

Abstract
The constraint of fitting a diverse repertoire of antigen specificities in a limited total population of lymphocytes results in the frequency of naive cells specific for any given antigen (defined as the precursor frequency) being below the limit of detection by direct measurement. We have estimated this precursor frequency by titrating a known quantity of antigen-specific cells into naive recipients. Adoptive transfer of naive antigen-specific T cell receptor transgenic cells into syngeneic nontransgenic recipients, followed by stimulation with specific antigen, results in activation and expansion of both donor and endogenous antigen-specific cells in a dose-dependent manner. The precursor frequency is equal to the number of transferred cells when the transgenic and endogenous responses are of equal magnitude. Using this method we have estimated the precursor frequency of naive CD8 T cells specific for the H-2Db–restricted GP33–41 epitope of LCMV to be 1 in 2 × 105. Thus, in an uninfected mouse containing ∼2-4 × 107 naive CD8 T cells we estimate there to be 100–200 epitope-specific cells. After LCMV infection these 100–200 GP33-specific naive CD8 T cells divide >14 times in 1 wk to reach a total of ∼107 cells. Approximately 5% of these activated GP33-specific effector CD8 T cells survive to generate a memory pool consisting of ∼5 × 105 cells. Thus, an acute LCMV infection results in a >1,000-fold increase in precursor frequency of DbGP33-specific CD8 T cells from 2 × 102 naive cells in uninfected mice to 5 × 105 memory cells in immunized mice.
Keywords: lymphocytic choriomeningitis virus, T cell antigen receptors, adoptive transfer, cellular immunity, CD8-positive T lymphocytes
Introduction
Two cardinal properties of the adaptive immune system are specificity and diversity; thus the immune system can mount exquisitely specific responses to a vast repertoire of possible antigens. As the immune system must accommodate this diversity within a relatively limited total population size there must be compromise between the repertoire of different antigens the immune system can effectively recognize and the number of precursors with any given specificity. Recent technical advances including the use of MHC class I tetramers complexed with defined viral peptide epitopes and measurement of the production of cytokines in response to stimulation with synthetic peptides have allowed CD8 T cell responses to be quantitated with great accuracy. However, neither method is sufficiently sensitive to measure the low frequency of naive antigen-specific T cells before expansion during the generation of an immune response (1–4).
The CD8 T cell response in C57Bl/6J mice to the Armstrong strain of lymphocytic choriomeningitis virus (LCMV) results in a rapid rise in the number of LCMV-specific CD8 T cells from below the threshold of detection (< ∼1,000 cells/mouse) to >107 antigen-specific CD8 T cells by 8 d after infection (3, 4). This expansion is followed by a death phase, during which there is approximately a 10-fold reduction in the number of LCMV-specific CD8 T cells from days 8 to 30, and thereafter the stable maintenance of an antigen-specific memory population. One of the immunodominant T cell responses elicited during this response is specific for the H-2Db–restricted glycoprotein (GP33–41) epitope of LCMV. Although analysis of the T cell receptors within this epitope-specific population has shown a heterogeneous usage of TCR Vβ gene segments (5, 6) adoptive transfer of excessive numbers of CD8 T cells from mice bearing a transgene encoding a TCR specific for this epitope (7), followed by challenge with LCMV, results in a highly restricted DbGP33 response using primarily the transgenic TCR Vβ8.1 and Vα2 TCR segments (8, 9).
In this paper we describe a novel approach for quantitating the precursor frequency of naive antigen-specific CD8 T cells in a polyclonal population. We adoptively transferred limiting numbers of naive antigen-specific TCR transgenic (TCR-Tg) CD8 cells (specific for the DbGP33 epitope) into nontransgenic syngeneic recipients and then measured the fraction of the epitope-specific response contributed by donor and endogenous cells after LCMV infection. In this quantitative-competitive system, adoptive transfer of increasing numbers of specific transgenic cells resulted in a greater fraction of the antigen-specific response arising from donor cells. If donor and endogenous epitope-specific cells have the same participation and proliferative properties, then the relationship between the number of transferred transgenic cells (T) and the fraction of the GP33 response which is transgenic (F) is given by:
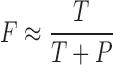 |
(1) |
where P is equal to the precursor frequency of endogenous epitope-specific cells. In the special case when the number of transferred cells equals the endogenous precursor frequency (i.e., T = P), half of the GP33-specific response will be generated by transgenic cells (F = 1/2). Using this experimental approach, we have measured the naive antigen-specific CD8 T cell precursor frequency in mice. Furthermore, we have verified that endogenous and donor transgenic epitope-specific cells have similar proliferative properties in response to antigenic stimulation in vivo and that transfer of exogenous cells does not significantly alter the magnitude or composition of the endogenous DbGP33 CD8 T cell response.
Materials and Methods
Animals and Virus.
6–8-wk-old female C56Bl/6J (B6) or P14 B6.D2-TgN (TCR-Tg) mice were obtained from The Jackson Laboratory. TCR-Tg mice were backcrossed to B6 background (>10 generations) at the Emory University animal facility before use. The Armstrong and clone-13 strains of LCMV were grown as previously described (10). The recombinant vaccinia virus expressing the GP33–41 epitope (VV-GP33) was generated and grown as described previously (11). For primary infections and generating immune mice, B6 mice were given 2 × 105 PFU of LCMV (Armstrong) intraperitoneally. For secondary infections, immune mice were given either 2 × 106 PFU of LCMV (clone-13) intravenously or 107 PFU of VV-GP33 intraperitoneally and assayed 5 d after infection. For chronic infection, naive B6 mice were given 2 × 106 PFU of LCMV (clone-13) intravenously.
Antibodies and MHC Tetramers.
Anti-CD8α (Lyt-2, clone 53–6.7), anti-Vα2 TCR (clone B20.1), and anti-Vβ8.1,8.2 TCR (clone MR5–2) antibodies as well as the panel of anti-Vβ antibodies were purchased from BD PharMingen. The anti-Db antibody (clone 28–14–8) was generated from the HB-27 hybridoma (American Type Culture Collection) and Fab fragments prepared using the Immunopure Fab preparation kit (Pierce Chemical Co.) in accordance with manufacturer's directions. H-2Db MHC class I tetramer complexes were refolded with synthetic GP33–41 peptide (KAVYNFATM) and prepared as previously described (3, 12). Biotinylated monomeric DbGP33 complexes were assembled into tetramers by the addition of allophycocyanin-conjugated streptavidin (Molecular Probes) in a 4:1 molar ratio.
Cells and Flow Cytometry.
Single cell suspensions of splenocytes were prepared as described previously (3). Peripheral blood lymphocytes were separated from whole blood over Histopaque®-1077 (Sigma-Aldrich) in accordance with manufacturer's recommendations. Cells were resuspended in FACS® buffer (PBS containing 2% bovine serum albumin and 0.2% sodium azide) and stained with the indicated reagents at a final concentration of 1 μg/ml for 30 min at 4°C. Cells were then washed twice in FACS® buffer, fixed in a 2% paraformaldehyde solution, and immediately acquired on a FACSCalibur™ flow cytometer (Becton Dickinson).
TCR Affinity Measurement by MHC Tetramer Fall-Off Assay.
Measurement of relative TCR affinity by a competitive tetramer fall-off assay was done as described previously (13). Briefly, 2 × 107 splenocytes from infected mice were stained with the DbGP33 tetramer and anti-CD8, -Vα2 antibodies as described above. After two washes, cells were incubated with or without unlabeled competitor anti-Db antibody Fab (clone 28–14–8f) at 2 mg/ml in FACS® buffer at 4°C. At the indicated times, 100-μl aliquots were removed and added to an equal volume of 2% PFA in PBS and immediately acquired on a FACSCalibur™ flow cytometer. Normalized total fluorescence less background staining was calculated using the following formula: NTFtime i = (% pos. × MFI)time i/(% pos. × MFI)time 0.
Adoptive Transfer of TCR-Tg Cells.
For cell transfers, single cell suspensions of splenocytes from naive P14 TCR-Tg transgenic mice, bearing TCR specific to the DbGP33 epitope of LCMV, were injected intravenously into the tails of recipient mice and infected 2 d after transfer. To account for differences in engraftment of such limited numbers of cells, splenocytes from naive P14 TCR-Tg transgenic mice were diluted in naive B6 splenocytes such that a constant number of total cells (2 × 106) were transferred into recipients receiving different doses of transgenic cells. For transfer of purified CD8+ populations, cells were positively selected by MACS separation (Miltenyi Biotec) in accordance with manufacturer's directions and purity determined (normally >90%) by flow cytometry before transfer.
Results and Discussion
Measurement of Precursor Frequency.
Adoptive transfer of an increasing number of naive TCR-Tg CD8 T cells into nontransgenic syngeneic recipients, followed by challenge with LCMV results in an increasing proportion of the DbGP33-specific response being contributed by donor cells (Fig. 1) . Donor TCR-Tg cells were designated as CD8 T cells which specifically bound the DbGP33 tetramer as well as expressed both the Vα2 (Fig. 1 A) and Vβ8.1 (Fig. 1 B) TCR gene segments. When no cells were transferred, <5% of cells that bound the DbGP33 tetramer used both the Vα2 and Vβ8.1 TCR gene segments. Because only a very small fraction (<5%) of the endogenous response had the same TCR Vα and Vβ gene usage as the donor transgenic cells, we were able to use Vα2+Vβ8.1+DbGP33+ CD8+ cells to follow the donor transgenic response. The transgenic proportion of the DbGP33 response increased with the number of cells transferred and at the highest doses, the response was dominated by donor transgenic cells (>98%). Similar results were seen when Thy1.1+ TCR-Tg cells were transferred into Thy1.2+ recipients using the Thy1 marker to distinguish between donor and host responses (data not shown).
Figure 1.

Adoptive transfer of increasing numbers of DbGP33-specific TCR Tg CD8+ T cells (Vα2+Vβ8.1+), followed by challenge with LCMV, results in an increasing proportion of the total epitope-specific response being contributed by donor cells. (A) Analysis of recipient mice after viral challenge. Contour plots are gated on CD8+ T cells and percentages indicate the relative proportions of DbGP33+ cells that are Vα2+ or Vα2−. Numbers above each plot indicate the number of TCR-Tg CD8+ T cells transferred. (B) Analysis of Vβ8.1 usage by epitope-specific cells. Histograms are gated on CD8+DbGP33+Vα2+ cells. Percentages under each histogram indicate the total Vα2+Vβ8.1+ portion of the DbGP33-specific CD8+ T cell response. (C) The donor TCR-Tg fraction of the epitope-specific response is plotted vs. the number of TCR-Tg cells engrafted. Open symbols circles represent mice that were given only TCR-Tg cells, while filled circles represent mice which were given TCR-Tg cells diluted in 2 × 106 naive polyclonal B6 splenocytes. The solid line is the best fit to the equation F = T/(T + P) while the dashed line indicates the dose of donor cells at which 50% of the DbGP33-specific response is endogenous.
Calculation of precursor frequency requires accurate estimation of the engraftment or “take” of these donor cells in recipient mice. We observed that ∼10% of total donor populations were recoverable from spleens of recipient mice 2 d after transfer (Fig. 2 A). In addition, splenic recovery did not differ after transfer of either polyclonal cells (measuring a congenic difference at the Thy1 locus) or cells from TCR-Tg mice. Using 10% as an estimate of the number of transferred cells which persist in the spleens of recipient mice, we plotted the transgenic fraction of the DbGP33-specific CD8 T cell response versus the number of donor transgenic cells engrafted (Fig. 1 C). The fraction of the DbGP33-specific response attributable to donor cells increased with the amount of cells transferred in a dose-dependent manner. The transgenic fraction of the DbGP33-specific response closely followed predicted values, namely fraction transgenic (F) is equal to the number of transgenic precursors transferred divided by the total number of precursors (T/[T+P]). This allowed us to estimate the number of endogenous DbGP33-specific precursors in the spleen either by the best fit to this equation or by the number of transferred transgenic cells at which half the response was transgenic. We thus estimated that there are on average 56 (range 46–65: >95% confidence interval) endogenous DbGP33-specific precursors per spleen in uninfected mice. Given that there are on average 107 naive CD8 T cells in the spleens of uninfected mice, this translates to an epitope-specific precursor frequency of 1 in 2 × 105.
Figure 2.

Engraftment of TCR-Tg and polyclonal B6 CD8+ T cells are similar. (A) Splenocytes from either TCR-Tg or B6 (Thy1.1+) mice were injected into naive, nonirradiated B6 (Thy1.2+) recipients. Percentages indicate the fraction of naive TCR-Tg or B6 donor CD8+ T cells recoverable from spleens of recipient mice 2 d after transfer. Each bar represents >10 mice and error bars indicate standard deviation from the mean. (B) To discount differences in survival of TCR-Tg vs. B6 cells, 108 purified polyclonal CD8+ cells from C57Bl/6 Thy1.1+ mice (which, given our estimates, contains ∼500 precursor cells) were mixed with 500 purified TCR-Tg Thy1.1+/Thy1.2+ CD8+ cells. These were then injected into naive B6 Thy1.2+ recipients. After challenge with LCMV, epitope-specific donor cells (Thy1.1+) from recipient mice were analyzed for Thy1.2 (TCR Tg only) expression. Percentages indicate the proportion of the CD8+DbGP33+Thy1.1+ response due to TCR-Tg (Thy1.2+) or polyclonal B6 (Thy1.2−) donor cells.
This estimate of precursor frequency depends upon several assumptions: accurate measurement of the engraftment or “take” of donor cells, similar proliferation of transgenic and polyclonal epitope-specific populations in response to antigenic stimulation, and that addition of increasing numbers of donor epitope-specific cells does not qualitatively change the recruitment from the endogenous epitope-specific pool. We have tested these assumptions directly and accounted for them in our estimate of precursor frequency.
Engraftment of Donor Cells.
We have defined engraftment or “take” as the percentage of total donor cells that are recoverable from spleens of recipient mice and have measured this to be 10% for large populations of either polyclonal or transgenic donor CD8 T cells. It is possible the number of donor cells surviving may (due to homeostatic mechanisms that limit the total number of naive CD8 T cells) diminish as increasing numbers of cells are transferred. To compensate for any variance due to the size of the transferred population, donor TCR-Tg cells were diluted in naive polyclonal cells such that in all cases, a constant number of total cells (2 × 106 splenocytes) were transferred. As shown in Fig. 1 C, the fraction of the DbGP33-specific CD8 T cell response attributable to transgenic donor cells was similar whether these were transferred alone (open symbols) or in the context of a larger population of polyclonal cells (filled symbols). Thus, our measurements demonstrate similar precursor frequency and engraftment of donor cells in recipient spleens regardless of the total number of cells transferred in these experiments. Also, it is unlikely that DbGP33-specific precursors in the donor polyclonal population made any significant contribution to the response as, given our estimate of precursor frequency, on average <1 antigen-specific cell is present in this population.
To directly compensate for any variance in the engraftment of donor transgenic cells into resident endogenous polyclonal populations, we transferred 108 polyclonal Thy1.1+/+ CD8 T cells (containing ∼500 DbGP33-specific precursors given our estimate) mixed with 500 epitope-specific Thy1.1+/Thy1.2+ TCR-Tg CD8 T cell precursors into naive irradiated Thy1.2+/+ recipient mice. These mice were then challenged with LCMV and their DbGP33-specific Thy1.1+ CD8 T cell response measured. If both the engraftment and proliferation of virus specific cells within these donor populations were similar, then roughly 50% of the DbGP33-specific CD8 Thy1.1+ T cell response would be due to each donor population. We observed a 2:1 ratio of polyclonal (Thy1.2−) to TCR-Tg (Thy1.2+) responders in challenged recipient mice (Fig. 2 B). Given the experimental constraints of transferring such a large number of CD8 T cells as well as genetic background differences in these strains, this indicates that our estimate of precursor frequency is off by at most a factor of 2. This would give an antigen-specific precursor frequency of 10−5 naive CD8 T cells in mice.
Transgenic and Endogenous GP33-specific Cells Have Similar Proliferative Properties.
In addition to accurate measurement of the donor cell “take,” our precursor measurement also depends on transgenic and endogenous DbGP33-specific CD8 T cells possessing similar proliferative properties during an immune response. To test the inherent proliferative “fitness” of transgenic and endogenous DbGP33-specific cells, we followed the increase in magnitude of each population and the ratio of endogenous to transgenic cells in individual mice during secondary responses to either LCMV or a vaccinia virus expressing the GP33 epitope (VV-GP33) (8, 11, 14). During recall responses in LCMV-rechallenged mice, both donor transgenic and endogenous DbGP33-specific CD8 T cells underwent expansion, with an approximately fivefold increase in frequency. A considerably greater expansion of both donor and endogenous populations (∼20-fold) was seen in mice rechallenged with VV-GP33. However, in both recall responses the ratio of endogenous to TCR-Tg donor DbGP33-specific CD8 T cells in individual mice did not change significantly during the secondary response compared with that seen before rechallenge (Fig. 3) . These data indicate that polyclonal and transgenic cells do not have any significant proliferative differences that would skew our estimate of precursor frequency and that donor and endogenous DbGP33-specific CD8 T cells behave similarly following antigenic stimulation in vivo.
Figure 3.

TCR-Tg and polyclonal B6 epitope-specific CD8+ T cells have similar proliferative properties. LCMV immune mice containing a mixture of TCR-Tg and endogenous DbGP33-specific CD8+ T cells were rechallenged with either LCMV (clone-13) (A) or a vaccinia virus expressing the GP33 epitope (VV-GP33) (B). PBMCs were analyzed for the frequency of DbGP33-specific cells (bar graphs, left axis) as well as the fraction of the DbGP33+ response from individual mice due to donor TCR-Tg cells (right axis) before (memory) and after (secondary) rechallenge. Graphs indicate mean frequency of antigen specific cells (>6 mice per group) and error bars indicate standard deviation from the mean.
Another potential factor that could affect the proliferation of donor and endogenous DbGP33-specific CD8 T cells is competition and/or differential participation in the immune response due to differences in the TCR affinity of these two populations. As the transgene encoding the P14 TCR is derived from a polyclonal response, the affinity of the transgenic TCR for its DbGP33 ligand must be contained within the spectrum of TCR affinities of the polyclonal DbGP33-specific response (7). We have measured the relative affinities of transgenic and polyclonal DbGP33-specific CD8 T cells in competitive tetramer fall-off assays (13, 15). The DbGP33-specific TCR-Tg cells have initial fall-off rates similar to that of the polyclonal population (see Fig. 5 A). Similar to published reports, the tetramer fall-off rate from P14 TCR transgenic cells proceeds with linear first-order decay kinetics. However, although the vast majority of endogenous polyclonal DbGP33-specific CD8 T cells follow similar kinetics, quantitative analysis of this population is complicated by its broad TCR diversity (5). As lower affinity cells are lost from the sample population during the course of the assay, the overall affinity of the remainder is increased (see Fig. 5 B). Thus, there may be a minor population of higher affinity endogenous DbGP33-specific CD8 T cells; precisely what one would predict if the narrow affinity of the transgenic TCR falls within a broader spectrum of polyclonal TCR with both higher and lower affinities.
Figure 5.

Transfer of increasing numbers of donor cells does not affect the TCR affinity or diversity of the endogenous DbGP33 response. TCR affinity. (A) Relative TCR affinity of P14 TCR transgenic (filled circles) versus polyclonal (open circles) DbGP33-specific CD8 T cells. For quantitative comparison of tetramer fall-off kinetics, data are shown as the (−) change in normalized total fluorescence (NTF, as described in Materials and Methods) over time. The mean (± SD) slope of each interval is equivalent to (NTFi − NTFi-1)/t where t is the length of the time interval. (B) Relative TCR affinity of endogenous DbGP33-specific CD8 T cells from mice whose DbGP33 response was completely due to endogenous cells (circles), 25–33% donor cells (triangles), or 66–75% donor cells (squares) were analyzed by tetramer fall-off assay as described. (C) TCR diversity. Endogenous DbGP33-specific CD8 T cells from mice containing either completely endogenous (open circles) or 50% transgenic DbGP33 cells (filled circles) were analyzed for TCR-Vβ usage using a panel of monoclonal antibodies. Shown is the percentage of the endogenous (Vα2−) DbGP33 response using each Vβ gene segment.
Antigen Availability and the Magnitude of the Endogenous Response.
Our measurement of precursor frequency may also have been affected by skewing of the endogenous response when increasing numbers of TCR-Tg cells are transferred (for example if competition for antigen affected recruitment of endogenous epitope-specific precursors). Thus, if the total number of precursors recruited into a given response were limited to a finite number, transfer of increasing numbers of donor cells may result in a partial participation by endogenous epitope-specific precursors. In the range where we have measured the endogenous precursor frequency, we observed little change in the magnitude of the endogenous DbGP33-specific response when low numbers of donor cells were transferred (Fig. 4 A). However, as transgenic cells became saturating, the endogenous response was effectively suppressed. Thus, in the range where we have measured precursor frequency antigen does not appear to be limiting, but as transgenic precursors become saturating, the viral infection is controlled faster and thus prevents recruitment from the endogenous pool.
Figure 4.

No suppression of the endogenous DbGP33-specific CD8+ T cell response due to limited antigen availability. (A) No suppression of the endogenous response was observed in the range of precursor frequency. Mice receiving different numbers of DbGP33-specific TCR Tg CD8+ T cells were challenged with LCMV and 7 d after infection, splenocytes were analyzed by flow cytometry. The magnitude of the endogenous polyclonal (open circles) and donor transgenic (filled circles) DbGP33-specific CD8+ response is shown per spleen versus the number of engrafted donor cells. (B) Increased antigen load results in precisely the same dose dependence of donor cells. Mice receiving different numbers of donor TCR-Tg cells were challenged with LCMV (ARM) (circles) or the more virulent LCMV (clone-13) (triangles). Plotted is the fraction of the response due to donor cells versus the number of donor cells engrafted.
We have directly determined the influence of antigen availability on our estimate of precursor frequency by increasing the viral burden and thus antigen load in recipient mice. Mice infected with the more virulent clone-13 strain of LCMV showed prolonged viremia and higher levels of DbGP33-specific CD8 T cell expansion in this system (data not shown). However, despite this larger expansion, we observed precisely the same dose dependence of transgenic cells in LCMV (clone-13) infected mice (measuring the transgenic fraction of the DbGP33 response) as in LCMV (Armstrong) infected mice (Fig. 4 B). Thus in either case, all available precursors were able to participate in the antiviral response indicting that our measurement of precursor frequency is not skewed by availability of antigen or suppression of the endogenous DbGP33 response. Also, increasing antigen load minimizes competition between cells with potentially different TCR affinities, further arguing against differences in participation between transgenic and polyclonal DbGP33-specific CD8 T cells due to TCR affinity.
Affinity and Diversity of the Endogenous Response.
Another factor that could potentially influence our estimate of precursor frequency is recruitment of a different subset of endogenous DbGP33-specific CD8 T cell precursors. We have directly measured both the affinity and diversity the endogenous responses in the range where we have measured precursor frequency. We observed no change in the affinity (as measured by the tetramer fall-off assay) of the endogenous DbGP33 CD8 T cell response (Vα2−) when an increasing proportion of the DbGP33 response was due to donor TCR-Tg cells (Fig. 5 B). Thus, endogenous DbGP33-specific CD8 T cell populations had precisely the same affinity regardless of whether 0%, ∼33%, or ∼66% of the response was contributed by donor cells. Additionally, we have compared the TCR diversity of the endogenous (Vα2−) DbGP33 CD8 T cells as increasing numbers of donor TCR-Tg cells were transferred. We observed no significant difference in the TCR diversity (in terms of TCR-Vβ gene segment usage) of the endogenous DbGP33 CD8 T cells in mice that received no transgenic cells or whose response was ∼50% transgenic (Fig. 5 C). Thus, in the range where we are measuring precursor frequency, we do not significantly affect the affinity or diversity of the endogenous DbGP33 CD8 T cell response.
Implications.
We have defined precursor frequency as the number of naive CD8 T cells specific for a given peptide-MHC combination in mice before antigenic challenge and have estimated this to between 10−5 and 2 × 10−5 or 50–100 cells/spleen. It has been estimated that an adult uninfected mouse contains between 2–4 × 107 naive CD8+ T cells. Based on this, we thus estimate that there are between 100–200 naive CD8 T cells specific for the DbGP33 epitope in an adult uninfected mouse. Certainly, an upper-bound estimate of the total number of antigen-specific CD8 T cells present would be equal to the 500 cells originally transferred.
As the response to a given epitope generally involves several different T cell clones the average precursor frequency per clone will be the precursor frequency per epitope divided by the number of clones (5, 6, 16–19). The total T cell clonal diversity has recently been estimated for humans (20) and mice (21). In mice at least 2 × 106 unique TCR were estimated. The reciprocal of this clonal diversity estimate in mice yields a clonal T cell frequency estimate of 2 × 10−6. This study also found a redundancy of ∼10 cells/unique TCR sequence (21). As it has been estimated that in a given epitope-specific T cell response ∼20 different clones participate (18, 19), this would indicate that on average 200 cells participate in an epitope-specific response, very similar to our estimate of the number of DbG33-specific precursors present in naive mice. Additionally, even though the clonal diversity in humans is increased only 10-fold, it has been estimated that there are ∼10,000-fold more CD8 T cells in humans compared with mice.
Although we have shown that the precursor frequency, defined as the total number of CD8 T cells specific for the DbGP33 epitope of LCMV, is 1 in 2 × 105, the precise relationship between this precursor frequency and the diversity of epitopes which could potentially be recognized is complex, dependent on both overlapping TCR specificities as well as the clonality of any given response. However the inverse of the precursor frequency provides a lower bound to the total number of epitopes that can be recognized, implying that at least 2 × 105 different epitopes may be recognized in mice.
In addition to obtaining an epitope-specific precursor estimate, this data also provides us with a more accurate estimate of the total number of divisions seen in LCMV-specific CD8 T cells during infection of mice. At the peak of the immune response to LCMV (8 d after infection), the number of DbGP33-specific CD8 T cells is roughly 107 (3, 4). Thus, there is a >50,000-fold increase in the number of epitope-specific CD8 T cells during the response to viral infection. Assuming there is very little death of antigen specific cells during this period (days 0–8), this also indicates that this cell population would have had to undergo on average >14 divisions. After the peak of this response, most of the virus-specific CD8 T cells undergo apoptosis, leaving behind a stable memory pool containing ∼5 × 105 DbGP33-specific CD8 T cells. Thus, in immune mice there is a ∼5,000-fold net increase in the number of antigen-specific cells contained in the memory CD8 T cell population.
The interrelationship between the specificity and diversity of T cell responses remain central to understanding the generation of immune responses to all pathogens. Estimating these parameters accurately is a first step in this process and facilitates a clearer understanding of the balance maintained in naive T cell repertoires versus the expansion of antigen-specific populations.
Acknowledgments
We would like to thank J. Miller, P.L. Mahar, and K. Madhavi-Krishna for technical assistance, and B.D. Evavold, E.J. Wherry, and J.M. Grayson for helpful discussion.
This work was supported by National Institutes of Health grants AI49334 (to R. Antia), AI42373 (to J.D. Altman), and AI30048 (to R. Ahmed). S.M. Kaech was supported in part by the Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation (DRG-1570).
References
- 1.Givan, A.L., J.L Fisher, M. Waugh, M.S. Ernstoff and P.K. Wallace. 1999. A flow cytometric method to estimate the precursor frequencies of cells proliferating in response to specific antigens. J. Immunol. Methods 230:99-112. [DOI] [PubMed] [Google Scholar]
- 2.Murali-Krishna, K., L.L. Lau, S. Sambhara, F. Lemonnier, J.D. Altman, and R. Ahmed. 1999. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science. 286:1377–1381. [DOI] [PubMed] [Google Scholar]
- 3.Murali-Krishna, K., J.D. Altman, M. Suresh, D.J. Sourdive, A.J. Zajac, J.D. Miller, J. Slansky, and R. Ahmed. 1998. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 8:177–187. [DOI] [PubMed] [Google Scholar]
- 4.Butz, E., and M.J. Bevan. 1998. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity. 8:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blattman, J.N., D.J.D. Sourdive, K. Murali-Krishna, R. Ahmed, and J.D. Altman. 2000. Evolution of the T cell repertoire during primary, memory, and recall responses to viral infection. J. Immunol. 165:6081–6090. [DOI] [PubMed] [Google Scholar]
- 6.Lin, M.Y., and R.M. Welsh. 1998. Stability and diversity of T cell receptor repertoire usage during lymphocytic choriomeningitis virus infection of mice. J. Exp. Med. 188:1993–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pircher, H., U.H. Rohrer, D. Moskophidous, R.M. Zinkernagel, and H. Hengartner. 1989. Lower receptor avidity required for thymic clonal deletion than for effector T-cell function. Nature. 351:482–485. [DOI] [PubMed] [Google Scholar]
- 8.Ehl, S., P. Klenerman, R.M. Zinkernagel, and G. Bocharov. 1998. The impact of variation in the number of CD8(+) T-cell precursors on the outcome of virus infection. Cell. Immunol. 189:67–73. [DOI] [PubMed] [Google Scholar]
- 9.Zimmerman, S., A. Prevost-Blondel, C. Blaser, and H. Pircher. 1999. Kinetics of the response of naïve and memory CD8 T cells to antigen: similarities and differences. Eur. J. Immunol. 29:284–290. [DOI] [PubMed] [Google Scholar]
- 10.Ahmed, R., and M.B.A. Oldstone. 1988. Organ-specific selection of viral variants during chronic infection. J. Exp. Med. 167:1719–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Most, R.G., K. Murali-Krishna, J.L. Whitton, C. Oseroff, J. Alexander, S. Southwood, J. Sidney, R.W. Chesnut, A. Sette, and R. Ahmed. 1998. Identification of Db- and Kb-restricted subdominant cytotoxic T-cell responses in lymphocytic choriomeningitis virus-infected mice. Virology. 240:158–167. [DOI] [PubMed] [Google Scholar]
- 12.Altman, J.D., P.A.H. Moss, P.J.R. Goulder, D.H. Barouch, M.G. McHeyzer-Williams, J.I. Bell, A.J. McMichael, and M.M. Davis. 1996. Phenotypic analysis of antigen-specific T lymphocytes. Science. 274:94–96. [DOI] [PubMed] [Google Scholar]
- 13.Savage, P.A., J.J. Boniface, and M.M. Davis. 1999. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity. 10:485–492. [DOI] [PubMed] [Google Scholar]
- 14.Moskophidis, D., F. Lechner, H. Pircher, and R.M. Zinkernagel. 1993. Virus persistence in acutely infected immunocompentent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 362:758–761. [DOI] [PubMed] [Google Scholar]
- 15.Sourdive, D.J., K. Murali-Krishna, J.D. Altman, A.J. Zajac, J.K. Whitmire, C. Pannetier, P. Kourilsky, B. Evavold, A. Sette, and R. Ahmed. 1998. Conserved T cell receptor repertoire in primary and memory CD8 T cell responses to an acute viral infection. J. Exp. Med. 188:71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Busch, D.H., I. Pilip and E.G. Pamer. 1998. Evolution of a complex T cell receptor repertoire during primary and recall bacterial infection. J. Exp. Med. 188:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maryanski, J.L., V. Attuil, P. Bucher, and P.R. Walker. 1999. A quantitative, single-cell PCR analysis of an antigen-specific TCR repertoire selected during an in vivo CD8 response: direct evidence for a wide range of clone sizes with uniform tissue distribution. Mol. Immunol. 36:745–753. [DOI] [PubMed] [Google Scholar]
- 18.Maryanski, J.L., C.V. Jongeneel, P. Bucher, J.L. Casanova, and P.R. Walker. 1996. Single-cell PCR analysis of TCR repertoires selected by antigen in vivo: a high magnitude CD8 response is comprised of very few clones. Immunity. 4:47–55. [DOI] [PubMed] [Google Scholar]
- 19.Yanagi, Y., A. Tishon, H. Lewicki, B.A. Cubitt, and M.B. Oldstone. 1992. Diversity of T-cell receptors in virus-specific cytotoxic T lymphocytes recognizing three distinct viral epitopes restricted by a single major histocompatibility complex molecule. J. Virol. 66:2527–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arstila, T.P., A. Casrouge, V. Baron, J. Even, J. Kanellopoulos, and P. Kourilsky. 2000. A direct estimate of the human alphabeta T cell receptor diversity. Science. 286:958–961. [DOI] [PubMed] [Google Scholar]
- 21.Casrouge, A., E. Beaung, S. Dalle, C. Pannetier, J. Kanellopoulos, and P. Kourilsky. 2000. Size estimate of the alphabeta TCR repertoire of naive mouse splenocytes. J. Immunol. 164:5782–5787. [DOI] [PubMed] [Google Scholar]