

Abstract
The α/β T cell receptor (TCR) HA1.7 specific for the hemagglutinin (HA) antigen peptide from influenza A virus is HLA-DR1 restricted but cross-reactive for the HA peptide presented by the allo-major histocompatibility complex (MHC) class II molecule HLA-DR4. We report here the structure of the HA1.7/DR4/HA complex, determined by X-ray crystallography at a resolution of 2.4 Å. The overall structure of this complex is very similar to the previously reported structure of the HA1.7/DR1/HA complex. Amino acid sequence differences between DR1 and DR4, which are located deep in the peptide binding groove and out of reach for direct contact by the TCR, are able to indirectly influence the antigenicity of the pMHC surface by changing the conformation of HA peptide residues at position P5 and P6. Although TCR HA1.7 is cross-reactive for HA presented by DR1 and DR4 and tolerates these conformational differences, other HA-specific TCRs are sensitive to these changes. We also find a dependence of the width of the MHC class II peptide-binding groove on the sequence of the bound peptide by comparing the HA1.7/DR4/HA complex with the structure of DR4 presenting a collagen peptide. This structural study of TCR cross-reactivity emphasizes how MHC sequence differences can affect TCR binding indirectly by moving peptide atoms.
Keywords: T cell receptor, MHC class II, cross-reactivity, antigen recognition, X-ray crystallography
Introduction
The MHC-restricted recognition of antigen peptides by TCRs is a central event in the cellular immunity against pathogens and in the immune surveillance of cancer cells. It is also directly involved in immunological diseases, such as autoimmune diseases, hypersensitive reactions, and alloreactive responses after organ transplantations. The recent crystal structures of TCRs in complex with peptide antigen presented by MHC class I and class II molecules identify atomic contacts between the TCR on one side and the antigen peptide and the MHC molecule on the other side and give a detailed picture of the structural diversity of this interaction (1–7). Although the different structures represent different biological situations (immune response against pathogens, positive selection in the thymus, alloreaction) no characteristic structural features attributable to the particular biological situation are obvious. The overall mode of TCR binding to pMHC is rather similar for all these structures. The extremely variable CDR3s are located over the center of the pMHC surface and make contacts with the antigen peptide as well as with the MHC α-helices, whereas the less variable CDR loops contact the termini of the antigen peptide and residues of the MHC helices adjacent to it (CDR1s) or exclusively contact the central parts of the MHC helices (CDR2s). Although in all these complexes TCRs bind to pMHC in a similar overall orientation and topology, the shapes and chemical properties of the interacting surfaces in these complexes are highly diverse and no conserved contacts between the CDR loops of the TCR and the pMHC exist that could determine this orientation (for a review, see references 8 and 9).
Based on the structures of the TCR–pMHCI complexes, recent mutational studies addressed the functional importance of the atomic contacts between the TCR and the pMHCI systematically by mutating residues of the TCR, antigen peptide, and MHC molecule involved in intermolecular contacts and correlating changes in protein conformation and thermodynamic and kinetic stability of the interaction with the outcome of the biological response (e.g., phosphorylation patterns of TCR and ZAP-70, cytolytic activity, cytokine production). These studies showed that (i) the outcome of the biological response correlated fairly well with kinetic properties of the TCR–pMHC interaction (for example, references 10–12; for a review, see references 13 and 14), (ii) the response does not correlate with structural changes at the interface (minor structural changes can have a dramatic effect on the biological response) (10, 15), and (iii) the importance of atomic contacts for functional binding can be similar for all contacts at the interface in some cases but vary strongly in others (16–18). Similar systematic experiments for TCRs bound to antigen peptide presented by MHC class II molecules have not been reported.
We recently solved the crystal structure of the human α/β TCR HA1.7 in complex with an immunodominant peptide epitope of hemagglutinin *(HA) (HA306–318: PKYVKQNTLKLAT) from influenza A virus H3N2 presented by the MHC class II molecule DR1 (4). HA is bound in an extended conformation deep in the binding groove of DR1 by a dozen hydrogen bonds between conserved DR1 residues of the α-helices and peptide main chain carbonyl and amide groups (19). Burying of the HA peptide side chains of Y308 (P1) in a deep pocket and of Q311 (P4), T313 (P6), L314 (P7), and L316 (P9) in more shallow pockets of DR1 establishes peptide binding specificity (20, 21). However, the HA peptide binds promiscuously and can be presented by most of the frequently occurring DR alleles. Due to differences in shape and chemical properties of the P4, P6, P7, and P9 pockets, HA residues at the corresponding positions are of varying importance for HA binding to these DR alleles, whereas Tyr308 at position P1 is always the dominant anchor residue (20, 22–24).
The interaction between the HA antigen peptide and TCR HA1.7 is dominated by charged interactions between the three lysine residues of HA at position P-1, P3, and P8 and glutamate and aspartate residues in CDR3α and CDR1β of TCR HA1.7 (4, 25). These charged interactions are conserved in the recognition of the HA peptide by other TCRs (21, 25–27). Contacts by TCR HA1.7 to these three lysines and to other residues that point upward from the DR1/HA surface (Val309 (P2), Asn312 (P5), and Leu314 (P7) are important for functional binding (21, 27–30).
Although TCR HA1.7 is DR1-restricted (DRA1, DRB1*0101), it is cross-reactive with HA peptide presented by the allo-MHC class II molecule HLA-DR4 (DRA1, DRB1*0401) (26, 31–33). However, other HA-specific TCRs are known that do not show this cross-reactive behavior (26, 34). To understand the basis of the cross-reactivity of HA1.7 and that of other TCRs we determined the X-ray structure of TCR HA1.7 in complex with DR4/HA at a resolution of 2.4 Å. The structure is very similar to the TCR HA1.7/DR1/HA structure. However, local conformational changes of HA peptide residues induced by MHC allelic differences are observed which obviously are tolerated by TCR HA1.7 but sensed by other TCRs. Comparison with the DR4/Col structure indicates a conformational difference in the DR4 molecule that depends on the sequence of the bound antigen peptide.
Materials and Methods
Protein Expression, Purification, and Crystallization.
Empty HLA-DR4 (DRA*0101, DRB1*0401) and HLA-DM were overexpressed from stable Schneider cell lines (35–37) and TCR HA1.7 with the HA306–318 antigen peptide linked to residue D1 of the TCR β-chain via an octapeptide linker was refolded from Escherichia coli inclusion bodies (4). The HA-HA1.7/DR4 complex was assembled by loading the HA peptide that is part of the p-TCR HA-HA1.7 onto empty DR4 with the help of the peptide exchange catalyst HLA-DM and purified as described previously (4). The addition of HLA-DM during complex formation increased the yield of the assembled complex by a factor of ∼4–10.
Crystallization, Structure Determination, and Refinement.
HA-HA1.7/DR4 crystals were obtained by streak seeding sitting drops of 1 μl of protein (10 mg/ml) and 1 μl of well solution (13% PEG 8000, 1 M NaCl, 100 mM Hepes, pH 7.0) with crystals of HA-HA1.7/DR1 after 12 h preequilibration of the drops at 18°C (4). Crystals are monoclinic, space group C2, with a = 143.8 Å, b = 73.3 Å, c = 123.0 Å, β = 108.5° and one complex molecule per asymmetric unit.
After briefly soaking the crystals in 20% glycerol, 16% PEG 8000, 1 M NaCl, 100 mM Hepes, pH 7.0, and flash cooling in liquid nitrogen, X-ray diffraction data were collected from a single cryocooled crystal (100K; 20.0–2.4 Å resolution) (see Table I) at the BIOCARS station 14-BM-C at the APS at Argonne National Laboratory using 1 Å wavelength X-rays and a Quantum4 CCD detector. Data processing was performed with HKL2000 (38).
Table I.
Crystallographic Data
| (A) Data Statistics | |
|---|---|
| Resolution (Å) | 20.0–2.4 (2.49–2.40) |
| No. unique reflections | 46705 |
| Multiplicity | 4.5 (4.2) |
| Completeness | 96.9 (96.7) |
| Average I/σI | 20.6 (4.3) |
| Rmerge (%) | 5.8 (33.0) |
| (B) Refinement statistics (20.0–2.4 Å) | |
| No. of reflections (free) | 43905 (2280) |
| Rwork (Rfree) | 0.208 (0.245) |
| Rmsd bonds (Å) | 0.007 |
| Rmsd angles (degree) | 1.38 |
| (C) Average B factors | |
| Protein atoms (Å2) | 45.6 |
| Sugars (Å2) | 83.7 |
| Water molecules (Å2) | 48.4 |
| Anisotropic B factors (Å2) | B11 = −6.19, B22 = −1.06, B33 = 7.25 |
| Bulk solvent correction | B = 38.4Å2, k = 0.35 e/Å3 |
Rrange = (Σhkl|l−<l>|)/(Σhkl<l>), ∀hkl ε {independent Miller indices}.
Rfree = (Σh|Fo−Fc|)/(ΣhFo), ∀h ε {free set, 5% of reflections}.
Rwork = (Σh|Fo−Fc|)/(ΣhFo), ∀h ε {working set}.
The structure of the complex was determined by molecular replacement using the program AMoRe (39) and the refined coordinates of the HA-HA1.7/DR1 complex (4) as search models. Solutions for the TCR HA1.7 and DR4/HA were found in independent rotation and translation searches (R = 31.4, corr. coeff. = 74.2). Rigid-body refinement of the individual domains of this model with CNS (40) yielded Rfree = 31.1% (Rwork = 29.5%) used all the data between 20 and 2.4 Å resolution.
To avoid model bias, sigma-A-weighted (2mFo-DFc) simulated annealing omit maps (composite omit maps) and (mFo-DFc) electron density maps (41) were used for model building and correction in O (42). For all residues that are different between DR4 and DR1 unambiguous electron density could be observed in the initial maps. For the calculation of the Rfree value during refinement the same reflections were set-aside in the test set as during the refinement of the HA-HA1.7/DR1 structure (4).
Simulated annealing torsion angle dynamics, positional, and individual B factor refinement was performed in CNS using a maximum likelihood (ML) target. A bulk solvent correction and anisotropic B-factor tensor were applied throughout the refinement. After the Rfree had dropped to 26.4% (Rwork = 23.1%) water molecules were added. The final model includes 813 of 855 residues and 305 water molecules. Weak or no electron density was observed for residues 105–113 of DR4β, residues 130–132 of the HA1.7 Cα domain, the last 10 residues of the Cα as well as the Cβ domain, for 0–3 residues at the N- or COOH termini of the different chains and for the octapeptide linker between the HA peptide and the HA1.7β chain (see Table I). Electron density corresponding to the N-linked carbohydrates at Asn78α, Asn118α, and Asn19β was observed, but only one (Asn78α, Asn19β) or two (Asn118α) monosaccharide units could be reliably modeled at each position.
Protein Database Code.
The coordinates have been deposited in the PDB under entry code 1J8H.
Results
Structural Overview over the Complex.
A stable complex between the α/β TCR HA1.7 and DR4/HA was prepared as described recently for the TCR HA1.7/DR1/HA complex, exploiting the strategy of flexibly linking the HA antigen peptide to the Vβ chain of the TCR (4). Crystals of the complex were of space group C2 and isomorphous to crystals of the TCR HA1.7/DR1/HA complex. The structure was solved by molecular replacement using the TCR HA1.7/DR1/HA complex as the search model and great care was taken to avoid model bias by the search model (Materials and Methods). For all allelic differences between DR4 and DR1 unambiguous electron density could be observed in the initial electron density maps. The structure was refined to a final R value of 0.208 and Rfree value of 0.245 at a resolution of 2.4 Å (Table I).
The structures of the TCR HA1.7/DR4/HA complex and the TCR HA1.7/DR1/HA complex (4) are very similar (RMSD for Cα atoms is 0.34 Å). The TCR HA1.7 binds similarly to DR4/HA and to DR1/HA with the Vα domain binding to the NH2-terminal half of the HA peptide and the center and COOH-terminal half of the β1 α-helix and the Vβ domain binding to the COOH-terminal half of the peptide and to the center and COOH-terminal half of the α1 α-helix (Fig. 1) . Only small conformational differences are observed in the polypeptide mainchain of the HA peptide and the MHC class II molecule around polymorphic residues (see below).
Figure 1.
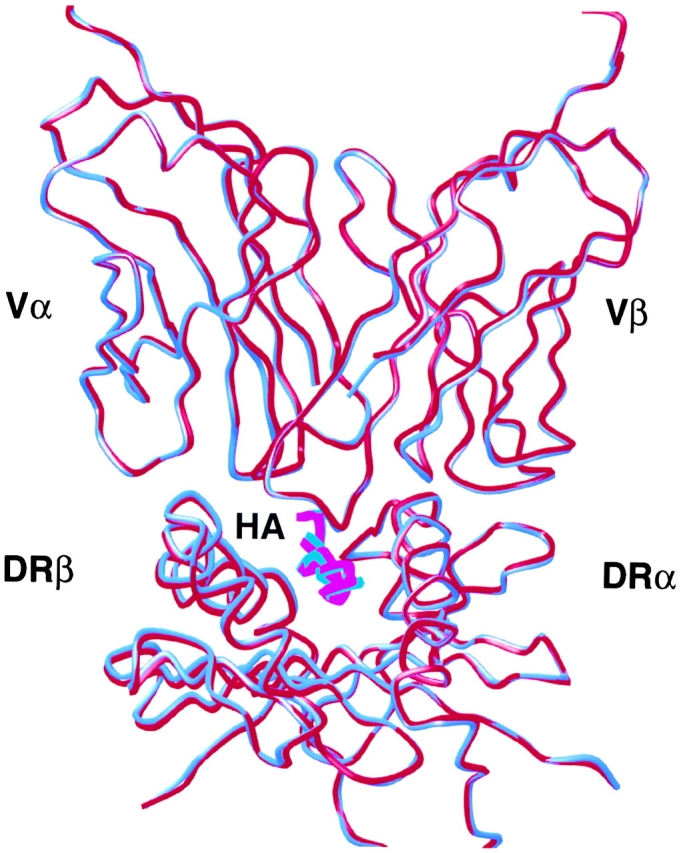
The structures of TCR HA1.7 bound to HLA-DR1 and HLA-DR4 are very similar. Superposition of the TCR V domains and the MHC class II antigen peptide binding groove of the TCR HA1.7/DR4/HA structure (blue) and the TCR HA1.7/DR1/HA structure (red; reference 4). The figure was created with MOLSCRIPT (reference 75).
Allelic Differences between DR1 and DR4 Determine the HA Peptide Conformation.
Although the allelic differences between DR1 and DR4 are not accessible for direct contact by the TCR, these residues nevertheless influence the antigenic properties of the pMHC complex by inducing conformational differences in the peptide antigen. There are 15 allelic differences in the DR β-chain between DR1 (DRB1*0101) and DR4 (DRB1*0401) (Fig. 2 A). Nine of the allelic differences are located in the β-strands that form the floor of the peptide binding groove and thereby, together with the allelic difference at position 71β in the center of the β1 α-helix, determine the differences in size and chemical properties of the P4, P6, P7, and P9 peptide binding pockets (19, 24, 35, 43) (Fig. 2 B). The remaining five polymorphic positions are located in the linker between the β1 and β2 domain and in the β2 domain itself, distant (at least 25 Å) from the interface with the TCR. Despite these allelic differences the HA peptide is bound in the same register in the DR4 peptide binding groove compared with the DR1/HA complex, but with some conformational differences. From residue P4 (Gln311) to P7 (Leu314) the HA peptide is bound slightly deeper in the groove of DR4 and with conformational differences in the peptide mainchain compared with HA bound to DR1. In addition, conformational differences are seen in the sidechains of residue P5 (Asn312) and P6 (Thr313) (Fig. 3) .
Figure 2.
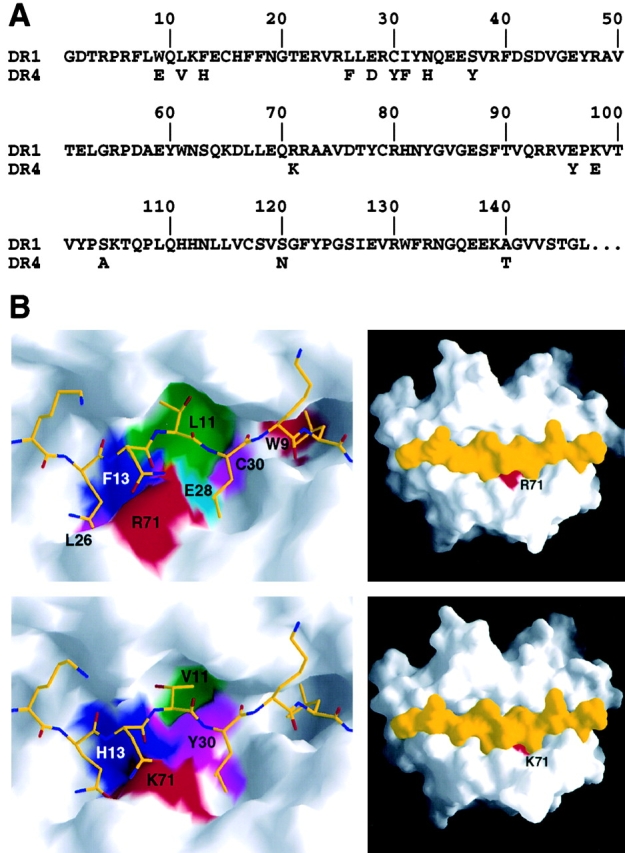
Allelic sequence differences between DR1 and DR4 are not accessible for direct contact by the TCR. (A) Sequence alignment of the DR1 (DRB1*0101) and DR4 (DRB1*0401) β-chains. For DR4 only the 15 residues differences are shown. (B) Allelic differences in the peptide-binding groove of DR1 (top row; reference 4) and DR4 (bottom row). Those residues that are different between DR1 and DR4 and map to the surface of the peptide-binding groove are colored. The HA peptide (yellow) is shown as a stick model in the partial view (left) and as a solvent accessible surface in the view of the whole peptide binding groove (right). Figures were prepared with GRASP (reference 76).
Figure 3.

DR1 and DR4 allelic sequence differences cause conformational differences in the HA peptide structure. Changes around HA peptide residue P6 Thr313 (A) and P5 Asn312 (B) when presented by DR1 (left; reference 4) and DR4 (right). The conformation of the HA antigen peptide (yellow) bound to DR1 and DR4 (gray) depends on polymorphic MHC residues (magenta) and influences recognition by the TCR (green) (Van-der-Waals contacts: dashed black lines; potential hydrogen bonds: dashed red lines). Side views into the peptide binding groove are shown with the HA peptide N- to COOH-terminal direction from right to left in A and left to right in B. To allow a better view into the peptide binding groove, the helix in front (α1 domain α-helix in A and β1 domain α-helix in B), which would otherwise block the view, are rendered transparent. Figures were prepared with MOLSCRIPT (reference 75) and Raster3D (reference 77).
Polymorphic residues around the P6 pocket influence the conformation of peptide residue P6 Thr (Fig. 3 A). In contrast to HA bound to DR1, the sidechain of P6 Thr points into the P6 pocket of DR4 due to a rotation of –148 degrees about the Cα-Cβ bond (Chi 1). This rotation leads to the formation of a hydrogen bond between the P6 Thr γ-hydroxyl group and His13β Nɛ2 atom (Fig. 3 A). Since this hydrogen-bonding partner is absent in DR1 (His13β is replaced by Phe) and the P6 pocket is shallower in DR1 (Val11β at the bottom of the P6 pocket is replaced by Leu), the Thr γ-hydroxyl group points upward toward the TCR (Fig. 3 A). In DR4, the whole P6 Thr residue is bound slightly deeper in the peptide-binding groove compared with DR1 (Cα of P6 Thr in this structure is ∼0.6 Å deeper in the groove compared with the DR1 structure). The TCR HA1.7 does not contact the P6 Thr sidechain in either case, but other TCR are sensitive to this difference (see below).
There is also a conformational difference in peptide residue P5 Asn. In the TCR HA1.7/DR4/HA structure the residue P5 Asn is slightly tipped and moved toward the β1 α-helix compared with the TCR HA1.7/DR1/HA structure (Fig. 3 B). The Cα, Cβ, and Cγ atoms of P5 Asn in the TCR HA1.7/DR4/HA structure are moved by 0.7 Å, 1.0 Å, and 1.3 Å, respectively, compared with the TCR HA1.7/DR1/HA structure (coordinate error from Luzzati plot: 0.3 Å). This location of the mainchain residues P4 to P6 (Gln311 to Thr313) of the antigen peptide is identical to the location of the mainchain residues P4 to P6 of the collagen peptide in the DR4/Col structure (35).
The difference is caused by residue Lys71β of the DR4 β-chain, which in order to form a hydrogen bond with the mainchain carbonyl group of peptide residue P5 Asn pulls the carbonyl further into the peptide binding groove and thereby tips and moves the whole residue toward the β1 α-helix. In DR1, the longer sidechain of Arg 71β can form the same hydrogen bond with the P5 mainchain carbonyl group in a more horizontal orientation (Fig. 3 B). The allelic differences of the DR residues 11β (L and V), 13β (F and H), 26β (L and F), and 28β (E and D) are also in the vicinity of this peptide conformational difference.
In HA1.7/DR4/HA the HA bound to DR4 lacks one hydrogen bond between Nδ2 of P5 Asn and the carbonyl oxygen of residue Thr97β of the TCR Vβ chain, relative to HA1.7/DR1/HA (Fig. 3 B), but has a new hydrogen bond to Oɛ1 of residue Gln70β of the DR4 β1 α-helix.
Differences in the Conformation of DR4 Bound to HA and Col Peptides.
The conformation of the DR4 peptide-binding groove depends on the sequence of the bound peptide. The conformation and location of the polypeptide main chain of the MHC class II peptide-binding groove is very similar for the structures of DR1/HA (19), TCR HA1.7/DR1/HA (4), and the TCR HA1.7/DR4/HA presented here. The RMSD of the MHC peptide binding groove of these structures is <0.5 Å and the largest difference in the Cα positions of the polypeptide mainchain is ∼1 Å (RMSD: DR1/HA and TCR HA1.7/DR1/HA: 0.38 Å; DR1/HA and TCR HA1.7/DR4/HA: 0.46 Å; TCR HA1.7/DR1/HA and TCR HA1.7/DR4/HA: 0.29 Å; calculated using residue 5 to 80 of DR1A and 5 to 90 of DR1B). However, the peptide-binding groove of DR4 in the DR4/Col structure (35) is considerably narrower, due to movements of the β1 α-helix near residues 62β to 70β of the DR4 β-chain, a region adjacent to the P4 and P7 pocket (Fig. 4) . Compared with DR4 in the TCR HA1.7/DR4/HA structure, the groove is 2 Å or 12% narrower (the distance between the CA atoms of residue 65α and 67β is 14.0 Å in DR4/Col and 16.0 Å in TCR HA1.7/DR4/HA).
Figure 4.

The sequence of the bound peptide determines the width of the DR4 peptide-binding groove. Differences between the structure of the β1 α-helix from residue 62β to 70β in DR4 presenting the HA peptide (yellow) and presenting the collagen II peptide, Col 1168–1180 (red, reference 35). The structure of DR4/HA was determined in complex with a TCR, while DR4/Col was not. The figure was prepared with MOLSCRIPT (reference 75).
Discussion
Cross-Reactivity of the HA1.7 TCR with the HA Peptide Bound to DR4 and to DR1.
The T cell line HA1.7 recognizes the HA306–318 antigen peptide from influenza A presented by HLA-DR1 (DRA*0101, DRB1*0101) and is also cross-reactive with the HA peptide presented by HLA-DR4 (DRA*0101, DRB1*0401) and other DR4 alleles (Table II) (31, 32, 44). We recently solved the structure of the HA1.7-TCR/DR1/HA complex (4) and compare the structure of the same TCR in complex with DR4/HA here. The recognition of a peptide/MHC pair by a TCR usually depends on the sequence of the antigen peptide and the MHC molecule at those positions that are directly contacted by the TCR (34, 45–49). Data also suggest that the antigenic properties of the pMHC surface can be modulated by residues not in direct contact by the TCR (34, 49–55) (for a review, see reference 56). The conformation and antigenicity of the bound peptide is not only strongly influenced by MHC residues that form the peptide binding pockets, but also by the peptide anchors that point into these pockets. Even subtle changes in these anchors can interfere with the T cell response (51). We have seen here how MHC and peptide residues not involved in direct contact by the TCR alter the antigenic properties of a peptide/MHC surface by the mutual influence of the conformation of the bound peptide and the MHC molecule.
Table II.
Activation of HA-specific T Cell Lines
In the structures of both TCR HA1.7 bound to DR1/HA and bound to DR4/HA, the TCR binds virtually identically to the pMHC surface and the low RMSD of the Cα atoms (0.32 Å) indicates that the overall structures of both complexes are very similar. However, small structural differences occur locally around MHC residues that differ between the DRB1*0101 and DRB1*0401 alleles. These residues are mainly located at the bottom of the peptide binding groove or in the β1 α-helix with the sidechain pointing into the peptide binding groove so that after binding of the HA peptide they are buried away from the TCR (Fig. 2 B). Despite the similarity at the pMHC surface, HA-specific T cell lines exist that are not cross-reactive for HA presented by DR1 and DR4 (Table II) (26, 34).
TCR Recognition of Small Structural Differences near P5 and P6, not Recognized by the HA1.7-TCR.
Although the HA peptide binds in the same register to DR1 and DR4, the center and COOH-terminal half of the peptide binds deeper in the groove and closer to the β1 α-helix of DR4. These slight differences are caused by allelic differences in the β-chain sequence, which are mostly located under peptide residues P5 to P9 (Fig. 3). Published T cell activity and mutagenesis data indicate that the differences in the sidechains of peptide residues P5 and P6, not recognized by the HA1.7-TCR, can be recognized by other TCR. (21, 34) (Fig. 3).
His13β is found in all DR4 alleles but is replaced by Phe in all DR1 alleles, resulting in a difference in the conformation of the P6 Thr sidechain (Fig. 3 A). TCR HA1.7 binds both DR4/HA and DR1/HA, tolerating this structural difference in the HA peptide, apparently because it does not make a contact to the P6 Thr sidechain. However, other TCRs are sensitive to this difference (34). 4/5 T cell lines examined by Fu and colleagues (34) were sensitive to changes around the P6 pocket. The HA-specific DR4-restricted TCRs, 3BC6.6, and KM1HA27 (34), are not cross-reactive with HA presented by DR1 (Table II). In these two cases, mutation of His13β at the bottom of the P6 pocket of DR4 to Phe, typically found at this position in DR1, interferes with T cell activation (Table II, and reference 34). This substitution in DR4 probably disrupts the P6 Thr-His13β hydrogen bond and results in a P6 Thr conformation similar to that seen with DR1 (Fig. 3 A). Based on the two HA1.7-TCR/DR/HA structures, we suggest that the lack of cross-reactivity to DR1 and DR4 HA complexes of the 3BC6.6 and KM1HA27 TCRs is based, at least partly, on sensing the conformational difference in the conformation of the P6 Thr sidechain. Tolerance to changes in peptide conformation and peptide sequence at positions that are not contacted by the TCR have been described earlier for other TCR/peptide pairs (6).
The additional polymorphism of residue 11β, which is located at the bottom of the P6 pocket and determines its depth, seems less likely to influence the conformation of the P6 Thr sidechain (Fig. 3 A). Residue 11β is Val in all DR4 alleles and Leu in DR1 alleles, so that the pocket is deeper in DR4. With a Leu at position 11β, as in DR1, the pocket is still deep enough to accommodate a Thr sidechain pointing into the pocket as in DR4.
The orientation of the Thr sidechain facing into the P6 pocket of DR4 and the formation of an additional hydrogen bond with His13β that should stabilize peptide binding is supported by experiments with peptide display libraries. These studies show that small sidechains with hydroxyl groups (Ser, Thr), i.e., those that are able to form a hydrogen bond with His13β, are preferred at peptide position P6 for binding to DR4, whereas Ala and Gly are preferred for binding to DR1 (24). A Thr at position P6 seems to be the second most important peptide residue for peptide binding to DR4 after the large hydrophobic sidechain at peptide position P1, whereas the P6 position is of less importance for peptide binding to DR1 (23, 57, 58).
A conformational difference was also observed in peptide residue P5 Asn (Fig. 3 B). When bound to DR4 the sidechain of P5 Asn is tipped toward and closer to the β1 α-helix. This difference is probably caused by the DR polymorphism at position 71β. Residue 71β is a Lys in DR4 (DRB1*0401) and an Arg in DR1 (DRB1*0101) and in both cases forms a hydrogen bond with the mainchain carbonyl of P5 Asn. Since in DR4 the Lys sidechain is shorter it apparently “pulls” P5 Asn further into the peptide-binding groove and closer to the β1 α-helix in order to form the hydrogen bond. Although this difference disrupts another hydrogen bond between the sidechain of P5 Asn and the TCR HA1.7 in the HA1.7-TCR/DR1/HA complex, the HA1.7-TCR is nevertheless cross-reactive, tolerating one less hydrogen bonded interaction. However, the conservative mutation of residue Lys 71β to Arg in DR4 (Table II) and the substitution seen in DR7 (Lys 71β to Arg) can interfere with the recognition by three out of five DR4- and DR7-restricted HA-specific T cell lines that were examined, (21, 34). With other peptide antigens, the effect of the substitution at position 71β of DR7 (Lys 71β to Arg) is less pronounced (55, 59). The two TCR that bind DR4/HA but not DR1/HA, 3BC6.6 and KM1HA27, both lose recognition of DR4 as the result of mutations in DR4 to DR1-like residues in the P5 pocket (Q70R or K71R)(Table II), suggesting that this small structural difference at P5 Asn is sensed by these TCR but not by the HA1.7-TCR.
The positively charged residue Lys/Arg at position 71β plus other residues in its vicinity, Leu67 — — Gln70 Lys/Arg71, collectively described as the shared epitope, have been implicated in being responsible for the susceptibility to rheumatoid arthritis in patients that carry the DRB1*0101, DRB1*0401, or DRB1*0404 allele (references 60 and 61; for a review, see reference 62).
The promiscuous binding of an antigen peptide to many different MHC class II molecules an their recognition by cross-reactive and cross-restricted T cells is not unique to the HA system. The P2 peptide from tetanus toxin (TT) shows a similar promiscuous binding to many different DR molecules. P2 TT peptide-specific T cells are known that are cross-reactive (less specific) and can recognize P2 TT in the context of many different DR alleles, whereas others are more specific (cross-restricted) and can recognize P2 TT only in the context of a particular MHC allele. This includes such T cells that can distinguish between P2 TT presented by DR1 and P2 TT presented by DR4 (59, 63), very similar to the case described above.
The Conformation of DR4 Depends on the Sequence of the Bound Antigenic Peptide.
In addition to the modulation of the TCR recognition of the HA peptide MHC class II residues in the peptide binding groove, we also observed a conformational change in DR4 that depends on the sequence of the bound peptide. The peptide binding groove of DR4 presenting the collagen peptide (35) is up to 2 Å smaller compared with the groove of DR4 presenting the HA peptide and bound by the TCR HA1.7. It is unlikely that these conformational differences are caused by the binding of the HA1.7 TCR since no structural changes were observed in DR1 upon binding the HA1.7 TCR (4, 19). Instead it appears that the width of the peptide-binding groove is determined by the sequence of the bound peptide. The sidechains of the collagen peptide in the peptide binding pockets in the DR4/Col structure are considerably smaller compared with the sidechains of HA peptide (P1: Met versus Tyr; P4: Asp versus Gln; P6: Ala versus Thr; P7: Ala versus Leu; P9: Gly versus Leu). The narrower peptide-binding groove in the DR4/Col structure can therefore be explained by a collapse of DR4 around those peptide binding pockets that are only partially occupied by peptide sidechains (35). The region of residue 62β to 70β which moves the most and the corresponding region in MHC class I molecules (148α to 155α) have been identified earlier as having a degree of conformational freedom (2, 64–68).
These effects may also be important for attempts to optimize peptides in vaccines for tighter binding to the MHC by changing anchor residues (69 and references cited therein), as those “optimized” peptide vaccines may have different conformations, and hence different antigenic properties when bound to the MHC molecule compared with the actual (original) antigen.
Subtle changes in the conformation or sequence of the pMHC molecule even in residues not directly contacted by the TCR can have a large effect on the biological response (34, 49–55, 66) (for a review, see reference 56). The fact that MHC residues that are buried deep in the binding groove can change the conformation of the bound peptide and thereby change the antigenicity of the pMHC surface may also be important in alloreactive T cell responses.
T cell alloreactivity, the major cause of organ transplant rejection and graft-versus-host disease, is the reaction of 1–10% of all T cells with nonself pMHC molecules against which they have not been previously negatively selected during development in the thymus (70). Mechanisms for the large T cell response in alloreactivity range from models where the TCR interaction is dominated by contacts with the allo-MHC to models where the TCRs interact mostly with the antigenic peptides (peptides that cannot be presented by self-MHCs but can be presented by the allo-MHC) (references 71 and 72; for a review, see references 73 and 74). The observation of different bound peptide conformations resulting from residues of the MHC molecule that are out of reach for the TCR, might also be important in the alloreactive T cell response. T cells that have been negatively selected or are not reactive against self-peptides presented by self-MHC molecules, may recognize those same peptides bound to an allo-MHC, where they could adopt different conformations, as seen in the HA peptide studied here. If the self- and allo-MHC molecule have identical residues on the part of the surface that is contacted by the TCR, so that contacts used during positive selection may be conserved, and differ mainly in residues buried in the peptide-binding groove, as in the case of DR1 and DR4, presented here, the opportunity for cross-reactivity might be enhanced.
This structural study of TCR cross-reactivity emphasizes that in addition to the peptide and MHC residues that are contacted by TCRs, the dependence of the conformation of the bound peptide and of the MHC molecule on peptide and MHC residues inaccessible to direct contact by the TCR plays an important role in determining T cell activation.
Acknowledgments
We thank K. Mahan and A. Haykov for excellent technical assistance, the staff at APS at Argonne National Laboratory beamline BIOCARS 14-BM-C for help with data collection, members of the Harrison/Wiley groups for assistance and discussion, R. Gaudet for reading the manuscript, Dr. D. Zaller (Merck) for the gifts of insect cell lines expressing both HLA-DR4 and DM, and Dr. W.C. Stallings (Monsanto) for sharing unpublished data on a DR4/peptide complex.
The research was supported by the National Institutes of Health and the Howard Hughes Medical Institute (HHMI). J. Hennecke was supported by fellowships from the Schweizerischer Nationalfonds and the Deutsche Forschungsgemeinschaft. D.C.Wiley was an investigator of the HHMI.
Footnotes
Abbreviations used in this paper: HA, hemagglutinin; TT, tetanus toxin.
References
- 1.Ding, Y.H., K.J. Smith, D.N. Garboczi, U. Utz, W.E. Biddison, and D.C. Wiley. 1998. Two human T cell receptors bind in a similar diagonal mode to the HLA-A2/Tax peptide complex using different TCR amino acids. Immunity. 8:403–411. [DOI] [PubMed] [Google Scholar]
- 2.Garboczi, D.N., P. Ghosh, U. Utz, Q.R. Fan, W.E. Biddison, and D.C. Wiley. 1996. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature. 384:134–141. [DOI] [PubMed] [Google Scholar]
- 3.Garcia, K.C., M. Degano, L.R. Pease, M. Huang, P.A. Peterson, L. Teyton, and I.A. Wilson. 1998. Structural basis of plasticity in T cell receptor recognition of a self peptide-MHC antigen. Science. 279:1166–1172. [DOI] [PubMed] [Google Scholar]
- 4.Hennecke, J., A. Carfi, and D.C. Wiley. 2000. Structure of a covalently stabilized complex of a human αβ T-cell receptor, influenza HA peptide and MHC class II molecule, HLA-DR1. EMBO J. 19:5611–5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reinherz, E.L., K. Tan, L. Tang, P. Kern, J. Liu, Y. Xiong, R.E. Hussey, A. Smolyar, B. Hare, R. Zhang, et al. 1999. The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science. 286:1913–1921. [DOI] [PubMed] [Google Scholar]
- 6.Reiser, J.-B., C. Darnault, A. Guimezanes, C. Gregoire, T. Mosser, A.-M. Schmitt-Verhulst, J.C. Fontecilla-Camps, B. Malissen, D. Housset, and G. Mazza. 2000. Crystal structure of a T cell receptor bound to an allogenic MHC molecule. Nat. Imm. 1:291–297. [DOI] [PubMed] [Google Scholar]
- 7.Teng, M.K., A. Smolyar, A.G. Tse, J.H. Liu, J. Liu, R.E. Hussey, S.G. Nathenson, H.C. Chang, E.L. Reinherz, and J.H. Wang. 1998. Identification of a common docking topology with substantial variation among different TCR-peptide-MHC complexes. Curr. Biol. 8:409–412. [DOI] [PubMed] [Google Scholar]
- 8.Garcia, K.C., L. Teyton, and I.A. Wilson. 1999. Structural basis of T cell recognition. Annu. Rev. Immunol. 17:369–397. [DOI] [PubMed] [Google Scholar]
- 9.Hennecke, J., and D.C. Wiley. 2001. T cell receptor-MHC interactions up close. Cell. 104:1–4. [DOI] [PubMed] [Google Scholar]
- 10.Ding, Y.H., B.M. Baker, D.N. Garboczi, W.E. Biddison, and D.C. Wiley. 1999. Four A6-TCR/peptide/HLA-A2 structures that generate very different T cell signals are nearly identical. Immunity. 11:45–56. [DOI] [PubMed] [Google Scholar]
- 11.Kalergis, A.M., N. Boucheron, M.A. Doucey, E. Palmieri, E.C. Goyarts, Z. Vegh, I.F. Luescher, and S.G. Nathenson. 2001. Efficient T cell activation requires an optimal dwell-time of interaction between the TCR and the pMHC complex. Nat. Immunol. 2:229–234. [DOI] [PubMed] [Google Scholar]
- 12.Kersh, G.J., E.N. Kersh, D.H. Fremont, and P.M. Allen. 1998. High- and low-potency ligands with similar affinities for the TCR: the importance of kinetics in TCR signaling. Immunity. 9:817–826. [DOI] [PubMed] [Google Scholar]
- 13.Davis, M.M., J.J. Boniface, Z. Reich, D. Lyons, J. Hampl, B. Arden, and Y.H. Chien. 1998. Ligand recognition by α-β T cell receptors. Annu. Rev. Immunol. 16:523–544. [DOI] [PubMed] [Google Scholar]
- 14.Lanzavecchia, A., G. Lezzi, and A. Viola. 1999. From TCR engagement to T cell activation: a kinetic view of T cell behavior. Cell. 96:1–4. [DOI] [PubMed] [Google Scholar]
- 15.Degano, M., K.C. Garcia, V. Apostolopoulos, M.G. Rudolph, L. Teyton, and I.A. Wilson. 2000. A functional hot spot for antigen recognition in a superagonist TCR/MHC complex. Immunity. 12:251–261. [DOI] [PubMed] [Google Scholar]
- 16.Manning, T.C., C.J. Schlueter, T.C. Brodnicki, E.A. Parke, J.A. Speir, K.C. Garcia, L. Teyton, I.A. Wilson, and D.M. Kranz. 1998. Alanine scanning mutagenesis of an αβ T cell receptor: mapping the energy of antigen recognition. Immunity. 8:413–425. [DOI] [PubMed] [Google Scholar]
- 17.Lee, P.U., H.R. Churchill, M. Daniels, S.C. Jameson, and D.M. Kranz. 2000. Role of 2C T cell receptor residues in the binding of self- and allo-major histocompatibility complexes. J. Exp. Med. 191:1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baker, B.M., R.V. Turner, S.J. Gagnon, D.C. Wiley, and W.E. Biddison. 2001. Identification of a crucial energetic footprint on the α1 helix of human histocompatibility leukocyte antigen (HLA)-A2 that provides functional interactions for recognition by Tax peptide/HLA-A2-specific T cell receptors. J. Exp. Med. 193:551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stern, L.J., J.H. Brown, T.S. Jardetzky, J.C. Gorga, R.G. Urban, J.L. Strominger, and D.C. Wiley. 1994. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature. 368:215–221. [DOI] [PubMed] [Google Scholar]
- 20.O'Sullivan, D., T. Arrhenius, J. Sidney, M.-F. Del Guercio, M. Albertson, M. Wall, C. Oseroff, S. Southwood, S.M. Colon, F.C. Gaeta, and A. Sette. 1991. On the interaction of promiscuous antigenic peptides with different DR alleles. Identification of common structural motifs. J. Immunol. 147:2663–2669. [PubMed] [Google Scholar]
- 21.Krieger, J.I., R.W. Karr, H.M. Grey, W.Y. Yu, D. O'Sullivan, L. Batovsky, Z.L. Zheng, S.M. Colon, F.C. Gaeta, J. Sidney, M. Albertson, M.-F. Del Guerico, R.W. Chestnut, and A. Sette. 1991. Single amino acid changes in DR and antigen define residues critical for peptide-MHC binding and T cell recognition. J. Immunol. 146:2331–2340. [PubMed] [Google Scholar]
- 22.Jardetzky, T.S., J.C. Gorga, R. Busch, J. Rothbard, J.L. Strominger, and D.C. Wiley. 1990. Peptide binding to HLA-DR1: a peptide with most residues substituted to alanine retains MHC binding. EMBO J. 9:1797–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sette, A., J. Sidney, C. Oseroff, M.F. del Guercio, S. Southwood, T. Arrhenius, M.F. Powell, S.M. Colon, F.C. Gaeta, and H.M. Grey. 1993. HLA DR4w4-binding motifs illustrate the biochemical basis of degeneracy and specificity in peptide-DR interactions. J. Immunol. 151:3163–3170. [PubMed] [Google Scholar]
- 24.Hammer, J., P. Valsasnini, K. Tolba, D. Bolin, J. Higelin, B. Takacs, and F. Sinigaglia. 1993. Promiscuous and allele-specific anchors in HLA-DR-binding peptides. Cell. 74:197–203. [DOI] [PubMed] [Google Scholar]
- 25.Yassine-Diab, B., P. Carmichael, F.E. L'Faqihi, G. Lombardi, S. Deacock, C. de Preval, H. Coppin, and R.I. Lechler. 1999. Biased T-cell receptor usage is associated with allelic variation in the MHC class II peptide binding groove. Immunogenetics. 49:532–540. [DOI] [PubMed] [Google Scholar]
- 26.Wedderburn, L.R., S.J.M. Searle, A.R. Rees, J.R. Lamb, and M.J. Owen. 1995. Mapping T cell recognition-the identification of a T cell receptor residue critical to the specific interaction with an influenza hemagglutinin peptide. Eur. J. Immunol. 25:1654–1662. [DOI] [PubMed] [Google Scholar]
- 27.Ostrov, D., J. Krieger, J. Sidney, A. Sette, and P. Concannon. 1993. T cell receptor antagonism mediated by interaction between T cell receptor junctional residues and peptide antigen analogues. J. Immunol. 150:4277–4283. [PubMed] [Google Scholar]
- 28.De Magistris, M.T., J. Alexander, M. Coggeshall, A. Altman, F.C. Gaeta, H.M. Grey, and A. Sette. 1992. Antigen analog-major histocompatibility complexes act as antagonists of the T cell receptor. Cell. 68:625–634. [DOI] [PubMed] [Google Scholar]
- 29.Alexander, J., K. Snoke, J. Ruppert, J. Sidney, M. Wall, S. Southwood, C. Oseroff, T. Arrhenius, F.C. Gaeta, S.M. Colon, et al. 1993. Functional consequences of engagement of the T cell receptor by low affinity ligands. J. Immunol. 150:1–7. [PubMed] [Google Scholar]
- 30.Snoke, K., J. Alexander, A. Franco, L. Smith, J.V. Brawley, P. Concannon, H.M. Grey, A. Sette, and P. Wentworth. 1993. The inhibition of different T cell lines specific for the same antigen with TCR antagonist peptides. J. Immunol. 151:6815–6821. [PubMed] [Google Scholar]
- 31.Eckels, D.D., T.W. Sell, S.R. Bronson, A.H. Johnson, R.J. Hartzman, and J.R. Lamb. 1984. Human helper T-cell clones that recognize different influenza hemagglutinin determinants are restricted by different HLA-D region epitopes. Immunogenetics. 19:409–423. [DOI] [PubMed] [Google Scholar]
- 32.Lamb, J.R., D.D. Eckels, P. Lake, J.N. Woody, and N. Green. 1982. Human T-cell clones recognize chemically synthesized peptides of influenza haemagglutinin. Nature. 300:66–69. [DOI] [PubMed] [Google Scholar]
- 33.Lamb, J.R., D.D. Eckels, M. Phelan, P. Lake, and J.N. Woody. 1982. Antigen-specific human T lymphocyte clones: viral antigen specificity of influenza virus-immune clones. J. Immunol. 128:1428–1432. [PubMed] [Google Scholar]
- 34.Fu, X.T., C.P. Bono, S.L. Woulfe, C. Swearingen, N.L. Summers, F. Sinigaglia, A. Sette, B.D. Schwartz, and R.W. Karr. 1995. Pocket 4 of the HLA-DR(α,β 1*0401) molecule is a major determinant of T cells recognition of peptide. J. Exp. Med. 181:915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dessen, A., C.M. Lawrence, S. Cupo, D.M. Zaller, and D.C. Wiley. 1997. X-ray crystal structure of HLA-DR4 (DRA*0101, DRB1*0401) complexed with a peptide from human collagen II. Immunity. 7:473–481. [DOI] [PubMed] [Google Scholar]
- 36.Mosyak, L., D.M. Zaller, and D.C. Wiley. 1998. The structure of HLA-DM, the peptide exchange catalyst that loads antigen onto class II MHC molecules during antigen presentation. Immunity. 9:377–383. [DOI] [PubMed] [Google Scholar]
- 37.Sloan, V.S., P. Cameron, G. Porter, M. Gammon, M. Amaya, E. Mellins, and D.M. Zaller. 1995. Mediation by HLA-DM of dissociation of peptides from HLA-DR. Nature. 375:802–806. [DOI] [PubMed] [Google Scholar]
- 38.Otwinowski, Z., and V. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. C.W. Carter and R.M. Sweet, editors. Academic Press, New York. 307–326. [DOI] [PubMed]
- 39.Navaza, J., and P. Saludjian. 1997. AMoRe: and automated molecular replacement program package. Methods Enzymol. 276:581–594. [DOI] [PubMed] [Google Scholar]
- 40.Brunger, A.T., P.D. Adams, G.M. Clore, W.L. Delano, P. Gros, R.W. Grossekunstleve, J.S. Jiang, J. Kuszewski, M. Nilges, N.S. Pannu, et al. 1998. Crystallography and NMR system-a new software suite for macromolecular structure determination. Acta Crystallogr. D54:905–921. [DOI] [PubMed] [Google Scholar]
- 41.Read, R.J. 1986. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. A42:140–149. [Google Scholar]
- 42.Jones, T.A., J.Y. Zou, S.W. Cowan, and M. Kjeldgaard. 1991. Improved methods for building protein models in electron density maos and the location of errors in these models. Acta. Crystallogr. A47:110–119. [DOI] [PubMed] [Google Scholar]
- 43.Brown, J.H., T.S. Jardetzky, J.C. Gorga, L.J. Stern, R.G. Urban, J.L. Strominger, and D.C. Wiley. 1993. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature. 364:33–39. [DOI] [PubMed] [Google Scholar]
- 44.Rothbard, J.B., R. Busch, V. Bal, J. Trowsdale, R.I. Lechler, and J.R. Lamb. 1989. Reversal of HLA restriction by a point mutation in an antigenic peptide. Int. Immunol. 1:487–495. [DOI] [PubMed] [Google Scholar]
- 45.Jorgensen, J.L., U. Esser, B. Fazekas de St. Groth, P.A. Reay, and M.M. Davis. 1992. Mapping T-cell receptor-peptide contacts by variant peptide immunization of single-chain transgenics. Nature. 355:224-230. [DOI] [PubMed] [Google Scholar]
- 46.Sant'Angelo, D.B., G. Waterbury, P. Preston-Hurlburt, S.T. Yoon, R. Medzhitov, S.C. Hong, and C.A. Janeway, Jr. 1996. The specificity and orientation of a TCR to its peptide-MHC class II ligands. Immunity. 4:367–376. [DOI] [PubMed] [Google Scholar]
- 47.Sun, R., S.E. Shepherd, S.S. Geier, C.T. Thomson, J.M. Sheil, and S.G. Nathenson. 1995. Evidence that the antigen receptors of cytotoxic T lymphocytes interact with a common recognition pattern on the H-2Kb molecule. Immunity. 3:573–582. [DOI] [PubMed] [Google Scholar]
- 48.Ehrich, E.W., B. Devaux, E.P. Rock, J.L. Jorgensen, M.M. Davis, and Y.H. Chien. 1993. T cell receptor interaction with peptide/major histocompatibility complex (MHC) and superantigen/MHC ligands is dominated by antigen. J. Exp. Med. 178:713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fu, X.T., T. Saibara, R.W. Karr, and E. Celis. 1996. Substitutions in the HLA-DR α chain differentially affect DR7-restricted T-cell recognition of rabies virus antigen. Hum. Immunol. 45:111–116. [DOI] [PubMed] [Google Scholar]
- 50.Reid, S.W., S. McAdam, K.J. Smith, P. Klenerman, C.A. O'Callaghan, K. Harlos, B.K. Jakobsen, A.J. McMichael, J.I. Bell, D.I. Stuart, and E.Y. Jones. 1996. Antagonist HIV-1 Gag peptides induce structural changes in HLA B8. J. Exp. Med. 184:2279–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kersh, G.J., M.J. Miley, C.A. Nelson, A. Grakoui, S. Horvath, D.L. Donermeyer, J. Kappler, P.M. Allen, and D.H. Fremont. 2001. Structural and functional consequences of altering a peptide MHC anchor residue. J. Immunol. 166:3345–3354. [DOI] [PubMed] [Google Scholar]
- 52.McMichael, A.J., F.M. Gotch, J. Santos-Aguado, and J.L. Strominger. 1988. Effect of mutations and variations of HLA-A2 on recognition of a virus peptide epitope by cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA. 85:9194–9198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shimojo, N., R.W. Anderson, D.H. Mattson, R.V. Turner, J.E. Coligan, and W.E. Biddison. 1990. The kinetics of peptide binding to HLA-A2 and the conformation of the peptide-A2 complex can be determined by amino acid side chains on the floor of the peptide binding groove. Int. Immunol. 2:193–200. [DOI] [PubMed] [Google Scholar]
- 54.Brett, S.J., K.B. Cease, C.S. Ouyang, and J.A. Berzofsky. 1989. Fine specificity of T cell recognition of the same peptide in association with different I-A molecules. J. Immunol. 143:771–779. [PubMed] [Google Scholar]
- 55.Karr, R.W., W. Yu, R. Watts, K.S. Evans, and E. Celis. 1990. The role of polymorphic HLA-DR β chain residues in presentation of viral antigens to T cells. J. Exp. Med. 172:273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jorgensen, J.L., P.A. Reay, E.W. Ehrich, and M.M. Davis. 1992. Molecular components of T-cell recognition. Annu. Rev. Immunol. 10:835–873. [DOI] [PubMed] [Google Scholar]
- 57.Hammer, J., C. Belunis, D. Bolin, J. Papadopoulos, R. Walsky, J. Higelin, W. Danho, F. Sinigaglia, and Z.A. Nagy. 1994. High-affinity binding of short peptides to major histocompatibility complex class II molecules by anchor combinations. Proc. Natl. Acad. Sci. USA. 91:4456–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hammer, J., B. Takacs, and F. Sinigaglia. 1992. Identification of a motif for HLA-DR1 binding peptides using M13 display libraries. J. Exp. Med. 176:1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karr, R.W., P. Panina-Bordignon, W.Y. Yu, and A. Lanzavecchia. 1991. Antigen-specific T cells with monogamous or promiscuous restriction patterns are sensitive to different HLA-DR β chain substitutions. J. Immunol. 146:4242–4247. [PubMed] [Google Scholar]
- 60.Barber, L.D., V. Bal, J.R. Lamb, R.E. O'Hehir, J. Yendle, R.J. Hancock, and R.I. Lechler. 1991. Contribution of T-cell receptor-contacting and peptide-binding residues of the class II molecule HLA-DR4 Dw10 to serologic and antigen-specific T-cell recognition. Hum. Immunol. 32:110–118. [DOI] [PubMed] [Google Scholar]
- 61.Hammer, J., F. Gallazzi, E. Bono, R.W. Karr, J. Guenot, P. Valsasnini, Z.A. Nagy, and F. Sinigaglia. 1995. Peptide binding specificity of HLA-DR4 molecules: correlation with rheumatoid arthritis association. J. Exp. Med. 181:1847–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wucherpfennig, K.W., and J.L. Strominger. 1995. Selective binding of self peptides to disease-associated major histocompatibility complex (MHC) molecules: a mechanism for MHC-linked susceptibility to human autoimmune diseases. J. Exp. Med. 181:1597–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Panina-Bordignon, P., A. Tan, A. Termijtelen, S. Demotz, G. Corradin, and A. Lanzavecchia. 1989. Universally immunogenic T cell epitopes: promiscuous binding to human MHC class II and promiscuous recognition by T cells. Eur. J. Immunol. 19:2237–2242. [DOI] [PubMed] [Google Scholar]
- 64.Madden, D.R., D.N. Garboczi, and D.C. Wiley. 1993. The antigenic identity of peptide-MHC complexes: a comparison of the conformations of five viral peptides presented by HLA-A2. Cell. 75:693–708. [DOI] [PubMed] [Google Scholar]
- 65.Fremont, D.H., W.A. Hendrickson, P. Marrack, and J. Kappler. 1996. Structures of an MHC class II molecule with covalently bound single peptides. Science. 272:1001–1004. [DOI] [PubMed] [Google Scholar]
- 66.Rudolph, M.G., J.A. Speir, A. Brunmark, N. Mattsson, M.R. Jackson, P.A. Peterson, L. Teyton, and I.A. Wilson. 2001. The crystal structures of K(bm1) and K(bm8) Reveal that subtle changes in the peptide environment impact thermostability and alloreactivity. Immunity. 14:231–242. [DOI] [PubMed] [Google Scholar]
- 67.Smith, K.J., S.W. Reid, D.I. Stuart, A.J. McMichael, E.Y. Jones, and J.I. Bell. 1996. An altered position of the α2 helix of MHC class I is revealed by the crystal structure of HLA-B*3501. Immunity. 4:203–213. [DOI] [PubMed] [Google Scholar]
- 68.Fremont, D.H., M. Matsumura, E.A. Stura, P.A. Peterson, and I.A. Wilson. 1992. Crystal structures of two viral peptides in complex with murine MHC class I H-2Kb. Science. 257:919–927. [DOI] [PubMed] [Google Scholar]
- 69.Hagmann, M. 2000. Computers aid vaccine design. Science. 290:80–82. [DOI] [PubMed] [Google Scholar]
- 70.Sherman, L.A., and S. Chattopadhyay. 1993. The molecular basis of allorecognition. Annu. Rev. Immunol. 11:385–402. [DOI] [PubMed] [Google Scholar]
- 71.Elliott, T.J., and H.N. Eisen. 1990. Cytotoxic T lymphocytes recognize a reconstituted class I histocompatibility antigen (HLA-A2) as an allogeneic target molecule. Proc. Natl. Acad. Sci. USA. 87:5213–5217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith, P.A., A. Brunmark, M.R. Jackson, and T.A. Potter. 1997. Peptide-independent recognition by alloreactive cytotoxic T lymphocytes (CTL). J. Exp. Med. 185:1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bevan, M.J. 1984. High determinant density may explain the phenomenon of alloreactivity. Immunol. Today. 161:767–775. [DOI] [PubMed] [Google Scholar]
- 74.Kranz, D.M. 2000. Incompatible differences: view of an allogeneic pMHC-TCR complex. Nat. Imm. 1:277–278. [DOI] [PubMed] [Google Scholar]
- 75.Kraulis, P.J. 1991. MOLSCRIPT-a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24:946–950. [Google Scholar]
- 76.Nicholls, A., K.A. Sharp, and B. Honig. 1991. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 11:281–296. [DOI] [PubMed] [Google Scholar]
- 77.Merritt, E.A., and M.E.P. Murphy. 1994. Raster3d version 2.0-a program for photorealistic molecular graphics. Acta. Crystallogr. D50:869–873. [DOI] [PubMed] [Google Scholar]