

Abstract
Cerebral malaria (CM) causes death in children and nonimmune adults. TNF-α has been thought to play a key role in the development of CM. In contrast, the role of the related cyto-kine lymphotoxin α (LTα) in CM has been overlooked. Here we show that LTα, not TNFα, is the principal mediator of murine CM. Mice deficient in TNFα (B6.TNFα−/−) were as susceptible to CM caused by Plasmodium berghei (ANKA) as C57BL/6 mice, and died 6 to 8 d after infection after developing neurological signs of CM, associated with perivascular brain hemorrhage. Significantly, the development of CM in B6.TNFα−/− mice was not associated with increased intracellular adhesion molecule (ICAM)-1 expression on cerebral vasculature and the intraluminal accumulation of complement receptor 3 (CR3)-positive leukocytes was moderate. In contrast, mice deficient in LTα (B6.LTα−/−) were completely resistant to CM and died 11 to 14 d after infection with severe anemia and hyperparasitemia. No difference in blood parasite burden was found between C57BL/6, B6.TNFα−/−, and B6.LTα−/− mice at the onset of CM symptoms in the two susceptible strains. In addition, studies in bone marrow (BM) chimeric mice showed the persistence of cerebral LTα mRNA after irradiation and engraftment of LTα-deficient BM, indicating that LTα originated from a radiation-resistant cell population.
Keywords: parasitic disease, protozoan infection, Plasmodium berghei, immunopathology, infection
Introduction
Cerebral malaria (CM) is a major cause of death in African children infected with Plasmodium falciparum (1). This serious neurological condition is characterized by the sequestration of parasitized erythrocytes in cerebral blood vessels (1). Because high levels of circulating TNFα are found in the serum of CM patients, this cytokine is thought to play an important role in CM (2, 3). This has been supported by reports of TNFα production in the brain during murine CM caused by Plasmodium berghei ANKA (4, 5). In this experimental model of CM, anti-TNFα antibodies (6) or soluble TNF receptor (TNFR; reference 7) have also been shown to attenuate the severity of symptoms and prevent subsequent death. In addition, mice deficient in TNFR2 (p75) are resistant to CM (8).
In both human and murine CM, the blockage of cerebral vessels by parasitized RBCs (pRBCs) is thought to be caused by TNFα-mediated up-regulation of adhesion molecules, such as intercellular adhesion molecule (ICAM)-1, on the endothelium (1, 6). In mice, this is also associated with the accumulation of activated leukocytes in the lumen of brain micro-vessels (6). TNFα is thought to mediate pathology by either direct cytolytic effects or induction of an inflammatory cascade involving mediators such as IFN-γ and nitric oxide (NO; reference 9). However, the critical role of TNFα in human CM has recently been questioned by the failure of TNFα-neutralizing reagents to decrease the incidence of clinical CM (10, 11).
Recently, lymphotoxin α (LTα), a related member of the TNF family, has been reported to have important, yet distinct, roles in various infectious and autoimmune disease models (12, 13). LTα forms a soluble homotrimer that along with TNFα is capable of binding both the TNFR1 and TNFR2. In addition, LTα can form a cell-bound heterotrimer with two LTβ molecules, which binds the LTβ receptor (14). Given the shared receptor usage of TNFα and LTα (14), and the potential cross-reactivity of anti-TNFα antibodies with LTα (15), it is possible that LTα, and not TNFα, is the principal mediator of CM. To test this hypothesis, C57BL/6 mice deficient in TNFα or LTα were infected with P. berghei ANKA pRBCs and their susceptibility to CM was compared. Our results clearly show that LTα, and not TNFα, is indeed the principal mediator of murine CM.
Materials and Methods
Mice.
C57BL/6 mice were purchased from Tuck and Co. and were housed under conventional conditions. Mice deficient in TNFα (B6.TNFα−/−) and both TNFα and LTα (B6.TNFα/LTα−/−; reference 16) were obtained from Bantin & Kingman, while those deficient in LTα−/− (B6.LTα−/−; reference 17) were obtained from The Jackson Laboratory. In some experiments, B6.LTα−/− mice generated from C57BL/6 mice embryonic stem cells (12) were purchased from Bantin & Kingman and used to confirm the resistance of LTα-deficient mice (17) to CM. B6.Ly5.1 mice were obtained from Charles River Laboratories (IFFA Credo). All mouse strains were bred at the London School of Hygiene and Tropical Medicine under barrier conditions. Mice used in all experiments were sex-matched and used at 6 wk of age. Chimeric mice were prepared by irradiating animals twice (48 h apart) with 5.5 Gy and then engrafting with 107 fresh bone marrow (BM) cells intravenously via the lateral tail vein within 2 h of the second radiation exposure. Mice were maintained on antibiotics for 4 wk after engraftment and infected with parasites 8 wk after receiving BM.
Parasites, Infection, and Disease Assessment.
P. berghei (ANKA) was obtained from Dr. N. Wedderburn (Royal College of Surgeons, London, UK) and used in all experiments after one in vivo passage in C57BL/6 mice. Mice were infected by injecting 104 pRBCs intravenous via the lateral tail vein, as described previously (18). Animals were monitored for neurological signs of CM, including convulsions, ataxia and paralysis. Parasite burden was determined from Giemsa stained blood smears, as described previously (18), and expressed as the percentage of pRBCs. Blood hemoglobin levels were determined when collecting blood using a Hemoglobin meter, according to the manufacturer's instructions (Buffalo Medical Specialities). RBCs were counted using a hemocytometer by diluting 2 μl of tail blood in 1 ml RPMI. Animals were monitored and killed by cervical dislocation when death was deemed inevitable, according to UK Home Office guidelines. Blood was harvested by cardiac puncture for serum isolation. Brain tissue was carefully removed and half was snap frozen on cork blocks in OCT embedding media solution (Raymond A. Lamb, Eastbourne, UK) and stored at –80°C until required for immunohistochemistry. The other half was stored in RNAlater (Ambion) for 24 h at 4°C, before freezing at –80°C until required for RNA extraction. Some tissue was fixed in formol saline (4% vol/vol; formaldehyde) for wax embedding and preparing tissue sections for hematoxylin and eosin staining.
Real-time Reverse Transcription PCR.
RNA was isolated from brain tissue using Tri Reagent (Sigma-Aldrich), and an RNeasy Mini Kit with on-column DNase digestion (QIAGEN), according to the manufacturers' instructions. RNA was reverse transcribed into cDNA as described previously (19, 20). Oligonucleotides (5′–3′) used for the specific amplification of LTα were AGGGGCCCAGGGACTCTCT (sense) and ACGATCCGTGCTTGCTCTC (antisense), and for amplification of the house keeping gene hypoxanthine-guanine phosphoribosyl transferase (HPRT) were GTTGGATACAGGCCAGACTTTGTTG (sense) and GATTCAACCTTGCGCTCATCTTAGGC (antisense). The number of LTα and HPRT cDNA molecules in each sample was calculated by real-time reverse transcription (RT)-PCR using QuantiTect SYBR green master mix (QIAGEN) and a LightCycler (Roche), according to the manufacturers' instructions. Standard curves were constructed with known amounts of LTα and HPRT cDNA, and the number of LTα molecules per 1,000 HPRT molecules in each sample was calculated.
Measurement of Serum IFN-γ and Nitrate Levels.
Serum samples were diluted 1:2 and IFN-γ levels measured by ELISA, as described previously (21). Serum nitrate levels were measured by reducing nitrate to nitrite and using Griess reagent, as described previously (22).
Immunohistochemistry on Cryostat Sections of the Brain
Antibodies used for histology included mouse anti-P. berghei polyclonal antibody (18), anti-complement receptor 3 (CR3) (5C6; reference 23), and anti-CD54 (ICAM-1; KAT-1; Serotec). Brain tissue responses were analyzed on 6 μm acetone-fixed cryostat tissue sections stained with the above primary antibodies and appropriate fluorescein-isothiocyanate (FITC)-conjugated secondary detection reagents according to the manufacturer's instructions (Vector Laboratories). Sections were mounted in Vectashield mounting media for fluorescence (Vector Laboratories), before analysis on an Axioplan fluorescent microscope (ZEISS). Images were recorded digitally using a MagnaFire Sp digital camera (Optronics). In some experiments, peroxidase-conjugated secondary reagents (Vector Laboratories) were used to detect ICAM-1 and CR3 expression in brain tissue. These sections were then used to count ICAM-1–positive vessels and CR3-positive cells in 25 consecutive microscope fields of view using a ×40 objective (final magnification ×400).
Statistics.
Statistical analysis was performed for IFN-γ and nitrate levels using Student's t test and for survival curves using Kaplan-Meier plots and Log Rank tests.
Results and Discussion
LTα-deficient Mice Do Not Die from CM.
To determine if LTα is involved in CM, we first tested for LTα production using LTα-specific primers in a real-time RT-PCR with mRNA isolated from the brains of C57BL/6 mice infected with P. berghei and showing signs of CM (Fig. 1 a). LTα mRNA was constitutively expressed in the mouse brain, and levels increased in mice with CM. Therefore, we sought to investigate further if this cytokine played a role in murine CM.
Figure 1.





In the absence of LTα the early death of P. berghei ANKA-parasitized C57BL/6 mice is prevented. (a) LTα mRNA is up-regulated in brains of C57BL/6 mice with CM, as indicated by real-time RT-PCR (n = 3 mice per group). (b) Survival curves of C57BL/6 (solid line), B6.TNFα−/− (overlapping C57BL/6 mice and indicated by arrow), B6.LTα−/− (dashed line), and B6.TNF/LTα−/− (dotted line) mice infected with P. berghei (ANKA). Results represent pooled data from three independent groups of mice (n = 5 to 11 per group) in experiments conducted on three separate occasions (total of n = 16–22 mice per group). Differences in survival between C57BL/6 or B6.TNFα−/− mice and mice deficient in LTα were statistically different (χ2 = 39.39, P < 0.0001). (c) Parasitemia over the course of P. berghei infection in C57BL/6 (filled circle), B6.TNFα−/− (open circle), B6.LTα−/− (filled triangle), and B6.TNF/LTα−/− (open triangle) mice. Arrow indicates the time point (day 7) that CM developed in C57BL/6 and B6.TNFα−/− mice. Data shown are from one experiment that is representative of the four performed. (d) Serum nitrate and (e) serum IFN-γ levels at day 7 and 12 postinfection (p.i.), as indicated. Statistically significant differences of P < 0.05 are indicated (*). Data shown are from one experiment that is representative of the three performed.
To distinguish the roles of TNFα and LTα in CM, we infected mice deficient in these cytokines (B6.TNFα−/− and B6.LTα−/− mice, respectively) with P. berghei (ANKA) and monitored animals for signs of CM. Strikingly, B6.TNFα−/− mice began to show neurological symptoms of CM, including ataxia, convulsions, and palsy, at the same time as infected C57BL/6 mice (days 6–8 after infection). Within 12–24 h of the onset of neurological symptoms, all C57BL/6 and B6.TNFα−/− mice had to be killed (Fig. 1 b). In stark contrast, B6.LTα−/− mice and those deficient in both TNFα and LTα (B6.TNF/LTα−/− mice) failed to show any neurological symptoms associated with CM. However, hemoglobin levels in B6.LTα−/− mice dropped from 14.5 –16 g/dL (at day 0 p.i.) to less than 4–6 g/dL (day 12 after infection), and RBC numbers decreased from ∼1010/ml to 1–3 × 109/ml. These mice died between days 11–15 after infection. Such decreases in hemoglobin and RBC numbers have previously been associated with death by complications arising from anemia (9). Importantly, when C57BL/6 and B6.TNFα−/− mice died, parasitemia ranged from 12–24% pRBCs and was similar for all mouse strains (Fig. 1 c). B6.TNF/LTα−/− mice have previously been shown to be resistant to CM, but this resistance was attributed to the absence of TNFα (24). These results clearly show that LTα, and not TNFα, is the primary mediator of murine CM, and that the development of CM is independent of blood parasitemia.
Both nitric oxide (NO) and IFN-γ have been reported to play important roles in the development of murine CM (9, 24). Serum nitrate (an indirect measure of NO) and IFN-γ levels were reduced in mice deficient in either TNFα or LTα, compared with C57BL/6 mice at the onset of CM symptoms in susceptible strains (Fig. 1, d and e, day 7 after infection). Subsequently, both IFN-γ and nitrate levels in the serum of B6.LTα−/− and B6.TNF/LTα−/− mice rose such that by day 12 after infection they were similar to the levels found in C57BL/6 mice. These data are in agreement with previous studies that show reduced NO and IFN-γ levels in P. berghei-infected B6.TNF/LTα−/− mice (24). These results suggest that both TNFα and LTα play a role in the production of NO and IFN-γ after infection with P. berghei. However, the development of CM in B6.TNFα−/− mice demonstrates that elevated serum NO and IFN-γ levels do not correlate with the development of CM.
Changes to the Cerebral Vasculature of TNF-α– and LTα-deficient Mice after P. berghei Infection.
Histological examination of brain sections revealed the presence of RBCs laden with malaria pigment (hemozoin) in C57BL/6 and B6.TNFα−/− mice at the time of death (Fig. 2 , a–d). Small vascular hemorrhages were also observed in these animals (Fig. 2, a–d), but not in animals deficient in LTα at the same time (Fig. 2, e and f). To determine if these anatomical features were associated with the presence of pRBCs in the brain, sections were stained with anti-P. berghei antibody (Fig. 3 a). Significant numbers of pRBCs were found in the cerebral blood vessels of C57BL/6 and B6.TNFα−/− mice at the time of death (Fig. 3 a), but not in B6.LTα−/− and B6.TNF/LTα−/− mice at the same time (day 7 after infection; Fig. 3 a), despite blood parasitemias being the same in all mouse strains at this time (Fig. 1 b). These data suggest that pRBC were being retained in cerebral blood vessels of mice that succumbed to CM.
Figure 2.
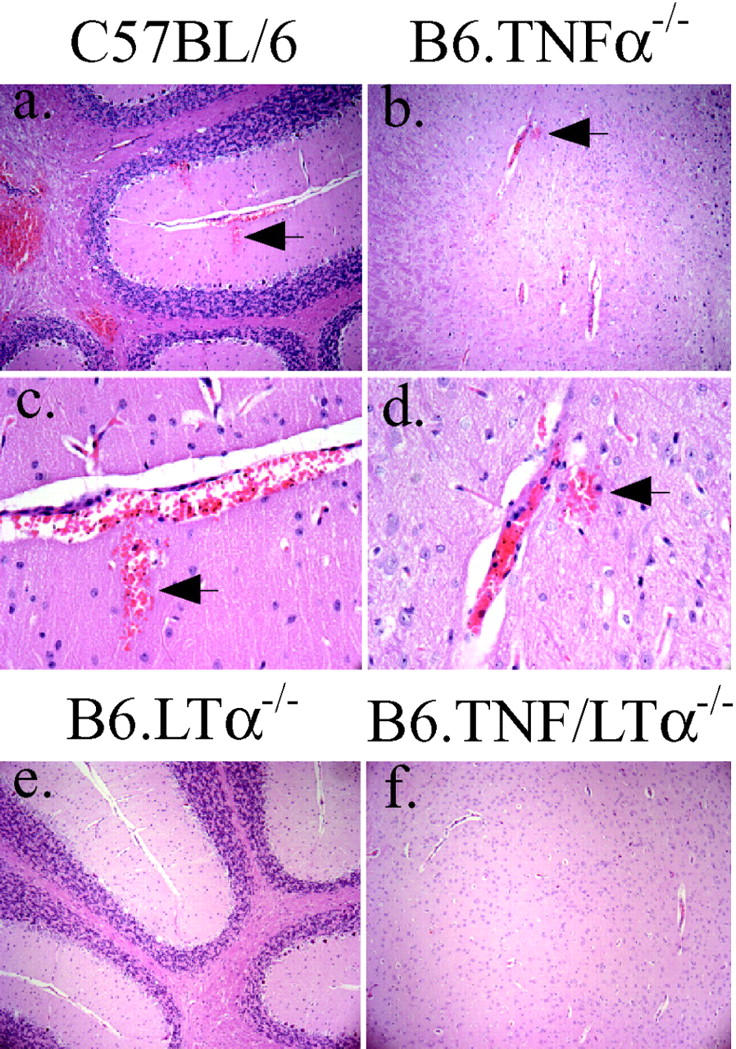
Perivascular hemorrhages are present in C57BL/6 and B6.TNFα−/− mice that develop CM. Sections (3 μM) were prepared from formol saline-fixed brain tissue and stained with hematoxylin and eosin. Magnifications of ×100 (a and b) and ×400 (c and d) are shown for C57BL/6 and B6.TNFα−/− mice that died with CM. Areas within the cerebellum (a, c, and e) and cerebrum (b, d, and f) are shown. Arrows highlight sites of perivascular hemorrhage. These hemorrhages were not observed in LTα-deficient mice (e and f; original magnification: ×100). These data are representative of material obtained in four experiments (at least 25 microscope fields from 12 or more samples from each mouse strain were examined).
Figure 3.

Comparative immunohistochemical analysis of brain sections taken from P. berghei-infected mice when C57BL/6 and B6.TNFα−/− mice developed CM (day 7 after infection). Brain cryosections (6 μM) were stained with (a) anti-P. berghei antibodies, (b) anti-ICAM-1, and (c), anti CR3 (5C6) mAbs with fluorescent secondary reagents. Original magnification is ×400. The samples are representative of material from four experiments.
Increased ICAM-1 expression is a feature of murine CM and is thought to mediate binding of leukocytes and possibly pRBCs to micro-vessels in the brain (25, 26). ICAM-1 expression on vascular endothelial cells in the brain was less in B6.TNFα−/− mice than in C57BL/6 mice (Fig. 3 b; 438 ± 22 ICAM-1+ vessels per 25 fields of view versus 894 ± 16 ICAM-1+ vessels per 25 fields of view, respectively), but increased when compared with expression in naive controls (Fig. 3 b; 245 ± 9 ICAM-1+ vessels per 25 fields of view). Expression of ICAM-1 was also reduced in B6.LTα−/− and B6.TNFα/LTα−/− mice at day 7 after (Fig. 3 b; 438 ± 23 ICAM-1+ vessels per 25 fields of view and 476 ± 23 ICAM-1+ vessels per 25 fields of view, respectively). Associated with the decreased ICAM-1 expression, in TNFα and LTα-deficient mice, there was also a decrease in the number of CR3-positive inflammatory cells found in the brain (Fig. 3 c; 100 ± 3, 427 ± 23, 160 ± 6, 165 ± 5, and 151 ± 7 CR3+ cells per 25 fields of view for naive C57BL/6 mice, and C57BL/6, B6.TNFα−/−, B6.LTα−/−, and B6.TNFα/LTα−/− mice at day 7 after infection, respectively). Together, these results indicate that TNFα and LTα are both required for optimal ICAM-1 up-regulation and leukocyte accumulation in the brain during murine CM. However, these data also show that these events do not necessarily predict the development of CM.
LTα Is Produced by a Radiation-resistant Cell Population.
To assess whether leukocytes accumulating in the brain were the source of LTα that mediated CM, BM chimeric mice were constructed. Congenic B6.Ly5.1 mice were irradiated and engrafted with BM from C57BL/6 mice or animals lacking TNFα, LTα, or both (all express the Ly5.2 CD45 allele). After engraftment, chimeric mice contained fewer than 2% Ly5.1-positive leukocytes in the spleen and peripheral blood (data not shown). After infection with P. berghei, chimeric mice engrafted with BM from C57BL/6 and B6.TNFα−/− mice died at the same time as C57BL/6 and B6.TNFα−/− mice (Fig. 4 a), and showed similar signs of CM. The leukocytes that accumulated in the brain in chimeric mice that developed CM were identified as being of donor origin by staining brain tissue with anti-Ly5.2 antibodies (data not shown). Surprisingly, chimeric mice engrafted with B6.LTα−/− BM died in the ensuing 24 h of CM (Fig. 4, a and c), in contrast to the prolonged survival and lack of CM in B6.LTα−/− mice. This observation indicates that a radiation-resistant cell population in the brain is the major source of LTα during murine CM. However, the fact that chimeric mice engrafted with BM from B6.TNF/LTα−/− mice survived a further 2–3 d (Fig. 4 a) and died showing few signs of CM indicates that either TNFα or LTα produced by leukocytes may contribute to the pathology induced by a LTα-producing, radiation-resistant cell population. LTα mRNA was found in the brains of all chimeric mice, including those engrafted with BM from LTα-deficient mice (Fig. 4 b), confirming that a radiation-resistant cell population was the source of LTα. No LTα mRNA molecules were ever detected by real-time RT-PCR in brain tissue from B6.LTα−/− mice (data not shown). Furthermore, when chimeric mice were prepared by irradiating B6.TNF/LTα−/− mice and engrafting with BM from C57BL/6, B6.TNFα−/−, or B6.LTα−/− mice, all mice failed to develop CM (Fig. 4 c), and died with anemia and hyperparasitemia (data not shown). The extended survival of irradiated B6.TNF/LTα−/− mice engrafted with BM from B6.TNFα−/− mice indicates a role for TNFα in the death of P. berghei-infected mice suffering from anemia and hyperparasitemia. Although the LTα-producing cell population is likely to be of nonhematopoietic origin, we cannot rule out the possibility that a long-lived, radiation-resistant, BM-derived cell population is a source of LTα in the brain. Similarly, we cannot yet rule out any involvement of immune defects associated with TNFα- and LTα-deficient mice in their different susceptibility to CM. Nevertheless, these data show that LTα produced by a radiation-resistant cell is the key factor for the development of murine CM.
Figure 4.



LTα is produced by a radiation-resistant cell population in the brain. (a) Survival curves of chimeric B6.Ly5.1 mice engrafted with BM from C57BL/6 (solid line), B6.TNFα−/− (overlapping C57BL/6 mice and indicated by arrow), B6.LTα−/− (dashed line), and B6.TNF/LTα−/− (dotted line) mice infected with P. berghei. Results represent pooled data from three independent groups of mice (n = 4–6 per group) in experiments conducted on three separate occasions (total of n = 12–16 mice per group). Differences in survival between B6.Ly5.1 mice engrafted with BM from C57BL/6 or B6.TNFα−/− mice and mice deficient in LTα were statistically different (χ2 = 7.01, P < 0.01), as was the difference between mice engrafted with BM from B6.LTα−/− and B6.TNF/LTα−/− mice (χ2 = 23.83, P < 0.0001). (b) LTα mRNA is detected by real-time RT-PCR in the brain of all chimeric B6.Ly5.1 mice following P. berghei infection (brain tissue collected at time of death). The source of engrafted BM is indicated. (c) Survival curve of chimeric B6.Ly5.1 mice engrafted with BM from C57BL/6, B6.TNFα−/− or B6.LTα−/− mice (all groups overlap; solid line), and chimeric B6.TNF/LTα−/− mice engrafted with BM from C57BL/6 (dotted line), B6.TNFα−/− (dashed line), or B6.LTα−/− (dashed and dotted line) mice (n = 4 per group). Differences in survival between B6.Ly5.1 mice engrafted with BM from C57BL/6 and B6.TNF/LTα−/− mice engrafted with BM from C57BL/6, B6.TNFα−/−, or B6.LTα−/− mice were statistically different (χ2 = 15, P < 0.0001), as was the increased survival time of B6.TNF/LTα−/− mice engrafted with BM from B6.TNFα−/− mice over all other groups (χ2 = 6.628, P < 0.01).
In summary, we have shown that LTα produced by a radiation-resistant cell population is the principal mediator of murine CM. Both endothelial cells that line the brain vasculature, microglial cells or astrocytes in the brain parenchyma are potential sources of LTα, as all are radiation-resistant (27, 28) and capable of producing inflammatory mediators (27, 29). Given the constitutive expression of LTα mRNA in the brain, it is likely that LTα acts in synergy with a parasite product or a parasite-induced host mediator. In fact, mice that die with CM accumulate pRBC in cerebral vessels (Fig. 3 a), despite having similar blood parasitemia to LTα-deficient mice (Fig. 1 c), potentially providing increased levels of these products in the brain. The identity of these factors is unknown, but a candidate parasite product for this role is glycosylphosphatidylinositol (GPI), the membrane anchor for various parasite surface antigens that has been shown to increase expression of inflammatory mediators by macrophages and vascular endotlial cells (30). The involvement of LTα in human CM is unknown, but results from this work indicate that it should be investigated.
Acknowledgments
We thank staff at the London School of Hygiene and Tropical Medicine Biological Services Unit for assistance in the breeding and maintenance of mouse colonies. We thank Dr. Martin Holland for assistance with real-time RT-PCR. We also thank Professor David Warhurst, Professor Eleanor Riley, and Dr. Fakhereldin Omer (London School of Hygiene and Tropical Medicine) for helpful advice.
This work was supported by the Wellcome Trust and the British Medical Research Council. C. Engwerda is a Wellcome Trust Career Development Fellow.
References
- 1.Miller, L.H., M.F. Good, and G. Milon. 1994. Malaria pathogenesis. Science. 264:1878–1883. [DOI] [PubMed] [Google Scholar]
- 2.Grau, G.E., T.E. Taylor, M.E. Molyneux, J.J. Wirima, P. Vassalli, M. Hommel, and P.H. Lambert. 1989. Tumor necrosis factor and disease severity in children with falciparum malaria. N. Engl. J. Med. 320:1586–1591. [DOI] [PubMed] [Google Scholar]
- 3.Kwiatkowski, D., A.V. Hill, I. Sambou, P. Twumasi, J. Castracane, K.R. Manogue, A. Cerami, D.R. Brewster, and B.M. Greenwood. 1990. TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet. 336:1201–1204. [DOI] [PubMed] [Google Scholar]
- 4.de Kossodo, S., and G.E. Grau. 1993. Role of cytokines and adhesion molecules in malaria immunopathology. Stem Cells. 11:41–48. [DOI] [PubMed] [Google Scholar]
- 5.Medana, I.M., N.H. Hunt, and G. Chaudhri. 1997. Tumor necrosis factor-alpha expression in the brain during fatal murine cerebral malaria: evidence for production by microglia and astrocytes. Am. J. Pathol. 150:1473–1486. [PMC free article] [PubMed] [Google Scholar]
- 6.Grau, G.E., L.F. Fajardo, P.F. Piguet, B. Allet, P.H. Lambert, and P. Vassalli. 1987. Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science. 237:1210–1212. [DOI] [PubMed] [Google Scholar]
- 7.Garcia, I., Y. Miyazaki, K. Araki, M. Araki, R. Lucas, G.E. Grau, G. Milon, Y. Belkaid, C. Montixi, W. Lesslauer, et al. 1995. Transgenic mice expressing high levels of soluble TNF-R1 fusion protein are protected from lethal septic shock and cerebral malaria, and are highly sensitive to Listeria monocytogenes and Leishmania major infections. Eur. J. Immunol. 25:2401–2407. [DOI] [PubMed] [Google Scholar]
- 8.Lucas, R., J.N. Lou, P. Juillard, M. Moore, H. Bluethmann, and G.E. Grau. 1997. Respective role of TNF receptors in the development of experimental cerebral malaria. J. Neuroimmunol. 72:143–148. [DOI] [PubMed] [Google Scholar]
- 9.Amani, V., A.M. Vigario, E. Belnoue, M. Marussig, L. Fonseca, D. Mazier, and L. Renia. 2000. Involvement of IFN-gamma receptor-medicated signaling in pathology and anti-malarial immunity induced by Plasmodium berghei infection. Eur. J. Immunol. 30:1646–1655. [DOI] [PubMed] [Google Scholar]
- 10.Kwiatkowski, D., M.E. Molyneux, S. Stephens, N. Curtis, N. Klein, P. Pointaire, M. Smit, R. Allan, D.R. Brewster, G.E. Grau, et al. 1993. Anti-TNF therapy inhibits fever in cerebral malaria. Q. J. Med. 86:91–98. [PubMed] [Google Scholar]
- 11.van Hensbroek, M.B., A. Palmer, E. Onyiorah, G. Schneider, S. Jaffar, G. Dolan, H. Memming, J. Frenkel, G. Enwere, S. Bennett, et al. 1996. The effect of a monoclonal antibody to tumor necrosis factor on survival from childhood cerebral malaria. J. Infect. Dis. 174:1091–1097. [DOI] [PubMed] [Google Scholar]
- 12.Riminton, S.D., H. Korner, D.H. Strickland, F.A. Lemckert, J.D. Pollard, and J.D. Sedgwick. 1998. Challenging cytokine redundancy: inflammatory cell movement and clinical course of experimental autoimmune encephalomyelitis are normal in lymphotoxin-deficient, but not tumor necrosis factor-deficient, mice. J. Exp. Med. 187:1517–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roach, D.R., H. Briscoe, B. Saunders, M.P. France, S. Riminton, and W.J. Britton. 2001. Secreted lymphotoxin-alpha is essential for the control of an intracellular bacterial infection. J. Exp. Med. 193:239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bazzoni, F., and B. Beutler. 1996. The tumor necrosis factor ligand and receptor families. N. Engl. J. Med. 334:1717–1725. [DOI] [PubMed] [Google Scholar]
- 15.Sheehan, K.C., N.H. Ruddle, and R.D. Schreiber. 1989. Generation and characterization of hamster monoclonal antibodies that neutralize murine tumor necrosis factors. J. Immunol. 142:3884–3893. [PubMed] [Google Scholar]
- 16.Korner, H., M. Cook, D.S. Riminton, F.A. Lemckert, R.M. Hoek, B. Ledermann, F. Kontgen, B. Fazekas de St Groth, and J.D. Sedgwick. 1997. Distinct roles for lymphotoxin-alpha and tumor necrosis factor in organogenesis and spatial organization of lymphoid tissue. Eur. J. Immunol. 27:2600–2609. [DOI] [PubMed] [Google Scholar]
- 17.De Togni, P., J. Goellner, N.H. Ruddle, P.R. Streeter, A. Fick, S. Mariathasan, S.C. Smith, R. Carlson, L.P. Shornick, J. Strauss-Schoenberger, et al. 1994. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 264:703–707. [DOI] [PubMed] [Google Scholar]
- 18.Hearn, J., N. Rayment, D.N. Landon, D.R. Katz, and J.B. de Souza. 2000. Immunopathology of cerebral malaria: morphological evidence of parasite sequestration in murine brain microvasculature. Infect. Immun. 68:5364–5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engwerda, C.R., S.C. Smelt, and P.M. Kaye. 1996. An in vivo analysis of cytokine production during Leishmania donovani infection in scid mice. Exp. Parasitol. 84:195–202. [DOI] [PubMed] [Google Scholar]
- 20.Banks, T.A., B.T. Rouse, M.K. Kerley, P.J. Blair, V.L. Godfrey, N.A. Kuklin, D.M. Bouley, J. Thomas, S. Kanangat, and M.L. Mucenski. 1995. Lymphotoxin-alpha-deficient mice. Effects on secondary lymphoid organ development and humoral immune responsiveness. J. Immunol. 155:1685–1693. [PubMed] [Google Scholar]
- 21.Kaye, P.M., and G.J. Bancroft. 1992. Leishmania donovani infection in scid mice: lack of tissue response and in vivo macrophage activation correlates with failure to trigger natural killer cell-derived gamma interferon production in vitro. Infect. Immun. 60:4335–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amante, F.H., and M.F. Good. 1997. Prolonged Th1-like response generated by a Plasmodium yoelii-specific T cell clone allows complete clearance of infection in reconstituted mice. Parasite Immunol. 19:111–126. [DOI] [PubMed] [Google Scholar]
- 23.Rosen, H. 1990. Role of CR3 in induced myelomonocytic recruitment: insights from in vivo monoclonal antibody studies in the mouse. J. Leukoc. Biol. 48:465–469. [DOI] [PubMed] [Google Scholar]
- 24.Rudin, W., H.P. Eugster, G. Bordmann, J. Bonato, M. Muller, M. Yamage, and B. Ryffel. 1997. Resistance to cerebral malaria in tumor necrosis factor-alpha/beta-deficient mice is associated with a reduction of intercellular adhesion molecule-1 up-regulation and T helper type 1 response. Am. J. Pathol. 150:257–266. [PMC free article] [PubMed] [Google Scholar]
- 25.Grau, G.E., P. Pointaire, P.F. Piguet, C. Vesin, H. Rosen, I. Stamenkovic, F. Takei, and P. Vassalli. 1991. Late administration of monoclonal antibody to leukocyte function-antigen 1 abrogates incipient murine cerebral malaria. Eur. J. Immunol. 21:2265–2267. [DOI] [PubMed] [Google Scholar]
- 26.Lucas, R., P. Juillard, E. Decoster, M. Redard, D. Burger, Y. Donati, C. Giroud, C. Monso-Hinard, T. De Kesel, W.A. Buurman, et al. 1997. Crucial role of tumor necrosis factor (TNF) receptor 2 and membrane-bound TNF in experimental cerebral malaria. Eur. J. Immunol. 27:1719–1725. [DOI] [PubMed] [Google Scholar]
- 27.Sedgwick, J.D., D.S. Riminton, J.G. Cyster, and H. Korner. 2000. Tumor necrosis factor: a master-regulator of leukocyte movement. Immunol. Today. 21:110–113. [DOI] [PubMed] [Google Scholar]
- 28.Gao, Z., V.C. McAlister, and G.M. Williams. 2001. Repopulation of liver endothelium by bone-marrow-derived cells. Lancet. 357:932–933. [DOI] [PubMed] [Google Scholar]
- 29.Pober, J.S., and R.S. Cotran. 1990. Cytokines and endothelial cell biology. Physiol. Rev. 70:427–451. [DOI] [PubMed] [Google Scholar]
- 30.Schofield, L., S. Novakovic, P. Gerold, R.T. Schwarz, M.J. McConville, and S.D. Tachado. 1996. Glycosylphosphatidylinositol toxin of Plasmodium up-regulates intercellular adhesion molecule-1, vascular cell adhesion molecule-1, and E-selectin expression in vascular endothelial cells and increases leukocyte and parasite cytoadherence via tyrosine kinase-dependent signal transduction. J. Immunol. 156:1886–1896. [PubMed] [Google Scholar]