

Abstract
Transforming growth factor (TGF)-β is the prototype in a family of secreted proteins that act in autocrine and paracrine pathways to regulate cell development and function. Normal cells typically coexpress TGF-β receptors and one or more isoforms of TGF-β, thus the synthesis and secretion of TGF-β as an inactive latent complex is considered an essential step in regula-ting the activity of this pathway. To determine whether intracellular activation of TGF-β results in TGF-β ligand–receptor interactions within the cell, we studied pristane-induced plasma cell tumors (PCTs). We now demonstrate that active TGF-β1 in the PCT binds to intracellular TGF-β type II receptor (TβRII). Disruption of the expression of TGF-β1 by antisense TGF-β1 mRNA restores localization of TβRII at the PCT cell surface, indicating a ligand-induced impediment in receptor trafficking. We also show that retroviral expression of a truncated, dominant-negative TβRII (dnTβRII) effectively competes for intracellular binding of active ligand in the PCT and restores cell surface expression of the endogenous TβRII. Analysis of TGF-β receptor–activated Smad2 suggests the intracellular ligand–receptor complex is not capable of signaling. These data are the first to demonstrate the formation of an intracellular TGF-β–receptor complex, and define a novel mechanism for modulating the TGF-β signaling pathway.
Keywords: receptor, trafficking, intracellular, signal-transduction, plasmacytoma
Introduction
Among the effects of TGF-β, the regulation of cell growth, cell death, differentiation, and genomic stability appear to be of particular importance (1, 2). The loss of responsiveness to TGF-β represents a significant step in the process of carcinogenesis (3), and several mechanisms underlying the development of TGF-β resistance have been identified (2). These mainly involve a disruption in either the expression or function of components of the TGF-β signaling pathway. TGF-β transduces signals via heteromeric complexes of the type I TGF-β (TβRI)*and type II TGF-β (TβRII) serine/threonine kinase receptors (4). After ligand binding to TβRII, TβRI is recruited into the complex and activated by TβRII-dependent phosphorylation, thereby enabling it to transduce signals through downstream mediators such as the Smad family of proteins (5).
The TGF-β ligand–receptor system has rapidly emerged as an important tumor suppressor pathway that acts to restrain cellular proliferation and to regulate differentiation (1). The first association between resistance to growth inhibition by TGF-β and lack of TβRII receptor expression was reported in retinoblastoma cells (6). Such loss of TβRII expression has since been reported in several types of human cancer, including small cell cancer of the lung (7), hepatoma (8), gastric (9), squamous cell (10), esophageal (11), and breast cancer (12). Known mechanisms underlying receptor down-regulation include mutations associated with the microsatellite instability phenotype (13), mutations of the TβRII gene promoter (14), transcriptional repression by the EWS-FLI1 oncogene (15), and DNA methylation of CpG islands in the TβRII promoter (16). Loss of TGF-β receptors at the cell surface has also been described in the absence of gross structural changes, mutations, or transcriptional repression, which suggests that alternative pathways of receptor deregulation must exist.
The role of the TGF-β ligands in disease pathogenesis is more complex. Most tumor cells retain the ability to express TGF-β and often secrete an active form of the ligand. When coupled with a resistance to the inhibitory effects of TGF-β, overexpression of this ligand by the malignant cell could confer a growth advantage through the suppression of immune surveillance (17), promotion of angiogenesis, and stimulation of stroma (18). This potential for TGF-β to exert pro-oncogenic effects in a context in which the tumor cell has an acquired defect in the TGF-β receptor system has been frequently observed in human cancer (19). The production of active TGF-β by plasma cell tumors (PCTs) in mice has been linked to immune dysfunction (20), including the inhibition of cytotoxic T lymphocytes (21). Immune suppression has also been linked to plasma cell production of active TGF-β in the setting of autoimmune disease (22). These studies in the MRL/lpr mouse not only implicate B cells and plasma cells as an important source of circulating active TGF-β, but also provide histochemical evidence that suggests the activation of TGF-β occurs within the plasma cell (23).
We have previously demonstrated that all PCTs that develop in pristane-primed mice not only secrete active TGF-β, but also uniformly lack the ability to bind exogenous TGF-β at the cell surface (24). In restoring surface TβRII, either by disrupting expression of TGF-β1 with antisense mRNA or by competing ligand binding with a truncated TβRII, we now reveal a novel mechanism whereby the pathologic production of active, intracellular TGF-β impedes receptor localization to the plasma membrane and precludes TGF-β signaling.
Materials and Methods
Cell Culture.
Plasmacytoma cell lines (MOPC315, BPC4, TEPC2027, TEPC 1165, and X24) and murine B lymphoma cell lines (CH31, P388, and 8498) were maintained in routine RPMI 1640 media with 10% fetal bovine serum (Biofluids). Recombinant IL-6 (PeproTech) was added at a concentration of 5 ng/ml for the maintenance of TEPC 1165. Mv1lu cells were maintained in DMEM containing 10% fetal bovine serum.
Constructs and Retroviral Infections.
To generate the TGF-β1 antisense vector, a neomycin-resistant gene containing an internal ribosomal entry site that forms a fusion RNA with antisense TGF-β1 RNA was subcloned into the BamHI site of the MFG vector (25). To produce retrovirus, the ABOSC packaging cell was transfected with 10 μg of either the sense or antisense plasmid and cotransfected with 2.5 μg of pCMV-VSV-G. Retroviral supernatant was collected at 24 h and applied directly to X24 cells. Stable antisense cell lines were selected using G418 and characterized by standard Northern blot analysis and ELISA (TGF-β1 Quantikine; R&D Systems).
To generate the dominant-negative TβRII vector (dnTβRII), a 567-bp fragment of the human TβRII (nucleotide positions 335–911), with a 5′ hemagglutinin (HA) tag inserted at bp 405, was cloned into the BamHI-EcoRI site of pcDNA3 (Invitrogen). The dnTβRII was transfected into TEPC 1165 and expression was determined by reverse transcription PCR with a forward primer specific to the HA tag (TF1: 5′ GATGTTCCTGATTATGCTAG 3′), and a reverse primer spanning nucleotides 734–759 of the TβRII cDNA (TF2: 5′ CATCAGAGCTACAGGAACACATGAAG 3′) that amplified a 350-bp region.
Immunohistochemistry.
Immunohistochemical analysis was performed as previously reported by Caver et al. (23). To demonstrate the presence of active intracellular TGF-β, 4-μm sections through PCT-containing peritoneal granulomas were fixed in formalin and embedded in paraffin. Slides were submerged in Trizma buffer solution (TBS) containing 0.1% Triton X-100 (Sigma-Aldrich) at room temperature for 15 min followed by TBS for 5 min, methanol for 2 min, and 0.6% (vol/vol) hydrogen peroxide in methanol for 30 min. Slides were subsequently washed at room temperature in methanol for 2 min, TBS for 5 min, and three times in TBS containing 0.1% (wt/vol) BSA for 3 min. After treatment with hyaluronidase (1 mg/ml in 100 mM sodium acetate, 0.85% [wt/vol] NaCl) for 30 min at 37°C, and three washes in TBS/0.1% BSA, slides were treated with an avidin-biotin block (Vectastain) for 15 min at room temperature, rinsed in TBS, and then blocked with 1% goat serum in TBS containing 0.5% BSA for 30 min at room temperature. Slides were incubated with 50 μg/ml biotinylated anti-TGFβ1 (1D11; R&D Systems) that reacts specifically with active and not latent TGF-β. The primary antibody was biotinylated with avidin-biotin reagent (Zymed Laboratories) according to the manufacturer's instructions. Slides were washed three times in TBS with 0.1% BSA and exposed to ABC complex (Vector Laboratories) followed by 0.05% diaminobenzidine and 0.1% hydrogen peroxide.
Cell Fractionation.
Membrane and cytosolic fractions were prepared according to the methods of Koli et al. (26). In brief, cells were washed with cold PBS, scraped into fractionation buffer (20 mM Tris-HCl [pH 7.4], 2 mM EDTA, 25 mM NaF, 1 mM DTT, 2 mM NaM04, 1 μg/ml aprotinin, and 1 μg/ml leupeptin) and sheared by repeatedly passing through a 26-gauge needle. After centrifugation at 100,000 g for 60 min, the soluble fraction (cytosol) was removed and the pellet was resuspended in fractionation buffer containing 0.8% Triton X-100 for 20 min at 4°C. The membrane fraction was cleared from insoluble material by centrifugation at 12,000 g for 15 min. Triton X-100 was added to the cytosol fraction to yield an 0.8% final concentration. The cytosol and membrane fractions were resolved on 8% SDS-PAGE gels (Novex) and immunoblotted with antibody to TβRII at 1 μg/ml followed by a 1:10,000 dilution of horseradish peroxidase–conjugated goat anti–rabbit secondary antibody. Blots were developed with Super Signal (Pierce Chemical Co.).
Purification and Immunoblotting of γ-Phosphate–linked ATP-Sepharose–purified Lysates.
TβRII was extracted from plasmacytoma cell lysates with γ-phosphate–linked ATP-Sepharose (Upstate Biotechnology), which selects for tyrosine and serine/threonine kinases. Eluted kinase-active supernatants were resolved on 8% SDS-PAGE gels (Novex) and immunoblotted with 1 μg/ml of C16 and a 1:10,000 dilution of horseradish peroxidase-conjugated goat anti–rabbit secondary antibody.
In Vitro Kinase Assay.
107 cells were washed in cold PBS and lysed in radioimmunoprecipitation assay (RIPA buffer). Lysates were pre-cleared and immunoprecipitated with 2 μg/ml of an anti-TβRII (N-terminal; Upstate Biotechnology). Complexes were captured with 50 μl of protein G–Sepharose for 1.5 h, and washed five times with RIPA and once with PAN (150 mM NaCl and 50 mM Pipes, pH 7.4). 5 μCi of 32P-γATP (3,000 Ci/Mmol; Amersham Pharmacia Biotech) in 50 μl of kinase buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 5 mM MgCl2, 5 mM MnCl2, 1 mM NaF, 1 mM NaVO4, and 1 mM DTT) was added to antibody-bound protein G beads and incubated at room temperature for 30 min. Beads were washed three times with RIPA and proteins were eluted in sample buffer, separated on a 10% SDS-PAGE gel (Novex), and immunoblotted with an antibody to full-length receptor (H567; Santa Cruz Biotechnology, Inc.).
Receptor Cross-linking Assay.
Analysis of cell surface TGF-β receptor expression by the cross-linking of 125I–TGF-β was performed as previously described (24). Where specified, murine PCTs were acid washed in 150 mM NaCl and 0.1% acetic acid according to a standard protocol for stripping ligand from cell surface receptor, as described by Zwaagstra et al. (27). For in vitro cross-linking analysis of intracellular receptor, cells were washed with cold PBS and lysed in 20 mM Tris-HCl (pH 7.4), 2 mM EDTA, 25 mM NaF, 1 mM DTT, 2 mM NaMo4, and 2 mM NaVO4 with protease inhibitors. After centrifugation at 100,000 g for 60 min, the soluble fraction (cytosol) was removed and the pellet was resuspended in fractionation buffer containing 0.8% Triton X-100 for 20 min at 4°C. Membrane fraction was clarified by centrifugation at 12,000 g for 15 min. Triton X-100 was also added to the cytosol fraction to a final 0.8% concentration. The cytosolic fraction was incubated for 2.5 h with 125I–TGF-β and then cross-linked with disuccinimidyl suberate (DSS) (Pierce Chemical Co.) added to a final concentration of 3 mM for the final 30 min. The reaction was quenched by the addition of 50 mM Tris-HCl, pH 7.4. Lysates were immunoprecipitated overnight with anti-TβRII (C16). Immunoprecipitates were captured by protein A–Sepharose beads, separated on a 4–20% PAGE gradient gel, and exposed to film.
Analysis of Endogenous, Intracellular TGF-β Ligand–Receptor Complex.
107 cells were washed three times in ice-cold wash buffer (RPMI 1640, 25 mM Hepes, pH 7.4), resuspended in 1 ml of the same buffer containing 3 mM DSS, and then incubated for 30 min at 4°C. The reaction was quenched with 50 mM Tris, pH 7.5. Cells were washed three times in cold sucrose buffer (250 mM sucrose, 10 mM Tris, pH 7.4, 1 mM EDTA) and lysed for 30 min at 4°C in RIPA buffer with protease inhibitors (Boehringer). Lysates were centrifuged at 4°C at 10,000 g in a tabletop Eppendorf centrifuge (model 5415C). Receptor immunoprecipitation was done overnight at 4°C with 1 μg/ml of C16 (Santa Cruz Biotechnology, Inc.). Sample buffer was added with 2-mercaptoethanol in case of immunoblotting with H567 (Santa Cruz Biotechnology, Inc.) and without mercaptoethanol in the case of 1D11 (Genzyme), and MCA797 (Serotec). Precipitates were run on a 4–20% SDS-PAGE gradient gel and transferred onto polyvinylidene difluoride membranes. Blots were incubated with 1 μg/ml of 1D11 or MCA979 diluted in PBS with 1% BSA or H567 diluted in TBST-milk overnight at 4°C. An additional incubation was done with a 1:10,000 dilution of either a goat anti–mouse or donkey anti–rabbit secondary antibodies. Blots were developed with Super Signal (Pierce Chemical Co.).
Analysis of Smad2.
Lysates of cells treated with 2.5 ng TGF-β1/ml in RPMI medium with 0.5% fetal bovine serum were separated on 8% Tris-glycine gels (Novex). Immunoblotting was performed with an anti-phoshoSmad2, rabbit polyclonal antibody (Upstate Biotechnology), followed by a 1:10,000 dilution of goat–anti rabbit secondary (Jackson ImmunoResearch Laboratories) and visualized with Super Signal (Pierce Chemical Co.).
Conditioned Media Preparation, Proliferation Assays, and Analysis of TGF-β Production.
The methods for the production of serum-free cell supernatants, measurement of DNA synthesis by [3H]thymidine, and TGF-β ELISAs and bioassays have been previously described (24). Quantikine TGF-β1 ELISA kits were purchased from R&D Systems.
Results and Discussion
PCTs Express Normal TβRII.
Inactivating mutations in TβRII have been described in human malignancies (28, 29). Therefore, to determine if there were any consistent mutations that would impair the processing, expression, or function of this receptor, we sequenced cloned segments of cDNA from TβRII mRNA. The sequencing of cDNAs from five PCTs (BPC4, two variants of MOPC 315, TEPC 1165, and X24) and from the spleen of normal BALB/c and C57BL6 mice, showed no consistent bp differences (These sequence data are available from GenBank/EMBL/DDBJ under accession no. AF406755). Any differences from the published sequence of TβRII (from NIH3T3 cells) were ascribed to differences in the genetic strain of the mouse (30).
Pristane-induced Plasmacytomas Express a Functional, Kinase-active TβRII.
TβRII receptor is a constitutively active serine threonine kinase and autophosphorylates its intracellular domain (31). To characterize the TβRII expressed in the PCT, whole cell lysates were also incubated with γ-phosphate–linked ATP-Sepharose, which selects for tyrosine and serine/threonine kinases. In Western analysis of γ–ATP-Sepharose–selected proteins with TβRII-specific antibodies, an ∼70-kD doublet band of TβRII is recognized in the kinase-enriched extracts of the positive controls (Fig. 1 A, lanes 1 and 6) and in each of the four PCTs (Fig. 1 A, lanes 2–5), which suggests that TβRII protein is indeed synthesized by the PCTs. To determine whether the TβRII of the murine PCT has kinase activity, cells from two representative lines (TEPC 1165 and X24) were incubated with 5 μCi of 32γATP. Immunoprecipitates of TβRII from lysates labeled with 32γATP resolved on an 8% SDS gel demonstrate autophosphorylation of the 70-kD TβRII receptor both in control lymphomas (Fig. 1 B, lanes 1 and 2) and in PCTs (Fig. 1 B, lanes 3 and 4). This suggests that the TβRII of the pristane-induced PCT is capable of autophosphorylation and therefore functional.
Figure 1.
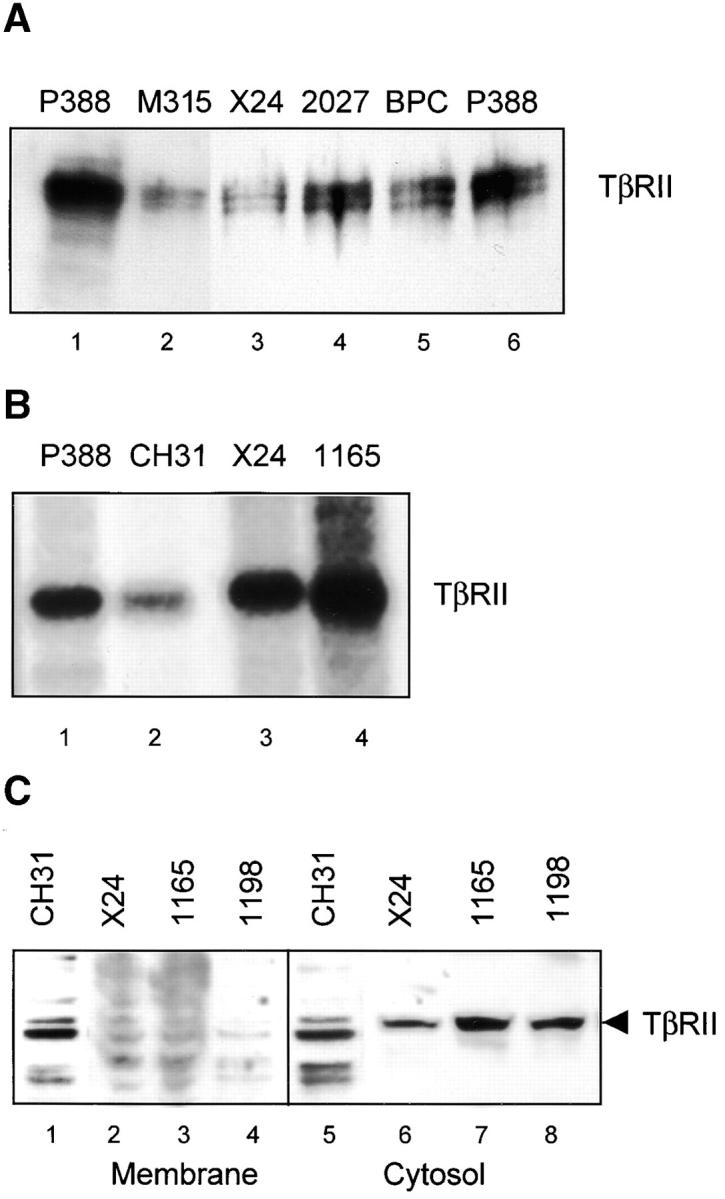
Murine PCTs contain active intracellular TβRII. (A) Affinity purification of TβRII from whole cell plasmacytoma lysates using γ-phosphate–linked ATP-Sepharose. Products were resolved on an 8% SDS-PAGE gel and immunoblotted with antibody to full-length TβRII. (B) Autophosphorylation of the 70-kD TβRII can be visualized when γ-ATP–labeled cell lysates of control lymphomas (lanes 1 and 2) and PCT cells (lane 3 and 4) are immunoprecipitated with anti-TβRII. (C) TβRII is not detected by Western in membrane fractions of PCTs. Membrane and cytosolic fractions were prepared from the control CH31 (lanes 1 and 5) and PCT cells (lanes 2–4 and 6–8) according to the methods described in Koli et al. (26) and as summarized in Materials and Methods.
TGFβ-II Receptor Protein Is Absent in Membrane Fraction but Present in the Cytosol.
Because the lack of ligand binding to surface receptors on the PCT is clearly not a consequence of a transcriptional or translational defect (24), we chose to investigate whether this defect represents a problem with localization of TβRII in the plasma membrane. We examined the membrane and cytosolic fractions of the PCTs for the presence of TβRII. Western blotting with an antibody specific for TβRII revealed abundant amounts of receptor protein in the cytosol (Fig. 1 C, lanes 6–8) but not in the membrane fractions (Fig. 1 C, lanes 2–4) of several PCTs. The control cell line CH31 (Fig. 1 C, lanes 1 and 5) shows presence of TβRII both in the membrane and cytosolic fractions. The relative absence of receptor protein in the membrane fractions of PCTs suggests that a defect in receptor trafficking underlies the inability to bind ligand at the surface of the PCT.
125I–TGF-β Does Not Bind to TβRII in Lysates of PCT Cells.
The absence of membrane TβRII can explain the inability to bind exogenous TGF-β at the cell surface. However, it should be possible to demonstrate the binding of 125I–TGF-β to the cytosolic receptor. Receptor cross-linking was performed with 125I–TGF-β and cytoplasmic lysates of TEPC 1165 and X24 to investigate whether the intracellular pool of TβRII would bind exogenous TGF-β1. Binding of 125I–TGF-β to TβRII was observed in lysates of control lymphoma cells (Fig. 2 , lanes 1 and 2) but not of PCT cells (Fig. 2 C, lanes 3 and 4). As expected, immunoprecipitation with an antibody specific for TβRI did not reveal the binding of 125I–TGF-β1 to TβRI (unpublished data), nor did anti-TβRII co-precipitate TβRI in this assay, supporting the model in which heteromers of TβRI and TβRII do not form in the presence of ligand until they are on the plasma membrane (32, 33). These data suggest that either TβRII expressed by PCTs is not available for ligand binding, or that a unique mechanism is responsible for the sequestration of TβRII within the PCT cell.
Figure 2.

Intracellular TβRII of the PCT does not bind exogenous ligand. Chemical cross-linking of 125I–TGF-β to cytoplasmic extracts from the positive control CH31 (lanes 1 and 2), followed by immunoprecipitation with TβRII-specific antibodies reveals ligand binding. In contrast, ligand binding to intracellular receptors could not be detected in cytoplasmic extracts of the parental TEPC 1165 (lanes 3 and 4).
Absence of Ligand–TβRII Complex on the PCT Cell Surface.
It is possible that the lack of 125I–TGF-β binding to TβRII in the PCT cell (24) could occur if receptors are occupied by secreted, endogenous ligand. We previously reported the secretion of active TGF-β by multiple PCT cell lines and now provide immunohistochemical evidence of active intracellular TGF-β in the PCT. Using the monoclonal antibody 1D11 that reacts specifically with active and not latent TGF-β, we evaluated sections through inflammatory granulomas for pristane-primed mice (Fig. 3 A). The specificity of the strong immunohistochemical staining in plasma cells was demonstrated by pre-blocking 1D11 with recombinant TGF-β1 (Fig. 3 B). Although Western analysis of membrane fractions indicates a true absence of TβRII at the PCT surface, we wanted to clearly exclude the possibility that the lack of 125I–TGF-β1 binding on whole PCT cells (Fig. 3 C, lane 2) is secondary to receptor occupancy at the cell surface by the active, endogenous ligand. We performed an acid wash on whole PCT cells, a technique that effectively strips ligand from cell surface receptors and allows for subsequent binding with 125I–TGF-β1 (16). The lack of binding of 125I–TGF-β1 after the acid wash of both X24 (Fig. 3 C, lane 3) and TEPC 1165 (unpublished data) confirms the true absence of cell surface localization of receptor. Cell membranes are not affected by transient exposure to low pH and 90–95% of TGF-β receptor–bound ligand is removed at low pH without affecting subsequent binding capacity (26). With a control-line cell line (Fig. 3 C, lane 4) we were also able to show that the acid wash of cells does not destroy ligand binding capacity of the receptor.
Figure 3.
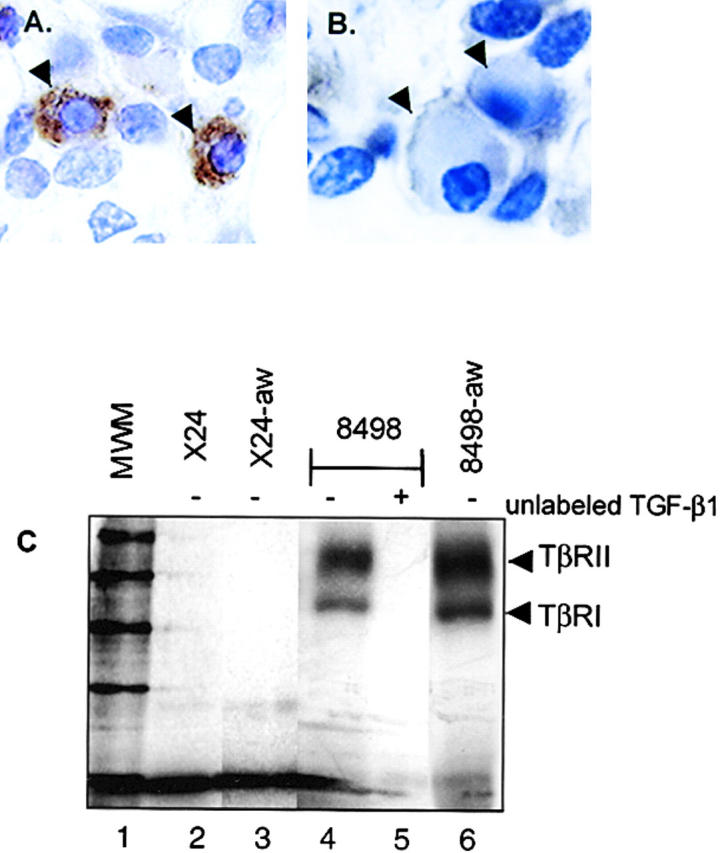
Active TGF-β is present in the PCT. (A) To demonstrate the presence of active intracellular TGF-β in the murine PCT in vivo, we prepared 4-μm sections through PCT-containing peritoneal granulomas that had been fixed in formalin and embedded in paraffin. Sections were processed by standard methods for histochemical analysis, as previously described (23) and incubated with a biotinylated monoclonal antibody (1D11) that reacts specifically with active and not latent TGF-β. (B) To demonstrate specificity, control sections were incubated with antibody that had been pre-blocked by incubation with recombinant TGF-β1. Arrowheads indicate plasma cells. (C) A ligand–receptor complex cannot be detected at the PCT cell surface. To strip receptor-bound ligand, whole cells were acid washed followed by incubation with 125I–TGF-β for detection of surface TβRII. There is no evidence of TβRII by receptor–ligand affinity labeling in PCTs X24 (lane 2) or 1165 (unpublished data), either before or after acid wash (X24 acid washed, lane 3). Transient exposure of whole cells to low pH does not destroy ligand binding capacity of cell surface receptor as shown with the control cell line (unwashed control, lanes 4 and 5; acid washed control, lane 6). The presence or absence of unlabeled TGF-β is indicated by − or +, respectively.
Production of Autocrine, Active TGF-β1 Precludes the Translocation of TβRII to the Plasma Membrane in the Murine Pristane–induced PCT.
To demonstrate the role of autocrine TGF-β1 in this PCT phenotype, we examined the effects of blocking endogenous TGF-β1 production through retroviral expression of antisense TGF-β1 (Fig. 4, A and B) . Antisense expression led to a 10-fold reduction of secreted TGF-β1 as determined by both ELISA and in a bioassay that evaluates the growth of the TGF-β1–sensitive Mv1Lu cell line in media conditioned by the PCT (120 pg/ml in sense control vs. 12.4 pg/ml in antisense line). Expression of sense has no effect on the pattern of ligand binding at the cell surface (Fig. 4 B, lane 5). However, the localization of TβRII at the cell surface is restored by the expression of antisense TGF-β1, as demonstrated by the binding of 125I–TGF-β1 to TβRII and TβRI on the antisense X24 PCT cells (Fig. 4 B, lane 3). The cross-linking pattern on antisense X24 is competed by excess unlabeled TGF-β1 (Fig. 4 B, lane 4) and is similar to that of the control B cell lymphoma CH31 (Fig. 4 B, lanes 1 and 2). The ability to detect cell surface TβRII in antisense X24 and not in cells expressing a control sense vector suggests that the effect is a direct consequence of blocking ligand production in the PCT.
Figure 4.
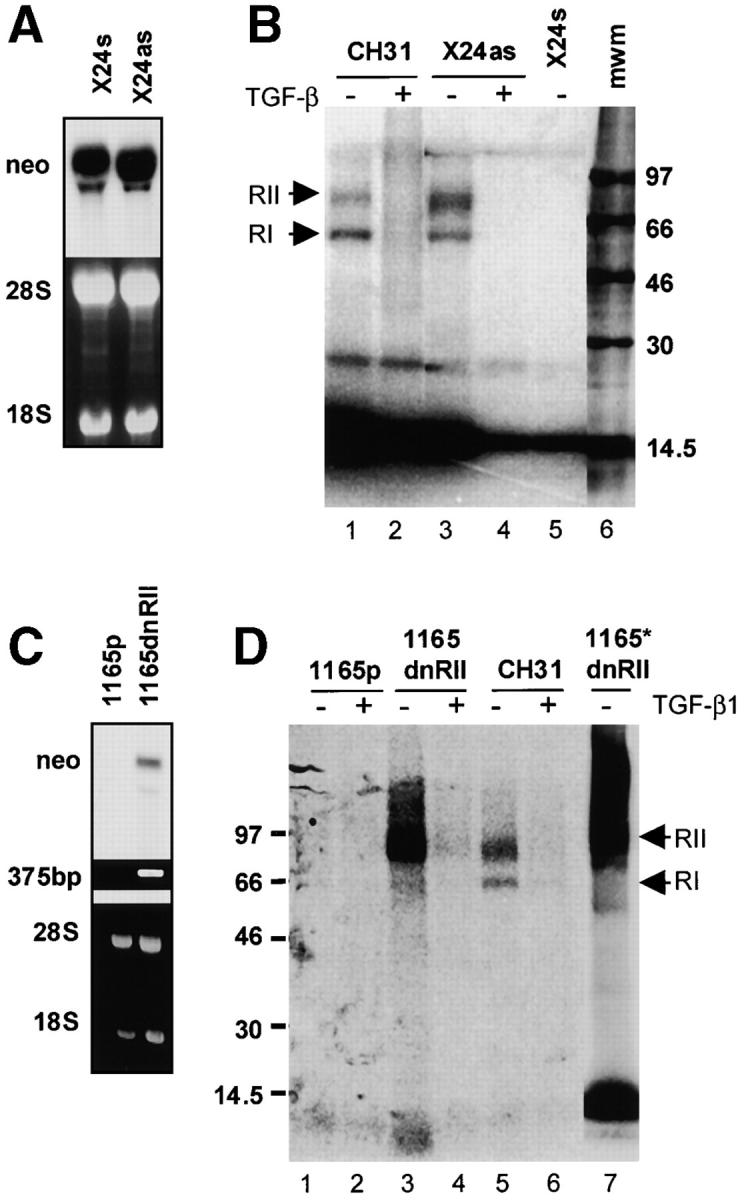
Restoration of cell surface expression of TβRII. (A and B) TGF-β1 anti-sense expression and disruption of TGF-β production restores the surface expression of TGF-β receptors (A, top). Northern analysis of sense and antisense TGF-β1 transfected cell lines with a 32P-labeled cDNA probe for neomycin (part of the bicistronic message) confirms the expression of sense and antisense TGF-β1 mRNA (A, bottom). Corresponding ethidium bromide–stained gel. (B) Ligand affinity cross-linking studies with 125I–TGF-β show a normal receptor complex in the control CH31 (lane 1), competed by unlabeled TGF-β (lane 2). The restoration of the cell surface expression of TβRII is obtained on transduction of the parental X24 with the antisense TGF-β1 (antisense X24, lanes 3 and 4). The binding and chemical cross-linking of 100 pM 125I–TGF-β to TβRII is competed by a 100-fold excess (1 nM) of unlabeled TGF-β1 and is indicated by +. (C and D) Transfection with a truncated TβRII restores surface localization of endogenous TβRII (C, top). Hybridization with a 32P-labeled cDNA neo probe demonstrates the expression of the dominant-negative TβRII mRNA in the PCT 1165 (C, bottom). Ethidium bromide–stained gel of a 375-bp amplimer specific to the HA-tagged TβRII. (D) Chemical cross-linking studies demonstrate the restoration of endogenous TβRII on the cell surface after transduction with the dominant-negative TβRII (1165dnRII). In vitro cross-linking of cytosolic fraction from 1165dnRII demonstrate the presence of free endogenous TβRII that is now available for binding to 125I–TGF-β (lane 7).
The data presented so far support a hypothesis in which the formation of active TGF-β1 within the PCT cell promotes direct intracellular interaction of ligand with TβRII, effectively trapping the receptor and preventing trafficking to the plasma membrane. If true, the restoration of cell surface localization might occur when the receptor is in relative excess of available ligand. To test this hypothesis, we infected a PCT line (TEPC 1165) with a retroviral vector expressing a dominant-negative version of the TGF-β type II receptor, dnTβRII, which lacks the kinase domain but has an intact extracellular binding domain and a transmembrane domain (34). Thus, the receptor is capable of binding ligand but is not capable of initiating signaling. We evaluated this line (1165dnRII) for the capacity to bind 125I–TGF-β1 (Fig. 4 D). The parental cell line TEPC 1165 (Fig. 4 D, lanes 1 and 2) consistently lack the ability to bind exogenous 125I–TGF-β1. However, the incubation of the dnTβRII-expressing 1165dnRII cells with 125I–TGF-β1 revealed ligand binding to endogenous receptor at the cell surface (immunoprecipitation of the receptor–ligand complex was performed with an antibody recognizing the C-terminus of wild-type TβRII, thereby distinguishing the endogenous receptor from the dnTβRII; Fig. 4 D, lanes 3 and 4). The ability of the dnTβRII to act as a decoy for endogenous TGF-β is also demonstrated by the ability of 125I–TGF-β1 to bind to TβRII in cytoplasmic extracts of the 1165dnTβRII cells (Fig. 4 D, lane 7). These results indicate that the lack of binding of 125I–TGF-β1 to the intracellular TβRII in the parental 1165 (Fig. 2, lanes 3 and 4) is a consequence of receptor binding to active intracellular TGF-β. When endogenous active TGF-β is sequestered by the dnTβRII in the PCT, there is a pool of endogenous TβRII that then becomes available for binding with exogenous ligand (Fig. 4 D, lane 7). Collectively, these data support the conclusion that autocrine, active intracellular TGF-β1 blocks the translocation of TβRII to the plasma membrane in the pristane-induced PCT.
Intracellular TGF-β Ligand–Receptor Complexes Are Present in the Murine Pristane–induced PCT.
To directly demonstrate the existence of an intracellular ligand–TβRII complex in the PCT, we performed an “intracellular” cross-linking assay by exposing intact PCT cells to the permeable membrane cross-linking reagent, DSS. Unlike traditional TGF-β receptor cross-linking studies that stabilize the interaction of cell surface receptor with 125I–TGF-β1, this experiment relies on the presence of active, endogenous TGF-β1 to cross-link with intracellular TβRII. Cross-linked PCT lysates were immunoprecipitated with a TβRII-specific antibody and immunoblotted with either of two distinct monoclonal antibodies specific for TGF-β1, or with a polyclonal antibody raised against the full-length type II receptor. Both anti–TGF-β1 antibodies clearly detected the existence of an identical intracellular ligand–receptor complex (Fig. 5 A, lanes 1 and 4) that was blocked by the pre-incubation of primary antibody with recombinant TGF-β1, and was not detected by incubation with the secondary antibody alone (unpublished data). The same complex was also present when immunoprecipitates were assayed by Western blotting with an antibody raised against the full-length TβRII (Fig. 5 A, lane 5). Because latent TGF-β does not bind TβRII, the demonstration of an intracellular ligand–receptor complex provides clear evidence that TGF-β is being activated intracellularly and is capable of binding TβRII.
Figure 5.
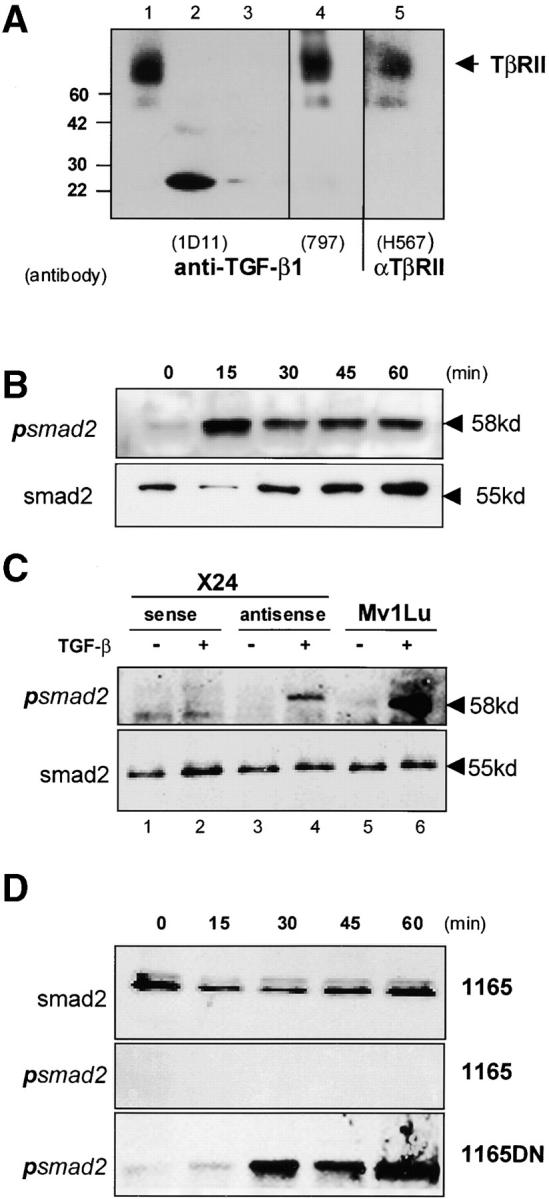
An intracellular TGF-β–TβRII complex. (A) Lysates of DSS-treated whole cells were immunoprecipitated with the anti-TβRII antibody (C16) followed by Western analysis with either antibodies to TGF-β1 (1D11, lanes 1–3, or MCA797, lane 4) or full-length TβRII (H567, lane 5). Each primary antibody detected the identical intracellular ligand–receptor complexes with recombinant TGF-β1 loaded as a control (500 ng, lane 2, and 50 ng, lane 3). (B) An ∼58-kD phosphorylated Smad2 band was induced by TGF-β treatment of the control lymphoma, CH31. (C) TGF-β–induced phosphorylation of Smad2 was detected in the PCT X24 containing the antisense expression vector (lanes 3 and 4), but not in the sense control line (lanes 1 and 2). Phosphorylation of the mink lung epithelial cell line (Mv1Lu) is shown as a positive control (lanes 5 and 6). (D) Similarly, TGF-β–induced phosphorylation of Smad2 was detected in the PCT 1165 containing the dnTβRII expression vector (1165dn, bottom). The result demonstrates the ability of TGF-β to initiate signaling once the endogenous receptor is localized to the plasma membrane.
The Intracellular TGF-β Ligand–Receptor Complexes Do Not Signal through Smad2.
The presence of an intracellular ligand–receptor complex raises an important question regarding the potential for signaling to occur in an “intracrine” fashion, outside the context of the plasma membrane. To address this question we looked for the presence of the receptor-activated phosphorylated Smad2 in lysates of both the parental PCTs and in the antisense and dnTβRII-expressing PCT cell lines (Fig. 5, C and D). No phosphorylated Smad2 was detected after the addition of exogenous ligand to the parental cell lines (Fig. 5, C and D), despite the fact that both X24 (unpublished data) and TEPC 1165 contain detectable levels of Smad2 protein. This suggests that the intracellular ligand–receptor complex is incapable of activating Smad2. More importantly, exogenous ligand could induce the phosphorylation of Smad2 not only in the control lymphoma (CH31; Fig. 5 B, top), but also in both the TGF-β1 antisense and dnTβRII-expressing PCT cell lines, in which we have restored receptor expression at the cell surface (Fig. 5, C and D).
It is worth noting that our use of the truncated dnTβRII has clearly given a result that might not be predicted based on conventional studies in which dominant-negative receptors have routinely been applied. Such dominant-negative constructs have invariably been expressed in cells that have an intact TGF-β signaling pathway. More importantly, the dnTβRII has never been introduced into a cell that spontaneously produces large amounts of active TGF-β or contains an intracellular pool of active TGF-β, such as that present within the PCT cell. However, the fact that one can induce phosphorylated Smad2 in a cell expressing the dnTβRII is not without precedent, as the expression of a similar dnTβRII in the Mv1Lu cell line blocks growth inhibition in response to TGF-β, but not the Smad-dependent induction of fibronectin or the plasminogen activator inhibitor (35). Regardless, it is clear that the endogenous TβRII synthesized by the PCT is capable of transducing a signal when localized in the plasma membrane.
It is also important to note that even upon the restoration of the endogenous receptor to the cell surface we were unable to restore sensitivity to TGF-β–mediated growth inhibition and apoptosis (unpublished data) in either the antisense X24 line or the 1165dn line. It has recently been demonstrated that a similar defect in the membrane localization of TGF-β receptors correlates with insensitivity to the growth inhibitory effects of TGF-β in human mammary epithelial tumors (36). However, this may not be the primary or principal defect underlying TGF-β resistance, especially in the PCT where deregulated expression of c-myc is invariant (37). As the repression of c-myc is critical for TGF-β–induced growth arrest (38), it is possible that our inability to couple Smad2 phosphorylation with either growth inhibition or apoptosis in the antisense X24 and 1165dn lines merely reflects an inability to suppress the expression of c-myc.
In contrast to the majority of growth factors, TGF-β is normally synthesized and secreted in a biologically latent form such that it is unable to bind to its cognate receptor, nor elicit a biological response (39). It is possible that genetic polymorphisms in the TGF-β ligands may result in the altered production and activation of TGF-β. Variants leading to increased circulating TGF-β have been described (40, 41) as well as domain-specific mutations of the TGF-β1 LAP that potentially result in the formation of a constitutively active TGF-β1 (42). We have sequenced the entire coding region for TGF-β1 in two PCTs (MOPC 315 and X24) and found no mutations. Another mechanism that might lead to the aberrant production of active TGF-β involves the increased production of proteases with the capacity to cleave the latent precursor. These include furin-like proteases (43) and matrix metalloproteinases (44, 45). In human myeloma there is significant production of matrix metalloproteinase (MMP)-9, MMP-2, and MMP-1 (46). So far there are no data regarding such protease activity in murine PCTs. This remains an area for future investigation. In this report, we provide the first evidence that the production of active TGF-β within a cell can disrupt autocrine TGF-β signaling via the formation of non-productive intracellular ligand–receptor complexes. In this model, an intracellular sequestration of TβRII by active TGF-β ligand prevents the receptor from trafficking to the cell surface (Fig. 6) . The data support the hypothesis that this novel mechanism underlies the consistent absence of TGF-β receptors on the surface of the pristane-induced PCT.
Figure 6.
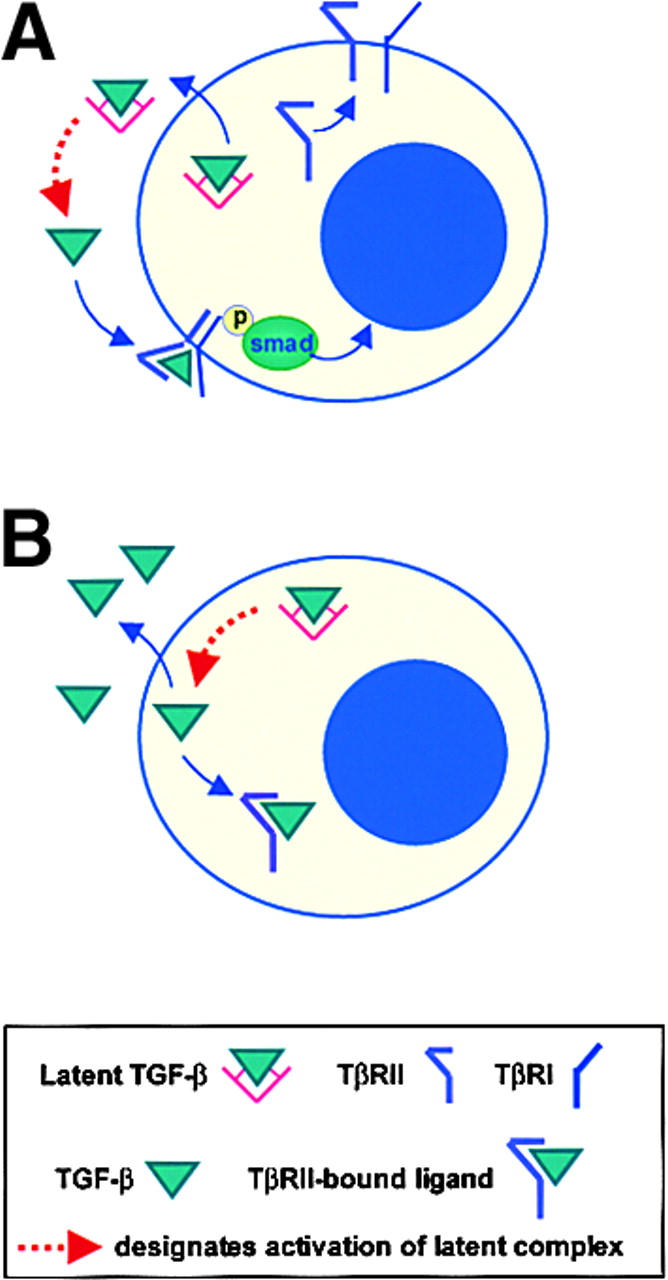
A model for the sequestration of TβRII inside the cell by active endogenous TGF-β1. TGF-β is typically secreted in a biologically latent form and therefore cannot bind to its cognate receptor, TβRII. (A) In a normal cell, the activation of TGF-β1 occurs outside the cell where it can bind cell surface TβRII. (B) In our plasmacytoma model, TGF-β1 is activated within the cell by a currently unknown mechanism and can readily bind TβRII, which consequently contributes to the observed loss of TGF-β receptor expression at the cell surface.
A role for intracellular ligand–receptor interactions in acquired defects in the membrane localization of growth factor receptors has been previously demonstrated in cells transformed by the viral oncoprotein v-sis (47, 48). In these studies, the loss of platelet-derived growth factor receptor at the cell surface was linked to internal activation of the receptor by the v-sis gene product. However, although intracellular autocrine loops have been described for v-sis and for other cytokines (49, 50), our data suggest that intracellular TGF-β ligand–receptor complexes are not capable of initiating signaling, at least through Smad2. This may be due to differences in the trafficking of TβRI and TβRII (33), such that ligand-bound TβRII may not be able to recruit TβRI unless both are present at the cell surface. Alternatively, it may reflect the inability of ligand-bound receptor complexes to recruit cell surface adaptor proteins such as Smad anchor for receptor activation, which are required for the phosphorylation of Smad2 (51). Studies focused on mechanisms of activation of the ligand in the pristane-induced PCT and the endocytic fate of ligand–receptor complexes are currently under investigation. In conclusion, these data not only support the concept that latency is a key step in regulating the TGF-β ligands, but also demonstrate how the activation of TGF-β within a cell can impact the TGF-β receptor expression, and therefore impact the responsiveness to both autocrine and paracrine sources of the cytokine.
Footnotes
Abbreviations used in this paper: dnTβRII, dominant-negative type II TGF-β receptor; DSS, disuccinimidyl suberate; HA, hemagglutinin; MMP, matrix metalloproteinase; PCT, plasma cell tumor; RIPA, radioimmunoprecipitation assay; TBS, Trizma buffer solution; TβRI, type I TGF-β receptor; TβRII, type II TGF-β receptor.
References
- 1.Markowitz, S.D., and A.B. Roberts. 1996. Tumor suppressor activity of the TGF-beta pathway in human cancers. Cytokine Growth Factor Rev. 7:93–102. [DOI] [PubMed] [Google Scholar]
- 2.Blobe, G.C., W.P. Schiemann, and H.F. Lodish. 2000. Role of transforming growth factor beta in human disease. N. Engl. J. Med. 342:1350–1358. [DOI] [PubMed] [Google Scholar]
- 3.Fynan, T.M., and M. Reiss. 1993. Resistance to inhibition of cell growth by transforming growth factor-beta and its role in oncogenesis. Crit. Rev. Oncog. 4:493–540. [PubMed] [Google Scholar]
- 4.Wrana, J.L., L. Attisano, R. Wieser, F. Ventura, and J. Massague. 1994. Mechanism of activation of the TGF-beta receptor. Nature. 370:341–347. [DOI] [PubMed] [Google Scholar]
- 5.Wrana, J., and T. Pawson. 1997. Signal transduction. Mad about SMADs. Nature. 388:28–29. [DOI] [PubMed] [Google Scholar]
- 6.Kimchi, A., X.F. Wang, R.A. Weinberg, S. Cheifetz, and J. Massague. 1988. Absence of TGF-beta receptors and growth inhibitory responses in retinoblastoma cells. Science. 240:196–199. [DOI] [PubMed] [Google Scholar]
- 7.Norgaard, P., L. Damstrup, K. Rygaard, M. Spang-Thomsen, and H. Skovgaard Poulsen. 1994. Growth suppression by transforming growth factor beta 1 of human small-cell lung cancer cell lines is associated with expression of the type II receptor. Br. J. Cancer. 69:802–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inagaki, M., A. Moustakas, H.Y. Lin, H.F. Lodish, and B.I. Carr. 1993. Growth inhibition by transforming growth factor beta (TGF-beta) type I is restored in TGF-beta-resistant hepatoma cells after expression of TGF-beta receptor type II cDNA. Proc. Natl. Acad. Sci. USA. 90:5359–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park, K., S.J. Kim, Y.J. Bang, J.G. Park, N.K. Kim, A.B. Roberts, and M.B. Sporn. 1994. Genetic changes in the transforming growth factor beta (TGF-beta) type II receptor gene in human gastric cancer cells: correlation with sensitivity to growth inhibition by TGF-beta. Proc. Natl. Acad. Sci. USA. 91:8772–8776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reiss, M., and E.B. Stash. 1990. High frequency of resistance of human squamous carcinoma cells to the anti-proliferative action of transforming growth factor beta. Cancer Commun. 2:363–369. [DOI] [PubMed] [Google Scholar]
- 11.Okamoto, A., W. Jiang, S.J. Kim, E.A. Spillare, G.D. Stoner, I.B. Weinstein, and C.C. Harris. 1994. Overexpression of human cyclin D1 reduces the transforming growth factor beta (TGF-beta) type II receptor and growth inhibition by TGF-beta 1 in an immortalized human esophageal epithelial cell line. Proc. Natl. Acad. Sci. USA. 91:1156–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arteaga, C.L., T. Carty-Dugger, H.L. Moses, S.D. Hurd, and J.A. Pietenpol. 1993. Transforming growth factor beta 1 can induce estrogen-independent tumorigenicity of human breast cancer cells in athymic mice. Cell Growth Differ. 4:193–201. [PubMed] [Google Scholar]
- 13.Grady, W.M., L.L. Myeroff, S.E. Swinler, A. Rajput, S. Thiagalingam, J.D. Lutterbaugh, A. Neumann, M.G. Brattain, J. Chang, S.J. Kim, et al. 1999. Mutational inactivation of transforming growth factor beta receptor type II in microsatellite stable colon cancers. Cancer Res. 59:320–324. [PubMed] [Google Scholar]
- 14.Munoz-Antonia, T., X. Li, M. Reiss, R. Jackson, and S. Antonia. 1996. A mutation in the transforming growth factor beta type II receptor gene promoter associated with loss of gene expression. Cancer Res. 56:4831–4835. [PubMed] [Google Scholar]
- 15.Hahm, K.B., K. Cho, C. Lee, Y.H. Im, J. Chang, S.G. Choi, P.H. Sorensen, C.J. Thiele, and S.J. Kim. 1999. Repression of the gene encoding the TGF-beta type II receptor is a major target of the EWS-FLI1 oncoprotein. Nat. Genet. 23:222–227. [DOI] [PubMed] [Google Scholar]
- 16.Ammanamanchi, S., S.J. Kim, L.Z. Sun, and M.G. Brattain. 1998. Induction of transforming growth factor-beta receptor type II expression in estrogen receptor-positive breast cancer cells through SP1 activation by 5-aza-2'-deoxycytidine. J. Biol. Chem. 273:16527–16534. [DOI] [PubMed] [Google Scholar]
- 17.Gorelik, L., and R.A. Flavell. 2001. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat. Med. 10:1118–1122. [DOI] [PubMed] [Google Scholar]
- 18.Yoo, Y.D., H. Ueda, K. Park, K.C. Flanders, Y.I. Lee, G. Jay, and S.J. Kim. 1996. Regulation of transforming growth factor-beta 1 expression by the hepatitis B virus (HBV) X transactivator. Role in HBV pathogenesis. J. Clin. Invest. 97:388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim, D.H., and S.J. Kim. 1996. Transforming growth factor-beta receptors: role in physiology and disease. J. Biomed. Sci. 3:143–158. [DOI] [PubMed] [Google Scholar]
- 20.Berg, D.J., and R.G. Lynch. 1991. Immune dysfunction in mice with plasmacytomas. I. Evidence that transforming growth factor-beta contributes to the altered expression of activation receptors on host B lymphocytes. J. Immunol. 146:2865–2872. [PubMed] [Google Scholar]
- 21.Weiskirch, L.M., Y. Bar-Dagan, and M.B. Mokyr. 1994. Transforming growth factor-beta-mediated down-regulation of antitumor cytotoxicity of spleen cells from MOPC-315 tumor-bearing mice engaged in tumor eradication following low-dose melphalan therapy. Cancer Immunol. Immunother. 38:215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowrance, J.H., F.X. O'Sullivan, T.E. Caver, W. Waegell, and H.D. Gresham. 1994. Spontaneous elaboration of transforming growth factor beta suppresses host defense against bacterial infection in autoimmune MRL/lpr mice. J. Exp. Med. 180:1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caver, T.E., F.X. O'Sullivan, L.I. Gold, and H.D. Gresham. 1996. Intracellular demonstration of active TGFbeta1 in B cells and plasma cells of autoimmune mice. IgG-bound TGFbeta1 suppresses neutrophil function and host defense against Staphylococcus aureus infection. J. Clin. Invest. 98:2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amoroso, S.R., N. Huang, A.B. Roberts, M. Potter, and J.J. Letterio. 1998. Consistent loss of functional transforming growth factor beta receptor expression in murine plasmacytomas. Proc. Natl. Acad. Sci. USA. 95:189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohashi, T., S. Boggs, P. Robbins, A. Bahnson, K. Patrene, F.S. Wei, J.F. Wei, J. Li, L. Lucht, and Y.Fei. 1992. Efficient transfer and sustained high expression of the human glucocerebrosidase gene in mice and their functional macrophages following transplantation of bone marrow transduced by a retroviral vector. Proc. Natl. Acad. Sci. USA. 89:11332–11336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koli, K.M., and C.L. Arteaga. 1997. Predominant cytosolic localization of type II transforming growth factor beta receptors in human breast carcinoma cells. Cancer Res. 57:970–977. [PubMed] [Google Scholar]
- 27.Zwaagstra, J.C., Z. Kassam, and M.D. O'Connor-McCourt. 1999. Down-regulation of transforming growth factor-beta receptors: cooperativity between the types I, II, and III receptors and modulation at the cell surface. Exp. Cell Res. 252:352–362. [DOI] [PubMed] [Google Scholar]
- 28.Massague, J. 2000. How cells read TGF-beta signals. Nat. Rev. Mol. Cell. Biol. 1:169–178. [DOI] [PubMed] [Google Scholar]
- 29.de Caestecker, M.P., E. Piek, and A.B. Roberts. 2000. Role of transforming growth factor-beta signaling in cancer. J. Natl. Cancer Inst. 92:1388–1402. [DOI] [PubMed] [Google Scholar]
- 30.Lawler, S., A.F. Candia, R. Ebner, L. Shum, A.R. Lopez, H.L. Moses, C.V. Wright, and R. Derynck. 1994. The murine type II TGF-beta receptor has a coincident embryonic expression and binding preference for TGF-beta 1. Development. 120:165–175. [DOI] [PubMed] [Google Scholar]
- 31.Wrana, J.L., L. Attisano, R. Wieser, F. Ventura, and J. Massague. 1994. Mechanism of activation of the TGF-beta receptor. Nature. 370:341–347. [DOI] [PubMed] [Google Scholar]
- 32.Gilboa, L., R.G. Wells, H.F. Lodish, and Y.I. Henis. 1998. Oligomeric structure of type I and type II transforming growth factor beta receptors: homodimers form in the ER and persist at the plasma membrane. J. Cell Biol. 140:767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wells, R.G., H. Yankelev, H.Y. Lin, and H.F. Lodish. 1997. Biosynthesis of the type I and type II TGF-beta receptors. Implications for complex formation. J. Biol. Chem. 272:11444–11451. [DOI] [PubMed] [Google Scholar]
- 34.Tang, B., K. de Castro, H.E. Barnes, W.T. Parks, L. Stewart, E.P. Bottinger, D. Danielpour, and L.M. Wakefield. 1999. Loss of responsiveness to transforming growth factor beta induces malignant transformation of nontumorigenic rat prostate epithelial cells. Cancer Res. 59:4834–4842. [PubMed] [Google Scholar]
- 35.Chen, R.H., R. Ebner, and R. Derynck. 1993. Inactivation of the type II receptor reveals two receptor pathways for the diverse TGF-beta activities. Science. 260:1335–1338. [DOI] [PubMed] [Google Scholar]
- 36.Lynch, M.A., T.A. Petrel, H. Song, T.J. Knobloch, B.C. Casto, D. Ramljak, L.M. Anderson, V. DeGroff, G.D. Stoner, R.W. Brueggemeier, et al. 2001. Responsiveness to transforming growth factor-beta (TGF-beta)-mediated growth inhibition is a function of membrane-bound TGF-beta type II receptor in human breast cancer cells. Gene Expr. 9:157–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Potter, M., and F. Wiener. 1992. Plasmacytomagenesis in mice: model of neoplastic development dependent upon chromosomal translocation. Carcinogenesis. 13:1681–1697. [DOI] [PubMed] [Google Scholar]
- 38.Staller, P., K. Peukert, A. Kiermaier, J. Seoane, J. Lukas, H. Karsunky, T. Moroy, J. Bartek, J. Massague, F. Hanel, et al. 2001. Repression of p15INK4b expression by Myc through association with Miz-1. Nat. Cell Biol. 3:392–399. [DOI] [PubMed] [Google Scholar]
- 39.Wakefield, L.M., D.M. Smith, T. Masui, C.C. Harris, and M.B. Sporn. 1987. Distribution and modulation of the cellular receptor for transforming growth factor-beta. J. Cell Biol. 105:965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Awad, M.R., A. El-Gamel, P. Hasleton, D.M. Turner, P.J. Sinnott, and I.V. Hutchinson. 1998. Genotypic variation in the transforming growth factor-beta1 gene: association with transforming growth factor-beta1 production, fibrotic lung disease, and graft fibrosis after lung transplantation. Transplantation. 66:1014–1020. [DOI] [PubMed] [Google Scholar]
- 41.Yamada, Y., T. Hosoi, F. Makimoto, H. Tanaka, Y. Seino, and K. Ikeda. 1999. Transforming growth factor beta-1 gene polymorphism and bone mineral density in Japanese adolescents. Am. J. Med. 106:477–479. [DOI] [PubMed] [Google Scholar]
- 42.Saito, T., A. Kinoshita, K. Yoshiura, Y. Makita, K. Wakui, K. Honke, N. Niikawa, and N. Taniguchi. 2001. Domain-specific mutations of a transforming growth factor (TGF)-beta 1 latency-associated peptide cause Camurati-Engelmann disease because of the formation of a constitutively active form of TGF-beta 1. J. Biol. Chem. 276:11469–11472. [DOI] [PubMed] [Google Scholar]
- 43.Leitlein, J., S. Aulwurm, R. Waltereit, U. Naumann, B. Wagenknecht, W. Garten, M. Weller, and M. Platten. 2001. Processing of immunosuppressive pro-TGF-beta 1,2 by human glioblastoma cells involves cytoplasmic and secreted furin-like proteases. J. Immunol. 166:7238–7243. [DOI] [PubMed] [Google Scholar]
- 44.Yu, Q., and I. Stamenkovic. 2000. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 45.Lee, C.G., R.J. Homer, Z. Zhu, S. Lanone, X. Wang, V. Koteliansky, J.M. Shipley, P. Gotwals, P. Noble, Q. Chen, et al. 2001. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor β1. J. Exp. Med. 194:809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barille, S., C. Akhoundi, M. Collette, M.P. Mellerin, M.J. Rapp, J.L. Harousseau, R. Bataille, and M. Amiot. 1997. Metalloproteinases in multiple myeloma: production of matrix metalloproteinase-9 (MMP-9), activation of proMMP-2, and induction of MMP-1 by myeloma cells. Blood. 90:1649–1655. [PubMed] [Google Scholar]
- 47.Aaronson, S.A., H. Igarashi, C.D. Rao, E. Finzi, T.P. Fleming, O. Segatto, and K.C. Robbins. 1986. Role of genes for normal growth factors in human malignancy. Princess Takamatsu Symp. 17:95–108. [PubMed] [Google Scholar]
- 48.Fleming, T.P., T. Matsui, C.J. Molloy, K.C. Robbins, and S.A. Aaronson. 1989. Autocrine mechanism for v-sis transformation requires cell surface localization of internally activated growth factor receptors. Proc. Natl. Acad. Sci. USA. 86:8063–8067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bejcek, B.E., D.Y. Li, and T.F. Deuel. 1989. Transformation by v-sis occurs by an internal autoactivation mechanism. Science. 245:1496–1499. [DOI] [PubMed] [Google Scholar]
- 50.Browder, T.M., C.E. Dunbar, and A.W. Nienhuis. 1989. Private and public autocrine loops in neoplastic cells. Cancer Cells. 1:9–17. [PubMed] [Google Scholar]
- 51.Tsukazaki, T., T.A. Chiang, A.F. Davison, L. Attisano, and J.L. Wrana. 1998. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell. 95:779–791. [DOI] [PubMed] [Google Scholar]