E.S. Schlaeger, and A. Bernard, editors. Editiones Roches, Basel. 76–91.
- 28.Karttunen, J., S. Sanderson, and N. Shastri. 1992. Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc. Natl. Acad. Sci. USA. 89:6020–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Kaer, L., P.G. Ashton-Rickardt, H.L. Ploegh, and S. Tonegawa. 1992. TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4–8+ T cells. Cell. 71:1205–1214. [DOI] [PubMed] [Google Scholar]
- 30.Ball, J.M., D.Y. Graham, A.R. Opekun, M.A. Gilger, R.A. Guerrero, and M.K. Estes. 1999. Recombinant Norwalk virus-like particles given orally to volunteers: phase I study. Gastroenterology. 117:40–48. [DOI] [PubMed] [Google Scholar]
- 31.Allsopp, C.E., M. Plebanski, S. Gilbert, R.E. Sinden, S. Harris, G. Frankel, G. Dougan, C. Hioe, D. Nixon, E. Paoletti, et al. 1996. Comparison of numerous delivery systems for the induction of cytotoxic T lymphocytes by immunization. Eur. J. Immunol. 26:1951–1959. [DOI] [PubMed] [Google Scholar]
- 32.Lo-Man, R., P. Rueda, C. Sedlik, E. Deriaud, I. Casal, and C. Leclerc. 1998. A recombinant virus-like particle system derived from parvovirus as an efficient antigen carrier to elicit a polarized Th1 immune response without adjuvant. Eur. J. Immunol. 28:1401–1407. [DOI] [PubMed] [Google Scholar]
- 33.Sedlik, C., A. Dridi, E. Deriaud, M.F. Saron, P. Rueda, J. Sarraseca, J.I. Casal, and C. Leclerc. 1999. Intranasal delivery of recombinant parvovirus-like particles elicits cytotoxic T-cell and neutralizing antibody responses. J. Virol. 73:2739–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rock, K.L. 1996. A new foreign policy: MHC class I molecules monitor the outside world. Immunol. Today. 17:131–137. [DOI] [PubMed] [Google Scholar]
- 35.Pfeifer, J.D., M.J. Wick, R.L. Roberts, K. Findlay, S.J. Normark, and C.V. Harding. 1993. Phagocytic processing of bacterial antigens for class I MHC presentation to T cells. Nature. 361:359–362. [DOI] [PubMed] [Google Scholar]
- 36.Gromme, M., F.G. Uytdehaag, H. Janssen, J. Calafat, R.S. van Binnendijk, M.J. Kenter, A. Tulp, D. Verwoerd, and J. Neefjes. 1999. Recycling MHC class I molecules and endosomal peptide loading. Proc. Natl. Acad. Sci. USA. 96:10326–10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Norbury, C.C., L.J. Hewlett, A.R. Prescott, N. Shastri, and C. Watts. 1995. Class I MHC presentation of exogenous soluble antigen via macropinocytosis in bone marrow macrophages. Immunity. 3:783–791. [DOI] [PubMed] [Google Scholar]
- 38.Sigal, L.J., and K.L. Rock. 2000. Bone marrow–derived antigen-presenting cells are required for the generation of cytotoxic T lymphocyte responses to viruses and use transporter associated with antigen presentation (TAP)-dependent and -independent pathways of antigen presentation. J. Exp. Med. 192:1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu, T., X. Zhou, C. Orvell, E. Lederer, H.G. Ljunggren, and M. Jondal. 1995. Heat-inactivated Sendai virus can enter multiple MHC class I processing pathways and generate cytotoxic T lymphocyte responses in vivo. J. Immunol. 154:3147–3155. [PubMed] [Google Scholar]
- 40.Schirmbeck, R., W. Bohm, K. Melber, and J. Reimann. 1995. Processing of exogenous heat-aggregated (denatured) and particulate (native) hepatitis B surface antigen for class I-restricted epitope presentation. J. Immunol. 155:4676–4684. [PubMed] [Google Scholar]
- 41.Rodriguez, A., A. Regnault, M. Kleijmeer, P. Ricciardi-Castagnoli, and S. Amigorena. 1999. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat. Cell Biol. 1:362–368. [DOI] [PubMed] [Google Scholar]
- 42.Heath, W.R., and F.R. Carbone. 2001. Cross-presentation, dendritic cells, tolerance and immunity. Annu. Rev. Immunol. 19:47–64. [DOI] [PubMed] [Google Scholar]
- 43.Huang, A.Y., A.T. Bruce, D.M. Pardoll, and H.I. Levitsky. 1996. In vivo cross-priming of MHC class I-restricted antigens requires the TAP transporter. Immunity. 4:349–355. [DOI] [PubMed] [Google Scholar]
- 44.Bennett, S.R., F.R. Carbone, F. Karamalis, J.F. Miller, and W.R. Heath. 1997. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J. Exp. Med. 186:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moser, M., and K.M. Murphy. 2000. Dendritic cell regulation of TH1-TH2 development. Nat. Immunol. 1:199–205. [DOI] [PubMed] [Google Scholar]
- 46.Pulendran, B., J. Banchereau, E. Maraskovsky, and C. Maliszewski. 2001. Modulating the immune response with dendritic cells and their growth factors. Trends Immunol. 22:41–47. [DOI] [PubMed] [Google Scholar]
- 47.Schlecht, G., C. Leclerc, and G. Dadaglio. 2001. Induction of CTL and nonpolarized Th cell responses by CD8alpha(+) and CD8alpha(−) dendritic cells. J. Immunol. 167:4215–4221. [DOI] [PubMed] [Google Scholar]
- 48.Pooley, J.L., W.R. Heath, and K. Shortman. 2001. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8− dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J. Immunol. 166:5327–5330. [DOI] [PubMed] [Google Scholar]
- 49.Ruedl, C., and M.F. Bachmann. 1999. CTL priming by CD8(+) and CD8(−) dendritic cells in vivo. Eur. J. Immunol. 29:3762–3767. [DOI] [PubMed] [Google Scholar]
- 50.del Hoyo, G.M., P. Martin, C.F. Arias, A.R. Marin, and C. Ardavin. 2002. CD8alpha(+) dendritic cells originate from the CD8alpha(−) dendritic cell subset by a maturation process involving CD8alpha, DEC-205, and CD24 up-regulation. Blood. 99:999–1004. [DOI] [PubMed] [Google Scholar]
- 51.Maraskovsky, E., K. Brasel, M. Teepe, E.R. Roux, S.D. Lyman, K. Shortman, and H.J. McKenna. 1996. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J. Exp. Med. 184:1953–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pulendran, B., J. Lingappa, M.K. Kennedy, J. Smith, M. Teepe, A. Rudensky, C.R. Maliszewski, and E. Maraskovsky. 1997. Developmental pathways of dendritic cells in vivo: distinct function, phenotype, and localization of dendritic cell subsets in FLT3 ligand-treated mice. J. Immunol. 159:2222–2231. [PubMed] [Google Scholar]