

Abstract
The 3β-hydroxysteroid dehydrogenase (3β-HSD) isoenzymes play a key role in cellular steroid hormone synthesis. Vaccinia virus (VV) also synthesizes steroid hormones with a 3β-HSD enzyme (v3β-HSD) encoded by gene A44L. Here we examined the effects of v3β-HSD in VV disease using wild-type (vA44L), deletion (vΔA44L), and revertant (vA44L-rev) viruses in a murine intranasal model. Loss of A44L was associated with an attenuated phenotype. Early (days 1–3) after infection with vΔA44L or control viruses the only difference observed between groups was the reduced corticosterone level in lungs and plasma of vΔA44L-infected animals. Other parameters examined (body weight, signs of illness, temperature, virus titres, the pulmonary inflammatory infiltrate, and interferon [IFN]-γ levels) were indistinguishable between groups. Subsequently, vΔA44L-infected animals had reduced weight loss and signs of illness, and displayed a vigorous pulmonary inflammatory response. This was characterized by rapid recruitment of CD4+ and CD8+ lymphocytes, enhanced IFN-γ production and augmented cytotoxic T lymphocyte activity. These data suggest that steroid production by v3β-HSD contributes to virus virulence by inhibiting an effective inflammatory response to infection.
Keywords: NK cell, CTL, glucocorticoid, virulence, immunomodulation
Introduction
The 3β-hydroxysteroid dehydrogenase/Δ5-Δ4isomerase (3β-HSD)* enzyme catalyses the conversion of Δ5-3β-hydroxysteroids to Δ4-3-ketosteroids, a reaction that is required for the biosynthesis of all classes of steroid hormones: progesterone, mineralocorticoids, glucocorticoids (GCs), androgens, and estrogens. The enzyme is expressed in classical steroidogenic tissues (adrenal cortex, ovaries, and testis) but also in the skin, liver, kidney, and lung (1–3). Multiple isoforms of 3β-HSD exist in human, rat and mouse tissues (4).
Steroids are important for the differentiation, growth, and physiology of many mammalian tissues and have multiple effects within the immune system. GCs are potent immunosuppressive and anti-inflammatory agents and are used during organ transplantation, autoimmune disease, and a broad spectrum of inflammatory diseases (5). Although the precise mechanisms underlying their immunosuppressive effects are unknown, GCs affect the immune system by modulating cytokine production, antigen presentation and the migration and cytotoxicity of immune cells (6–8).
Steroid hormones such as GCs affect bacterial, protozoan, and viral infections (9). Sex hormones modulate immune responses and are implicated in sex-associated susceptibilities to infectious agents, such as coxsackievirus (10, 11) and hepatitis B virus (12, 13). Another example is the greater severity of variola virus infections in pregnant women than in men or nonpregnant women (14). This could be mimicked by the administration of steroids such as cortisone to variola virus-infected macaque monkeys (15). Cortisone administration also increased the severity of the primary inoculation lesion and delayed healing in guinea pigs (16) and rabbits (17) infected with vaccinia virus (VV). Early leukocyte infiltration was also reduced in rabbits inoculated with VV (17).
Poxviruses express several proteins that inhibit components of the host immune system including IFNs, complement, cytokines, and chemokines (18). VV strain Western Reserve (WR) gene A44L encodes an intracellular 3β-HSD that contributes to virulence after intranasal (19, 20) or intradermal (21) infection of mice. This report examines the role of the A44L protein in VV disease and viral clearance using wild-type (vA44L), deletion (vΔA44L), and revertant (vA44L-rev) viruses in a murine intranasal model. We show that early during infection loss of A44L is associated with reduced levels of corticosterone, the natural murine GC, in the plasma and lungs of vΔA44L-infected mice, but the body weights, signs of illness, inflammatory response, and body temperature were unaltered. Subsequently, there was a more rapid recruitment of CD4+ and CD8+ lymphocytes, enhanced IFN-γ production and augmented CTL activity in the lungs of vΔA44L-infected mice. These data suggest that enhanced steroid production by the A44L protein contributes to immunosuppression during VV infection.
Materials and Methods
Cells and Viruses.
BS-C-1, CV-1, and TK− 143 cells and VV strains were grown as described previously (22). VV strain WR was partially purified through sucrose cushions as described (23).
Construction of Recombinant Viruses.
A VV WR mutant lacking 86% of the A44L gene (formerly vJM2, renamed vΔA44L) was described previously (19). A revertant virus (vA44L-rev) was isolated by reinserting the A44L gene into its natural locus within vΔA44L. A 2318-bp HincII fragment containing the VV WR A44L gene and flanking regions was cloned into the SmaI site of pUC119, forming pJM5. This plasmid was transfected into vΔA44L-infected cells and mycophenolic acid-resistant viruses were isolated as described (24). These were grown on hypoxanthine guanine phosphoribosyltransferase negative D980R cells in the presence of 6-thioguanine and a plaque representing a revertant virus (A44L-rev) was identified.
3β-HSD Assay.
CV-1 cells were mock-infected or infected in triplicate with VV at 10 PFU/cell. At 10 h pi 3β-HSD activity was measured by conversion of [3H]-pregnenolone to [3H]-progesterone as previously described (19).
Assay for Virus Virulence.
Female, 6–8-wk-old BALB/c mice were inoculated intranasally under general anesthesia with 104 or 105 PFU of VV in PBS on day 0. Each day, mice were weighed and monitored for signs of illness, and those having lost >25% of their original body weight were killed. To determine virus titers in organs, mice were killed and their lung, brains, livers, and spleens were removed, dounce homogenized, frozen, and thawed three times and sonicated. The titer of infectious virus was determined by plaque assay on BS-C-1 cells. All in vivo experiments were repeated at least once and data shown are representative of these multiple experiments.
Recovery of Bronchoalveolar Lavage and Lung Cells.
To obtain bronchoalveolar lavage (BAL) fluid mice were killed and their lungs were inflated five times with 1 ml of PBS containing 10 U/ml of heparin through a blunted 23-gauge needle inserted into the trachea. BAL was centrifuged (3,000 rpm, 10 min) and the supernatant was frozen at –20°C until analysis of cytokines by ELISA. BAL cells were treated with Tris-NH4Cl (0.14 M NH4Cl in 17 mM Tris, adjusted to pH 7.2) to lyse erythrocytes, washed twice, and resuspended in cold RPMI 1640 medium containing 10% FBS. BAL cells were centrifuged onto glass slides, dried, and stained with hematoxylin and eosin for differential cell counts. Single cell suspensions of lung cells were prepared by sieving lungs through a 100-μm nylon mesh followed by hypotonic lysis of erythrocytes. Cell viability was assessed using trypan blue exclusion.
Cytotoxic Assays.
NK cell cytotoxicity and VV-specific CTL activity in lung cell suspensions was assayed in 51Cr-release assays. NK-mediated lysis was tested on YAC-1 cells while P815 cells (H-2d, mastocytoma) were used as targets for VV-specific CTL lysis. Prior to labeling with Na2 51CrO4 (150 μCi per 3 × 106 cells), P815 cells were mock-infected or infected with VV WR at 10 PFU/cell for 2 h at 37°C. Uninfected YAC-1 cells were labeled as above. Serial dilutions of effector cells were incubated in triplicate cultures with either noninfected or VV-infected target cells in 100 μl of RPMI 1640 containing 10% FBS in 96-well V-bottomed plates at 37°C in 5% CO2. After 4 h (YAC-1) or 6 h (P815) cells were pelleted and 50 μl of the supernatant was transferred to a Lumaplate-96 (Packard Instrument Co.) and counted using a Packard Microplate Scintillation counter. The percentage of specific 51Cr release was calculated as: specific lysis = [(experimental release – spontaneous release)]/(total detergent release – spontaneous release)] × 100. The spontaneous release values were always <10% of total lysis.
In some experiments CD8+ cells were depleted from lung cell suspensions by incubation at 37°C with an anti-CD8 mAb (clone 3.115 [25]) in the presence of human complement. Analysis by flow cytometry demonstrated selective depletion of the CD8+ cell population. Depleted cells were added to cytotoxicity assays without adjustment for the reduced cell number.
ELISA for Cytokines.
IFN-γ, IL-10, and IL-4 in BAL fluid and culture supernatants was quantified using OptEIA kits from BD Biosciences. Cytokine concentrations were calculated from a standard curve and expressed as ng/ml.
Flow Cytometric Analysis of Cell Surface and Intracellular Antigens.
Single cell suspensions of BAL cells were blocked with 10% normal rat serum and 0.5 μg of Fc block (BD Biosciences) in FACS® buffer (PBS containing 0.1% bovine serum albumin and 0.1% sodium azide) on ice for 20 min. Cells were stained with FITC anti-CD4, Tri-color anti-CD8 and PE-labeled anti-CD3 or isotype antibody controls (all from Caltag). The distribution of cell surface markers was determined on a FACScan™ flow cytometer with CELLQuest™ software (Becton Dickinson). A lymphocyte gate was used to select at least 20,000 events.
To detect intracellular cytokines, 106 lung cells/ml were stimulated with 50 ng/ml PMA (Sigma-Aldrich), 500 ng/ml ionomycin (Calbiochem) in the presence of 10 μg/ml brefeldin A (Sigma-Aldrich) for 5 h at 37°C. Cells were washed with FACS® buffer and stained with Tri-color anti-CD4 and FITC anti-CD8 for 30 min on ice and then fixed for 30 min at room temperature with 2% paraformaldehyde in PBS. Samples were permeabilized with 0.5% saponin in FACS® buffer for 10 min. PE-conjugated anti-mouse IFN-γ (clone XMG1.2; BD Biosciences) was added for a further 30 min at room temperature and the cells were washed once with 0.5% saponin in FACS® buffer and twice in FACS® buffer alone. Cells were analyzed on a Becton Dickinson flow cytometer collecting data on at least 20,000 lymphocytes.
Corticosterone Levels in Plasma and Lung.
To guard against fluctuations due to circadian rhythm, samples were obtained between 9:00 and 10:00 a.m. Mice were killed by cervical dislocation and exanguinated within 4 min of disturbance. Blood was collected in EDTA-coated tubes on ice and was then centrifuged at 3,000 rpm for 10 min. Plasma was collected and stored at –20°C. Lungs were removed immediately after exanguination, washed once in PBS, and placed on ice. Tissue was homogenized and extracted in 2 ml of methanol. Corticosterone levels in plasma and lung extracts were determined by radioimmunoassay using a rat corticosterone 3H kit (ICN Pharmaceuticals). For lung extracts, excess methanol was evaporated and the dried pellet was dissolved in 0.5 ml of the steroid buffer supplied with the kit. Corticosterone levels were determined using a standard curve and expressed as ng/ml for plasma or ng/g of lung tissue.
Results
The gene encoding 3β-HSD in VV WR was originally called SalF7L (19) but is hereafter called A44L using VV strain Copenhagen nomenclature (26). Deletion of A44L from VV WR abrogated v3β-HSD activity and caused virus attenuation in vivo (19). To explore further the role of 3β-HSD in VV virulence we constructed a revertant virus (vA44L-rev) in which the A44L gene was reinserted into the vΔA44L mutant. This virus served as a control to ensure that any phenotypic differences of vΔA44L were due to loss of the A44L protein and not due to mutations elsewhere in the genome. Analysis of wild-type (vA44L), vΔA44L, and vA44L-rev viruses by PCR confirmed that the genomes were as predicted. Furthermore, no differences were found in the rate of replication or the final virus titer observed after infection of BS-C-1 cells with each of the viruses, whether a high (10 PFU per cell) or low (0.01 PFU per cell) multiplicity of infection was used (unpublished data). The plaque morphology formed by each virus on BS-C-1 cells was also indistinguishable (unpublished data).
Next, expression of 3β-HSD was measured in vitro by the conversion of [3H]-pregnenolone to [3H]-progesterone. As seen in Fig. 1 A, vA44L- and vA44L-rev–infected CV-1 cells exhibited 3β-HSD activity but mock- and vΔA44L-infected cells did not. In addition, all 8 strains of VV tested converted [3H]-pregnenolone to [3H]-progesterone, indicating v3β-HSD is conserved in VVs (Fig. 1 B).
Figure 1.
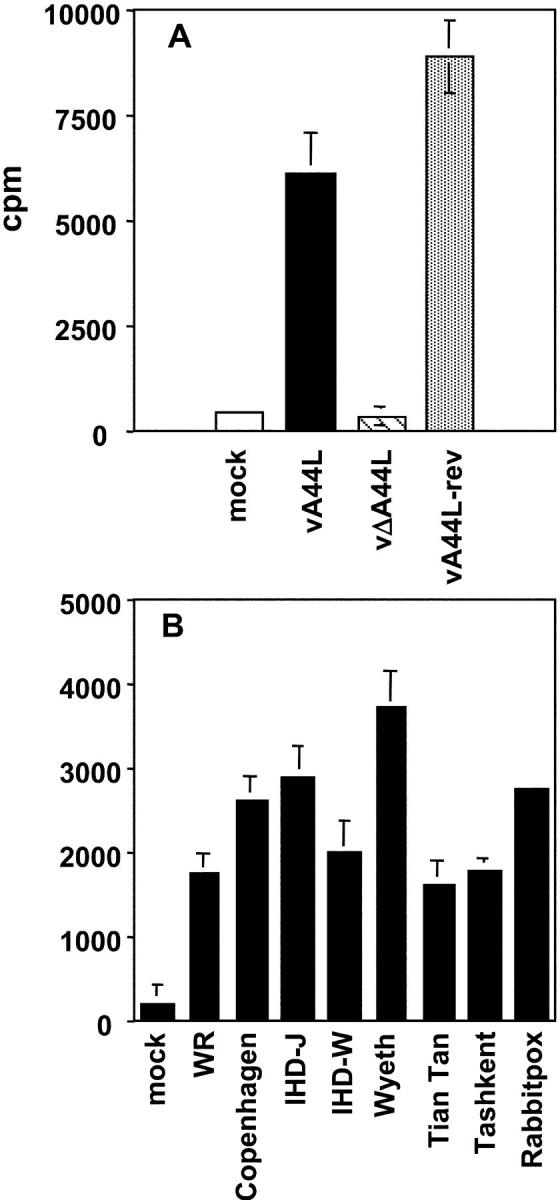
3β-HSD activity in VV-infected cells. CV-1 cells were mock-infected or infected at 10 PFU/cell with (A) vA44L, vΔA44L or A44L-rev, or (B) different VV strains, and assayed for 3β-HSD activity at 8–10 h p.i. All infections were performed in triplicate and results are expressed as mean ± SEM. The background values from nonenzymatic conversion to progesterone (measured in ethanol-fixed monolayers) were subtracted from each value.
Deletion of A44L Attenuates VV Strain WR In Vivo.
The role of the A44L protein in VV infection was explored using a murine intranasal model. Mice infected with vA44L, vΔA44L, or vA44L-rev were weighed and assessed for signs of illness (ruffled fur, reduced mobility, and tachypnea) daily. Fig. 2 demonstrates the attenuated phenotype of vΔA44L relative to both vA44L and vA44L-rev viruses. Mice infected with vA44L or vA44L-rev lost over 20% of body weight during the infection (Fig. 2 A) and displayed severe signs of illness (Fig. 2 B). In contrast, mice infected with vΔA44L lost significantly less weight, recovered more rapidly, and displayed very mild signs of illness. There was no significant difference between the weight loss profiles of mice infected with vA44L or vA44L-rev indicating that the attenuation seen with vΔA44L is due to loss of the A44L gene.
Figure 2.
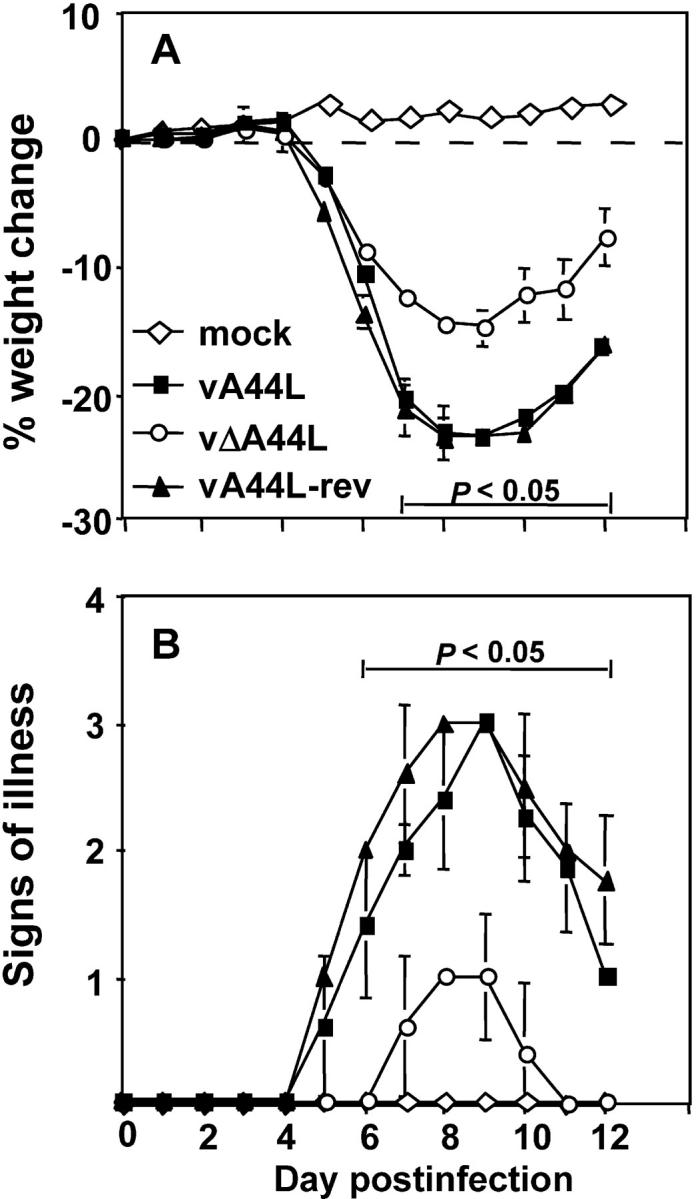
Deletion of A44L attenuates VV WR infection in a murine intranasal model. Groups of five BALB/c mice were mock-infected (⋄) or infected with 104 PFU of vA44L (▪), vΔA44L (○), or A44L-rev (▴). (A) Mice were weighed daily and results are expressed as the mean percentage weight change of each group ± SEM compared with the weight immediately before infection. (B) Animals were monitored daily for signs of illness, scored from 1 to 4. Data are expressed as the mean ± SEM from five mice. P values were determined using the Student's t test and indicate the mean % weight changes or signs of illness of mice infected with vΔA44L that were significantly different from both those of mice infected with vA44L or A44L-rev.
VV WR gene B15R encodes a soluble IL-1β receptor (27) that blocks IL-1β–induced fever (28). To determine if A44L affected body temperature during VV infection, rectal temperatures were taken daily after infection with vA44L, vΔA44L, or vA44L-rev viruses. No differences were noted in the body temperatures of animals infected with these viruses (unpublished data) indicating that A44L affects virulence through an alternative mechanism(s).
Virus Replication and Spread In Vivo.
As the lung is the site of primary infection in the intranasal model, we assayed lung homogenates for infectious virus after infection with vA44L, vΔA44L, or vA44L-rev. As shown in Fig. 3 A, all three viruses had similar titers at 1 and 3 d pi, indicating that A44L is not critical for the initial rounds of replication in the lung. However, by the time peak titers of virus occurred at 7 d pi the level of infectivity in the lungs of vA44L and vA44L-rev–infected mice was significantly greater than that in mice infected with vΔA44L. These observations were consistent in multiple experiments (n = 4). By day 12, infectious virus could be detected in the lungs of only 1/5 vΔA44L-infected animals compared with 5/5 in each of the other groups.
Figure 3.
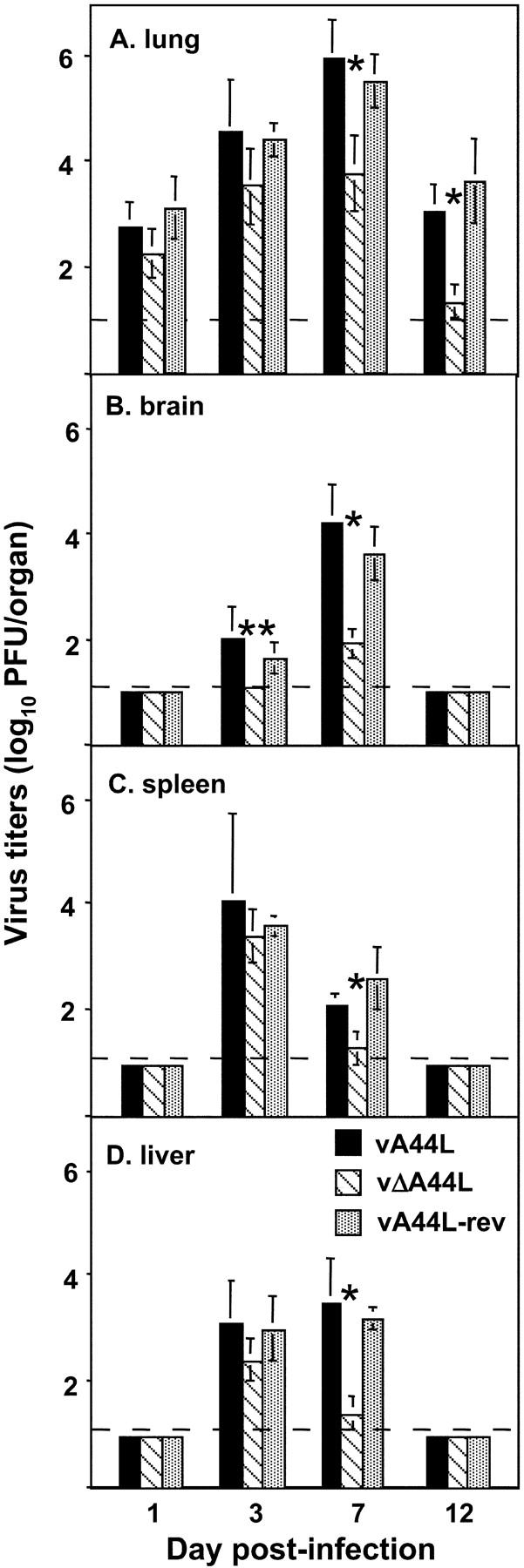
Titers of vA44L, vΔA44L, and A44L-rev in the lungs (A), brains (B), spleens (C), and livers (D). Groups of five mice were infected intranasally with 104 PFU of VV and the lungs, brains, spleens, and livers were harvested on the days indicated. Organs were homogenized and stored at –70°C and the titer of infectious virus was determined by plaque assay on BS-C-1 cells. Virus titers are expressed as mean log10 PFU per organ, with SEM. The broken line indicates the minimum detection limit of the plaque assay. Columns marked with an asterisk represent virus titers from vΔA44L-infected mice that were significantly different to those from both vA44L- and vA44L-infected animals. *, P < 0.05, **, P < 0.02.
WR is a neurovirulent strain of VV that was derived by repeated passage in suckling mouse brain (29). In the murine intranasal model, VV strain WR infection is accompanied by extensive respiratory infection and virus dissemination to multiple organs (30, 31). To examine the ability of the three viruses to spread and replicate in extrapulmonary sites, homogenates of brain, spleen, and liver were assayed for infectious virus. Virus titers were below detection levels in all organs at day 1 pi, however by day 3 infectious virus was recovered from brain, spleen, and liver of all mice infected with vA44L or vA44L-rev, and from the spleen and livers of all vΔA44L-infected animals (Fig. 3, B, C, and D). Infectivity titers were similar in all groups at this time, except in the brain of vΔA44L-infected mice, where virus was recovered from only 1/5 animals. By day 7, virus titers were reduced in all organs examined from vΔA44L-infected mice compared with controls, indicating that while this virus is capable of in vivo spread, it is cleared more rapidly from the brain, liver, spleen, and lungs compared with vA44L and vA44L-rev viruses. Virus was still present in lungs of VV-infected animals 12 d pi; however, it had been cleared from all other organs examined by this time.
To investigate if vA44L, vΔA44L, and vA44L-rev could replicate to similar levels in murine cells in vitro, cultures of primary fibroblasts were prepared from the kidneys of naive BALB/c mice (Materials and Methods). The replication kinetics of the three viruses were indistinguishable during either single-step (10 PFU/cell) or multistep (0.01 PFU/cell) growth curves (unpublished data); the latter conditions are particularly sensitive to accentuating small differences in growth between viruses. Our findings that (a) the rate of virus replication and the maximum virus titer obtained in lungs after infection with vA44L, vΔA44L, and vA44L-rev was not statistically different over the first three days of infection (Fig. 3 A), and (b) that there was no difference in the replicative capacity of the viruses in a primary mouse culture, indicated that the difference in virulence between these viruses was probably due to effects of v3β-HSD on host antiviral mechanisms rather than altered replicative ability.
Analysis of BAL Cells from Lungs of VV-infected Mice.
The swifter clearance of virus after infection with vΔA44L suggested a more effective antiviral host response against this virus and therefore the cellular inflammatory response was assessed. BALs were performed at various times pi and the numbers of viable cells were determined. Few cells were recovered from BAL of mock-infected animals or 1 d pi with 104 PFU of VV, indicating there were only low levels of recruitment to the lung in the early phase of VV infection (Fig. 4 A). BAL cell numbers increased over time with peak numbers 12 d pi with vA44L or vA44L-rev, and 7 d pi with vΔA44L. Significantly more cells were recovered from vΔA44L-infected mice at day 7 pi compared with other VV-infected groups.
Figure 4.

Characterization of BAL cell suspensions from mice infected with 104 PFU of vA44L, vΔA44L, or vA44L-rev. BAL cells were recovered, counted, and stained to determine the numbers of (A) total cells, (B) macrophages, and (C) lymphocytes from mock-infected and VV-infected mice. Columns represent the mean cell yield per mouse ± SEM from groups of 4–5 mice. Columns marked with an asterisk represent mean cell numbers recovered from vΔA44L-infected mice that were significantly different (*, P < 0.05) to those of both vA44L and vA44L-rev-infected mice. (D and E) At 7 d pi, BAL cells were recovered, stained for expression of CD3, CD4, and CD8 and analyzed by flow cytometry. Lymphocytes were identified by their characteristic FSC/SSC profile and by expression of CD3. Data shown are the mean percentage of BAL cells ± SEM from 4–5 individual mice, and are representative of two independent experiments.
Cytospins of BAL cells were stained with hematoxylin and eosin for differential cell counts (Fig. 4, B and C). Uninfected mouse BAL cells were composed almost entirely of macrophages, with some (<10%) lymphocytes noted. The majority of inflammatory cells recruited to the lung during the course of VV infection were macrophages (Fig. 4 B) and lymphocytes (Fig. 4 C), with granulocytes representing less than 5% of total cells at all time points tested (unpublished data). The enhanced recruitment of lymphocytes to the lungs of vΔA44L-infected mice at day 7 correlated with the more rapid clearance of this virus from the lungs (Fig. 3 A). Notably, by day 12 pi with vΔA44L the levels of total cells, macrophages, and lymphocytes in BALs were all lower than on day 7, whereas with vA44L and vA44L-rev the cell numbers were higher on day 12 than day 7.
A striking feature of the cellular inflammatory response to vΔA44L was the early recruitment of lymphocytes (Fig. 4 C). Therefore, flow cytometric analysis was used to examine lymphocyte subsets in BAL at day 7 pi. A greater percentage of both CD4+ (Fig. 4 D) and CD8+ (Fig. 4 E) lymphocytes were observed in BAL from vΔA44L-infected mice compared with animals infected with vA44L or vA44L-rev. Less than 5% of BAL cells were DX5+ NK cells or B220+ B lymphocytes at this time (unpublished data). These data show that by day 7 pi with vΔA44L more cells were recruited to the lungs and a higher proportion of these were CD4+ and CD8+ T cells.
Analysis of IFN-γ Production in the Lungs of VV-infected Mice.
Fig. 4 indicates an increased presence of CD4+ and CD8+ T cells within the lungs of vΔA44L-infected mice; however, functional analysis of these cells was required. IFN-γ plays an important role in antiviral host defense following infection of mice with VV or other poxviruses (32–34). Given the enhanced recruitment of T cells to the lungs of vΔA44L-infected mice and the ability of both CD4+ and CD8+ T lymphocytes to produce antiviral cytokines, the level of IFN-γ in BAL fluids was determined (Fig. 5 A). IFN-γ levels in BAL fluids from mock-infected animals were below the detection limit of the assay. Low levels of IFN-γ were detected in lavage 3 d pi and these increased markedly at days 7 and 10 pi. Significantly higher levels of IFN-γ were detected from vΔA44L-infected mice compared with animals infected with control viruses at day 7 pi. IFN-γ levels at day 10 were also generally higher in mice infected with vΔA44L (Fig. 5 A). Levels of IL-10 or IL-4 in BAL fluid were below the detection limit of their respective ELISA assays at each time point tested (unpublished data).
Figure 5.
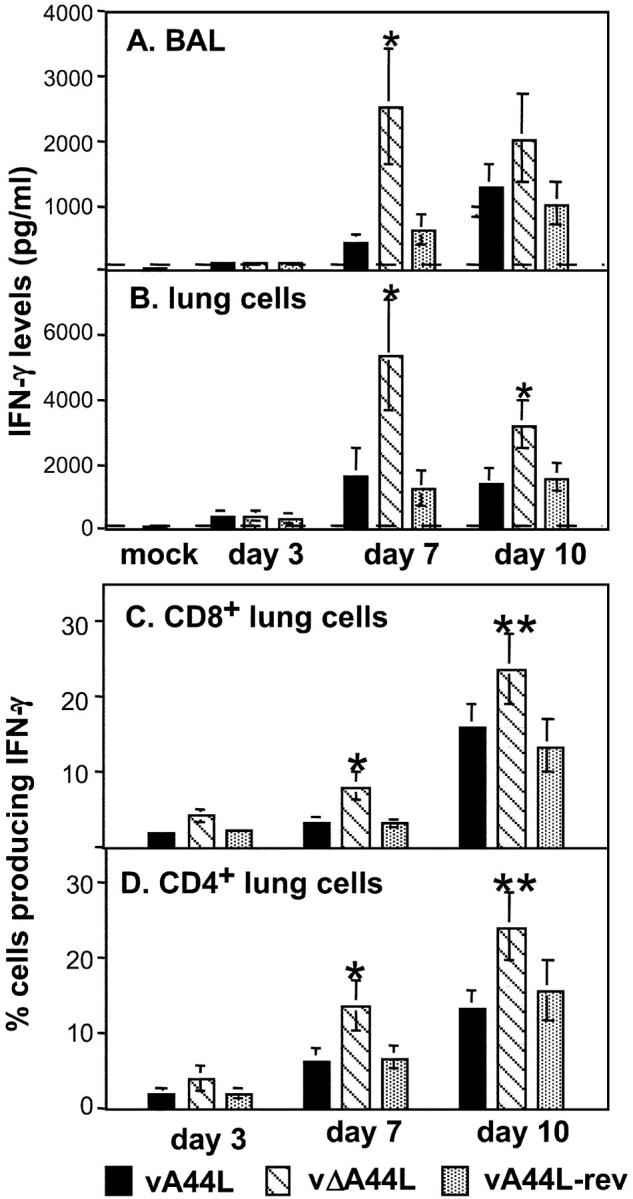
Production of IFN-γ in the lungs of VV-infected mice. Groups of 4–6 BALB/c mice were mock-infected or infected with 104 PFU of vA44L, vΔA44L, or vA44L -rev. At days 3, 7, and 10 mice were killed, BALs were performed and single cell suspensions were prepared from lung tissue. (A) Levels of IFN-γ in BAL fluids of VV-infected mice. BAL fluid was centrifuged and the level of IFN-γ present in the supernatant was determined by ELISA. Values represent the mean, with SEM, from two groups (n = 3/group). The dashed line represents the detection limit of the IFN-γ ELISA (50 pg/ml). (B) IFN-γ production from lung cells. Lung cell suspensions were stimulated with PMA and ionomycin for 5 h as described in Materials and Methods. Cells were pelleted and level of IFN-γ present in the supernatant was determined by ELISA. Values represent the mean ± SEM, from two groups (n = 3/group). The dashed line represents the detection limit of the IFN-γ ELISA (50 pg/ml). (C and D) Intracellular production of IFN-γ by lung lymphocytes from mice 7 d after intranasal VV. Lung cells were stimulated with PMA and ionomycin for 4 h; brefeldin A was added to retain cytokines in the cytoplasm. Cells were stained with FITC-labeled anti-CD8, APC-labeled anti-CD4, and after permeabilization using saponin, with PE-labeled anti–IFN-γ before analysis by three-color flow cytometry. Shown are the percentages of CD8+ (C) or CD4+ (D) T cells producing IFN-γ. Values are averaged from two groups (n = 3/group). The frequency of IL-4-producing cells was below the detection limit (<2%) and is not shown. *, P < 0.05, **, P < 0.02.
Next, IFN-γ production was determined after culturing lung cell suspensions in the presence of PMA and ionomycin for 5 h at 37°C. IFN-γ levels were very low in lung cell supernatants from mock-infected mice (Fig. 5 B). A modest increase was observed in supernatants from lung cells 3 d pi, and levels were elevated further from lung cells harvested days 7 and 10 pi. Consistent with IFN-γ levels observed in BAL, PMA-stimulated lung cells from vΔA44L-infected mice produced significantly more IFN-γ than those from animals infected with vA44L or vA44L-rev on days 7 and 10 pi (Fig. 5 B).
The elevated IFN-γ production detected in BAL fluids and lung cell suspensions from vΔA44L-infected mice suggested that the additional lymphocytes recruited to the lungs of these animals were producing IFN-γ. Intracellular staining of lung lymphocytes for IFN-γ on days 3, 7, and 10 pi demonstrated that the proportion of both CD4+ and CD8+ lymphocytes positive for IFN-γ was higher in the lungs of vΔA44L-infected mice at days 7 and 10 pi (Fig. 5 C, D). Only a very low proportion of CD4+ or CD8+ cells (<5%) stained for IL-4 or IL-10 in any of the groups (unpublished data).
Analysis of Cytolytic Activity of Lymphocytes Isolated from VV-infected Lungs.
To assess the effect of VV 3β-HSD on VV-specific CTL activity, the cytotoxic activity of cells from lung cell suspensions was examined against VV-infected or uninfected P815 cells (Fig. 6) . At day 5, primary CTL activity was very low in lung cell suspensions from all infected animals (< 10%, unpublished data); however, by day 7 significant CTL activity was detected against VV-infected targets (Fig. 6 A). The cytolytic activity of lung lymphocytes from vΔA44L-infected mice was greater than from mice infected with vA44L or vA44L-rev, and this trend was also observed using lung cells from animals 10 d pi. (Fig. 6 B). All lung cell suspensions showed very weak cytotoxic activity against uninfected P815 cells (less than 10% at effector:target ratio of 100:1). Furthermore, no significant cytotoxic activity was observed against the NK-sensitive YAC-1 cell line (unpublished data).
Figure 6.

Virus-specific CTL activity in the lung of VV-infected mice. Groups of six mice were infected with 104 PFU of vA44L (▪), vΔA44L (○), or vA44L-rev (▴). At days 7 (A and C) and 10 (B and D) mice were killed and lung cell suspensions were prepared. Specific lysis of WR-infected P815 cells was assessed by 51Cr-release assay. Data are expressed as the mean percent specific lysis ± SEM from two groups of three mice plotted against the lung cell:target ratio (A and B) or the CD8+ lung cell:target ratio (C and D). Lysis of uninfected P815 cells by day 7 and day 10 effector cell populations was always <10% at an effector:target ratio of 100:1 (unpublished data).
The increase in cytotoxic activity of lung cells from vΔA44L-infected mice may represent increased recruitment of CTL effectors or an increased activation state of the cells. To confirm that CD8+ T lymphocytes were mediating CTL activity, day 7 lung cells from VV-infected mice were treated with complement plus a mAb to murine CD8. This treatment abrogated virtually all CTL activity (undepleted = 56% specific lysis, complement alone = 46%, complement plus anti-CD8 = 3%, at effector:target ratio of 100:1).
The numbers of CD8+ lymphocytes in the lung cell suspensions were determined by flow cytometry, and used to compare CTL activity between lung cells based on a CD8+ lung cell:target ratio (Fig. 6, C and D). The enhanced lysis observed using lung cells from vΔA44L-infected mice is partly explained by the higher relative numbers of CD8+ T cells in this compartment (7.4 ± 2.2%, 13.6 ± 2.0%, and 8.0 ± 1.9% of total lung cells at day 7, and 14.5 ± 3.8%, 25.1 ± 4.7%, and 13.5 ± 3.0% of total lung cells at day 10 from mice infected with vA44L, vΔA44L and vA44L-rev, respectively). However, CTL from vΔA44L-infected mice also show an enhanced level of lysis on a per cell basis consistent with an enhanced activation state of virus-specific CTL within the T cell population (Fig. 6, C and D).
Levels of Corticosterone in Plasma and Lungs of VV-infected Mice.
Data presented so far have demonstrated that the vΔA44L mutant is markedly attenuated in the murine intranasal model, and that after an initial period of 3–4 d when infection with each virus was indistinguishable, the attenuated phenotype of the vΔA44L mutant was accompanied by a more vigorous cellular inflammatory response in the lungs of infected mice. As the A44L encodes an active 3β-HSD and GCs and other steroids possess important immunomodulatory functions, the levels of corticosterone were determined in the plasma and lungs of VV-infected mice (Fig. 7, A and B) . Corticosterone is released after stress and as part of the acute phase response to infection, therefore we determined levels only at times when all indicators of disease (and therefore stress) were similar between groups. At days 1, 2, and 4 virus titers were similar in the lungs of vA44L-, vΔA44L-, or vA44L-rev–infected mice (Fig. 7 C), and no differences were observed in visible signs of illness, weight loss, body temperature, or the number of inflammatory cells and IFN-γ levels recovered from the BAL of infected mice (unpublished data). Corticosterone levels were, however, significantly lower in plasma at days 1 and 2 (Fig. 7 A) and in lung extracts at days 2 and 4 (Fig. 7 B) from vΔA44L-infected mice. After day 4 any differences in corticosterone levels between the groups would be difficult to interpret because the attenuated phenotype of vΔA44L is apparent and therefore stress levels might vary between groups. The enhanced levels of endogenous GCs observed in vA44L and vA44L-rev–infected mice suggest that A44L may have a direct effect upon local and systemic steroid levels during VV infection.
Figure 7.
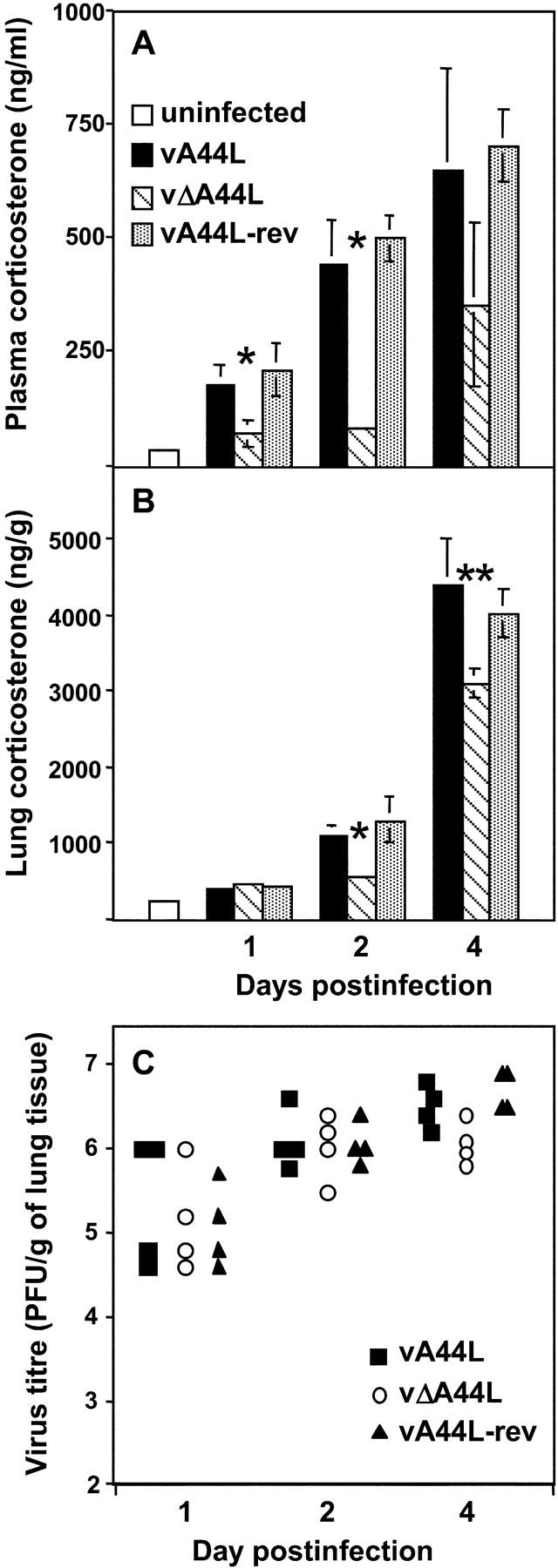
Corticosterone levels in plasma and lungs after intranasal infection with VV. Corticosterone levels were measured in (A) plasma and (B) lung extracts collected from BALB/c mice under low stress conditions (samples were obtained within 4 min of handling) at the indicated times after intranasal infection with 105 PFU of vA44L, vΔA44L, or A44L-rev. Lung extracts were prepared as described in Materials and Methods. Data represent mean ± SEM of four or five mice per time point and are expressed as ng/ml of plasma or as ng/g of lung tissue. Columns marked with an asterisk represent corticosterone levels from vΔA44L-infected mice that were significantly different to those from vA44L- and vA44L-rev-infected mice. *, P < 0.05, **, P < 0.02. (C) Titers of infectious virus in the lungs of mice after infection with 105 PFU of VV. Virus titers were determined by plaque assay on BS-C-1 cells and are expressed as PFU/g of lung tissue.
Discussion
VV and other poxviruses encode many proteins that interfere with specific components of the immune system. Here we have examined the biological significance of the A44L protein from VV strain WR by comparing the virulence of wild-type WR (vA44L), a mutant lacking 86% of the A44L ORF (vΔA44L), and a revertant virus where the A44L ORF was reinserted into its original locus (vA44L-rev). The results demonstrate that A44L is an important virulence factor of VV and has potent immunosuppressive properties.
First, this study confirmed that deletion of A44L from VV strain WR reduced virus virulence following intranasal infection. Previously, it was reported that mice infected intranasally with vΔA44L showed reduced mortality and relatively mild weight loss compared with those infected with parental strain WR (19). In the current study, we also report that vΔA44L-infected mice showed milder signs of illness and do not develop ruffled fur, reduced mobility, and tachypnea that are characteristic of VV-induced pneumonia (Fig. 2 B). In contrast, Sroller et al. (20) reported that loss of A44L caused only a modest reduction in virulence. In the current study the inclusion a revertant virus strengthens our conclusion that A44L is a virulence factor in the murine intranasal model, as it is in the murine intradermal model (21).
Second, this study demonstrated that although (a) vA44L, vΔA44L, and vA44L-rev viruses replicated to similar levels in primary murine fibroblasts, (b) in repeated experiments virus titers in the lungs of vA44L, vΔA44L, and vA44L-rev-infected animals were equivalent on days 1–3 pi, and (c) all 3 viruses were capable of spread and replication in secondary organs (brain, liver, spleen), the vΔA44L virus was cleared more rapidly. These findings indicate that A44L is not critical for the initial rounds of virus replication in the host and that the difference between the viruses probably reflects host antiviral mechanisms. The rapid clearance of vΔA44L coincided with an early influx of inflammatory leukocytes, in particular T lymphocytes, into the lungs. Functional analysis demonstrated that IFN-γ production and VV-specific CTL activity were enhanced in these lymphocytes. This phenotype was consistent with the A44L protein interfering with aspects of the proinflammatory host response.
The VV WR A44L gene encodes a 3β-HSD enzyme (19). The mammalian 3β-HSD enzyme catalyzes the conversion of Δ5-3β-hydroxysteroids to Δ4-3-ketosteroids, a reaction that is required for the biosynthesis of all classes of steroid hormones. Therefore, the most likely role for A44L in VV pathogenesis is in the production of steroid hormones such as GCs that are immunosuppressive and are used widely in the therapy of inflammatory, autoimmune, and allergic diseases (5). GCs have the potential to affect multiple aspects of the antiviral immune response. Previous studies established that restraint stress (RST) reduced the accumulation of leukocytes in the lungs of influenza virus-infected mice and this response was dependent upon elevated serum GCs as treatment with the GC receptor antagonist RU486 restored cellular infiltration (35). RST suppression of influenza virus-specific production of cytokines such as IL-2 and IFN-γ was also reversed by RU486 treatment (36). By analogy, our findings that infection of mice with vΔA44L was associated with enhanced leukocyte infiltration (Fig. 4) and IFN-γ production (Fig. 5) in the lung is consistent with A44L-mediated enhancement of GC levels in vivo following infection with vA44L and vA44L-rev viruses.
GC release through the hypothalamic-pituitary-adrenal (HPA) axis occurs as part of circadian rhythm (37) and is induced by additional stimuli including physical or cognitive stress (38) and cytokine responses to bacterial LPS (39, 40) or viruses such as murine cytomegalovirus (41). Intranasal infection of mice with VV led to increased levels of corticosterone, a GC, in plasma and in the lung compared with uninfected mice (Fig. 7). Moreover, there was a significant increase in plasma and lung corticosterone levels in vA44L- and vA44L-rev–infected mice compared with animals infected with vΔA44L (Fig. 7) at times when all other indicators of disease (weight loss, signs of illness, body temperature, recovery of inflammatory cells from BAL, IFN-γ levels, and virus titres) were similar between groups. Later time points were not examined because any differences in corticosterone levels could be due to either the A44L protein or the more severe illness and stress caused by viruses expressing this protein.
A striking feature of the inflammatory response to vΔA44L was the rapid recruitment of leukocytes to the lungs, in particular CD4+ and CD8+ T lymphocytes with the potential to produce IFN-γ (Fig. 5) and CD8+ CTLs (Fig. 6), indicating that A44L can suppress at least two important components of the antiviral host response. MHC class I–restricted, CD8+ CTL and IFN-γ play an important role in the resolution of acute infection by poxviruses such as VV (32, 42, 43). VV is very sensitive to IFN-γ in vitro (44) and IFN-γ production is critical for recovery of mice after VV infection (32, 33, 42, 45). Poxviruses encode several intracellular and extracellular proteins to interfere with the production or action of IFN-γ; however, the VV IFN-γ receptor secreted from infected cells does not neutralize mouse IFN-γ (46–49). CD8+ CTLs are key mediators of viral clearance by cytolysis of virus-infected cells and the secretion of cytokines such as IFN-γ and TNF-α. Interestingly, GCs also suppress CTL activity (50).
A44L is nonessential for virus replication in vitro; however, it is quite well conserved amongst members of the poxvirus family. All 8 VV strains tested expressed 3β-HSD activity (Fig. 1 B) and similar activity has been described in cells infected with VV strains Praha, LIVP, and MVA (20) as well as the avipoxviruses fowlpox and canarypox (51) and a fish iridiovirus (52). The gene is also highly conserved in other sequenced orthopoxviruses (camelpox, ectromelia, cowpox, and monkeypox) and other poxvirus genera (suipoxvirus, molluscipoxvirus, and yatapoxvirus) suggesting the v3β-HSD plays an important role in poxvirus biology. Interestingly, despite the presence of a closely related gene in several variola strains, in each case the gene is disrupted by mutation and is not predicted to encode an active 3β-HSD (53–57). Deletion of the v3β-HSD from VV did not enhance antibody responses to infection but the cell-mediated immune responses were not examined (20).
It is apparent that VV and other poxviruses possess multiple genes that contribute to virulence by subverting the host immune response or by altering virus dissemination and/or tropism. Data presented here indicate that A44L is immunosuppressive and interferes with the early events in the host cellular response to VV infection, thus contributing to protracted virus replication and improved virus dissemination within the host. The removal of this gene from poxvirus vaccines, such as modified vaccinia virus Ankara (MVA), is likely to enhance vaccine immunogenicity, particularly T cell responses to infection.
Acknowledgments
The authors wish to thank Dr. Fiona Culley and Dr. Jonothan Dodd for assistance and helpful discussion.
This work was supported by a programme grant from The Wellcome Trust. P.C. Reading was a Howard Florey Fellow and G.L. Smith is a Wellcome Trust Principal Research Fellow.
J.B. Moore's present address is Millennium Pharmaceuticals, Inc., 640 Memorial Dr., Cambridge, MA 02139.
Footnotes
Abbreviations used in this paper: BAL, bronchoalveolar lavage; GC, glucocorticoid; 3β-HSD, 3β-hydroxysteroid dehydrogenase; VV, vaccinia virus; WR, Western Reserve.
References
- 1.Labrie, F., J. Simard, V. Luu-The, G. Pelletier, A. Belanger, Y. Lachance, H.F. Zhao, C. Labrie, N. Breton, Y. de Launoit, et al. 1992. Structure and tissue-specific expression of 3 beta-hydroxysteroid dehydrogenase/5-ene-4-ene isomerase genes in human and rat classical and peripheral steroidogenic tissues. J. Steroid Biochem. Mol. Biol. 41:421–435. [DOI] [PubMed] [Google Scholar]
- 2.Pelletier, G., E. Dupont, J. Simard, V. Luu-The, A. Belanger, and F. Labrie. 1992. Ontogeny and subcellular localization of 3 beta-hydroxysteroid dehydrogenase (3 beta-HSD) in the human and rat adrenal, ovary and testis. J. Steroid Biochem. Mol. Biol. 43:451–467. [DOI] [PubMed] [Google Scholar]
- 3.Martel, C., M.H. Melner, D. Gagne, J. Simard, and F. Labrie. 1994. Widespread tissue distribution of steroid sulfatase, 3 beta-hydroxysteroid dehydrogenase/delta 5-delta 4 isomerase (3 beta-HSD), 17 beta-HSD 5 alpha-reductase and aromatase activities in the rhesus monkey. Mol. Cell. Endocrinol. 104:103–111. [DOI] [PubMed] [Google Scholar]
- 4.Morel, Y., F. Mebarki, E. Rheaume, R. Sanchez, M.G. Forest, and J. Simard. 1997. Structure-function relationships of 3 beta-hydroxysteroid dehydrogenase: contribution made by the molecular genetics of 3 beta- hydroxysteroid dehydrogenase deficiency. Steroids. 62:176–184. [DOI] [PubMed] [Google Scholar]
- 5.Boumpas, D.T., G.P. Chrousos, R.L. Wilder, T.R. Cupps, and J.E. Balow. 1993. Glucocorticoid therapy for immune-mediated diseases: basic and clinical correlates. Ann. Intern. Med. 119:1198–1208. [DOI] [PubMed] [Google Scholar]
- 6.Boumpas, D.T., F. Paliogianni, E.D. Anastassiou, and J.E. Balow. 1991. Glucocorticosteroid action on the immune system: molecular and cellular aspects. Clin. Exp. Rheumatol. 9:413–423. [PubMed] [Google Scholar]
- 7.Schwiebert, L.M., L.A. Beck, C. Stellato, C.A. Bickel, B.S. Bochner, R.P. Schleimer, and L.A. Schwiebert. 1996. Glucocorticosteroid inhibition of cytokine production: relevance to antiallergic actions. J. Allergy Clin. Immunol. 97:143–152. [DOI] [PubMed] [Google Scholar]
- 8.Ashwell, J.D., F.W. Lu, and M.S. Vacchio. 2000. Glucocorticoids in T cell development and function. Annu. Rev. Immunol. 18:309–345. [DOI] [PubMed] [Google Scholar]
- 9.Kass, E.H., and M. Finland. 1953. Adrenocortical hormones in infection and immunity. Annu. Rev. Microbiol. 7:361–388. [DOI] [PubMed] [Google Scholar]
- 10.Huber, S.A., and B. Pfaeffle. 1994. Differential Th1 and Th2 cell responses in male and female BALB/c mice infected with coxsackievirus group B type 3. J. Virol. 68:5126–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huber, S.A., J. Kupperman, and M.K. Newell. 1999. Hormonal regulation of CD4(+) T-cell responses in coxsackievirus B3-induced myocarditis in mice. J. Virol. 73:4689–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sherman, M., K.M. Peltekian, and C. Lee. 1995. Screening for hepatocellular carcinoma in chronic carriers of hepatitis B virus: incidence and prevalence of hepatocellular carcinoma in a North American urban population. Hepatology. 22:432–438. [PubMed] [Google Scholar]
- 13.McMahon, B.J., S.R. Alberts, R.B. Wainwright, L. Bulkow, and A.P. Lanier. 1990. Hepatitis B-related sequelae. Prospective study in 1400 hepatitis B surface antigen-positive Alaska native carriers. Arch. Intern. Med. 150:1051–1054. [DOI] [PubMed] [Google Scholar]
- 14.Rao, A.R., I. Prahlad, M. Swaminathan, and A. Lakshmi. 1963. Pregnancy and smallpox. J. Indian Med. Assoc. 40:353–363. [PubMed] [Google Scholar]
- 15.Rao, A.R., M.S. Sumkumar, S. Kamalakshi, T.V. Paramasivam, T.A. Parasuraman, and M. Shantha. 1968. Experimental variola in monkeys. I. Studies on disease enhancing property of cortisone in smallpox. A preliminary study. Indian J. Med. Res. 56:1855–1865. [PubMed] [Google Scholar]
- 16.Kligman, F. 1951. The effect of cortisone on the pathologic responses of guinea pigs infected cutaneously with fungi, viruses and bacteria. J. Lab. Clin. Med. 37:615–620. [PubMed] [Google Scholar]
- 17.Bugbee, L.M., A.A. Like, and R.B. Stewart. 1960. The effects of cortisone on intradermally induced vaccinia infection in rabbits. J. Infect. Dis. 106:166–173. [DOI] [PubMed] [Google Scholar]
- 18.Alcamí, A., and U.H. Koszinowski. 2000. Viral mechanisms of immune evasion. Trends Microbiol. 8:410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore, J.B., and G.L. Smith. 1992. Steroid hormone synthesis by a vaccinia enzyme: a new type of virus virulence factor. EMBO J. 11:1973–1980 [published erratum at 11:3490]. [DOI] [PMC free article] [PubMed]
- 20.Sroller, V., L. Kutinova, S. Nemeckova, V. Simonova, and V. Vonka. 1998. Effect of 3-beta-hydroxysteroid dehydrogenase gene deletion on virulence and immunogenicity of different vaccinia viruses and their recombinants. Arch. Virol. 143:1311–1320. [DOI] [PubMed] [Google Scholar]
- 21.Tscharke, D.C., P.C. Reading, and G.L. Smith. 2002. Dermal infection with vaccinia virus reveals roles for virus proteins not seen using other inoculation routes. J. Gen. Virol. 83:1977–1986. [DOI] [PubMed] [Google Scholar]
- 22.Alcamí, A., J.A. Symons, P.D. Collins, T.J. Williams, and G.L. Smith. 1998. Blockade of chemokine activity by a soluble chemokine binding protein from vaccinia virus. J. Immunol. 160:624–633. [PubMed] [Google Scholar]
- 23.Mackett, M., G.L. Smith, and B. Moss. 1985. The construction and characterization of vaccinia virus recombinants expressing foreign genes. DNA Cloning: A Practical Approach. D.M. Glover, editor. IRL Press, Oxford. 191–211.
- 24.Falkner, F.G., and B. Moss. 1990. Transient dominant selection of recombinant vaccinia viruses. J. Virol. 64:3108–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarmiento, M., A.L. Glasebrook, and F.W. Fitch. 1980. IgG or IgM monoclonal antibodies reactive with different determinants on the molecular complex bearing Lyt 2 antigen block T cell-mediated cytolysis in the absence of complement. J. Immunol. 125:2665–2672. [PubMed] [Google Scholar]
- 26.Goebel, S.J., G.P. Johnson, M.E. Perkus, S.W. Davis, J.P. Winslow, and E. Paoletti. 1990. The complete DNA sequence of vaccinia virus. Virology. 179:247–266. [DOI] [PubMed] [Google Scholar]
- 27.Alcamí, A., and G.L. Smith. 1992. A soluble receptor for interleukin-1 beta encoded by vaccinia virus: a novel mechanism of virus modulation of the host response to infection. Cell. 71:153–167. [DOI] [PubMed] [Google Scholar]
- 28.Alcamí, A., and G.L. Smith. 1996. A mechanism for the inhibition of fever by a virus. Proc. Natl. Acad. Sci. USA. 93:11029–11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bronson, L.H., and R.F. Parker. 1941. The neutralization of vaccine virus by immune serum: titration by the intracerebral inoculation of mice. J. Bacteriol. 41:56–57. [Google Scholar]
- 30.Turner, G.S. 1967. Respiratory infection of mice with vaccinia virus. J. Gen. Virol. 1:399–402. [DOI] [PubMed] [Google Scholar]
- 31.Williamson, J.D., R.W. Reith, L.J. Jeffrey, J.R. Arrand, and M. Mackett. 1990. Biological characterization of recombinant vaccinia viruses in mice infected by the respiratory route. J. Gen. Virol. 71:2761–2767. [DOI] [PubMed] [Google Scholar]
- 32.Huang, S., W. Hendriks, A. Althage, S. Hemmi, H. Bluethmann, R. Kamijo, J. Vilcek, R.M. Zinkernagel, and M. Aguet. 1993. Immune response in mice that lack the interferon-gamma receptor. Science. 259:1742–1745. [DOI] [PubMed] [Google Scholar]
- 33.Dalton, D.K., S. Pitts-Meek, S. Keshav, I.S. Figari, A. Bradley, and T.A. Stewart. 1993. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 259:1739–1742. [DOI] [PubMed] [Google Scholar]
- 34.Muller, U., U. Steinhoff, L.F. Reis, S. Hemmi, J. Pavlovic, R.M. Zinkernagel, and M. Aguet. 1994. Functional role of type I and type II interferons in antiviral defense. Science. 264:1918–1921. [DOI] [PubMed] [Google Scholar]
- 35.Hermann, G., F.M. Beck, and J.F. Sheridan. 1995. Stress-induced glucocorticoid response modulates mononuclear cell trafficking during an experimental influenza viral infection. J. Neuroimmunol. 56:179–186. [DOI] [PubMed] [Google Scholar]
- 36.Dobbs, C.M., N. Feng, F.M. Beck, and J.F. Sheridan. 1996. Neuroendocrine regulation of cytokine production during experimental influenza viral infection: effects of restraint stress-induced elevation in endogenous corticosterone. J. Immunol. 157:1870–1877. [PubMed] [Google Scholar]
- 37.Dhabhar, F.S., A.H. Miller, M. Stein, B.S. McEwen, and R.L. Spencer. 1994. Diurnal and acute stress-induced changes in distribution of peripheral blood leukocyte subpopulations. Brain Behav. Immun. 8:66–79. [DOI] [PubMed] [Google Scholar]
- 38.Khansari, D.N., A.J. Murgo, and R.E. Faith. 1990. Effects of stress on the immune system. Immunol. Today. 11:170–175. [DOI] [PubMed] [Google Scholar]
- 39.Fong, Y., K.J. Tracey, L.L. Moldawer, D.G. Hesse, K.B. Manogue, J.S. Kenney, A.T. Lee, G.C. Kuo, A.C. Allison, S.F. Lowry, et al. 1989. Antibodies to cachectin/tumor necrosis factor reduce interleukin 1 beta and interleukin 6 appearance during lethal bacteremia. J. Exp. Med. 170:1627–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Imura, H., and J. Fukata. 1994. Endocrine-paracrine interaction in communication between the immune and endocrine systems. Activation of the hypothalamic-pituitary-adrenal axis in inflammation. Eur. J. Endocrinol. 130:32–37. [DOI] [PubMed] [Google Scholar]
- 41.Ruzek, M.C., A.H. Miller, S.M. Opal, B.D. Pearce, and C.A. Biron. 1997. Characterization of early cytokine responses and an interleukin (IL)-6- dependent pathway of endogenous glucocorticoid induction during murine cytomegalovirus infection. J. Exp. Med. 185:1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruby, J., and I. Ramshaw. 1991. The antiviral activity of immune CD8+ T cells is dependent on interferon-gamma. Lymphokine Cytokine Res. 10:353–358. [PubMed] [Google Scholar]
- 43.Ramsay, A.J., J. Ruby, and I.A. Ramshaw. 1993. A case for cytokines as effector molecules in the resolution of virus infection. Immunol. Today. 14:155–157. [DOI] [PubMed] [Google Scholar]
- 44.Melkova, Z., and M. Esteban. 1994. Interferon-gamma severely inhibits DNA synthesis of vaccinia virus in a macrophage cell line. Virology. 198:731–735. [DOI] [PubMed] [Google Scholar]
- 45.Karupiah, G., R.V. Blanden, and I.A. Ramshaw. 1990. Interferon-γ is involved in the recovery of athymic nude mice from recombinant vaccinia virus/interleukin 2 infection. J. Exp. Med. 172:1495–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alcamí, A., and G.L. Smith. 2002. The vaccinia virus soluble interferon-gamma receptor is a homodimer. J. Gen. Virol. 83:545–549. [DOI] [PubMed] [Google Scholar]
- 47.Alcamí, A., and G.L. Smith. 1995. Vaccinia, cowpox, and camelpox viruses encode soluble gamma interferon receptors with novel broad species specificity. J. Virol. 69:4633–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mossman, K., C. Upton, R.M. Buller, and G. McFadden. 1995. Species specificity of ectromelia virus and vaccinia virus interferon-gamma binding proteins. Virology. 208:762–769. [DOI] [PubMed] [Google Scholar]
- 49.Symons, J.A., D.C. Tscharke, N. Price, and G.L. Smith. 2002. A study of the vaccinia virus interferon-gamma receptor and its contribution to virus virulence. J. Gen. Virol. 83:1953–1964. [DOI] [PubMed] [Google Scholar]
- 50.Schleimer, R.P., A. Jacques, H.S. Shin, L.M. Lichtenstein, and M. Plaut. 1984. Inhibition of T cell-mediated cytotoxicity by anti-inflammatory steroids. J. Immunol. 132:266–271. [PubMed] [Google Scholar]
- 51.Skinner, M.A., J.B. Moore, M.M. Binns, G.L. Smith, and M.E. Boursnell. 1994. Deletion of fowlpox virus homologues of vaccinia virus genes between the 3 beta-hydroxysteroid dehydrogenase (A44L) and DNA ligase (A50R) genes. J. Gen. Virol. 75:2495–2498. [DOI] [PubMed] [Google Scholar]
- 52.Baker, M.E., and R. Blasco. 1992. Expansion of the mammalian 3 beta-hydroxysteroid dehydrogenase/plant dihydroflavonol reductase superfamily to include a bacterial cholesterol dehydrogenase, a bacterial UDP-galactose-4-epimerase, and open reading frames in vaccinia virus and fish lymphocystis disease virus. FEBS Lett. 301:89–93. [DOI] [PubMed] [Google Scholar]
- 53.Aguado, B., I.P. Selmes, and G.L. Smith. 1992. Nucleotide sequence of 21.8 kbp of variola major virus strain Harvey and comparison with vaccinia virus. J. Gen. Virol. 73:2887–2902. [DOI] [PubMed] [Google Scholar]
- 54.Massung, R.F., J.J. Esposito, L.-I. Liu, J. Qi, T.R. Utterback, J.C. Knight, L. Aubin, T.E. Yuran, J.M. Parsons, V.N. Loparev, et al. 1993. Potential virulence determinants in terminal regions of variola smallpox virus genome. Nature. 366:748–751. [DOI] [PubMed] [Google Scholar]
- 55.Massung, R.F., L.I. Liu, J. Qi, J.C. Knight, T.E. Yuran, A.R. Kerlavage, J.M. Parsons, J.C. Venter, and J.J. Esposito. 1994. Analysis of the complete genome of smallpox variola major virus strain Bangladesh-1975. Virology. 201:215–240. [DOI] [PubMed] [Google Scholar]
- 56.Shchelkunov, S.N., R.F. Massung, and J.J. Esposito. 1995. Comparison of the genome DNA sequences of Bangladesh-1975 and India-1967 variola viruses. Virus Res. 36:107–118. [DOI] [PubMed] [Google Scholar]
- 57.Shchelkunov, S.N., A.V. Totmenin, V.N. Loparev, P.F. Safronov, V.V. Gutorov, V.E. Chizhikov, J.C. Knight, J.M. Parsons, R.F. Massung, and J.J. Esposito. 2000. Alastrim smallpox variola minor virus genome DNA sequences. Virology. 266:361–386. [DOI] [PubMed] [Google Scholar]