

Abstract
Pathogenic mechanisms relevant to rheumatoid arthritis occur in the mouse model of collagen-induced arthritis (CIA). Cytosolic phospholipase A2α (cPLA2α) releases arachidonic acid from cell membranes to initiate the production of prostaglandins and leukotrienes. These inflammatory mediators have been implicated in the development of CIA. To test the hypothesis that cPLA2α plays a key role in the development of CIA, we backcrossed cPLA2α-deficient mice on the DBA/1LacJ background that is susceptible to CIA. The disease severity scores and the incidence of disease were markedly reduced in cPLA2α-deficient mice compared with wild-type littermates. At completion of the study, >90% of the wild-type mice had developed disease whereas none of the cPLA2α-deficient mice had more than one digit inflamed. Furthermore, visual disease scores correlated with severity of disease determined histologically. Pannus formation, articular fibrillation, and ankylosis were all dramatically reduced in the cPLA2α-deficient mice. Although the disease scores differed significantly between cPLA2α mutant and wild-type mice, anti-collagen antibody levels were similar in the wild-type mice and mutant littermates. These data demonstrate the critical role of cPLA2α in the pathogenesis of CIA.
Keywords: inflammation, autoimmunity, rheumatoid arthritis, lipid mediators, gene targeting
Introduction
The murine model of collagen-induced arthritis (CIA)*has been used extensively to increase our understanding of autoimmune-mediated arthritis and identify potential new therapeutic agents to treat rheumatoid arthritis. A series of reports have suggested that cytosolic phospholipase A2α (cPLA2α), which releases arachidonic acid from the plasma membrane to initiate the production of prostaglandins, leukotrienes, and platelet-activating factor (PAF; 1, 2) might be pivotal in the development of CIA. Mice deficient in 5-lipoxygenase–activating protein (FLAP), a protein required for the conversion of arachidonic acid to leuko-trienes, developed less severe disease than wild-type animals despite generating antibodies to type II collagen (3). Similarly, the severity of the disease was dramatically reduced when a potent leukotriene B4 receptor antagonist was used (4). In both cases, the decrease in disease severity due to leukotriene antagonism was maintained when IL-1 was added to potentiate inflammation and accelerate disease. The role of prostaglandins in arthritis is supported by studies in the CIA model using cyclooxygenase (COX)-2–deficient mice. COX-2 gene deletion reduced the incidence and severity of CIA compared with both the wild-type and COX-1–deficient mice. The COX-2–deficient mice showed reduced anti-collagen type II antibody levels. The COX-2–deficient mice remained resistant to disease when the humoral response was bypassed through passive transfer of anti-collagen II antibody (5). In another passive transfer experiment, mice that are deficient for prostaglandin E2 receptor (EP)4 showed reduced disease in comparison to wild-type, EP1, EP2, and EP3 receptor–deficient mice (6). Thus, the role of COX-2 in CIA appears to be mediated at least in part by prostaglandin E2 acting through the EP4. However, there are no published reports of selective COX-2 inhibitors showing benefit in the CIA model. A subset of nonsteroidal antiinflammatory drugs, which inhibit both COX-1 and COX-2, are efficacious in the CIA model if administered before onset of disease (7). Interestingly, several laboratories have reported that combined inhibition of both leukotriene and prostaglandin production is more effective at inhibiting disease progression (8, 9). Additionally, a PAF receptor antagonist has been reported to reduce the clinical and histopathological signs of disease in CIA (10). Thus, given the role of prostaglandins, leukotrienes, and PAF in animal models of arthritis, we anticipated that cPLA2α would be essential for the development of CIA.
Although studies with cPLA2α-deficient mice strongly support the central role of this phospholipase A2 in prostaglandin, leukotriene, and PAF production (1, 2), cPLA2α is a member of a superfamily of PLA2 enzymes including cPLA2β and cPLA2γ (11, 12), calcium-independent PLA2 (13), and numerous low molecular weight PLA2s (14). Type II secretory PLA2 originally isolated from synovial fluid is present at elevated levels in the synovial lining of rheumatoid arthritis patients (15). Thus, PLA2 other than cPLA2α could conceivably be important in the pathogenesis of CIA. To assess the role of cPLA2α in the CIA model, we established the cPLA2α-deficient mice on the susceptible DBA1/LacJ strain and studied the development of disease in cPLA2α−/− and cPLA2α+/+ littermates.
Materials and Methods
Mice.
The Institutional Animal Care and Use Committee approved all animal procedures. cPLA2α-deficient DBA/1LacJ mice were obtained by crossing cPLA2α-deficient C57BL/6 mice (1) with the CIA-susceptible DBA/1LacJ mice for eight generations. DBA/1LacJ mice were originally obtained from The Jackson Laboratory. The cPLA2α+/− DBA/1LacJ mice were then intercrossed to generate mice homozygous for the cPLA2α mutation (cPLA2α−/−) and mice homozygous for the wild-type cPLA2α allele (cPLA2α+/+) for CIA experiments. In the CIA experiments, female mice 10–15 wk of age were used. The mice were maintained at a maximum of five per cage and were fed mouse breeder chow and allowed access to water ad libitum. In each experiment, mutant and wild-type mice were age matched. The mice were crossed and bred at Taconic Farms and maintained for the CIA experiments in the animal unit at Wyeth Research, Cambridge, MA. All animals were routinely serologically tested and were negative for common pathogens.
Genotype Analysis by PCR.
The genotype of mice was determined by PCR using 100–200 ng genomic DNA prepared from tail biopsies as template. The nucleotide sequence of the primers was 5′-TTCTCTGGTGTGATGAAGGC-3′ (cPLAF), 5′-AAACTGACTGTAGCATCACAC-3′ (cPLAR), 5′-ATCGCCTTCTTGACGAGTTC-3′ (NeoF2). The primer set of NeoF2 and cPLAR amplified a 570-bp fragment for the neo allele and the set of cPLAF and cPLAR yielded a 224-bp fragment specific for the wild-type allele. The reaction was performed using a Taq polymerase (GIBCO BRL) with 30 cycles of amplification (45 s at 94°C, 1 min at 55°C, and 1 min plus 10 s autoextension at 72°C).
Induction and Assessment of Arthritis.
Chicken collagen type II (CII; Sigma-Aldrich) was dissolved in 0.01 N acetic acid and emulsified in an equal volume of CFA containing 1 mg/ml heat-killed Mycobacterium tuberculosis (Sigma-Aldrich). Arthritis was induced by the initial immunization with 200 μg/100 μl emulsion by an intradermal injection in the base of the tail. A boost 21 d later with an aqueous solution of 200 μg/100 μl CII was administered intraperitoneally. On day 49, 28 d after the boost, 0.3 μg murine rIL-1α diluted in PBS containing 1 mg/ml BSA was administered subcutaneously. Individual experiments contained at least 12 female cPLA2 −/− and cPLA2 +/+ DBA/1LacJ mice per group and all experiments were performed twice. Mice were scored weekly, beginning 3 wk after primary CII immunization, for signs of developing arthritis. The severity of the arthritis was assessed using a visual scoring system. Each paw was scored on a graded scale from 0 to 3: 0, normal paw; 1, swelling and/or redness of one toe or finger joint; 2, swelling of two or more toes or joints, or increased swelling; 3, severe swelling and/or ankylosis throughout the entire paw. Each paw was graded and the four scores were added such that the maximal score per mouse was 12. On day 81, blood was collected for anti-collagen II ELISA testing and paws were collected for histopathology.
Histological Techniques.
For histological processing, paws were fixed in phosphate buffer containing 10% formaldehyde and decalcified in sodium citrate. Paws were processed by routine methods to paraffin blocks. Specimens were sectioned at 6 μm and stained with hematoxylin and eosin according to the manufacturer's protocol (Sigma-Aldrich). The sections were evaluated for the degree of synovial hyperplasia, inflammatory cell infiltrate, cartilage damage, pannus formation, bone erosion, fibrillation, and ankylosis. The severity of the disease in the joint sections was graded using a scoring system from 0 to 5: 0, within normal limits; 1, minimal; 2, mild; 3, moderate; 4, marked; 5, severe. The severity score for a paw was weighted based on the number of joints within a paw receiving a specific score. Each paw was graded and the score for four paws were added such that the maximal score per mouse was 20.
Anti–Type II Collagen Antibody ELISA.
IgG antibody levels against the immunogen were measured by standard ELISA methodology using peroxidase-conjugated secondary antibody and substrate ABTS. Serum dilutions, 1/1,000, were chosen after preliminary assays. The optical density was measured at 405 nm using a Spectramax Plus 384 plate reader (Molecular Devices Corporation). The anti–type II collagen concentrations were determined by reference to standard curves of murine IgG, IgG1, IgG2a, or IgG2b (Southern Biotechnology Associates, Inc.).
Statistical Analysis.
Data are presented as the mean ± SEM. Clinical and histopathological scores and serum anti-CII IgG levels were analyzed with Student's t test. Incidence of mice that developed disease was analyzed with Fisher's exact test. P values <0.05 were considered significant.
Results
cPLA2α-deficient Mice Show Reduced Severity and Incidence of CIA Compared with Wild-type Littermate Mice.
To directly explore the pathophysiological role of cPLA2α in arthritis, we backcrossed cPLA2α-deficient mice for at least eight generations into the CIA-susceptible DBA/1LacJ mouse strain (1, 16). The arthritic symptoms in cPLA2α-deficient DBA/1LacJ mice and wild-type littermates were studied after immunization with CII on day 0 and a boost with CII on day 21. Mice were examined weekly after the boost for signs of developing arthritis. The severity of the arthritis was assessed using a visual scoring system. The disease severity scores and the incidence of disease were markedly reduced in cPLA2α-deficient mice compared with wild-type littermates (Fig. 1, A and B) . By day 48, only 8% of the cPLA2α−/− mice had a score >1 compared with 89% of the wild-type mice (Fig. 1 B). To test whether we could accelerate disease in the cPLA2α−/− mice, IL-1α was administered to all mice on day 49 (17). Although the severity of disease was increased in the wild-type littermates, the cPLA2α-deficient mice were unaffected by the inflammatory challenge. By the end of the study on day 77, 94% of the wild-type animals had a disease severity >1 whereas none of the animals in the cPLA2α−/− group had >1 digit inflamed.
Figure 1.

Comparison of CIA symptoms in female wild-type and cPLA2α-deficient mice. (A) Disease severity scores of arthritis for each group of animals (n = 12 for cPLA2α−/− and n = 17 for cPLA2α+/+). Significant differences to the wild-type group are marked with ** for P < 0.01 and * for P < 0.05 (two-tailed t test). (B) Percentage of female, wild-type, and cPLA2α-deficient mice that developed arthritis in >1 digit. Of the cPLA2α-deficient mice that developed arthritis, the disease was limited to 1 digit or less. In comparison, 16 out of 17 wild-type mice developed arthritis in >1 digit. Significant differences to the wild-type group are marked with *** for P < 0.0001, ** for P < 0.001, and * for P < 0.05 (Fisher's exact test). The results are from a single representative experiment.
Histological Features of Immunized cPLA2α Mutant and Wild-type Littermate Mice.
An observer unaware of the genotype of the animals scored the histopathology of both front and rear paws. The severity of disease as determined by the histological features correlated with the observed visual scores (Fig. 2 A). None of the cPLA2α−/− mice that were classified as nonarthritic had any evidence of arthritis upon histological examination. Therefore, the histological analysis of the paws confirmed the visual scoring results. The joints of wild-type mice frequently showed severe pathology with cartilage erosion, synovial inflammation, and formation of invasive pannus (Fig. 2 B). New bone formation was also evident in late stage disease (unpublished data).
Figure 2.

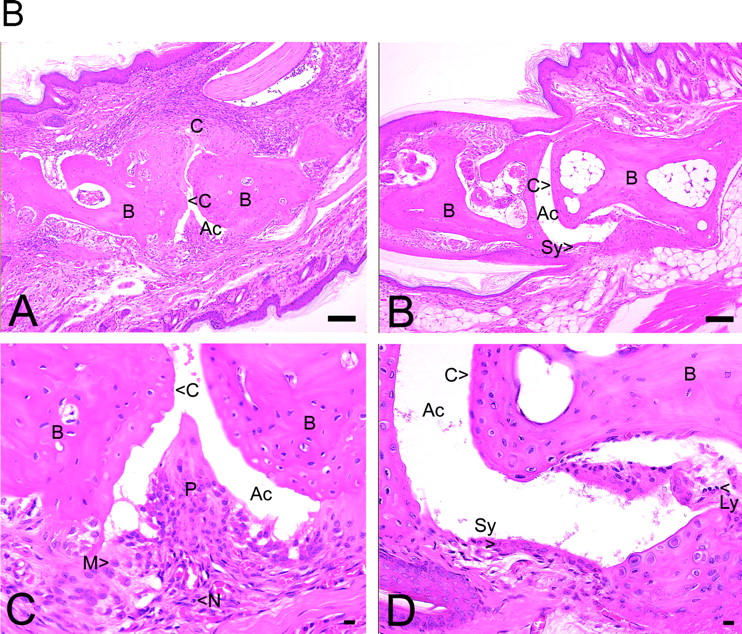
(A) Summary of histopathological grades for synovial hyperplasia, inflammatory cells, pannus formation, ankylosis, and fibrillation of all joints of cPLA2α-deficient and wild-type mice. Significant differences to the wild-type group are marked with *** for P < 0.0001, ** for P < 0.001, and * for P < 0.01 (two-tailed t test). Concerning ankylosis, t test was not applicable because all cPLA2α-deficient mice scored zero. (B) Histopathology of hematoxylin and eosin–stained joints of front paws of cPLA2α-deficient and wild-type mice. In A, the joints of wild-type mice most frequently showed severe pathology with cartilage erosion, synovial inflammation, and formation of invasive pannus (P). (B) The pannus (see close-up in C) was comprised of a mixture of monocyte/macrophages (M) and neutrophils (N). In B, the majority of examined joints of cPLA2α-deficient mice were normal in appearance, with smooth intact articular cartilage. In D, the close up of B showed few infiltrating cells, e.g., lymphocytes (Ly). Ac, articular cavity; B, bone. original magnification: ×10 for A and B, bar = 0.1 mm; ×40 for C and D, bar = 0.01 mm.
Pannus formation, fibrillation of the articular surface, and eventual ankylosis are hallmarks of rheumatoid arthritis. Mild to moderate pannus (76% of mice) and fibrillation of the articular surface (47% of mice) were common in the cPLA2α+/+ mice. In contrast, none of the cPLA2α−/− mice were observed to have more than minimal pannus formation or fibrillation of the articular cartilage. Similarly, ankylosis was observed in 24% of the cPLA2α+/+ mice but in none of the cPLA2α−/− mice.
Immune Response Against CII in cPLA2α-deficient DBA/1LacJ Mice.
High levels of anti-CII antibodies accompany the development of the disease (16). To investigate if the reduced severity and incidence of arthritis in cPLA2α mutant mice was due to the lack of an antibody response to type II collagen, the anti-CII–specific levels of IgG in the serum were determined at the termination of the CIA experiment. Anti-CII antibody levels were similar in wild-type mice and in cPLA2α-deficient littermates and did not correlate with disease severity (Fig. 3 A). Although scores of clinical and pathological analyses were segregated mostly by genotypes of mice, IgG concentrations overlapped completely (Fig. 3 B).
Figure 3.

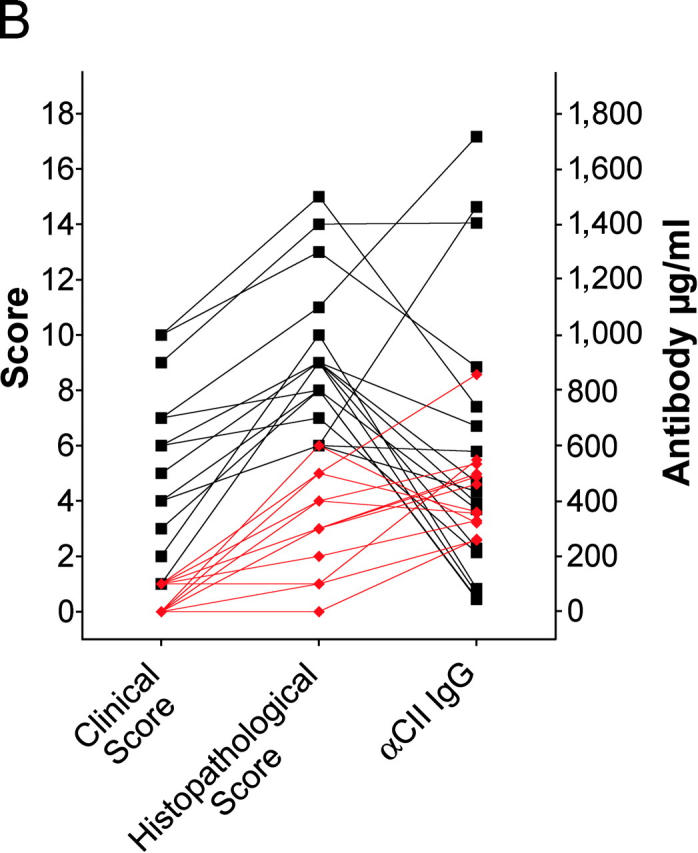
(A) Anti–type II collagen (CII) IgG, IgG1, IgG2a, and IgG2b levels in serum of CII-immunized mice. No significant difference was observed by genotype. (B.) Correlation of clinical scores, histopathological scores, and serum anti-CII total IgG levels. Wild-type and cPLA2α-deficient mice are represented by black squares and red diamonds, respectively. Data from the same mouse are connected by lines and some symbols overlap. Scores of synovial hyperplasia were chosen to represent histopathological evaluations.
Discussion
This study is the first to demonstrate the critical role of cPLA2α as a key player in the pathogenesis of CIA. cPLA2α−/− mice show markedly reduced severity and incidence of disease in CIA (Fig. 1) and these results were confirmed by histopathological evaluation (Fig. 2). The results observed with the cPLA2α−/− mice are similar to those seen with FLAP−/− mice. The reduced levels of infiltrating cells in the joints of both the cPLA2α−/− and FLAP−/− mice are consistent with the absence of leukotriene B4-mediated chemotaxis (3). In addition, both gene-deleted mice remained resistant to disease when IL-1α was added to mice 28 d after the boost to accelerate disease. The amount of anti–collagen type II IgG antibodies in cPLA2α mutant mice was not different from the amount in wild-type littermates (Fig. 3). These data are also consistent with the results obtained with FLAP-deficient mice but appear to differ with data from the COX-2–deficient mice. In the COX-2−/− mice total anti–collagen type II Ig levels were dramatically reduced (∼10-fold), but anti–collagen type II IgG1 and IgG2 isotypes were only reduced by approximately twofold (5). Both the amount of anti-collagen total IgG levels and IgG subclasses did not differ significantly between cPLA2α−/− and wild-type mice. When the defect in the humoral response was bypassed in the passive transfer model, the COX-2−/− and EP4−/− mice remained resistant to disease (5, 6). Thus, the collective studies using cPLA2α−/−, COX-2−/−, EP4−/−, and FLAP−/− mice suggest that both prostaglandins and leukotrienes are important in the pathogenesis of CIA and that inhibitors of cPLA2α will be beneficial for individuals with rheumatoid arthritis due to the simultaneous inhibition of multiple lipid mediators. Although no cPLA2α inhibitor is currently available for clinical use, nutritional studies have been performed where the omega-3 polyunsaturated fatty acids, eicosapenteanoic acid, and docosahexaenoic acid have shown clinical benefit in numerous placebo-controlled studies of rheumatoid arthritis (18, 19). The omega–3 acids may act by reducing the arachidonic acid content of the phospholipid membrane and thus decrease the ability of cells to generate arachidonyl-derived proinflammatory mediators. This would be analogous to blocking the release of arachidonic acid through the use of a cPLA2α antagonist. The eicosapenteanoic and docosahexaenoic acids could exert additional antiinflammatory effects by serving as the precursor to resolvins, which are newly reported antiinflammatory mediators generated through the action of aspirin-acetylated COX-2 (20).
The role of cPLA2α extends beyond arthritis. For example, cPLA2α has been shown to be essential for osteoclast formation and LPS-induced bone resorption (21). This effect of cPLA2α might be mediated by prostaglandin E2 because the EP4 is required for bone resorption and osteoclast formation (22). Thus, cPLA2α may play a role in bone resorption associated with osteoporosis as well as rheumatoid arthritis. These data support that a prospective cPLA2α inhibitors may have the potential activity of a disease-modifying antirheumatic drug. Furthermore, the importance of cPLA2α has been demonstrated in models of respiratory disease including anaphylaxis, bronchial hyperreactivity, adult respiratory distress syndrome, and pulmonary fibrosis using the cPLA2α−/− mice (1, 23, 24). A reduction in infarct size has also been observed after cerebral ischaemia (2). Consequently, cPLA2α is an ideal target for a potent selective inhibitor for the treatment of numerous diseases, including rheumatoid arthritis.
Acknowledgments
The authors thank Wen Zhang and Qiuna Bi for expert technical assistance and Dr. Andre-Jean Lambert for help and advice with the evaluation of the histopathology.
This work was supported in part by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to. T. Shimizu), and ONO Medical Research Foundation (to N. Uozumi).
Footnotes
Abbreviations used in this paper: CIA, collagen-induced arthritis; CII, chicken collagen type II; COX, cyclooxygenase; cPLA2α, cytosolic phospholipase A2α; EP, prostaglandin E2 receptor; FLAP, 5-lipoxygenase–activating protein; PAF, platelet-activating factor.
References
- 1.Uozumi, N., K. Kume, T. Nagase, N. Nakatani, S. Ishii, F. Tashiro, Y. Komagata, K. Maki, K. Ikuta, Y. Ouchi, et al. 1997. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature. 390:618–622. [DOI] [PubMed] [Google Scholar]
- 2.Bonventre, J.V., Z. Huang, M.R. Taheri, E. O'Leary, E. Li, M.A. Moskowitz, and A. Sapirstein. 1997. A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 390:622–625. [DOI] [PubMed] [Google Scholar]
- 3.Griffiths, R.J., M.A. Smith, M.L. Roach, J.L. Stock, E.J. Stam, A.J. Milici, D.N. Scampoli, J.D. Eskra, R.S. Byrum, B.H. Koller, et al. 1997. Collagen-induced arthritis is reduced in 5-lipoxygenase–activating protein-deficient mice. J. Exp. Med. 185:1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffiths, R.J., E.R. Pettipher, K. Koch, C.A. Farrell, R. Breslow, M.J. Conlyn, M.A. Smith, B.C. Hackman, D.J. Wimberly, A.J. Milici, et al. 1995. Leukotriene B4 plays a critical role in the progression of collagen-induced arthritis. Proc. Natl. Acad. Sci. USA. 92:517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myers, L.K., A.H. Kang, A.E. Postlethwaite, E.F. Rosloniec, S.G. Morham, B.V. Shilopov, S. Goorha, and L.R. Ballou. 2000. The genetic ablation of cyclooxygenase 2 prevents the development of autoimmune arthritis. Arthritis Rheum. 43:2687–2693. [DOI] [PubMed] [Google Scholar]
- 6.McCoy, J.M., J.R. Wicks, and L.P. Audoly. 2002. The role of prostaglandin E2 receptors in the pathogenesis of rheumatoid arthritis. J. Clin. Invest. 110:651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cannon, G.W., S. McCall, B.C. Cole, M.M. Griffiths, L.A. Radov, and J.R. Ward. 1990. Effects of indomethacin, cyclosporin, cyclophosphamide, and placebo on collagen-induced arthritis of mice. Agents and Actions. 29:315–323. [DOI] [PubMed] [Google Scholar]
- 8.Griswold, D.E., L.M. Hillegass, P.C. Meunier, M.J. DiMartino, and N. Hanna. 1988. Effect of inhibitors of eicosanoid metabolism in murine collagen-induced arthritis. Arthritis Rheum. 31:1406–1412. [DOI] [PubMed] [Google Scholar]
- 9.Nickerson-Nutter, C.L., and E.D. Medvedeff. 1996. The effect of leukotriene synthesis inhibitors in models of acute and chronic inflammation. Arthritis Rheum. 39:515–521. [DOI] [PubMed] [Google Scholar]
- 10.Palacios, I., R. Miguelez, O. Sanchez-Pernaute, S. Gutierrez, J. Egido, and G. Herrero-Beaumont. 1999. A platelet activating factor receptor antagonist prevents the development of chronic arthritis in mice. J. Rheumatol. 26:1080–1086. [PubMed] [Google Scholar]
- 11.Song, C., X.J. Chang, K.M. Bean, M.S. Proia, J.L. Knopf, and R.W. Kriz. 1999. Molecular characterization of cytosolic phospholipase A2-β. J. Biol. Chem. 274:17063–17067. [DOI] [PubMed] [Google Scholar]
- 12.Underwood, K.W., C. Song, R.W. Kriz, X.J. Chang, J.L. Knopf, and L.L. Lin. 1998. A novel calcium–independent phospholipase A2, cPLA2-γ, that is prenylated and contains homology to cPLA2. J. Biol. Chem. 273:21926–21932. [DOI] [PubMed] [Google Scholar]
- 13.Tang, J., R.W. Kriz, N. Wolfman, M. Shaffer, J. Seehra, and S.S. Jones. 1997. A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. J. Biol. Chem. 272:8567–8575. [DOI] [PubMed] [Google Scholar]
- 14.Dennis, E.A. 1997. The growing phospholipase A2 superfamily of signal transduction enzymes. Trends Biochem. Sci. 22:1–2. [DOI] [PubMed] [Google Scholar]
- 15.Jamal, O.S., P.G. Conaghan, A.M. Cunningham, P.M. Brooks, V.F. Munro, and K.F. Scott. 1998. Increased expression of human type IIa secretory phospholipase A2 antigen in arthritic synovium. Ann. Rheum. Dis. 57:550–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wooley, P.H., H.S. Luthra, J.M. Stuart, and C.S. David. 1981. Type II collagen–induced arthritis in mice. I. Major histocompatibility complex (I region) linkage and antibody correlates. J. Exp. Med. 154:688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hom, J.T., A.M. Bendele, and D.G. Carlson. 1988. In vivo administration with IL-1 accelerates the development of collagen-induced arthritis in mice. J. Immunol. 141:834–841. [PubMed] [Google Scholar]
- 18.Calder, P.C., and R.B. Zurier. 2001. Polyunsaturated fatty acids and rheumatoid arthritis. Curr. Opin. Clin. Nutr. Metab. Care. 2:115–121. [DOI] [PubMed] [Google Scholar]
- 19.James, M.J., R.A. Gibson, and L.G. Cleland. 2000. Dietary polyunsaturated fatty acids and inflammatory mediator production. Am. J. Clin. Nutr. 71:343S–348S. [DOI] [PubMed] [Google Scholar]
- 20.Serhan, C.N., S. Hong, K. Gronert, S.P. Colgan, P.R. Devchand, G. Mirick, and R.L. Moussignac. 2002. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 196:1025–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyaura, C., M. Inada, C. Matsumoto, T. Ohshiba, N. Uozumi, T. Shimizu, and A. Ito. 2003. An essential role of cytosolic phospholipase A2α in prostaglandin E2–mediated bone resorption associated with inflammation. J. Exp. Med. 197:1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyaura, C., M. Inada, T. Suzawa, Y. Sugimoto, F. Ushikubi, A. Ichikawa, S. Narumiya, and S. Tatsou. 2000. Impaired bone resorption to prostaglandin E2 in prostaglandin E receptor EP4-knockout mice. J. Biol. Chem. 275:19819–19823. [DOI] [PubMed] [Google Scholar]
- 23.Nagase, T., N. Uozumi, S. Ishii, K. Kume, T. Izumi, Y. Ouchi, and T. Shimizu. 2000. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nat. Immunol. 1:42–46. [DOI] [PubMed] [Google Scholar]
- 24.Nagase, T., N. Uozumi, S. Ishii, Y. Kita, H. Yamamoto, E. Ohga, Y.Y. Ouchi, and T. Shimizu. 2002. A pitoval role of cytosolic phospholipase A2 in bleomycin-induced pulmonary fibrosis. Nat. Med. 8:480–484. [DOI] [PubMed] [Google Scholar]