

Abstract
The hypothesis that FasL expression by tumor cells may impair the in vivo efficacy of antitumor immune responses, through a mechanism known as ‘Fas tumor counterattack,’ has been recently questioned, becoming the object of an intense debate based on conflicting results. Here we definitely show that FasL is indeed detectable in the cytoplasm of melanoma cells and its expression is confined to multivesicular bodies that contain melanosomes. In these structures FasL colocalizes with both melanosomal (i.e., gp100) and lysosomal (i.e., CD63) antigens. Isolated melanosomes express FasL, as detected by Western blot and cytofluorimetry, and they can exert Fas-mediated apoptosis in Jurkat cells. We additionally show that melanosome-containing multivesicular bodies degranulate extracellularly and release FasL-bearing microvesicles, that coexpress both gp100 and CD63 and retain their functional activity in triggering Fas-dependent apoptosis of lymphoid cells. Hence our data provide evidence for a novel mechanism potentially operating in Fas tumor counterattack through the secretion of subcellular particles expressing functional FasL. Such vesicles may form a sort of front line hindering lymphocytes and other immunocompetent cells from entering neoplastic lesions and exert their antitumor activity.
Keywords: melanoma, apoptosis, T cells, melanosome, microvesicles
Introduction
Despite the existence of specific T lymphocytes recognizing tumor cells is now well established, a clear understanding of the functional status of these cells in vivo, and their impact on tumor growth has been so far elusive. In contrast, several factors have been described that potentially contribute to tumor cell evasion of the immune response (1), suggesting that potential antitumor activity of immune effectors may be impaired by mechanisms that ultimately result in tumor progression.
One of the strategies of tumor immune escape is represented by the acquisition of FasL expression that may enable cancer cells to deliver death signals to activated Fas-positive T lymphocytes (2, 3). FasL is a transmembrane type II protein belonging to the TNF protein superfamily, that plays a pivotal role in the induction of receptor (Fas)-mediated apoptosis. In fact, Fas–FasL interaction regulates important immune functions such as downmodulation of immunological responses, T cell activation–induced cell death, clonal downsizing, and control of peripheral tolerance to self-antigens (4). Apart from immunocompetent cells, FasL expression is additionally well documented in organs such as eye, central nervous system, and testis where it is thought to contribute to the maintenance of these immune privileged sites (4). Due to the functional properties of this molecule, it has been thus hypothesized that tumor cells could take advantage of FasL expression for mediating apoptosis of antitumor-activated T cells. Many authors have indeed shown that tumor cells of different histotypes (melanoma, colon, breast, esophageal cancers) do acquire FasL expression as detected both in vivo and in vitro (3). Additionally, such expression has been reported to negatively correlate with patient prognosis when evaluated on breast, ovarian, liver cancers, and melanoma specimens (3, 5, 6).
Although intriguing, this hypothesis is currently under evaluation, animating a debate based on controversial issues, such as the specificity of the different antibodies available for FasL detection or the usage of nonintron spanning primers for FasL detection by RT-PCR. The presence of infiltrating FasL-positive lymphocytes in tumor lesions has been additionally hypothesized to interfere with ex vivo FasL staining (7–10).
However, the recent use of specific reagents for the detection of FasL has indeed confirmed the expression of FasL in some tumor cell types (9, 11). Moreover, the concomitant presence of apoptotic T cells in FasL-expressing tumors (12, 13) and the negative prognostic role of FasL expression by neoplastic cells (5, 6, 14) suggest a potential efficacy of tumor counterattack in influencing in vivo tumor progression.
A relevant mechanism of FasL trafficking, occurring in NK cells and CTL, has been recently reported as involving specific intracellular transport of FasL on lysosomal-like vesicles that are unidirectionally polarized on the membrane of NK and T cells (15, 16) and a mechanism based on the release of FasL-bearing microvesicles (MVs)* has been also described to occur in FasL transfected cells (17). Indeed, melanoma cells contain specialized granules (melanosomes), which are the site of synthesis and storage of melanin and related molecules. These organelles have been clearly demonstrated to share phenotypic and functional features with lysosome vesicles and lytic granules (18). Additionally, a secretory pathway has been described in melanoma cells that involves multivesicular bodies (MVB) containing MVs which express tumor antigens such as MART-1 and tyrosinase (19). The physiologic fate of these vesicles is to be injected by melanocytes into keratinocytes in order to transfer melanin pigments.
On the basis of these data, we have reevaluated the role of FasL in tumor immune escape, taking into account the possibility that melanoma cells may share FasL expression pathway and intracellular trafficking with immunocompetent cells. Expression of FasL in melanoma cells was thus studied by investigating FasL localization in cytoplasmic organelles of various human melanoma cell lines and its possible implication in the counterattack of melanoma against immune cells. Here we report that melanoma cells do express FasL intracellularly, with a localization confined to MVB that contains melanosomes. FasL-bearing MVs can be released extracellularly, retaining their FasL expression and proapoptotic activity on Fas-sensitive lymphoid cells.
Materials and Methods
Antibodies and FACS® Analysis.
Cellular expression of FasL was evaluated after 24-h treatment with 10 μM KB8301 (4-[N-hydroxyamino]-2R-isobutyl-3S-methylsuccinyl)-L-3-[5,6,7,8-tetrahydro-1-naphthyl] alanine-N-methylamide; BD PharMingen), a metalloproteinase inhibitor known to block soluble FasL release (20). Wild-type and FasL-transfected 293 cells (transiently transfected using a DNA plasmid provided by H. Eibel, Clinical Research Unit for Rheumatology, University Hospital Freiburg, Germany) were used as negative and positive controls, respectively. After mechanical detachment, cells were stained with anti-FasL mAbs (clone G247 and clone NOK-1; both from BD PharMingen), recognizing two different epitopes of the FasL molecule, and with FITC-conjugated goat anti–mouse IgG (Biosource International). As negative control, an isotype-matched mAb followed by the FITC-conjugated anti–mouse IgG was used. Intracellular staining for detection of FasL was performed by previous permeabilization of melanoma cells with 70% methanol (at 4°C for 5 min) followed by incubation with anti-FasL G247 and FITC-conjugated anti–mouse IgG. Fluorescence was then analyzed by FACSCalibur™ flow cytometer (Becton Dickinson) and CELLQuest™ software (Becton Dickinson). Staining of melanosomes and MVs was additionally performed with anti–lysosome-associated membrane protein (LAMP)-2, anti-CD63 (both from BD PharMingen), and anti-gp100 (Immunotech) mAbs, followed by incubation with FITC goat anti–mouse IgG (Biosource International). An irrelevant isotype-matched mAbs (anti–TCR-αβ; Becton Dickinson), followed by the FITC-conjugated anti–mouse IgG, was used as negative control. Melanosomes and MVs were resuspended in 0.1 μ-filtered PBS (100 μl per sample) and primary mAb were added at a concentration of 10–30 μg/ml. After 1 h at 4°C, samples were then incubated with FITC-conjugated anti–mouse IgG (10 μg/ml) for 30 min at 4°C. Labeled melanosome and microvesicle suspensions were then diluted to 300 μl with filtered PBS and samples were analyzed by FACSCalibur™ and CELLQuest™ software. Melanosomes were detected as granules of the approximate mean size of 1 μm, while MVs had a size range of 100–600 nm, relative to standard beads of 6 μm size (CaliBRITE beads; Becton Dickinson).
Immunocytochemistry.
Human melanoma cells were attached to poly-l-lysine-covered glass chamber slides (Labtek) or spun onto glass slides (Shandon), as appropriate, fixed with 70% ethanol (EtOH) 10 min at 4°C, and stained by immunocytochemistry for anti-FasL (clones G247 and NOK-1) using the alkaline phosphatase anti-alkaline phosphatase (Dako) method or the peroxidase-anti-peroxidase (PAP) (Dako) method, in single and double staining, as appropriate (21). In single staining, cells were counterstained with Mayer's haematoxylin.
Electron and Immunoelectron Microscopy.
For transmission electron microscopy, melanoma cells, fixed with 2.5% glutaraldehyde, and postfixed in 1% OsO4, were dehydrated in alcohol and embedded in epoxy resin. For immunoelectron microscopy cells were fixed, after two washes in 0.1 M cacodylate buffer, with 3% paraformaldehyde in 0.1 M cacodylate buffer for 2 h. Then, they were embedded in 2% agar Noble in H2O, at 37°C. For subsequent embedding, the samples were dehydrated with increasing concentrations of EtOH. Infiltration of the samples was performed with increasing concentrations of Lowicryl HM20 resin (Taab) in 100% EtOH and finally in pure Lowicryl. The polymerization was performed in beem capsules by indirect UV irradiation (360 nm) at 4°C. For FasL immunolocalization in postembedding procedure, thin sections, collected on gold grids, were treated for 5 min with PBS containing 0.15% (wt/vol) glycin. After washing by quickly floating the grids on PBS drops containing 0.1% (wt/vol) BSA (Sigma-Aldrich), the sections were incubated overnight at 4°C with mAb G247 (1:10 diluted). After washing by floating the grids on PBS drops containing 0.1% (wt/vol) BSA for 1 h at room temperature, the sections were preincubated with PBS 0.1% (wt/vol) BSA for 10 min, washed in PBS containing 0.1% (wt/vol) BSA, and then labeled with a goat anti–mouse IgG 10-nm gold conjugate (1:10 diluted; Sigma-Aldrich) for 20 min, then washed in PBS containing 0.1% BSA for 20 min at room temperature. As negative control, sections were incubated with an irrelevant isotype-matched mAb or with a goat anti–mouse IgG gold alone.
For ultrathin cryosections cells were fixed with 4% paraformaldehyde plus 0.1% glutaraldehyde in PBS, pH 7.4, for 2 h at 4°C, washed, and embedded in 2% agarose low melting point (LMP) that was solidified on ice. Agarose blocks were infused with 2.3 M sucrose in PBS for 3 h at 4°C, frozen in LN2, and cryosectioned following the method by K.T. Tokuyasu (22), as described previously (23). Ultrathin cryosections were collected using sucrose and methyl-cellulose and incubated with specific mAb, then revealed with protein A-gold conjugates of different size (10 or 20 nm, as appropriate). Finally, ultrathin cryosections were stained with the solutions of 2% methylcellulose and 0.4% uranyl acetate before EM examination.
For electron microscopy of the isolated microvescicles, droplets of RPMI 1640 with suspended membranes from 70,000 g pellets were fixed with 2% paraformaldehyde in PBS and put on thin carbon film-coated grids for TEM observation and air-dried. FasL labeling was performed by the immunonegative stain technique. Briefly, the grids were incubated at 4°C overnight with the anti–human FasL mAb, G247, diluted 1:10, in PBS containing 1.0% BSA (Sigma-Aldrich). After washing with PBS, the grids were incubated with anti–mouse IgG-gold conjugate (Sigma-Aldrich; average diameter of gold particles 5 nm), and diluted 1:10 in PBS at room temperature for 1 h. Negative controls were performed by incubating samples with anti–mouse IgG2a or with immunoconjugate alone (data not shown). After washing with PBS, grids were negatively stained with 2.0% aqueous phosphotungstic acid for 2 min. Samples were examined with a Philips 208 transmission electron microscope (FEI Company).
Immunoprecipitation.
Adherent melanoma cells (106) were surface-labeled with sulfo-N-hydroxysuccinimide-biotin according to the manufacturer's instructions (Amersham Pharmacia Biotech). After extensive washing, the cells were lysed with lysing buffer (1% Triton X-100, 150 mM NaCl, 1 mM Tris-HCl, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 30 μg/ml KB8301).
Proteins corresponding to 106 cells equivalent were used for each specific immunoprecipitation. After preclearing with control mouse IgG, the supernatant was specifically immunoprecipitated using 2 μg/ml of NOK-1, G247 (BD PharMingen), W6.32 (an anti–HLA-class I monomorphic determinant), or control mouse IgG1. After washing with lysing buffer, the eluates were subjected to SDS-PAGE and electroblotted. The membrane was then incubated with strepavidin-HPR and developed with ECL detection system (Amersham Pharmacia Biotech).
Supernatants, Total Cell Lysates, Cytoplasmic-enriched Preparations and Isolation of Melanosomes, and MVs.
Supernatants from melanoma cell lines were harvested from 72 h confluent melanoma cell cultures, concentrated with centrifuge filter devices, Centricon (Millipore), with a cut off of 50 kD to enrich for soluble molecules <50 kD, aliquoted, and stored at −80°C. Jurkat cells (4 × 105 cells per milliliter) were stimulated with 50 μg/ml PHA for 5 min; after PHA removal by centrifugation and washing, they were resuspended in complete medium and cultured at 37°C for 1 h (16). Supernatants from resting or activated cells were collected, concentrated, and stored as described previously.
Total cell lysates were obtained lysing cells by an hypotonic solution (10 mM hepes, pH 6.9, 10 mM KCl, 0.3% aprotinin, and 0.1 mM PMSF), extensively homogenizing using a Potter-Elvehjem homogenizer and centrifuging the suspension for 10 min at 600 g. Total cell lysates were obtained from confluent melanoma cell cultures or from Jurkat cells, activated as described previously. Cytoplasm-enriched preparations were prepared by differential centrifugation starting from the homogenate. After 3 min at 1,000 g at 4°C, the supernatant containing cytoplasma, cytoskeleton and membrane fractions was centrifuged at 11,000 g for 30 min at 4°C in order to separate the cytoplasma (supernatant) from cell surface membranes and cytoskeleton (pellet).
Isolation of melanosomes was performed as described previously (24). Briefly, the homogenate from 109 melanoma cells resuspended in a solution of 0.25 M sucrose, underwent serial centrifugations (600 g for 10 min × 2; 11,000 g for 10 min × 2). The sediment was then suspended in 0.25 M sucrose. The suspension was layered over 1.7 M sucrose and centrifuged at 37,000 g for 1 h using the swing-out SW 41 rotor of the Beckman ultracentrifuge (Beckman Coulter). The sediment was suspended in 0.25 M sucrose, layered over 2.0 M sucrose, and centrifuged again at 37,000 g for 1 h. The obtained sediment contained isolated melanosomes.
Microvesicles from melanoma and resting/PHA-activated Jurkat cells were separated according to Martinez-Lorenzo et al. (16). After centrifugation at 10,000 g for 30 min at 4°C, the obtained supernatant was further ultracentrifugated at 100,000 g for 18 h at 4°C. The pellet, containing isolated MVs, was then resuspended in appropriate medium according to following treatments. Quantification of total proteins contained in melanosome or microvesicle preparations was evaluated by Lowry assay (Bio-Rad Laboratories) on organelle lysates. The mean protein recovery was 0.98 mg (range 0.28–1.7) for melanosomes, and 0.75 mg (range 0.31–1.2) for exosome-derived vesicles.
Western Blot Analysis.
Supernatants, total cell lysates, and subcellular fractions (cytoplasma, melanosomes, and microvesicles) from melanoma and Jurkat cells were analyzed for FasL protein expression by Western blot analysis. Isolated melanosomes and microvesicles were resuspended in lysis buffer (1% Triton X-100, 0.1% SDS in 0.1 M Tris/HCL, pH 7) with the addition of the 10 μM matrix metalloproteinase inhibitor KB8301 (BD PharMingen). After protein determination using the Lowry assay (Bio-Rad Laboratories), 75 μg of total protein of the different preparations were boiled for 6 min in SDS sample buffer containing β-mercaptoethanol, separated on a 10% SDS-PAGE, and then electroblotted onto a PVDF membrane. As a positive control 1 ng of rFasL (Alexis) was used. The membrane was stained using 2 μg/ml anti–human FasL mAb G247, developed with a secondary mouse anti–human IgG conjugated to horseradish peroxidase (BD Transduction Laboratories), and detected by SuperSignal detection system (Pierce Chemical Co.).
To further check purity of melanosome and microvesicle preparations, the following additional molecules were analyzed in Western blot analysis under reducing conditions: gp100 (on a 15% SDS-PAGE gel) protein; and markers specific for endoplastic reticulum, Golgi, mitochondria, and plasma membrane. The following mAb were used: anti–human gp100/pmel17 (clone HMB45; Dako); BiP/GRP78 (BD Transduction Laboratories); GM130 (BD Transduction Laboratories); mitochondria (clone MAB1273; Chemicon International, Inc.); HLA-class I (L31; provided by P. Giacomini, Istituto Regina Elena, Rome, Italy); and anti–caveolin-1 (Santa Cruz Biotechnology, Inc.).
To exclude any possible contamination, isolated microvesicles from FBS and from RPMI supplemented with 10% FBS were also analyzed for FasL expression in Western blot analysis, but the protein was never detected (data not shown).
Analysis of Apoptotic Activity.
Quantitative assessment of apoptosis was performed by cytofluorimetric analysis of propidium iodide (PI)-stained cells as described previously (25) with minor modifications and by annexin V binding assay. The occurrence of apoptosis was also monitored by Western blot analysis of caspase-8 activation. Briefly, Jurkat cells (106 cells per milliliter) were seeded in 24-well plates in 10% FBS RPMI 1640. rFasL at the concentration of 50–100 ng/ml or different concentrations of melanosomes or microvesicles were added. For assessing Fas-involvement, cells were preincubated for 30 min at 42°C with different concentrations of anti-Fas mAb (ZB4; Upstate BioTechnology). Apoptosis was analyzed after 15–18-h incubation. For analysis of PI-staining, PBS-washed cells were suspended in hypotonic solution containing 10 μg/ml PI, 0.1% sodium citrate, 0.1% Triton X-100, and RNase A (66 U/ml), and incubated 4 h at 4°C. The red fluorescence of individual nuclei was measured with a FACSCalibur™ flow cytometer (Becton Dickinson). Annexin V binding was measured using the Annexin V-Biotin Apoptosis Detection kit (Oncogene Research Products) according to manufacturer's instructions. After the final incubation in binding buffer containing FITC-streptavidin (Amersham Pharmacia Biotech) and PI (0.6 μg/ml), cells were incubated on ice and immediately analyzed by flow cytometry. Caspase-8 activation was monitored by the appearance of the activated form of 40 kD, using Western blot analysis. Proteins were separated on SDS-PAGE and immunoblotted. The nitrocellulose membrane was then incubated overnight at 4°C with a polyclonal Ab to caspase-8 (BD PharMingen). A rabbit antiactin Ab was used as control for loading. Ab binding was detected by enhanced chemiluminescence (Amersham Pharmacia Biotech).
For detecting lymphocyte FasL-mediated apoptosis induced by tumor cells, melanoma cells (at 105 cells per well, in U-bottomed 96-well plates) were cultured in 10% FBS RPMI 1640 for 2 h at 37°C, for allowing cell adhesion. 51[Cr]-labeled Jurkat cells (at 103 cells per well), in the presence or absence of neutralizing anti-Fas mAb ZB4 (50 ng/ml), were then added as target cells and the plates were incubated for 16 h 0.50 μl per well of supernatant were harvested, counted, and killing was calculated as percentage of lysis (26). Comparable results were obtained using targets labeled with 3[H]-TdR or 125IdU (data not shown). Melanoma supernatant was harvested from 72 h confluent melanoma cell cultures, concentrated with centrifuge filter devices, Centricon (Millipore), with a cut off of 50 kD to enrich for soluble molecules <50 kD, aliquoted, and stored at −80°C. Different dilutions of supernatant (starting from 1:2) were added to the cytotoxic assay. rFasL (at 100 ng/ml) was also used as positive control.
Results
FasL Intracellular Expression in Melanoma Cells.
To study the expression of FasL in melanoma cells, we first investigated the presence of this molecule at melanoma cell surface level. To avoid significant FasL release by metalloproteases, cells were pretreated with the metalloprotease inhibitor KB8301, which has been shown to effectively inhibit FasL shedding from cell membrane (20). As reported in Fig. 1 A, none of the melanoma lines examined expressed detectable levels of FasL molecules on their cell surface, as evaluated by staining with NOK-1 mAb, known to specifically recognize FasL (9, 27, 28). As positive control, a population of FasL-positive cells could be identified by the same Ab in FasL-gene transfected but not in wild-type 293 cells (Fig. 1 A). Comparable data were obtained with another specific anti-FasL mAb (G247) (9, 27, 28, and data not shown). The absence of detectable levels of FasL on the cell surface was confirmed by immunoprecipitation of cell surface biotin-labeled proteins using both G247 mAb and NOK-1. As shown in Fig. 1 B (lanes 2 and 3), no specific band compatible with FasL molecule could be precipitated from melanoma cell membranes. The same gel developed by the isotype IgG1 control mAb failed to show any band (Fig. 1 B, lane 4). As control of cell surface labeling and immunoprecipitation procedures, HLA-class I molecules were immunoprecipitated by W6.32 mAb and appeared as a 44-kD band (Fig. 1 B, lane 1). To further rule out the possibility that FasL could be present on melanoma cell surface and be functionally active, despite the undetectability by cytometry and immunoprecipitation analyses, we evaluated the ability of melanoma cells to directly induce apoptosis of Fas-positive lymphoid cells. No significant apoptosis could be detected by incubating melanoma with Jurkat cells which, on the contrary, underwent substantial cell death in the presence of rFasL (Fig. 1 C). The possibility that FasL undetectability could be caused by massive shedding from cell membrane was further investigated by analyzing melanoma cell supernatants for the presence of soluble FasL (Fig. 1 D). Concentrated supernatants from melanoma cell lines were analyzed by Western blot analysis with G247 mAb and showed no positivity for soluble FasL, which was on the contrary detected in supernatants from PHA-activated Jurkat cells as a 27-kD band. The absence of detectable levels of sFasL was also confirmed by ELISA (data not shown). The same supernatants from melanoma did not show any cytotoxic activity when incubated with Jurkat cells (Fig. 1 C), confirming the absence of any functional proapoptotic molecule.
Figure 1.


FasL cannot be detected on melanoma cell membrane and in culture supernatant. (A) Cytofluorimetric analysis. Melanoma and transiently FasL-gene transfected 293 cells (control) were stained with anti-FasL NOK-1 mAb (dark area), or with an isotype-matched IgG (clear area), after 24 h-treatment with KB8301 10 μM; fluorescence was analyzed by FACSCalibur™ and CELLQuest™ software; data refer to three representative melanoma lines, from a panel of 10 tested. (B) Immunoprecipitation of membrane proteins: cell surface biotin-labeled cells of Me15392 were immunoprecipitated with G247 (lane 2) and NOK-1 (lane 3) anti-FasL mAb, with the isotype-matched murine IgG1 negative control (lane 4), or with the anti–HLA-class I mAb W6.32, as positive control. Cell lysates were analyzed in Western blot analysis with Streptavidin-HPR. A specific band (44 kD) is detectable only in lysates immunoprecipitated with W6.32 (lane 1). (C) Proapoptotic activity of melanoma cells. 51[Cr]-labeled Jurkat cells (at 103cells per well), in the presence or absence of neutralizing anti-Fas mAb ZB4 (50 ng/ml), were incubated for 16 h with either melanoma cells (105cells per well), rFasL (100 ng/ml), or concentrated supernatant from 501mel culture (dilution 1:2). Killing was calculated as percentage of lysis. (D) Western blot analysis of FasL expression in supernatants from melanoma cell cultures. Melanoma supernantants were harvested from 72 h confluent cell cultures and concentrated by Centricon filter devices (cut-off 50 kD) (lanes 2 and 3). Concentrated supernatant from resting (lane 4) or PHA-activated (lane 5) Jurkat cells were additionally used as negative and positive controls, respectively. rFasL was also included (lane 1). Western blot analysis was stained with the anti-FasL G247 mAb. A 27-kD band, corresponding to soluble FasL, could be detected in supernatant from PHA-activated Jurkat cells.
Immunocytochemical analysis was then performed in different melanoma cell lines to evaluate possible intracellular expression of FasL, using G247 mAb. The results of immunocytochemistry clearly demonstrated that FasL was expressed intracellularly by all the melanoma cell lines examined (data in Fig. 2 refer to three melanoma lines, representative of a panel of 10 lines tested). Fig. 2 shows that the intensity of the staining for FasL varied among melanomas and that the staining was invariably localized in defined endocytic compartments, markedly represented in melanoma cells. Intracellular FasL expression was also confirmed by flow cytometry performed in the same melanoma cell lines after permeabilization with 70% methanol (Fig. 3 A).
Figure 2.

Immunocytochemical analysis of FasL expression in human melanoma cells. FasL expression in three representative melanoma lines (Me15392, 501mel, and Me30966, in A, B, and C, respectively), from a panel of 10 tested, is shown. Note (i) the striking localization of the FasL staining on well-defined cytoplasmic vesicles and (ii) the differential quantitative expression among the three cell lines. Immunophenotyping of fixed cells was performed using the PAP method. AEC was used as chromogen and Mayer's haematoxylin for the counterstaining. Original magnification: 2,500×.
Figure 3.


FasL intracellular localization in melanoma cells. (A) Cytofluorimetric analysis: melanoma cells were permeabilized with 70% methanol for 5 min on ice and subsequently stained with the anti-FasL G247 mAb (dark area) or with an isotype-matched IgG (clear area). Fluorescence was analyzed by FACSCalibur™ and CELLQuest™ software; data refer to three representative melanoma lines, from a panel of 10 tested. (B and C) Western blot detection of FasL in melanoma total cell lysates. Melanoma or PHA-activated Jurkat total cell lysates were stained by the anti-FasL G247 mAb. A significant positivity was detected in melanoma cell lysates (panel B, lanes 2–3), resembling the pattern observed in PHA-activated Jurkat cells (panel C, lane 2). Three major bands were detected in both melanoma and Jurkat cell lysates, corresponding to a molecular weight ranging between 40 and 33 kD and likely representing differently glycosylated forms of FasL molecule. (D) Western blot detection of FasL in cytoplasmic fractions of melanoma cells. Cytoplasmic-enriched preparations from melanoma cells were obtained as described in Materials and Methods. Western blot was stained with G247 mAb. Significant positivity (with bands ranging between 40 and 33 kD, with a predominance of the 40-kD band) was observed in both melanoma lines (lanes 2 and 3).
To validate cytochemical and cytofluorimetric data, the presence of FasL was further evaluated in the total lysate from melanoma cells. Indeed, Western blot analysis with the G247 mAb revealed a significant positivity (Fig. 3 B, lanes 2 and 3), resembling the pattern observed in PHA-activated Jurkat cells (Fig. 3 C, lane 2). Three major bands were detected in both melanoma and Jurkat cell lysates, corresponding to a molecular weight ranging between 40 and 33 kD (as compared with rFasL, estimated molecular weight 35 kD). This pattern, which is typically observed in Western blot analysis with G247 mAb (27), likely represents differently glycosylated forms of FasL molecule. Similar positivity was observed when cytoplasm-enriched preparations purified by differential centrifugations of total melanoma cell lysate were tested (Fig. 3 D, lanes 2 and 3). However, a clear predominance of the 40-kD band could be detected in cytoplasmic preparations as compared with total melanoma cell lysates, possibly due to a higher concentration of the mature, glycosylated form of FasL.
Altogether, these data strongly indicate that the expression of FasL molecules in these tumor cells is mostly confined to the intracellular compartment.
Immunoelectron microscopy analysis was used to further investigate FasL subcellular localization in human melanoma cells. The results showed that FasL immunolabeling was clearly confined to MVB (Fig. 4 A), specialized cellular compartments displaying internal membrane-bound vesicles (29). Within the MVB, FasL was expressed by vesicles that contain melanin in various stages of maturation (Fig. 4 B), suggesting the melanosomal origin of these structures. With the aim of further evaluating the nature of the FasL-expressing vesicles, we performed double-labeling experiments on cryosectioned cells. The results showed that FasL positive vesicles coexpressed both markers of lysosomes (i.e., CD63) and melanosomes (i.e., gp100). Fig. 4 C shows that MVB of melanoma cells are double labeled for FasL and CD63, while Fig. 4 D reports both coexpression of FasL and gp100 by MVB and the absence of staining in other vesicular structures, such as endoplasmic reticulum (ER). Figs. 4 E shows a colocalization of CD63 and gp100 in MVB, that was consistent with the costaining of FasL with either CD63 or gp100. Fig. 4 F is a higher magnification of a MVB showing the staining for FasL and gp100 in this structure. This set of results strongly indicates that FasL is localized in MVs coexpressing lysosome and melanosome markers and that its expression can be detected in MVB of melanoma cells.
Figure 4.

Immunoelectron microscopic analysis of FasL expression in human melanoma cells. (A and B) Immunoelectron microscopy obtained by Lowicryl HM20 resin embedment (see Materials and Methods) of melanoma cells (bars, 1 μm). (A) A multivesicular body containing melanosomes at different state of maturation labeled for FasL. (B) Magnification of the squared area in A, showing the specific FasL labeling on light and dark organelles corresponding to early (low melanin levels) and late (high melanin levels) melanosome differentation state, respectively. (C–F) Immunoelectron microscopy pictures obtained by ultrathin cryosections of melanoma cells (see Materials and Methods) (bars, 0.1 μm). (C) Double immunolabeling of FasL (large golds, 20 nm; arrow) and CD63 (small golds, 10 nm; arrowhead); (D) of FasL (large golds, 20 nm; arrow) and gp100 (small golds, 10 nm; arrowhead); small arrowheads indicate the ER free of gold particle staining; (E) double immunolabeling of CD63 (large golds, 20 nm; arrow) and gp100 (small golds, 10 nm; arrowhead); (F) higher magnification of a multivesicular body double stained for FasL (large golds, 20 nm; arrow) and gp100 (small golds, 10 nm; arrowhead).
FasL Expression and Apoptotic Activity of Purified Melanosomes.
On the basis of the immunoelectron microscopy data, we thus evaluated the expression of FasL on melanosomes purified from human melanoma cells. Western blot analysis was performed using proteins derived from melanosomal preparations isolated by sucrose gradient from the same melanoma lines previously tested for FasL intracellular expression. Staining with G247 mAb revealed the presence in melanosome preparations of the 40–33-kD band pattern (Fig. 5 A, lanes 2–4), similar to that detectable in melanoma cell lysates and in PHA-activated Jurkat cells (Fig. 3). These results confirmed the presence of FasL molecule in melanosome granules from melanoma cells, as previously suggested by immunocytochemistry and immunoelectron microscopy. Controls performed to check purity of melanosome preparations revealed the expression of the melanosomal marker gp100, and no significant contamination with Golgi, mitochondria, or plasma-membrane markers (Fig. 5 B). As described previously (30), the ER marker Bip/GRP78 was detectable in melanosome preparations (Fig. 5 B) and could not be eliminated by standard separation procedures. However, being ER-negative for FasL (Fig. 4 D), the presence of these subcellular contaminant are unlikely to contribute to FasL detection in melanosome preparations.
Figure 5.

FasL expression in melanosomes purified from melanoma cells. (A) Western blot analysis of purified melanosome preparations. Western blot analysis was performed using proteins derived from melanosomal preparations purified from melanoma lines by sucrose gradient. Staining with G247 mAb revealed the presence in melanosome preparations of the 40–33 kD band pattern (lane 3–5). (B) Purity controls of melanosome preparations. Controls performed to check purity of melanosome preparations revealed the expression of the melanosomal marker gp100, and no significant contamination with Golgi, mitochondria, or plasma-membrane markers. The ER marker Bip/GRP78 was detectable in melanosome preparations. (C) Cytofluorimetric analysis of FasL expression on purified melanosomes. Melanosomes were stained with anti-FasL NOK-1, anti-gp100, anti LAMP-2, and anti-CD63 mAbs, followed by incubation with FITC goat anti–mouse IgG (dark areas); an irrelevant isotype-matched Ab plus the FITC-conjugated goat anti–mouse IgG were used as negative control (clear areas). Fluorescence was analyzed by FACSCalibur™ and CELLQuest™ software. Melanosomes were detected as granules of the approximate mean size of 1 μm, relative to standard beads of 6 μm size.
Cytofluorimetric analysis was then performed to further characterize the antigenic profile, including FasL expression, of purified melanosomes. As shown in Fig. 5 C, melanosomes were positive for FasL as detected by NOK-1 mAb, thus suggesting a possible membrane localization of FasL in such organelles, along with the melanosomal molecule gp100, and the lysosomal markers LAMP-2 and CD63.
Then, we performed experiments aimed at evaluating the functional role of FasL expressed in melanosomes. To investigate the possible proapoptotic activity of melanoma-derived melanosomes, purified preparations were tested for induction of Fas-mediated apoptosis in FasL-sensitive Jurkat cells. Indeed, melanosomes induced significant and dose-dependent apoptosis of Jurkat cells, as detected by cytofluorimetric analysis with propidium and annexin staining (Fig. 6 A). The observed apoptosis was indeed FasL-mediated, since it could be significantly inhibited by preincubation of target cells with different concentrations of the anti-Fas mAb ZB4 (Fig. 6 A and B). ZB4-mediated inhibition of apoptosis appeared to be more effective with rFasL as compared with melanosomes, suggesting the presence in melanosome preparations of a higher concentration either of bioactive FasL or bystander proteins, possibly impairing Ab binding efficiency. In any case, these results provide evidence for the presence of functionally active FasL molecules in melanosomes derived from melanoma cells.
Figure 6.

Induction of Fas-mediated apoptosis by purified melanosomes. (A) Jurkat cells were incubated overnight in the presence of rFasL (100 ng/ml), or different doses of melanosomes (total protein concentration: 0.35 mg/ml), purified from 501mel melanoma line as described in Materials and Methods. Cells were incubated alone or after treatment with ZB4 mAb (50 ng/ml) and harvested 18 h later. Apoptosis was then evaluated by propidium and annexin staining and FACS®analysis and expressed as percentage of apoptotic cells. (B) Jurkat cells were pretreated with different concentrations of ZB4 and incubated with 100 μl per well melanosomes. Apoptosis was then evaluated by propidium and annexin staining and FACS®analysis.
Release of MVs by Melanoma Cells and their Apoptotic Activity.
Previous studies have shown that a secretion of MVs, or exosome-like particles, underlies the formation of MVB in various cells types (29). Thus, we performed a series of experiments aimed at evaluating the possible secretion of MVs expressing FasL by human melanoma cells. A preliminary analysis by immunocytochemistry showed that human melanoma cells cultured in chamber-slides exerted a straightforward polarization of FasL staining on their interdigitated structures (Fig. 7 A). Further observations suggested a secretory behavior of the FasL-bearing MVs (Fig. 7 B) and in some cases revealed FasL-positive MVB-like structures with a clear secretory behavior, together with the presence of clusters of FasL-positive vesicles 8–10 mm away from the plasma membrane of melanoma cells (Fig. 7 C). Immunocytochemistry is not suitable for the demonstration of the MVB secretory behavior, nor for the identification of exosome-like particles. Thus, we performed immunoelectron microscopy in order to define both MVB secretory activity and the presence of FasL-bearing vesicles in human melanoma cells. Fig. 8 A and B clearly show that FasL-immunolabeled MVs degranulate from MVB of melanoma cells in the extracellular environment. Fig. 8 B also shows that the ER beneath the plasma membrane does not stain for FasL.
Figure 7.

Immunocytochemical analysis of MVs released by melanoma cells. (A) A cluster of human melanoma cells adherent on glass chamber slides, showing a high level of FasL expression (as detected by G247 mAb), polarized on cellular filopodia and interdigitations (arrowheads). (B) Higher magnification of the FasL stainings on the tip of a melanoma cell interdigitation, showing both the unidirectional secretory behavior (arrowhead) and the marked polar concentration (arrow) of the FasL staining. (C) High electronic magnification of a defined field of a melanoma cell where a cluster of FasL-positive vesicles is under degranulation and a group of three isolated FasL-positive vesicles is detectable 8–10 μm away from the cell membrane (arrow). Immunophenotyping of fixed cells was performed using the PAP method; AEC was used as chromogen and Mayer's haematoxylin for the counterstaining. Final original magnifications: (A) 1,000×; (B) 2,500×; and (C) 3,000×.
Figure 8.
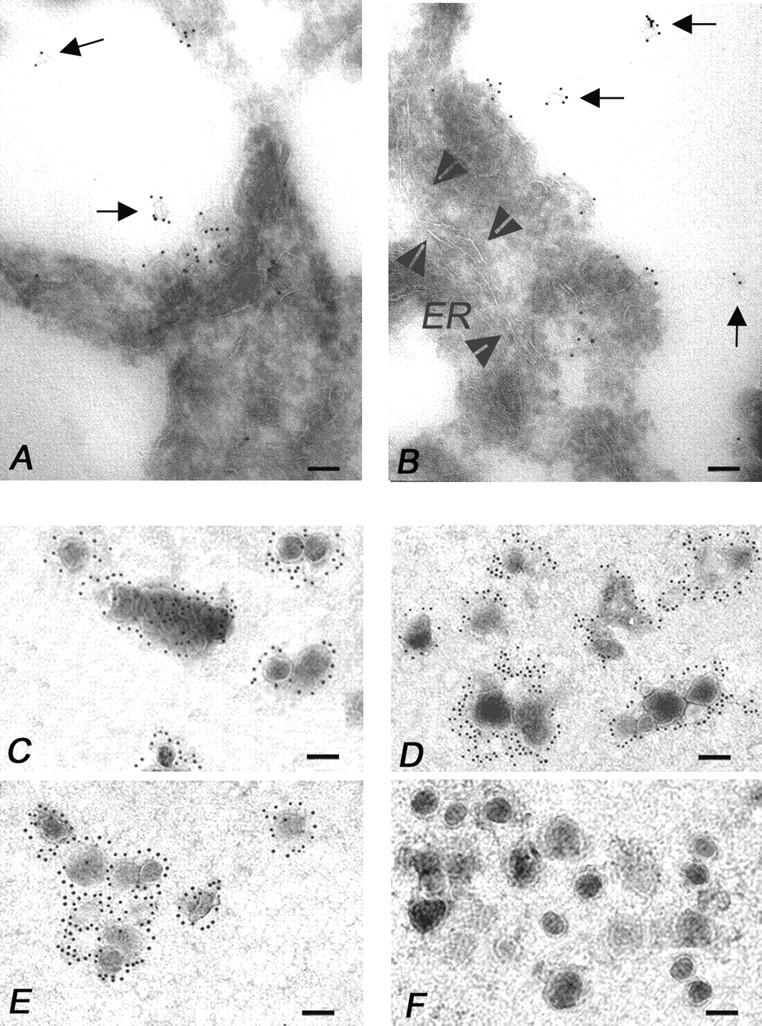
Immunoelectron microscopy analysis of multivesicular bodies, secretory vesicles, and isolated MVs from melanoma cells. (A and B) Immunoelectron microscopy on ultrathin cryosections of human melanoma cells (see Materials and Methods). (A) Note the clear FasL immunolabeling of exosome-like particles (arrows) degranulating in the extracellular environment by a multivesicular body. In B, multiple degranulation sites are shown with FasL immunolabeled exosome-like vesicles (arrows) scattered into the extracellular environment. Arrowheads outline a wide ER vesicle (ER) that appears negative for the FasL immunolabeling. (C–F) Electron microscopy of the 100,000 g pellet immunogold labeled for (C) FasL, (D) CD63, (E) gp100, and (F) Golgi, respectively. The pellet was composed of 100–200 nm MVs, often detected as aggregates, showing abundant FasL, CD63, and gp100 immunolabeling, while they do not stain for Golgi markers (bars, 0.1 μm).
These particles, as well as their secretory pathway, highly resembled those described in various immunocompetent cells (15, 16). Thus, we performed experiments aimed at obtaining purified preparations of MVs from melanoma cell supernatant. Immunoelectron microscopy analysis of the pellets obtained by sequential ultracentrifugation of melanoma cell culture supernatant revealed the presence of vesicles of 100–200 nm in size showing a marked positivity for FasL (Fig. 8 C), CD63 (Fig. 8 D), and gp100 (Fig. 8 E) stainings, being negative for the Golgi immunolabeling (Fig. 8 F). The vesicles, often detectable as aggregates, were morphologically similar to those contained in the MVB and excreted extracellularly (Fig. 8 A and B).
Analysis of MVs isolated from melanoma cell supernatant, as performed by Western blot, showed that FasL was indeed detectable (Fig. 9 A, lanes 4, 6, and 7) in such preparations, with an ∼35-kD band, highly resembling the one observed in MVs from PHA-activated Jurkat cells (Fig. 9 A, lane 2).
Figure 9.


FasL expression on MVs purified from melanoma cell supernatant. (A) Western blot analysis. Analysis of MVs isolated from melanoma cell supernatant by serial ultracentrifugations, as performed by Western blot and staining with G247 mAb. An ∼35-kD band can be observed in MVs from melanoma lines (lanes 4, 6, and 7), resembling the one observed in MVs from PHA-activated Jurkat cells (lane 2). Only MV from PHA-activated, and not from resting Jurkat cells (lane 3) showed positivity for FasL, suggesting that this molecule unlikely derived from FBS used for cell culture. (B) Purity controls of microvesicle preparations. Controls performed to check purity of microvesicle preparations from melanoma cell supernatant revealed the expression of the melanosomal marker gp100, and no significant contamination with Golgi, ER, or mitochondria. (C) Cytofluorimetric analysis of FasL expression on purified MVs. MVs from 501mel line were stained with anti-FasL NOK-1, anti-gp100, anti–LAMP-2, and anti-CD63 mAbs, followed by incubation with FITC goat anti–mouse IgG (dark areas); an irrelevant isotype-matched Ab plus the FITC-conjugated goat anti–mouse IgG were used as negative control (clear areas). Fluorescence was analyzed by FACSCalibur™ and CELLQuest™ software. MVs were detected as granules with a size range of 100–600 nm, relative to standard beads of 6 μm size.
It should be noted that FasL expressed in microvesicle preparations did derived from melanoma and activated Jurkat cells, respectively, and not from FBS present in culture media. In fact, only MVs isolated from PHA-activated Jurkat cells showed strong positivity for FasL, while no positivity was detected in MVs isolated from nonactivated cells (Fig. 9 A, lane 3), maintained under the same culture conditions (i.e., in 10% FBS). Further analysis of melanoma-derived MVs showed the concomitant expression in these preparations of the melanosomal marker gp100 (Fig. 9 B) and the absence of any ER, Golgi, and mitochondria markers (Fig. 9 B). These vesicles resulted positive for class I HLA expression, as expected (19), but showed no positivity for caveolin, which was however detectable in purified melanoma cell membranes (data not shown).
On particulate, ultracentrifugable fractions from melanoma cell supernatants we additionally performed FACS® analysis, which confirmed the presence of MVs (with a diameter ≥ 100 nm) expressing FasL, in addition to melanosomal and lysosomal antigens (gp100, LAMP-2, and CD63) (Fig. 9 C).
Experiments were finally performed to assess the proapoptotic activity of the MVs by exploiting the same approach used for melanosomes. The results clearly showed that purified MVs from melanoma cells expressed bioactive FasL and induced ZB4-blockable, thus Fas-mediated apoptosis of Jurkat cells, as detected by propidium and annexin staining (Fig. 10 A). However, the observed apoptosis was significantly lower than that mediated by MVs derived from PHA-activated Jurkat cell supernatants, suggesting a less efficient activity in melanoma as compared with immune competent cells. Specificity of Jurkat apoptosis induced by melanoma-derived MVs was further confirmed by the dose-dependent inhibition obtained in the presence of the anti-Fas mAb ZB4 (Fig. 10 B). An additional evidence for the induction of a specific apoptotic pathway was provided by the analysis of caspase-8 activation, that showed the appearance of the cleaved activated form of 40 kD after incubation with both melanoma-derived MVs and isolated melanosomes, undetectable though when Jurkat cells were pretreated with ZB4 (Fig. 10 C).
Figure 10.


Apoptotic activity of MVs isolated from melanoma cell supernatant. (A) Jurkat cells were incubated overnight in the presence of rFasL (100 ng/ml), or 50 μl/ml MVs isolated from resting Jurkat cells, PHA-activated Jurkat cells or 501mel cells, purified as described in Materials and Methods (protein concentrations: 13.3, 15, and 5.1 mg/ml, respectively). Target cells were used with or without pretreatment with ZB4 mAb (50 ng/ml) and harvested 18 h later. Evaluation of apoptosis induction as detected by FACS® analysis for propidium and annexin staining. (B) Jurkat cells were pretreated with different concentrations of ZB4 and incubated with rFasL (100 ng/ml) or 50 μl per well MVs from 501mel line. Apoptosis was then evaluated by propidium and annexin staining and FACS®analysis. (C) Western blot analysis of caspase-8 cleavage. Control loading is shown by actin. Jurkat cells, with (Jurkat/ZB4) or without preexposure to ZB4 mAb (50 ng/ml), were incubated overnight with different apoptotic stimuli: lane 1, Jurkat; 2, Jurkat/ZB4; 3, Jurkat + rFasL (100 ng/ml); 4, Jurkat/ZB4 plus rFasL; 5, Jurkat plus melanosomes (100 μl/ml, protein concentration 3.5 mg/ml); 6, Jurkat/ZB4 plus melanosomes; 7, Jurkat plus 501mel MVs (50 μl/ml, protein concentration 5.1 mg/ml); Jurkat/ZB4 plus 501mel MVs. Induction of apoptosis was detected as a 40-kD band, representing the cleaved activated form of activated caspase-8. No band is detectable when Jurkat cells were pretreated with ZB4.
These functional data suggest that melanoma cells can release MVs which retain the functional and phenotypic pattern observed in cytoplasmatic melanosomes and can exert FasL-dependent proapoptotic activity on Fas-sensitive target cells.
Discussion
In this study, we describe a novel pathway of tumor counterattack through the secretion by melanoma cells of FasL-positive MVs, highly supporting the role of FasL in tumor escape from immune effector cells. The results of our study show that (i) FasL is indeed expressed in the cytoplasm of human melanoma cells; (ii) its expression is confined to MVB where FasL is coexpressed with both melanosomal and lysosomal markers; (iii) purified melanosomes are able to induce a Fas-mediated apoptosis in Fas-sensible human T cells; and (iv) melanosomes may undergo a secretory pathway through MVB, which also release MVs expressing FasL, melanosomal, and lysosomal markers and inducing Fas-mediated T cell apoptosis. These results clearly conciliate the opposite views on the FasL-mediated cancer counterattack, since they provide a comprehensive explanation to the controversial issue on FasL expression by tumor cells. In fact, we confirm the absence of detectable FasL expression at cell membrane level and the lack of proapoptotic activity mediated by whole melanoma cells, as reported by some authors (7, 8). At the same time, however, we provide an explanation for other results showing the presence of detectable levels of FasL in the intracellular compartment of tumor cells (10) and the prognostic role of FasL in tumor progression (5, 6, 9, 14).
The expression of FasL in lysosome-related vesicles or other secretory granules has been indeed reported as a feature of immunocompetent cells (15, 16). However, the MVB of melanoma cells showed some peculiarities, such as the presence of melanosome-like electrondense vesicles, secreted in the extracellular environment as FasL-positive vesicles detectable by both immunocytochemistry and immunoelectron microscopy. Microvescicles can be separated from melanoma cell culture supernatant and are able to trigger Fas-mediated apoptosis of sensitive human lymphoid target cells. Notably, these MVs displayed lysosomal features in terms of expression of lysosomal antigens such as CD63 and LAMP-2 and showed morphological features similar to those described in immunocompetent cells (15, 16). On the basis of our data, we cannot rule out the possibility that vesicles of a size inferior to the detection limit of cytofluorimetric analysis (presently classified as ‘exosomes’) could be also present in our melanoma supernatants. In fact, the evidence of MVB formation in melanoma cells, together with the information from immunoelectron microscopy on supernatant ultracentrifugates, suggest that exosome-like particles, known to be released by melanoma cells (19) and by other cell types (29, 31), may contribute to the phenomenon of microvesicle-induced FasL-mediated apoptosis here described.
The release of membrane-associated FasL through secretion of intracellular organelles represents an effective mechanism for mediating apoptosis, given the higher efficiency in triggering Fas-mediated apoptosis of membrane-linked FasL as compared with its soluble form (32) and the evidence that conversion of membrane FasL to its soluble form leads to downregulation of the proapoptotic activity (33). A vectorial transport of FasL has been recently described on lysosomal-like vesicles in NK and T cells (15, 16). On the basis of these data it has been hypothesized that this targeting of FasL to the secretory lysosomes is specific to cells of hematopoietic lineage, as a sort of tight regulatory mechanism of FasL expression in cells that are committed to kill target cells through the Fas-mediated pathway (15). This is further supported by studies showing that FasL is stored in the secretory granules of monocytes (34). However, it is well known that melanosomes, as well as lytic granules of NK cells and phagosomes, belong to the family of the lysosomal-like organelles, expressing the same lysosomal-associated antigens (18). We describe here FasL-expressing melanosome-like organelles secreted by human melanoma cells as vesicles able to deliver a Fas-mediated cell death to Fas-sensible lymphoblastoid cells. These data suggest that melanoma cells may share with NK cells, and probably with monocytes, (i) a similar pathway of FasL targeting to lysosomal-like particles and (ii) a similar secretory pathway of these particles mediating a Fas-mediated cell death that does not necessarily imply a cell-to-cell contact. It seems hence conceivable that secretion of FasL-expressing MVs can be a more efficient mechanism in inducing apoptosis of Fas-expressing cells, and that this mechanism may well operate in physiologic (NK, CTL, monocytes, dendritic cells) as well as in aberrant (tumor cells) conditions. In fact, it has been suggested that the cytoplasmic tail of FasL expressed in hematopoietic cells contains a sorting motif that targets the protein to secretory lysosomes. Additionally, this specialized motif appears to be shared by other members of the TNF family (e.g., CD40L; reference 35). On the basis of these considerations it has been further speculated that the accumulation of proteins in the secretory lysosomes may represent a generalized mechanism for the control of the release of proteins involved in the regulation of the immune response. Since tumor cells may lack such type of tight control, it is thus conceivable that melanoma cells could mediate a continuous release and/or degranulation of FasL-bearing vesicles that indiscriminately kill all the Fas-sensible cells encountered in the extracellular environment. This phenomenon would have no consequences on melanoma cells, being these cells insensitive to Fas triggering, and therefore refractory to the autocrine FasL-vesicles-induced cell death (26, 36, 37). On the basis of our data, it is still unclear whether the induction of lymphocyte death by FasL-bearing MVs could be a specific feature of melanoma cells or it could be shared by other tumors. In fact, although in some histological types (e.g., colon carcinoma) this molecule has been located exclusively in the cytoplasm, other tumors, such as squamous cell carcinoma, have been clearly shown to stably express FasL at the plasma membrane level (11).
Our data support the relevance of FasL-mediated apoptosis in tumor evasion from immunological control. Other mechanisms can play a significant role in reducing the in vivo efficacy of CTL response to melanoma antigens (1) and antitumor T cell subpopulations resistant to FasL-induced apoptosis have been identified at least in vitro (26). However, this functional pathway of inducing lymphocyte apoptosis through the release of FasL-positive MVs may indeed play a significant role in eliminating the most effective component of the antitumor T cell response in vivo.
While we cannot rule out the possibility that FasL-bearing granules could instead deliver proinflammatory signals, as indeed described in other experimental systems (38, 39), it is likely, given the natural history of neoplastic disease, that this mechanism may contribute in tilting the balance toward an immunosuppressive environment at tumor site. Thus, we believe that future studies aimed at understanding the mechanism responsible for this phenomenon, together with the identification of therapeutical strategies for specifically inhibiting tumor exocytosis, could contribute to ameliorate immunological control of tumor growth in cancer patients.
Acknowledgments
The authors wish to express their gratitude to Drs. Giovina Ruberti and Giuliana Papoff (Institute of Cell Biology, National Research Council, Rome, Italy), and Hermann Eibel (Clinical Research Unit for Rheumatology, University Hospital Freiburg, Germany) for providing reagents and for their invaluable support and discussion. We also thank Agata Cova, Gloria Sovena, Nives Carenini, and Anna Maria Zaccheddu for the skillful technical work, and Dr. Simona Galuzzi for manuscript editing.
This work was supported by grants from AIRC (Associazione Italiana per la Ricerca sul Cancro), Milano, Italy and from the Italian Ministry of Health, Rome, Italy.
G. Andreola and L. Rivoltini contributed equally to this work.
Footnotes
Abbreviations used in this paper: ER, endoplasmic reticulum; EtOH, ethanol; LAMP, lysosome-associated membrane protein; MVB, multivesicular bodies; MV, microvesicle; PAP, peroxidase-anti-peroxidase; PI, propidium iodide.
References
- 1.Marincola, F.M., E.M. Jaffee, D.J. Hicklin, and S. Ferrone. 2000. Escape of human solid tumors from T-cell recognition: molecular mechanisms and significance. Adv. Immunol. 74:181–273. [DOI] [PubMed] [Google Scholar]
- 2.Hahne, M., D. Rimoldi, M. Schroter, P. Romero, M. Schreier, L.E. French, P. Schneider, T. Bornand, A. Fontana, D. Lienard, et al. 1996. Melanoma cell expression of Fas(Apo-1/CD95) ligand: implications for tumor immune escape. Science. 274:1363–1366. [DOI] [PubMed] [Google Scholar]
- 3.O'Connell, J., M.W. Bennett, G.C. O' Sullivan, J.K. Collins, and F. Shanahan. 1999. The Fas counterattack: cancer as a site of immune privilege. Immunol. Today. 20:46–52. [DOI] [PubMed] [Google Scholar]
- 4.Siegel, R.M., F.K. Chan, H.J. Chun, and M.J. Lenardo. 2000. The multifaceted role of Fas signaling in immune cell homeostasis and autoimmunity. Nat. Immunol. 1:469–474. [DOI] [PubMed] [Google Scholar]
- 5.Reimer, T., C. Herrnring, D. Koczan, D. Richter, B. Gerber, D. Kabelitz, K. Friese, and H.J. Thiesen. 2000. FasL:Fas ratio - a prognostic factor in breast carcinomas. Cancer Res. 60:822-828. [PubMed] [Google Scholar]
- 6.Soubrane, C., R. Mouawad, E.C. Antoine, O. Verola, M. Gil-Delgado, and D. Khayat. 2000. A comparative study of Fas and Fas-ligand expression during melanoma progression. Br. J. Dermatol. 143:307–312. [DOI] [PubMed] [Google Scholar]
- 7.Restifo, N.P. 2001. Countering the “counterattack” hypothesis. Nat. Med. 7:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Restifo, N.P. 2000. Not so Fas: re-evaluating the mechanisms of immune privilege and tumor escape. Nat. Med. 6:493–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O'Connell, J., A. Houston, M.W. Bennett, G.C. O'Sullivan, and F. Shanahan. 2001. Immune privilege or inflammation? Insights into the Fas ligand enigma. Nat. Med. 7:271–274. [DOI] [PubMed] [Google Scholar]
- 10.Favre-Felix, N., A. Fromentin, A. Hammann, E. Solary, F. Martin, and B. Bonnotte. 2000. Cutting edge: the tumor counterattack hypothesis revisited: colon cancer cells do not induce T cell apoptosis via the Fas (CD95, APO-1) pathway. J. Immunol. 164:5023–5027. [DOI] [PubMed] [Google Scholar]
- 11.Gastman, B.R., Y. Atarshi, T.E. Reichert, T. Saito, L. Balkir, H. Rabinowich, and T.L. Whiteside. 1999. Fas ligand is expressed on human squamous cell carcinomas of the head and neck, and it promotes apoptosis of T lymphocytes. Cancer Res. 59:5356–5364. [PubMed] [Google Scholar]
- 12.Okada, K., K. Komuta, S. Hashimoto, S. Matsuzaki, T. Kanematsu, and T. Koji. 2000. Frequency of apoptosis of tumor-infiltrating lymphocytes induced by Fas counterattack in human colorectal carcinoma and its correlation with prognosis. Clin. Cancer Res. 6:3560–3564. [PubMed] [Google Scholar]
- 13.Bennett, M.W., J. O'Connell, G.C. O'Sullivan, C. Brady, D. Roche, J.K. Collins, and F. Shanahan. 1998. The Fas counterattack in vivo: apoptotic depletion of tumor-infiltrating lymphocytes associated with Fas ligand expression by human esophageal carcinoma. J. Immunol. 160:5669–5675. [PubMed] [Google Scholar]
- 14.Munakata, S., T. Enomoto, M. Tsujimoto, Y. Otsuki, H. Miwa, H. Kanno, and K. Aozasa. 2000. Expression of Fas ligand and other apoptosis-related genes and their prognostic significance in epithelial ovarian neoplasms. Br. J. Cancer. 82:1446–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bossi, G., and G.M. Griffiths. 1999. Degranulation plays an essential part in regulating cell surface expression of FasL in T cells and natural killer cells. Nat. Med. 5:90–96. [DOI] [PubMed] [Google Scholar]
- 16.Martínez-Lorenzo, M.J., A. Anel, S. Gamen, I. Monleón, P. Lasierra, L. Larrad, A. Piñeiro, M.A. Alava, and J. Naval. 1999. Activated human T cells release bioactive Fas Ligand and APO2 ligand in microvesicles. J. Immunol. 163:1274–1281. [PubMed] [Google Scholar]
- 17.Jodo, S., S. Xiao, A. Hohlbaum, D. Strehlow, A. Marshak-Rothstein, and S.T. Ju. 2001. Apoptosis-inducing membrane vesicles. A novel agent with unique properties. J. Biol. Chem. 276:39938–39944. [DOI] [PubMed] [Google Scholar]
- 18.Dell'Angelica, E.C., C. Mullins, S. Caplan, and J.S. Bonifacino. 2000. Lysosome-related organelles. FASEB J. 14: 1265–1278. [DOI] [PubMed] [Google Scholar]
- 19.Wolfers, J., A. Lozier, G. Raposo, A. Regnault, C. Thery, C. Masurier, C. Flament, S. Pouzieux, F. Faure, T. Tursz, et al. 2001. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 7:297–303. [DOI] [PubMed] [Google Scholar]
- 20.Kayagaki, N., A. Kawasaki, T. Ebata, H. Ohmoto, S. Ikeda, S. Inoue, K. Yoshino, K. Okumura, and H. Yagita. 1995. Metalloproteinase-mediated release of human Fas ligand. J. Exp. Med. 182:1777–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parlato, S., A.M. Giammarioli, M. Logozzi, F. Lozupone, P. Matarrese, F. Luciani, M. Falchi, W. Malorni, and S. Fais. 2000. CD95 (APO-1/Fas) linkage to the actin cytoskeleton through ezrin in human T lymphocytes: a novel regulatory mechanism of the CD95 apoptotic pathway. EMBO J. 19:5123–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tokuyasu, K.T. 1973. A technique for ultracryotomy of cell suspensions and tissues. J. Cell Biol. 57:551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Torrisi, M.R., M. Gentile, G. Cardinali, M. Cirone, C. Zompetta, L.V. Lotti, L. Frati, and A. Faggioni. 1999. Intracellular transport and maturation pathway of human herpesvirus 6. Virology. 257:460–471. [DOI] [PubMed] [Google Scholar]
- 24.Menon, I.A., and H.F. Haberman. 1974. Isolation of melanin granules. Methods Enzymol. 31:389–394. [DOI] [PubMed] [Google Scholar]
- 25.Spinozzi, F., E. Agea, O. Bistoni, A. Travetti, G. Migliorati, R. Moraca, I. Nicoletti, C. Riccardi, F.P. Paoletti, R. Vaccaro, and A. Bertotto. 1995. T lymphocytes bearing the γδ T cell receptor are susceptible to steroid-induced programmed cell death. Scand. J. Immunol. 41:504–508. [DOI] [PubMed] [Google Scholar]
- 26.Rivoltini, L., M. Radrizzani, P. Accornero, P. Squarcina, C. Chiodoni, A. Mazzocchi, C. Castelli, P. Tarsini, V. Viggiano, F. Belli, et al. 1998. Human melanoma-reactive CD4+ and CD8+ CTL clones resist Fas ligand-induced apoptosis and use Fas/Fas ligand-independent mechanisms for tumor killing. J. Immunol. 161:1220–1230. [PubMed] [Google Scholar]
- 27.Papoff, G., G. Stassi, R. De Maria, C. Giordano, A. Galuzzo, M. Bagnasco, G. Ruberti, and R. Testi. 1998. Constitutive expression of FasL in thyrocytes. Technical comments. Science. 279:2015a. [Google Scholar]
- 28.FASL (CD178). 2001. Available at http://www.ncbi.nlm.nih. gov/PROW/guide/333879674_g.htm.
- 29.Denzer, K., M.J. Kleijmeer, H.F. Heijnen, W. Stoorvogel, and H.J. Geuze. 2000. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J. Cell Sci. 113:3365–3374. [DOI] [PubMed] [Google Scholar]
- 30.Kushimoto, T., V. Basrur, J. Valencia, J. Matsunaga, W.D. Vieira, V.J. Ferrans, J. Muller, E. Appella, and V.J. Hearing. 2001. A model for melanosome biogenesis based on the purification and analysis of early melanosomes. Proc. Natl. Acad. Sci. U.S.A. 98:10698–10703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raposo, G., H.W. Nijman, W. Stoorvogel, R. Liejendekker, C.V. Harding, C.J. Melief, and H.J. Geuze. 1996. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 183:1161–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka, M., T. Itai, M. Adachi, and S. Nagata. 1998. Downregulation of Fas ligand by shedding. Nat. Med. 4:31–36. [DOI] [PubMed] [Google Scholar]
- 33.Schneider, P., N. Holler, J.L. Bodmer, M. Hahne, K. Frei, A. Fontana, and J. Tschopp. 1998. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J. Exp. Med. 187:1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kiener, P.A., P.M. Davis, B.M. Rankin, S.J. Klebanoff, J.A. Ledbetter, G.C. Starling, and W.C. Liles. 1997. Human monocytic cells contain high levels of intracellular Fas ligand: rapid release following cellular activation. J. Immunol. 159:1594–1598. [PubMed] [Google Scholar]
- 35.Henn, V., J.R. Slupsky, M. Grafe, I. Anagnostopoulos, R. Forster, G. Muller-Berghaus, and R.A. Kroczek. 1998. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 391:591–594. [DOI] [PubMed] [Google Scholar]
- 36.Irmler, M., M. Thome, M. Hahne, P. Schneider, K. Hofmann, V. Steiner, J.L. Bodmer, M. Schroter, K. Burns, C. Mattmann, et al. 1997. Inhibition of death receptor signals by cellular FLIP. Nature. 388:190–195. [DOI] [PubMed] [Google Scholar]
- 37.Raisova, M., M. Bektas, T. Wieder, P. Daniel, J. Eberle, C.E. Orfanos, and C.C. Geilen. 2000. Resistance to CD95/Fas-induced and ceramide-mediated apoptosis of human melanoma cells is caused by a defective mitochondrial cytochrome c release. FEBS Lett. 473:27–32. [DOI] [PubMed] [Google Scholar]
- 38.Seino, K., N. Kayagaki, K. Okumura, and H. Yagita. 1997. Antitumor effect of locally produced CD95 ligand. Nat. Med. 3:165–170. [DOI] [PubMed] [Google Scholar]
- 39.Owen-Schaub, L.B., K.L. van Golen, L.L. Hill, and J.E. Price. 1998. Fas and Fas ligand interactions suppress melanoma lung metastasis. J. Exp. Med. 188:1717–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]