

Abstract
Spatiotemporal aspects of filovirus entry and release are poorly understood. Lipid rafts act as functional platforms for multiple cellular signaling and trafficking processes. Here, we report the compartmentalization of Ebola and Marburg viral proteins within lipid rafts during viral assembly and budding. Filoviruses released from infected cells incorporated raft-associated molecules, suggesting that viral exit occurs at the rafts. Ectopic expression of Ebola matrix protein and glycoprotein supported raft-dependent release of filamentous, virus-like particles (VLPs), strikingly similar to live virus as revealed by electron microscopy. Our findings also revealed that the entry of filoviruses requires functional rafts, identifying rafts as the site of virus attack. The identification of rafts as the gateway for the entry and exit of filoviruses and raft-dependent generation of VLPs have important implications for development of therapeutics and vaccination strategies against infections with Ebola and Marburg viruses.
Keywords: Filovirus, Ebola, rafts, budding, VLP
Introduction
The filoviruses Ebola (EBOV) and Marburg (MBGV) are two of the most pathogenic viruses in humans and nonhuman primates (1) which cause a severe hemorrhagic fever (2). The mortality rates associated with infections of Ebola or Marburg virus reach as high as 70–80% (1, 2). Although natural outbreaks have been geographically restricted so far, limited knowledge of the mechanisms of pathogenicity, potential of aerosol transmission (3), unknown natural reservoir, and lack of immunological and pharmacological therapeutic measures pose a challenge to classification of the public health threat of Marburg and Ebola viruses. Despite recent advances in vaccine development in certain animal models (4–7), substantial obstacles need to be overcome before such vaccines could qualify for human clinical trials (8). Efforts to develop therapeutics against Ebola and Marburg have been hampered, in part, by limited knowledge of the mechanism of action of viral proteins and the poor understanding of the process of filovirus entry and budding at the molecular level. Understanding the nature of interactions between filoviruses and the host, both at the cellular and organism levels, is essential for successful development of efficacious prophylactic and therapeutic measures.
Both entry and release of enveloped virus particles are dependent on an intimate interaction with components of the cellular membranes. While the plasma membrane was initially envisioned as a fluid, randomly arranged lipid bilayer with incorporated proteins, recent advances demonstrate that this important cellular barrier is more sophisticated and dynamic than portrayed by the original simplistic models. Cholesterol-enriched regions in the lipid bilayer have been recently defined that adopt a physical state referred to as liquid-ordered phase (lo) displaying reduced fluidity and enhanced ability for lateral and rotational mobility (9, 10). These low density, detergent-insoluble microdomains, known as lipid rafts, accommodate a selective set of molecules such as gangliosides, glycosphingolipids, glycosylphosphatidylinositol (GPI) anchored proteins, and signaling proteins such as Src family kinases, T and B cell receptors, and phospholipase C (9, 11–14). By virtue of these unique biochemical and physical properties, lipid rafts function as specialized membrane compartments for channeling certain external stimuli into specific downstream pathways (15, 16), act as platforms in cell–cell interactions (17, 18), and have also been implicated in membrane trafficking (10, 19). Lipid rafts are believed to perform such diverse functions by providing a specialized microenvironment in which the relevant molecules for the initiation of the specific biological processes are partitioned and concentrated (11). Such compartmentalization may help the signals achieve the required threshold at the physiological concentrations of the stimuli. Partitioning in lipid rafts can also be perceived as a measure to perform functions in a more specific and economic manner while keeping distinct pathways spatially separated.
Several lines of evidence suggest a role for cholesterol-enriched lipid rafts in host-pathogen interactions. Cholesterol has been shown to play a critical role for the entry of mycobacterium into macrophages (20). Multiple components of influenza virus (21), measles virus (22), and HIV (23, 24) have been shown to localize to lipid rafts. These lipid platforms have also been implicated in the budding of HIV and influenza virus (21, 23). Therefore, rafts, as tightly regulated specialized domains, may represent a coordination site for the intimate interactions of viral proteins required for the assembly and budding process. While involvement of rafts in virus entry has been postulated (25), supporting data on this issue have been reported only for HIV infection of certain T cell lines (26).
Here, using a variety of biochemical and microscopic approaches, we demonstrate the compartmentalization of Ebola and Marburg viral proteins in lipid rafts during viral assembly and budding. Our findings also show that the entry of filoviruses into cells is dependent on functional rafts. Thus, this study provides a deeper understanding of the molecular mechanisms of filovirus pathogenicity at the cellular level and suggests raft integrity and/or raft components as potential targets for therapeutic interventions. We also report, for the first time, the raft-dependent formation of Ebola-based, genome-free, virus-like particles (VLPs)*, which resemble live EBOV in electron micrographs. Such VLPs, besides being a research tool, could be potentially useful as vaccines against filovirus infections, or as vehicles for the delivery of a variety of antigens artificially targeted to the rafts.
Materials and Methods
Plasmids, Transfections, Western Blot Analysis, and GM1 Blot.
cDNAs encoding EBOV-Zaire glycoproteins (GPs) and VP40 as well as MBGV Musoke GP were cloned in pWRG7077 mammalian expression vector. 293 T cells were transfected using a calcium phosphate transfection kit (Edge Biosystems) according to manufacturer's instructions. Western blot analysis was performed using as primary antibodies anti-EBOVGP mAb 13F6 (27), anti-Marburg GP mAb (5E2), anti-EBOV-VP40 mAb, or a guinea pig anti-Marburg antibody, followed by blotting with HRP-conjugated secondary antibodies and signals were detected by enhanced chemiluminescence. GM1 was detected in lysates or immunoprecipitates by spotting on a nitrocellulose membrane after boiling in SDS, followed by blocking of the membranes and blotting with HRP-conjugated CTB and detection by ECL.
Preparation of Detergent Insoluble Fractions and Lipid Rafts.
Lipid rafts were prepared after lysing the cells in lysis buffer containing 0.5% Triton X-100 as described previously (13). Raft and soluble fractions were then analyzed by immunoblotting. In some experiments (see Fig. 3 A), detergent-insoluble fractions were extracted without ultracentrifugation as described previously (24). Briefly, cells were pelleted and lysed in 0.5% Triton X-100 lysis buffer. After removing the lysate (soluble fraction), the pellet was washed extensively and SDS sample buffer added to the pellet (insoluble fraction). Soluble and insoluble fractions were analyzed by SDS-PAGE and immunoblotting.
Figure 3.
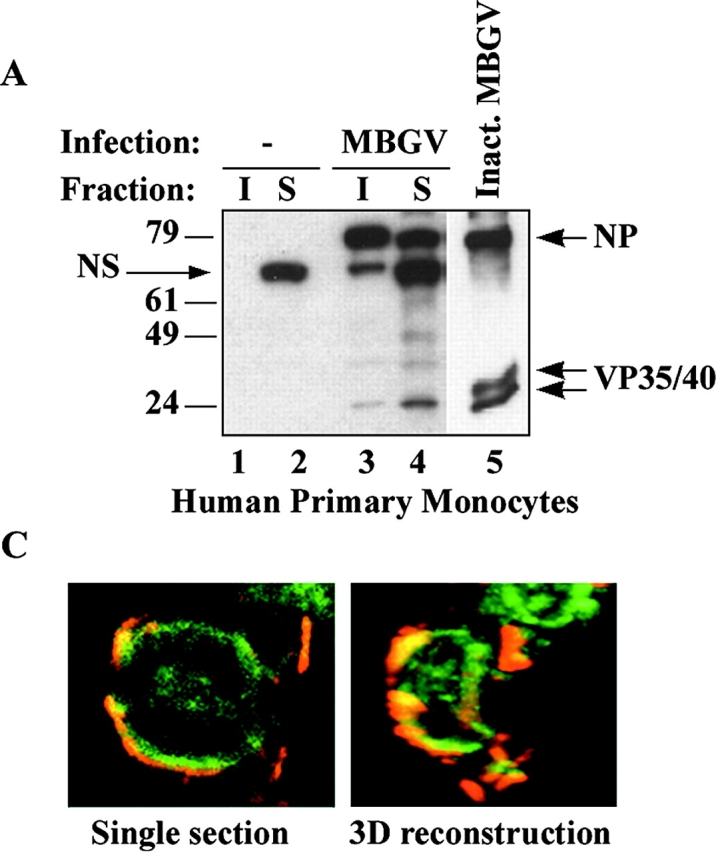

Localization of filovirus proteins in lipid rafts in infected cells. (A) Primary human monocytes were infected with MBGV. After 24 h cells were lysed in 0.5% Triton X100 and detergent-soluble (S) and -insoluble (I) fractions were separated by centrifugation, samples were irradiated (2 × 106 R), and analyzed by immunoblotting with a guinea pig anti-MBGV antibody to detect viral proteins NP and VP35/VP40 (lanes 3 and 4); lanes 1 and 2, uninfected control; lane 5, inactivated MBGV (1 μg). NS, nonspecific band. (B) HepG2 hepatocytes were infected with EBOV-Zaire, lysed, irradiated (6 × 106 R), and rafts (R) and soluble (S) fractions were prepared by ultracentrifugation 24 h after infection. Ebola GP and VP40 were detected by immunoblotting. (C) Ebola-infected Vero E6 cells were irradiated (4 × 106 R), fixed and stained for Ebola virus (red) and GM1 (green) at 4°C and imaged by confocal microscopy. (Left panel) single section; (right panel), 3-D reconstruction of the compiled data.
Cell Culture, Infections, Virus, and VLP Purification.
PBMCs were isolated by density centrifugation through Ficoll-Paque (Amersham Pharmacia Biotech) according to the manufacturer's instructions. PBMCs were cultured in RPMI/10% FBS for 1 h at 37°C and 5% CO2 after which nonadherent cells were removed. HEPG2 cells (American Type Culture Collection) were cultured to confluency with complete RPMI 1640 before use. Monocyte-derived macrophages, HEPG2 cells, and Vero-E6 cells were infected at a multiplicity of infection of 1 with either Ebola-Zaire or Marburg Musoke virus for 50 min at 37°C, 5% CO2. Nonadsorbed virus was removed from cells by washing monolayers twice with PBS followed by addition of fresh complete medium for an additional 24–48 h. Purification and inactivation of Marburg virus was performed as described previously (7). Briefly, Vero-E6 cells were infected with MBGV and supernatant was harvested 6–7 d after infection. The medium was clarified and virus concentrated by polyethylene glycol precipitation. After centrifugation at 10,000 g for 30 min, pellets were resuspended in Tris buffer, and layered atop 20–60% sucrose gradients and centrifuged at 38,000 rpm for 4 h. The visible virus band was collected. Samples were inactivated by irradiation (107 R, 60Co source) and tested for absence of infectivity in cell culture before use. For preparation of VLPs, supernatants were collected 60 h after transfection, overlaid on 30% sucrose, and ultracentrifuged at 26,000 rpm for 2 h. Pelleted particulate material was recovered in PBS and analyzed by immunoblotting or electron microscopy. As a further purification step, in some experiments, this particulate material was loaded on a step gradient consisting of 80%, 40%, and 30% sucrose. After 2-h centrifugation at 26,000 rpm, the VLPs were recovered from the interface of 80% and 40% sucrose layers.
Plaque Assays.
Infectious Ebola and Marburg virions were enumerated using a standard plaque assay as described previously (6). Briefly, culture supernatants were serially diluted in EMEM. 100 μl of each dilution were added to wells of Vero-E6 cells in duplicate. Virus was allowed to adsorb for 50 min. Wells were then overlaid with 1× EBME and 0.5% agarose. Plates were incubated at 37°C, 5% CO2 at which time a second overlay of 1× EBME/0.5% agarose and 20% neutral red was added to each well, incubated for an additional 24 h and plaques were counted.
Cell Staining and Confocal Microscopy.
293T cells were stained with indicated antibodies to viral proteins followed by Alexa-647 conjugated secondary antibodies (Molecular Probes). Rafts were visualized by staining of GM1 with Alexa-488 conjugated CTB and in some experiments with rhodamine-conjugated CTB (see Fig. 2 B). Staining was performed on live cells on ice for 20 min. Cells were then washed with PBS, fixed in 3% paraformaldehyde, washed, and mounted on microscopy slides. Images were collected using the Bio-Rad Laboratories Radiance 2000 system attached to a Nikon E600 microscope. Alexa-488 immunostain was excited using 488 nm light from a Krypton-Argon laser and the emitted light was passed through an HQ515/30 filter. Fluorescence from the Alexa-647 dye was excited by 637 nm light from a red diode laser and collected after passing through an HQ660LP emission filter. The lasers were programmed to scan over successive focal planes (0.25–0.5 μm intervals) at 50 lines per second. Lasersharp software was used to control the confocal system and to reconstruct individual focal planes into three-dimensional renderings.
Figure 2.

Colocalization of filovirus GPs with GM1 on intact cells. (A) 293T cells were transfected with the indicated GP, and stained at 4°C with Alexa488-CTB (green) and anti-GP mAb followed by Alexa-647–conjugated anti–mouse antibodies (red), cells were fixed and imaged using confocal microscopy. Colocalization is represented by yellow appearance in the overlay (right panels). A 3-D reconstruction of the compiled data from 25 sections of an Ebo-GP–transfected cell is also shown. (B) 293T cells were concurrently stained at 4°C with Alexa-488–conjugated anti-TrfR antibody (green) and rhodamine-CTB (red), fixed and analyzed by confocal microscopy. No colocalization between these two molecules was observed, evident by the lack of yellow appearance in the overlay. Two representative cells are shown.
Electron Microscopy.
Portions of particulate material were applied to 300-mesh, nickel electron microscopy grids precoated with formvar and carbon, treated with 1% glutaraldehyde in PBS for 10 min, rinsed in distilled water, and negatively stained with 1% uranyl acetate. For immunoelectron microscopy, fractions were processed as described previously for fluid specimens (28). Briefly, fractions were applied to grids and immersed for 45 min in dilutions of mAbs against EBOV GP. Normal mouse ascetic fluid was tested in parallel. Grids were washed with the TRIS buffer and incubated for 45 min with goat anti–mouse IgG labeled with 10-nm gold spheres (Ted Pella, Inc.). Grids were washed in PBS, and fixed in 1% glutaraldehyde. After fixation, grids were rinsed in drops of distilled water and negatively stained with 1% uranyl acetate. For preembedment staining, cells were stained with anti-Ebola GP mAb followed by gold anti–mouse Ab, fixed with 2% glutaraldehyde in Millonig's buffer (pH 7.4) for 1 h, and postfixed in 1% uranylacetate, dehydrated, and embedded in POLY/BED 812 resin (Polysciences). Resin was allowed to polymerize for 16 h at 60°C, ultrathin sections (∼80 nm) were cut, placed on 200-mesh copper electron microscopy grids, and negatively stained. Stained grids were examined with a JEOL 1200 EX transmission electron microscope at 80 kV.
Online Supplemental Material.
The cells shown in supplemental Videos 1–4 were stained, fixed, and imaged using confocal microscopy as described above and in the video legends. 3-D reconstructions were created using Lasersharp software (Bio-Rad Laboratories). Montage videos of successive optical planes (supplemental Videos 1 and 3) and 3-D rotational views (supplemental Videos 2 and 4) were created using Metamorph imaging software (Universal Imaging Corp.).
Results
Association of Filovirus GPs with Lipid Rafts.
Targeting of membrane-spanning proteins to lipid rafts is commonly governed by dual acylation of cysteine residues at the cytosolic end of the transmembrane domains (24, 29). The filovirus envelope GPs contain such acylation signals in their transmembrane domains (1) and palmitoylation of Ebola GP has been recently reported (30). By transient expression of the filovirus envelope GPs in 293T cells and subsequent extraction of rafts by sucrose gradient ultracentrifugation (13), we examined whether these GPs localize to lipid rafts. As shown in Fig. 1 A and B, a significant fraction of Ebola and Marburg GPs were found to reside in rafts. In contrast, an Ebola GP, mutated at cysteine residues 670 and 672 (Ebo-GPC670/672A), the putative palmitoylation sites, failed to localize to the rafts (Fig. 1 B). Lipid rafts are highly enriched in ganglioside M1 (GM1) which can be detected by its specific binding to cholera toxin B (CTB) (31, 32). As a control for the quality of raft preparations, we analyzed the soluble and raft fractions for the presence of GM1 by spot blots using HRP-conjugated CTB and demonstrated that GM1 was exclusively found in the raft fractions (Fig. 1 A and B, bottom panels). The association of GPs with detergent insoluble fraction was dependent on cholesterol since pretreatment with methyl-β-cyclodextrin (MβCD), a drug that depletes the membrane of cholesterol (33), resulted in almost complete removal of Ebola GP from rafts (Fig. 1 C, top panel). As a further control, we showed that transferrin receptor (TrfR), a molecule excluded from rafts (31), was only found in the soluble fraction (Fig. 1 C, bottom panel). To confirm the raft localization of Ebola and Marburg GP on intact cells, we also performed confocal laser microscopy on 293T cells that were transfected with Ebola or Marburg GP and costained with anti-GP antibodies and CTB. As shown in Fig. 2 A, a substantial portion of both of the GPs were found to colocalize with GM1 in large patches on the plasma membrane, confirming the raft association of both GPs on intact cells. Videos visualizing 25 sections through the cells, as well as three-dimensional (3-D) reconstruction of the cells by compiling data from these sections are available as supplemental data on the web (Videos 1 and 2). Confocal microscopy again showed that the membrane domains visualized by CTB staining were devoid of the raft excluded TrfR (Fig. 2 B).
Figure 1.
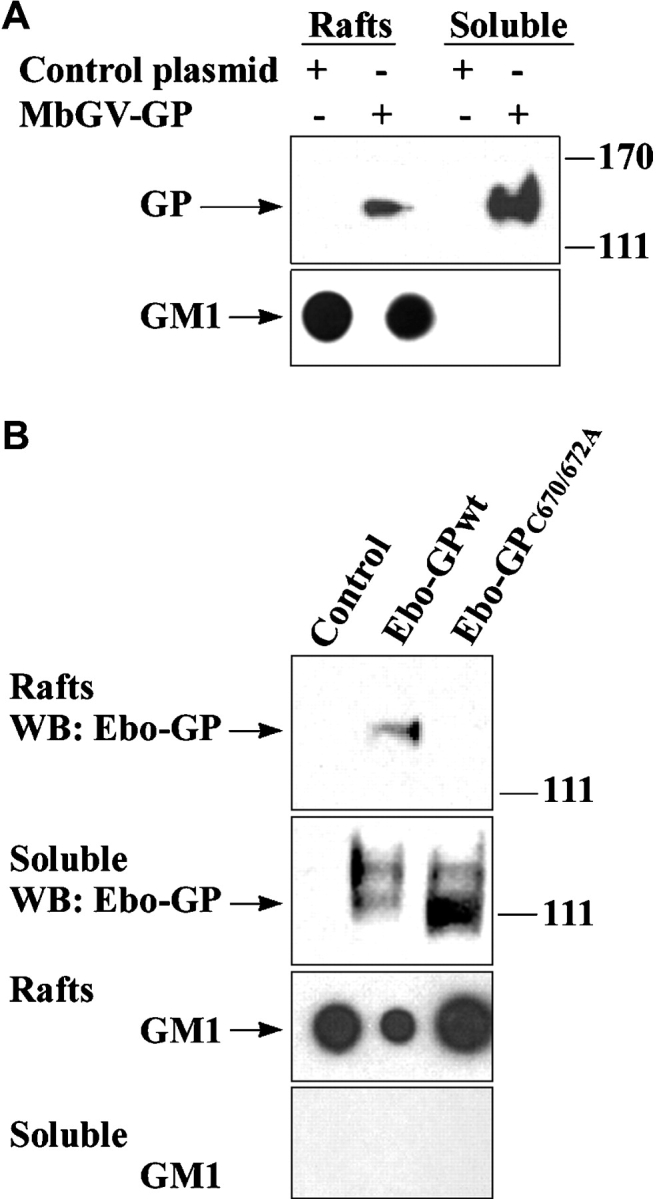

Localization of filovirus GPs in lipid rafts. 293T cells were transfected with Marburg GP (A), Ebo-GPwt, or Ebo-GPC670/672A (B), or a control plasmid, rafts were prepared by ultracentrifugation and GP was detected by immunoblotting. GM1 was detected by blotting with HRP-CTB in the corresponding fractions spotted on a nitrocellulose membrane, as a control for the quality of raft preparation. (C) 48 h after transfection of 293T cells with Ebola GP, a portion of cells were treated for 20 min with 10 mM methyl-β-cyclodextrin (MβCD) and another portion was left untreated. Raft and soluble fractions were prepared and analyzed by immunoblotting for GP (top panel) and for the raft-excluded protein TrfR (bottom panel).
Filoviral Proteins Associate with Lipid Rafts in Cells Infected with Live Virus.
Two of the primary target cells of filoviruses are monocyte/macrophages and hepatocytes (1). Thus, to examine the localization of EBOV and MBGV proteins with respect to lipid rafts during infection with live virus, primary human monocytes, HepG2 hepatocytes, and also Vero-E6 cells (commonly used to propagate filoviruses) were used as target cells. Human monocytes were infected with the Musoke strain of MBGV, after 24-h detergent-insoluble and detergent-soluble fractions were separated by centrifugation (24). As shown in Fig. 3 A, a major portion of viral proteins was detected in the detergent-insoluble fraction (I) 24 h after infection. We then performed similar experiments with HepG2 cells, infected with EBOV-Zaire95, and prepared lipid rafts by sucrose gradient ultracentrifugation. Similar to Marburg, Ebola VP40, and GP were detected mainly in lipid rafts 24 h after infection of HepG2 hepatocytes (Fig. 3 B). To further confirm the accumulation of filovirus proteins in lipid rafts in intact cells, Vero-E6 cells, infected with EBOV, were fixed, irradiated, and costained with anti-Ebola antibody and CTB. As shown in Fig. 3 C, we observed a striking colocalization of viral proteins with the lipid rafts in intact Ebola-infected cells (Videos 3 and 4), suggesting that viral proteins assemble at lipid rafts during the course of viral replication.
Ebola and Marburg Virions Incorporate the Raft Molecule GM1 during Budding.
To determine whether the virus was released through lipid rafts, we analyzed EBOV from culture supernatants of infected cells for the presence of the raft marker GM1. Enveloped viruses bud as virions surrounded by the portion of the plasma membrane at which assembly takes place (34). Release of virions through lipid rafts would therefore result in incorporation of raft-associated molecules in the viral envelope, thus identifying virus budding from the rafts. As shown in Fig. 4 A, EBOV immunoprecipitated with anti-Ebola GP antibody from supernatant of infected Vero-E6 cells contained readily detectable levels of GM1. We also analyzed inactivated Marburg virus that had been purified by ultracentrifugation for the incorporation of GM1 and demonstrated that GM1 was detectable in MBGV (Fig. 4 B, bottom panel). In contrast, the raft-excluded protein TrfR was not incorporated in Marburg virions (Fig. 4 B, middle panel). Taken together, these data strongly suggested that both viruses exit cells through lipid rafts.
Figure 4.


Incorporation of GM1 in released filovirus virions. (A) Ebola virus was immunoprecipitated from supernatant of infected Vero-E6 cells (lane 2), or uninfected cells as control (lane 1), using anti-GP mAb. After irradiation (2 × 106 R), a fraction of immunoprecipitate (IP) was spotted on nitrocellulose membrane and blotted with HRP-conjugated CTB to detect GM1 (bottom panel). Another portion of the IP was analyzed by SDS-PAGE and immunoblotting with anti-GP mAb (top panel). (B) MBGV (1 μg), prepared by ultracentrifugation and inactivated by radiation (107 R), was analyzed for the presence of GM1, TrfR, and GP in a similar fashion. As control, rafts and soluble fractions from untransfected 293T cells were used.
Release of GM1-containing Particles by Ectopic Expression of Ebola Proteins.
To further test the hypothesis that filoviruses assemble and bud via lipid rafts, we transiently expressed viral proteins and searched for GM1-containing virus-like particles (VLPs). Several viral proteins have been shown to support the formation of VLPs (35–37). In transfected 293T cells, Ebola GPwt, GPC670/672A, and VP40 were readily detected in cell lysates when each protein was expressed individually (Fig. 5 A, panels 1 and 3, lanes 2, 3, and 4). However, when VP40 and GP were coexpressed, little GP and almost no VP40 were found associated with the cells 60 h after transfection (Fig. 5 A, panels 1 and 3, lane 5). To examine the viral proteins released from the cells, culture supernatants were cleared of cells, and particulate material was purified by ultracentrifugation >30% sucrose cushion. As shown in Fig. 5 A (panels 2 and 4, lanes 2–4), large amounts of GPwt and lesser quantities of GPC670/672A or VP40 were detected in the particulate material from the supernatants of singly transfected cells. Interestingly, coexpression of GPwt and VP40, directed the majority of both proteins into the supernatant (Fig. 5 A, panels 2 and 4, lane 5). Next, we tested if the released particles incorporated the raft-associated molecule GM1. Anti–Ebo-GP immunoprecipitates from the supernatants of the cells transfected with GPwt or GPwt+VP40, but not GPC670/672A, contained GM1 (Fig. 5 A, panel 5), suggesting that the release of these particles takes place through the rafts. We performed a second step of purification on these particles using a sucrose step gradient to separate the virus-like particles from the cell debris. The low-density fraction floating between 40 and 80% sucrose was then analyzed by Western blot analysis. As shown in Fig. 5 B, these particles contained GM1 but totally excluded TrfR, further confirming the release of particles through lipid rafts.
Figure 5.


Release of Ebola GP and VP40 as GM1-containing particles. (A) 293T cells were transfected with the indicated plasmids, supernatants were cleared from floating cells by centrifugation, and particulate material was pelleted through 30% sucrose by ultracentrifugation. The individual proteins were detected in the cell lysates and in the particulate material from supernatant by immunoblotting (IB). A fraction of cleared supernatant was subjected to immunoprecipitation using anti-GP mAb and analyzed for the presence of GM1 (bottom panel) as described in the legend to Fig. 1. (B) The particulate material from cells transfected with GP+VP40 was further purified on a sucrose step gradient and the low density fraction was analyzed for the presence of VP 40 (top panel), TrfR (middle panel), and GM1 (bottom panel). Rafts and soluble fractions from untransfected 293T cells were used as control.
Particles Formed by EBOV GP and VP40 Display the Morphological Characteristics of Ebola Virus.
We determined the composition and morphology of these particles by examination of the purified particulate material using electron microscopy. Interestingly, most of the particles obtained from the supernatants of the cells cotransfected with GPwt and VP40 displayed a filamentous morphology strikingly similar to filoviruses (Fig. 6 A and B) (28, 38). In contrast, the material obtained from cells transfected with GPwt, GPC670/672A, or VP40 only contained small quantities of membrane fragments, likely released during cell death (data not shown). The VLPs generated by GP and VP40 were released at a high efficiency. Typically, we achieve a titer of 0.5–1.0 × 106 VLPs/ml 2–3 d after transfection. The VLPs have a diameter of 50–70 nm and are 1–2 μm in length (Fig. 6). This is similar to the length range of Ebola virions found in cell culture supernatants after in vitro infection (28). The shorter diameter of VLPs (as compared with 80 nm for EBOV) may be due to the lack of ribonucleoprotein complex. We observed all types of morphologies described for filoviruses such as branched, rod-, U-, and 6-shaped forms (1, 28) among these particles (Fig. 6). In addition, the VLPs were coated with 5–10 nm surface projections or “spikes” (Fig. 6), characteristic for EBOV (1, 28). Immunogold staining of the VLPs with anti-Ebola GP antibodies demonstrated the identity of the spikes on the surface of the particles as Ebola GP (Fig. 6 B). To visualize the process of the release of the VLPs, cells transfected with GP and VP40 were analyzed by electron microscopy after preembedment immunogold staining. Fig. 6 C shows a typical site of VLP release, where a large number of particles that stain for GP exit through a small region of the plasma membrane (indicated by arrows). These sites of VLP release have an average diameter of ∼1 μm. Given the incorporation of GM1 in the VLPs (Fig. 5) these particle-releasing platforms most likely represent coalesced lipid raft domains.
Figure 6.


Electron microscopic analysis of VLPs generated by EBOV GP and VP40. Particles obtained by ultracentrifugation of the supernatants of 293T cells transfected with Ebola GP+VP40 were negatively stained with uranyl-acetate to reveal the ultrastructure (A), or stained with anti-EBOV-GP mAb followed by Immunogold rabbit anti–mouse Ab (B), and analyzed by electron microscopy. The length of each particle is indicated in μm. (C) 293T cells transfected with Ebola GP+VP40 were immunogold-stained for Ebola GP, fixed, cut, and analyzed by electron microscopy. The picture depicts a typical site of VLP release from the cells, indicated by arrows. A magnification of the site of VLP release is also shown to better visualize the gold staining on the particles.
Entry of EBOV Is Dependent on the Integrity of Lipid Rafts.
Having established a critical role for lipid rafts in virus release, we sought to investigate if filoviruses utilize the same gateway for entry. To examine the role of lipid rafts in filovirus entry, the effects of raft-disrupting agents filipin and nystatin on Ebola infection were explored. Brief treatment of cells with filipin (0.2 μg/ml, 30 min) before infection resulted in a significant inhibition of EBOV infection evident by reduced viral titer 48 h after infection (Fig. 7) . Similar results were also obtained with another cholesterol-destabilizing agent nystatin (Fig. 7). This effect was not due to a general cytotoxic effect by the drugs as cells were shown to be viable by trypan blue exclusion (data not shown). To rule out the possibility of a persistent effect of this brief drug treatment on the viral replication, we let an aliquot of the cells recover in medium (for 4 h) after filipin treatment before infecting them with EBOV. As shown in Fig. 7 (Filipin→recovery), these cells could produce large amounts of virus, ruling out the possibility of late effects of the drug on viral replication. In fact, in cells recovered from raft disruption the infection was even more efficient. This might be due to a synchronizing effect by reorganization of the microdomains resulting in a more efficient entry of the virus into a larger number of cells. We also considered the possibility that raft disruption may interfere with virus attachment rather than entry. However, titration of the virus recovered after the 50 min binding showed that same amount of EBOV had bound to both treated and control cells (data not shown). Taken together, these data suggest that lipid rafts play a critical role in the entry stage of Ebola infection.
Figure 7.

Inhibition of Ebola infection by raft-disrupting agents filipin and nystatin. Vero E6 cells were left untreated or treated for 30 min with 0.2 μg/ml of filipin or 100 U/ml of nystatin at 37°C, washed extensively with PBS, and infected with EBOV at an MOI of 1. As a control for lack of general toxicity and persistent effect on viral replication, upon treatment with Filipin, cells were washed, and incubated in medium for 4 h before infection with EBOV (Filipin→recovery). After 48 h supernatants were harvested and viral titers determined by plaque assay.
Discussion
Our findings demonstrate that filoviruses utilize lipid rafts as a platform for budding from the cells. We documented this phenomenon in reconstruction experiments and in the process of live virus infections. Both after transient expression of filovirus GPs and in EBOV and MBGV infected cells, we observed large patches of envelope GPs in association with lipid rafts (Figs. 2 and 3). Our results also demonstrate that the released virions incorporate the raft-associated molecule GM1, but not TrfR, a protein excluded from lipid rafts (31). Using electron microscopy on cells transfected with Ebola GP and matrix protein VP40, we also demonstrate the site of release of EBOV-like particles to be localized in a small area of the plasma membrane ∼1 μm wide (Fig. 6 C). Therefore, such patches of rafts appear to represent the site of filovirus assembly and budding. Electron microscopic studies show that virus budding at the plasma membrane requires an accumulation of viral components including nucleocapsid, matrix, and envelope GP in an orchestrated manner, concurrent with structural changes in the plasma membrane (39). This process is dependent on a precise coordination of the involved components (40). Thus, compartmentalization of viral assembly in a specialized microdomain, such as rafts, with its ordered architecture and selective array of molecules may increase the efficiency of virus budding and decrease the frequency of release of defective, noninfectious particles.
Besides acting as a coordination site for viral assembly, rafts may have a profound impact on viral pathogenicity as well as host immune response to viruses. Transfer of the incorporated molecules with signaling capabilities into newly infected cells may affect the intracellular biochemical processes in favor of a more efficient viral replication. Furthermore, selective enrichment of certain proteins such as adhesion molecules can affect the efficiency of viral entry and possibly virus tropism. Incorporation of GPI-anchored proteins in the viral envelope such as inhibitors of complement pathway CD55 and CD59, which have been detected in HIV virions (41), may help the virus evade the complement-mediated lysis.
An important aspect of our study is the generation of genome-free filovirus-like particles. Our biochemical data show that the VLPs incorporate both Ebola GP and matrix protein VP40, as well as raft-associated ganglioside M1, similar to the results obtained with live virus infections (Fig. 4). A striking morphological similarity between these VLPs and live filoviruses was observed in electron microscopic studies (Fig. 6). These findings have several important implications. While several viral matrix proteins support the formation of VLPs, Ebola VP40 seems to be unique in that it requires the expression of envelope GP for efficient formation of particles. Recently, Timmins et al. reported that a small fraction of transfected VP40 could be detected in culture supernatants in association with filamentous particles (42). While we detected VP40 in the supernatants of transfected 293T cells, electron microscopic analysis revealed that the protein was associated with unstructured membrane fragments. In multiple experiments, filamentous particles were only observed when both VP40 and GP were concurrently expressed. These findings imply that the driving force for the assembly and release of EBOV may be the interaction between GP and matrix protein, as suggested previously (1). Ebola VP40 has an NH2-terminal and a COOH-terminal domain, the latter being involved in membrane localization (43). Removal of most of the COOH-terminal domain induces hexamerization of the protein, the multimeric form believed to be involved in viral assembly (44). While our data show that the majority of VP40 is membrane associated, we were unable to detect VP40 in the rafts when expressed independently (data not shown). Our attempts to detect VP40 in the lipid rafts in the presence of GP was hampered by the efficient release of the proteins in the supernatants resulting in hardly detectable cellular levels of VP40 (Fig. 3). However, given the incorporation of GM1 in the VP40-containing VLPs, it is reasonable to speculate that a transient association of VP40 with lipid rafts takes place in the cells. It is possible that association of VP40 with GP drives VP40 into the rafts. Since a fraction of GPs is outside the rafts (Fig. 1), probably in a dynamic exchange with the rafts, this pool of GPs might be involved in the initial interaction with VP40. This interaction and subsequent movement to the rafts may, at the same time, induce a conformational change in VP40 resulting in dissociation of the COOH-terminal domain from the nonraft membrane and thus removing the constraints on the formation of VP40 hexamers required for viral assembly. Detailed studies are underway to test this model. In this regard, the successful generation of VLPs by ectopic expression of viral proteins provides a safe approach for the study of the steps involved in filovirus assembly and budding without the restrictions of biosafety level-4 laboratories.
VLPs could be an excellent vehicle for antigen delivery, thus useful as a vaccination strategy (45, 46). Different types of recombinant HIV-1 virus-like particles have been shown to not only trigger the induction of neutralizing antibodies but also induce HIV-specific CD8+ CTL responses in preclinical studies (46). Therefore, VLPs are capable of mobilizing different arms of the adaptive immune system. Given the importance of both cellular and humoral immune response for protection against Ebola (27, 47), filovirus-based VLPs, alone or in combination with DNA vaccination, may represent a good vaccine candidate. We are currently investigating the capability of Ebola and Marburg VLPs to elicit an immune response. Another potential use of VLPs is in the delivery of foreign antigens. Parvovirus-like particles have been engineered to express foreign polypeptides, resulting in the production of large quantities of highly immunogenic peptides, and to induce strong antibody, Th cell, and CTL responses (46). Given the compartmentalized release of VLPs through rafts, artificial targeting of antigens to lipid rafts by introduction of dual acylation signals may result in their enrichment in filovirus-based VLPs, providing a potential novel strategy for delivery of a variety of antigens.
VLPs are also valuable research tools. Mutational analysis of the proteins involved in filovirus release can be performed using VLP formation as a quick read-out. Our VLPs express the envelope GP in addition to the matrix protein and can therefore be also used for detailed study of the steps involved in the fusion and entry of EBOV and MBGV by circumventing the restrictions of working under biosafety level-4 conditions.
Most enveloped viruses use a specific interaction between their GPs and cell surface receptors to initiate the attachment to cells and subsequent fusion. Organization of viral receptors in the ordered environment of lipid rafts may facilitate the virus binding through its multimeric GP, promote lateral assemblies at the plasma membrane required for productive infections, concentrate the necessary cytosolic and cytoskeletal components, and enhance the fusion process by providing energetically favorable conditions. It is intriguing that the HIV receptor CD4 (14), its coreceptor CXCR4 (26), as well as molecules favoring HIV infection such as glycosphingolipids (9, 48), and CD44 (17, 49) all reside in lipid rafts. Our data suggest that filoviruses use lipid rafts as a gateway for entry into cells. This may relate to the presence of the filovirus receptor(s) in these microdomains. Recently, it has been demonstrated that folate receptor-α can function as a cellular cofactor for filoviruses (50). Interestingly, folate receptor-α is a GPI-anchored protein shown to reside in rafts (51). Thus, rafts may be crucial for viral entry by concentrating the receptor for filovirus GPs. Our finding that disruption of lipid rafts can interfere with filovirus entry suggests that the integrity of these compartments or their molecular components may be potential therapeutic targets against Ebola and Marburg infections. Further characterization of the raft composition during host-virus interaction, for instance by proteomic analysis, will help to identify such potential targets.
In summary, our findings shed new light on the molecular mechanisms involved in filovirus entry as well as assembly and budding. Much deeper understanding of these mechanisms is needed for successful design of efficacious therapeutic and vaccination strategies. However, identification of rafts as a gateway for cellular trafficking of Ebola and Marburg viruses and generation of Ebola VLPs are important steps toward this goal.
Acknowledgments
We thank Drs. Dimiter Dimitrov and Robert Blumental (National Cancer Institute) for critical review of the manuscript, Dr. Mary Kate Hart for anti-Ebola GP antibodies, Dr. Kevin Anderson for anti-VP40 antibody, and Dr. Tim Nelle for Ebola VP40 cDNA. We are also in debt to Ms. Denise Braun, Ms. Kathleen Kuehl, and Ms. Diane Negley for excellent technical help.
Footnotes
The online version of this article contains supplemental material.
Abbreviations used in this paper: GP, glycoprotein; GPI, glycosylphosphatidylinositol; TrfR, transferrin receptor; VLP, virus-like particle.
References
- 1.Feldmann, H., and H.D. Klenk. 1996. Marburg and Ebola viruses. Adv. Virus Res. 47:1–52. [DOI] [PubMed] [Google Scholar]
- 2.Johnson, K.M., J.V. Lange, P.A. Webb, and F.A. Murphy. 1977. Isolation and partial characterisation of a new virus causing acute haemorrhagic fever in Zaire. Lancet. 1:569–571. [DOI] [PubMed] [Google Scholar]
- 3.Jaax, N., P. Jahrling, T. Geisbert, J. Geisbert, K. Steele, K. McKee, D. Nagley, E. Johnson, G. Jaax, and C. Peters. 1995. Transmission of Ebola virus (Zaire strain) to uninfected control monkeys in a biocontainment laboratory. Lancet. 346:1669–1671. [DOI] [PubMed] [Google Scholar]
- 4.Sullivan, N.J., A. Sanchez, P.E. Rollin, Z.Y. Yang, and G.J. Nabel. 2000. Development of a preventive vaccine for Ebola virus infection in primates. Nature. 408:605–609. [DOI] [PubMed] [Google Scholar]
- 5.Vanderzanden, L., M. Bray, D. Fuller, T. Roberts, D. Custer, K. Spik, P. Jahrling, J. Huggins, A. Schmaljohn, and C. Schmaljohn. 1998. DNA vaccines expressing either the GP or NP genes of Ebola virus protect mice from lethal challenge. Virology. 246:134–144. [DOI] [PubMed] [Google Scholar]
- 6.Hevey, M., D. Negley, P. Pushko, J. Smith, and A. Schmaljohn. 1998. Marburg virus vaccines based upon αvirus replicons protect guinea pigs and nonhuman primates. Virology. 251:28–37. [DOI] [PubMed] [Google Scholar]
- 7.Hevey, M., D. Negley, J. Geisbert, P. Jahrling, and A. Schmaljohn. 1997. Antigenicity and vaccine potential of Marburg virus glycoprotein expressed by baculovirus recombinants. Virology. 239:206–216. [DOI] [PubMed] [Google Scholar]
- 8.Burton, D.R., and P.W. Parren. 2000. Fighting the Ebola virus. Nature. 408:527–528. [DOI] [PubMed] [Google Scholar]
- 9.Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature. 387:569–572. [DOI] [PubMed] [Google Scholar]
- 10.Brown, D.A., and E. London. 1998. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14:111–136. [DOI] [PubMed] [Google Scholar]
- 11.Brown, D.A., and E. London. 2000. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 275:17221–17224. [DOI] [PubMed] [Google Scholar]
- 12.Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. 1:31–39. [DOI] [PubMed] [Google Scholar]
- 13.Aman, M.J., and K.S. Ravichandran. 2000. A requirement for lipid rafts in B cell receptor induced Ca2+ flux. Curr. Biol. 10:393–396. [DOI] [PubMed] [Google Scholar]
- 14.Xavier, R., T. Brennan, Q. Li, C. McCormack, and B. Seed. 1998. Membrane compartmentation is required for efficient T cell activation. Immunity. 8:723–732. [DOI] [PubMed] [Google Scholar]
- 15.Cheng, P.C., A. Cherukuri, M. Dykstra, S. Malapati, T. Sproul, M.R. Chen, and S.K. Pierce. 2001. Floating the raft hypothesis: the roles of lipid rafts in B cell antigen receptor function. Semin. Immunol. 13:107–114. [DOI] [PubMed] [Google Scholar]
- 16.Janes, P.W., S.C. Ley, A.I. Magee, and P.S. Kabouridis. 2000. The role of lipid rafts in T cell antigen receptor (TCR) signalling. Semin. Immunol. 12:23–34. [DOI] [PubMed] [Google Scholar]
- 17.Viola, A., S. Schroeder, Y. Sakakibara, and A. Lanzavecchia. 1999. T lymphocyte costimulation mediated by reorganization of membrane microdomains. Science. 283:680–682. [DOI] [PubMed] [Google Scholar]
- 18.Moran, M., and M.C. Miceli. 1998. Engagement of GPI-linked CD48 contributes to TCR signals and cytoskeletal reorganization: a role for lipid rafts in T cell activation. Immunity. 9:787–796. [DOI] [PubMed] [Google Scholar]
- 19.Verkade, P., and K. Simons. 1997. Robert Feulgen Lecture 1997. Lipid microdomains and membrane trafficking in mammalian cells. Histochem. Cell Biol. 108:211–220. [DOI] [PubMed] [Google Scholar]
- 20.Gatfield, J., and J. Pieters. 2000. Essential role for cholesterol in entry of mycobacteria into macrophages. Science. 288:1647–1650. [DOI] [PubMed] [Google Scholar]
- 21.Scheiffele, P., A. Rietveld, T. Wilk, and K. Simons. 1999. Influenza viruses select ordered lipid domains during budding from the plasma membrane. J. Biol. Chem. 274:2038–2044. [DOI] [PubMed] [Google Scholar]
- 22.Manie, S.N., S. Debreyne, S. Vincent, and D. Gerlier. 2000. Measles virus structural components are enriched into lipid raft microdomains: a potential cellular location for virus assembly. J. Virol. 74:305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen, D.H., and J.E. Hildreth. 2000. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J. Virol. 74:3264–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rousso, I., M.B. Mixon, B.K. Chen, and P.S. Kim. 2000. Palmitoylation of the HIV-1 envelope glycoprotein is critical for viral infectivity. Proc. Natl. Acad. Sci. USA. 97:13523–13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dimitrov, D.S. 2000. Cell biology of virus entry. Cell. 101:697–702. [DOI] [PubMed] [Google Scholar]
- 26.Manes, S., G. del Real, R.A. Lacalle, P. Lucas, C. Gomez-Mouton, S. Sanchez-Palomino, R. Delgado, J. Alcami, E. Mira, and A.C. Martinez. 2000. Membrane raft microdomains mediate lateral assemblies required for HIV-1 infection. EMBO Rep. 1:190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson, J.A., M. Hevey, R. Bakken, S. Guest, M. Bray, A.L. Schmaljohn, and M.K. Hart. 2000. Epitopes involved in antibody-mediated protection from Ebola virus. Science. 287:1664–1666. [DOI] [PubMed] [Google Scholar]
- 28.Geisbert, T.W., and P.B. Jahrling. 1995. Differentiation of filoviruses by electron microscopy. Virus Res. 39:129–150. [DOI] [PubMed] [Google Scholar]
- 29.Zhang, W., R.P. Trible, and L.E. Samelson. 1998. LAT palmitoylation: its essential role in membrane microdomain targeting and tyrosine phosphorylation during T cell activation. Immunity. 9:239–246. [DOI] [PubMed] [Google Scholar]
- 30.Ito, H., S. Watanabe, A. Takada, and Y. Kawaoka. 2001. Ebola virus glycoprotein: proteolytic processing, acylation, cell tropism, and detection of neutralizing antibodies. J. Virol. 75:1576–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harder, T., P. Scheiffele, P. Verkade, and K. Simons. 1998. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 141:929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heyningen, S.V. 1974. Cholera toxin: interaction of subunits with ganglioside GM1. Science. 183:656–657. [DOI] [PubMed] [Google Scholar]
- 33.Christian, A.E., M.P. Haynes, M.C. Phillips, and G.H. Rothblat. 1997. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 38:2264–2272. [PubMed] [Google Scholar]
- 34.Simons, K., and H. Garoff. 1980. The budding mechanisms of enveloped animal viruses. J. Gen. Virol. 50:1–21. [DOI] [PubMed] [Google Scholar]
- 35.Porter, D.C., L.R. Melsen, R.W. Compans, and C.D. Morrow. 1996. Release of virus-like particles from cells infected with poliovirus replicons which express human immunodeficiency virus type 1 Gag. J. Virol. 70:2643–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delchambre, M., D. Gheysen, D. Thines, C. Thiriart, E. Jacobs, E. Verdin, M. Horth, A. Burny, and F. Bex. 1989. The GAG precursor of simian immunodeficiency virus assembles into virus-like particles. EMBO J. 8:2653–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomsen, D.R., A.L. Meyer, and L.E. Post. 1992. Expression of feline leukaemia virus gp85 and gag proteins and assembly into virus-like particles using the baculovirus expression vector system. J. Gen. Virol. 73:1819–1824. [DOI] [PubMed] [Google Scholar]
- 38.Murphy, F.A., G. van der Groen, S.G. Whitfield, and J.V. Lange. 1978. Ebola and Marburg Virus Morphology and Taxonomy. 1st edition. S.R. Pattyn, editor. Elsevier, Amsterdam. pp. 1–61.
- 39.Dubois-Dalcq, M., and T.S. Reese. 1975. Structural changes in the membrane of vero cells infected with a paramyxovirus. J. Cell Biol. 67:551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garoff, H., R. Hewson, and D.J. Opstelten. 1998. Virus maturation by budding. Microbiol. Mol. Biol. Rev. 62:1171–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saifuddin, M., T. Hedayati, J.P. Atkinson, M.H. Holguin, C.J. Parker, and G.T. Spear. 1997. Human immunodeficiency virus type 1 incorporates both glycosyl phosphatidylinositol-anchored CD55 and CD59 and integral membrane CD46 at levels that protect from complement-mediated destruction. J. Gen. Virol. 78:1907–1911. [DOI] [PubMed] [Google Scholar]
- 42.Timmins, J., S. Scianimanico, G. Schoehn, and W. Weissenhorn. 2001. Vesicular release of ebola virus matrix protein VP40. Virology. 283:1–6. [DOI] [PubMed] [Google Scholar]
- 43.Dessen, A., V. Volchkov, O. Dolnik, H.D. Klenk, and W. Weissenhorn. 2000. Crystal structure of the matrix protein VP40 from Ebola virus. EMBO J. 19:4228–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruigrok, R.W., G. Schoehn, A. Dessen, E. Forest, V. Volchkov, O. Dolnik, H.D. Klenk, and W. Weissenhorn. 2000. Structural characterization and membrane binding properties of the matrix protein VP40 of Ebola virus. J. Mol. Biol. 300:103–112. [DOI] [PubMed] [Google Scholar]
- 45.Johnson, J.E., and W. Chiu. 2000. Structures of virus and virus-like particles. Curr. Opin. Struct. Biol. 10:229–235. [DOI] [PubMed] [Google Scholar]
- 46.Wagner, R., Y. Shao, and H. Wolf. 1999. Correlates of protection, antigen delivery and molecular epidemiology: basics for designing an HIV vaccine. Vaccine. 17:1706–1710. [DOI] [PubMed] [Google Scholar]
- 47.Wilson, J.A., and M.K. Hart. 2001. Protection from Ebola virus mediated by cytotoxic T lymphocytes specific for the viral nucleoprotein. J. Virol. 75:2660–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hug, P., H.M. Lin, T. Korte, X. Xiao, D.S. Dimitrov, J.M. Wang, A. Puri, and R. Blumenthal. 2000. Glycosphingolipids promote entry of a broad range of human immunodeficiency virus type 1 isolates into cell lines expressing CD4, CXCR4, and/or CCR5. J. Virol. 74:6377–6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dukes, C.S., Y. Yu, E.D. Rivadeneira, D.L. Sauls, H.X. Liao, B.F. Haynes, and J.B. Weinberg. 1995. Cellular CD44S as a determinant of human immunodeficiency virus type 1 infection and cellular tropism. J. Virol. 69:4000–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan, S.Y., C.J. Empig, F.J. Welte, R.F. Speck, A. Schmaljohn, J.F. Kreisberg, and M.A. Goldsmith. 2001. Folate receptor-α is a cofactor for cellular entry by Marburg and Ebola viruses. Cell. 106:117–126. [DOI] [PubMed] [Google Scholar]
- 51.Nichols, B.J., A.K. Kenworthy, R.S. Polishchuk, R. Lodge, T.H. Roberts, K. Hirschberg, R.D. Phair, and J. Lippincott-Schwartz. 2001. Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J. Cell Biol. 153:529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]